

Abstract
Carboxy-terminal Src kinase (Csk) is a negative regulator of Src family kinases, which play pivotal roles in controlling cell adhesion, migration, and cancer progression. To elucidate the in vivo role of Csk in epithelial tissues, we conditionally inactivated Csk in squamous epithelia using the keratin-5 promoter/Cre-loxP system in mice. The mutant mice developed apparent defects in the skin, esophagus, and forestomach, with concomitant hyperplasia and chronic inflammation. Histology of the mutant epidermis revealed impaired cell–cell adhesion in basal cell layers. Analysis of primary keratinocytes showed that the defective cell–cell adhesion was caused by cytoskeletal remodeling via activation of the Rac1 pathway. Mutant keratinocytes also showed elevated expression of mesenchymal proteins, matrix metalloproteinases (MMPs), and the proinflammatory cytokine TNF-α. Inhibition of the expression of TNF-α and MMP9 by the anti-inflammatory reagent FK506 could cure the epidermal hyperplasia, suggesting a causal link between inflammation and epidermal hyperplasia. These observations demonstrate that the Src/Csk circuit plays crucial roles in development and maintenance of epithelia by controlling cytoskeletal organization as well as phenotypic conversion linked to inflammatory events.
Keywords: Csk, hyperplasia, K5 promoter, squamous epithelia, Src family kinase
Introduction
The Src family kinases (SFKs) are non-receptor tyrosine kinases, originally identified as proto-oncogene products (Hunter and Sefton, 1980; Jove and Hanafusa, 1987), which play pivotal roles in various cellular signaling pathways involved in cell growth, differentiation, adhesion, and migration (Brown and Cooper, 1996). In epithelial cells, Src has been implicated in regulation of the epithelial–mesenchymal transition (EMT), a phenotypic conversion from polarized epithelia to motile mesenchymal cells, which occurs during embryonic development, as well as in cancer progression (Avizienyte and Frame, 2005; Huber et al, 2005). During EMT processes, Src has been proposed to play multiple roles, including activation of the STAT3 pathway (Yu and Jove, 2004), promotion of E-cadherin endocytosis and degradation (Matsuyoshi et al, 1992; Behrens et al, 1993; Fujita et al, 2002), induction of matrix metalloproteinases (MMPs) (Hamaguchi et al, 1995; Hsia et al, 2003), and activation of cell adhesion proteins such as focal adhesion kinase (FAK) (Brown and Cooper, 1996). However, the in vivo function of endogenous Src still remains to be evaluated. In metastatic human epithelial cancers, such as colon, breast, bladder, and stomach cancers, the kinase activity and/or amount of Src protein is frequently upregulated in the absence of somatic mutations in the src gene locus (Talamonti et al, 1993; Irby and Yeatman, 2000). The elevated Src activity is thought to contribute to cancer metastasis by promoting the EMT (Frame, 2002). These observations suggest that Src activity is strictly regulated to maintain normal epithelial organization and suppress cancer progression.
Generally, SFK activity is negatively regulated by phosphorylation at the C-terminal regulatory tyrosine (Y529 in human c-Src) by another cytoplasmic tyrosine kinase, the carboxy-terminal Src kinase (Csk) (Nada et al, 1991; Okada et al, 1991). The phosphorylated form of SFK adopts a catalytically inactive conformation due to intramolecular interactions (Xu et al, 1997). In response to certain extracellular stimuli, SFKs become activated by interacting with adaptor proteins that disrupt the inactive conformation or by dephosphorylation at the regulatory site. As SFK function can be controlled by overexpression of Csk (Chow et al, 1993; Suzuki et al, 1998), it is thought that the phosphorylation status at the critical site would define the sensitivity of SFK to extracellular stimuli. In epithelial cells, SFK activation by a dominant negative Csk disrupts cell–cell interactions and enhances cell motility; conversely, SFK inactivation by active Csk restores the epithelial features of these cells (Rengifo-Cam et al, 2004). Furthermore, adenovirus-mediated introduction of Csk into cancer cells suppresses their metastatic activity in vivo (Nakagawa et al, 2000). These observations suggest that the Csk-mediated negative regulation of SFK is involved in controlling the epithelial phenotype as well as the metastatic potential. However, little is known about the in vivo events controlled by Csk in epithelial cells.
To elucidate the in vivo role of Csk, we previously generated Csk-knockout mice that were developmentally arrested at mid-gestation, potentially as a result of defects in neural development (Imamoto and Soriano, 1993; Nada et al, 1993). However, mutant mice in which csk was conditionally disrupted, specifically in immature thymocytes, showed antigen receptor-independent development and T-cell lineage selection (Schmedt et al, 1998). Moreover, the disruption of csk in granulocytes caused hyper-responsiveness to pathogens and aberrant cell adhesion (Thomas et al, 2004). These findings suggest that Csk plays essential roles in regulation of the development and/or function of the nervous and immune systems. However, the role of Csk in other tissues, especially in epithelia, from which the majority of human cancers are derived, remains to be defined.
In this study, we generated mutant mice in which Csk was conditionally inactivated in basal cells of squamous epithelia using the keratin-5 (K5) promoter in a Cre-loxP system. The mutant mice developed apparent defects in skin, esophagus, and forestomach, with concomitant chronic inflammation and epithelial hyperplasia. Analysis of the epidermal tissues and the primary keratinocytes in these mutant mice reveals that Csk is crucial for the development and maintenance of squamous epithelia by controlling cytoskeletal organization and phenotypic conversion linked to inflammatory events.
Results
Generation of K5-Cre Cskflox/flox mice
To generate mutant mice in which Csk was inactivated in the basal cells of squamous epithelia, csk-flox (cskflox) mice (Schmedt et al, 1998) were crossbred with Keratin5-Cre (K5-Cre) transgenic mice (Tarutani et al, 1997). The K5 promoter directs gene expression in the basal cell layers of various squamous epithelial tissues including skin, esophagus, forestomach, and uterus (data not shown). Cre-mediated recombination of the cskflox locus causes gene inactivation through deletion of exons 9 and 10, which encode part of the kinase domain of Csk (Schmedt et al, 1998). Genotyping of cskflox revealed that csk disruption occurred in the epidermis but not in the liver (Figure 1A). Consistent with the loss of Csk protein, undifferentiated mutant keratinocytes showed a decrease in phosphorylation of Src Y529 (a negative regulatory site) and an increase in phosphorylation at Src Y418 (an autophosphorylation site) (Figure 1B). A similar phosphorylation pattern was also obtained for Src from differentiated keratinocytes (see Figure 6B). These results indicate that SFKs are constitutively activated in the K5 csk-knockout (K5 csk-KO) cells. As reported previously (Harris et al, 1999), downregulation of activated Src protein was also observed in these mutant cells (Figure 1B).
Figure 1.
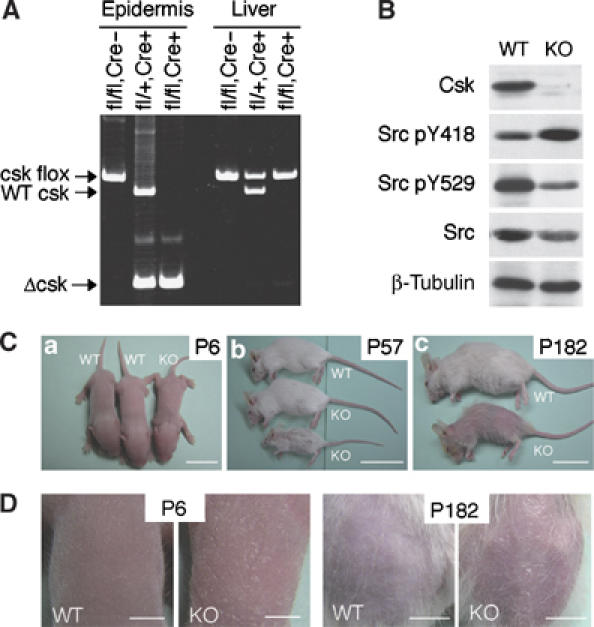
Phenotype of K5 csk-KO mice. (A) Genotyping of K5 csk-KO mice. Genomic DNA was prepared from epidermis or liver from cskflox/flox mice (fl/fl, Cre−), K5-Cre, cskflox/+ mice (fl/+, Cre+), or K5 Cre, cskflox/flox mice (fl/fl, Cre+), and genotyped by PCR. (B) Keratinocytes were cultured from wild-type (WT) and K5 csk-KO (KO) newborn mice, and whole-cell lysates of undifferentiated cells were subjected to immunoblotting with antibodies to the indicated proteins and phosphorylation sites. (C) Gross appearance of KO and WT littermates at P6 (a), P57 (b), and P182 (c). Scale bars: a, 1 cm, b and c, 5 cm. (D) Skin of KO and WT littermates at P6 and P182. At P182, hair was removed to reveal the appearance of the skin. Scale bars: P6, 0.3 cm, P182, 1 cm.
Figure 6.
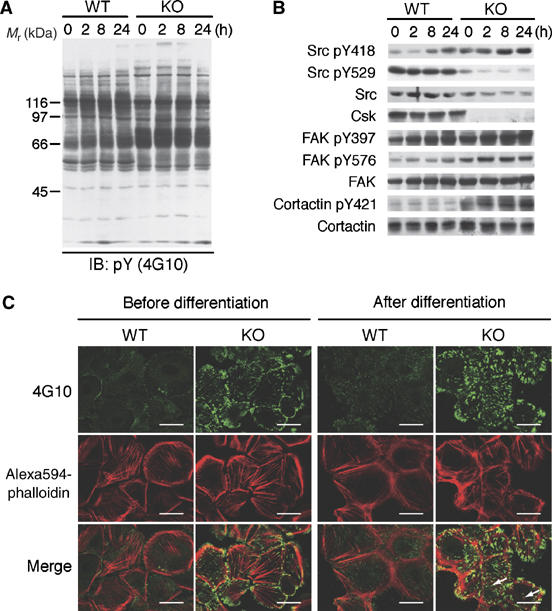
Cellular events in K5 csk-KO keratinocytes. (A) Wild-type (WT) and K5 csk-KO (KO) keratinocytes were induced to differentiate for the indicated periods, and the cell lysates were subjected to immunoblotting with the 4G10 antibody. (B) The samples used in (A) were probed with antibodies to the indicated phosphorylation sites and proteins. (C) WT and KO keratinocytes were costained with 4G10 (green) and Alexa594-phalloidin (red) before and after differentiation (8 h). Merged images are also shown. Podosome-like punctate structures are indicated by arrows. Scale bars: 5 μm.
Phenotypes of K5 csk-KO mice
K5 csk-KO mice were born normally, but exhibited weak, flaky skin, and sparse hair, even during the lactation stage (postnatal day 6 (P6)) (Figure 1Ca and left panels in Figure 1D). Some mutant mice showed severe growth retardation during early development and died within 5 months (Figure 1Cb); others showed growth retardation at later stages and died within 1 year. The growth retardation may be associated with the defects in esophagus and forestomach (Supplementary Figure 1). In aged mutant mice (P182), the amount of hair was substantially reduced, and spontaneous dermatitis became apparent in the skin over the entire body (Figure 1Cc and right panels in Figure 1D).
Hematoxylin–eosin (HE) staining of dorsal skin sections revealed no apparent defects in young mutant mice (P29), with the exception of decreased numbers of hair follicles (Figure 2Aa and d), but aged mutant mice (P218) developed dramatic alterations in epidermal organization, accompanied by apparent epidermal hyperplasia (Figure 2Ab and e). With age, the hyperplasia gradually spread over the entire skin and the thickness of stratified cell layer was greatly increased (Figure 2B). Furthermore, the mutant mice exhibited chronic inflammation. In the dermis of adult mutant mice, there were increase in the number of fibroblasts (Figure 2Af), T lymphocytes (Figure 2Cc), and macrophages (Figure 2Cd), but not neutrophils (Figure 2Cg). The inflammation indicated by lymphocyte infiltration became evident at approximately P20, before the onset of hyperplasia (Figure 2D). The mutant mice also had similar defects in their esophagus and forestomach (Supplementary Figure 1A). The esophagus epithelia of mutant mice (P208) exhibited apparent hyperplasia in the basal layers, and the protrusion of rete ridges into submucosae was evident (Supplementary Figure 1B). These findings suggest that Csk inactivation broadly influences the organization of squamous epithelia.
Figure 2.
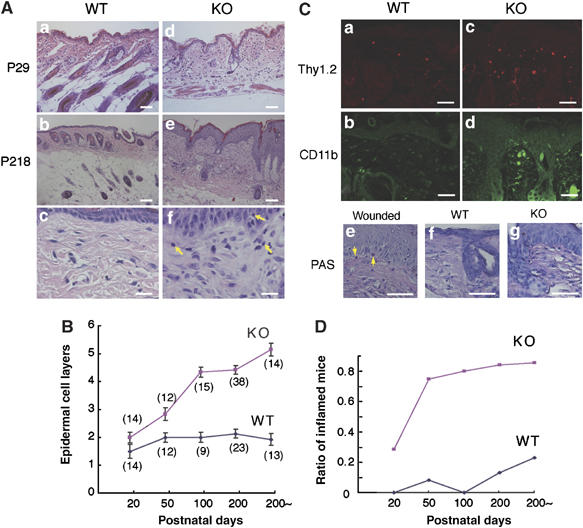
Histology of the dorsal skin and esophagus. (A) HE-stained skin sections of K5 csk-KO mice at P29 (d) and P218 (e), and those of corresponding wild-type littermates (a and b). Scale bars: 20 μm. Higher magnification views of (b) and (e) are shown in (c) and (f), respectively. Yellow arrows indicate infiltrating lymphocytes. Scale bars: 3 μm. (B) The numbers of epidermal cell layers in dorsal skin sections were counted at the indicated stages. Data are means±s.d. The numbers of mice examined are shown in parentheses. (C) Skin sections of wild-type and mutant mice were stained for T lymphocytes (a and c: Thy1.2-positive) and macrophages (b and d: CD11b-positive). Neutrophils were stained using PAS. Wounded skin was stained as a positive control. Scale bars: 10 μm. (D) Inflammation was quantified by the presence of lymphocytes infiltrating the dermis for the same mice used in (B). The ratios of inflamed mice to the total number of mice examined are indicated.
Histology of K5 csk-KO epidermis
To characterize the defects in K5 csk-KO epidermis, dorsal skin sections (P208) were immunostained for keratinocyte differentiation markers (K1, K5, and involucrin) and a cell proliferation marker (K6). In normal epidermis, K5 is detected in the basal cell layer (Figure 3B), and K1 and involucrin (Inv) are expressed in the squamous cell layer (Figure 3C and D). In the mutant epidermis, however, K5 was detected in multiple layers and K1 was detected in the uppermost of the K5-expressing layers, indicating that differentiation of K5-expressing basal cells proceeds, but is delayed in the mutant epidermis (Figure 3H–J). Wild-type mice showed K6 expression only in proliferating cells, such as the outer root sheath of hair follicles, whereas the mutant mice exhibited strong K6 expression in the basal and squamous cell layers (Figure 3E and K). These results suggest that the defects in K5 csk-KO mice are associated with delayed differentiation and hyperproliferation of epidermal basal cells.
Figure 3.
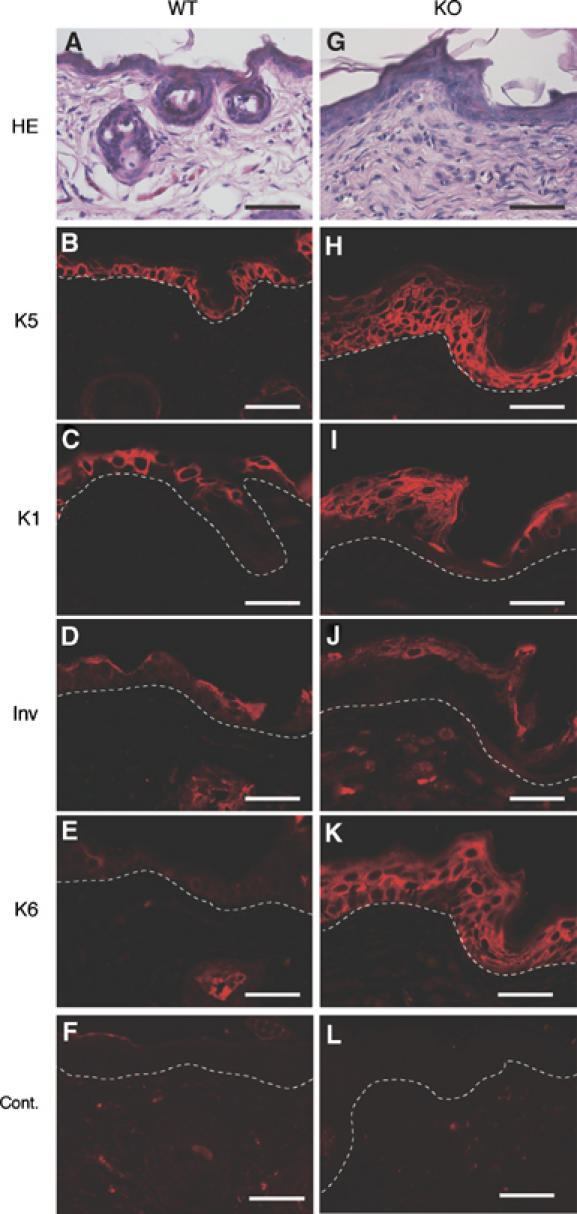
Immunohistochemical analysis of the dorsal skin of K5 csk-KO mice. Dorsal skin sections from wild-type littermates (A–F) and K5 csk-KO mice (G–L) were stained with HE (A and G) and subjected to immunofluorescent staining with antibodies to marker proteins: K5 (B and H), K1 (C and I), involucrin (Inv; D and J), and K6 (E and K). Staining of serial sections is shown. Control staining (without primary antibody) gave no significant signal (Cont.). The dotted white line shows the epidermal–dermal border. Scale bars: 10 μm.
Dorsal skin sections were immunostained for β-catenin, a component of adherence junctions, to examine the defects in epithelia specific cell–cell adhesion. In wild-type skin, there was clear β-catenin staining at cell–cell contacts in the basal monolayers, showing the stable formation of adherence junctions (Figure 4Aa). In the mutant skin, however, β-catenin staining was detected diffusely in the cytoplasm of growing K5-expressing cells, and relatively clear staining was observed at the cell–cell contacts of cells located in the upper layers (Figure 4Ab). Similar staining patterns were obtained for other adhesion-related proteins including E-cadherin, desmoglein3, and plakoglobin (data not shown). In the lactation stages (P6-P10), however, clear β-catenin staining at cell–cell contacts was not observed in the skin of either wild-type or mutant mice, probably because most of the epithelia is in a growth stage (Supplementary Figure 2).
Figure 4.
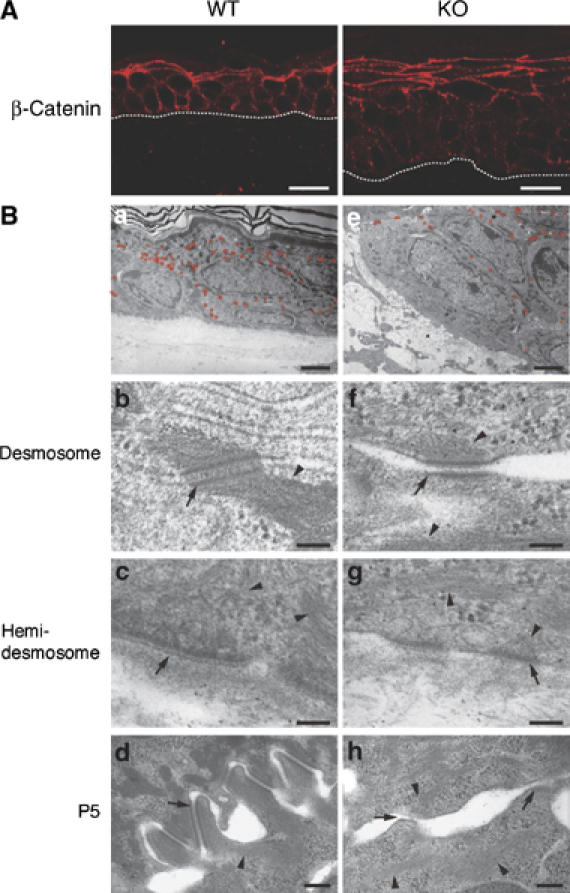
Cell adhesion structures in the dorsal skin of K5 csk-KO mice. (A) Dorsal skin sections from wild-type littermates (a) and K5 csk-KO mice (b) were subjected to immunofluorescent staining with anti-β-catenin to observe adherence junctions. The dotted white line represents the epidermal–dermal border. Scale bars: 5 μm. (B) Electron microscopic analysis of dorsal skin sections from wild-type littermates (a–d) and K5 csk -KO mice (e–h). Desmosomal structures are indicated by small red circles in (a) and (e). Scale bars: 2 μm. Higher magnification (× 60 000) images of a desmosome (b and f) and a hemidesmosome (c and g) are shown. Scale bar: 0.1 μm. Higher magnification images of dorsal skin sections at P5 are shown in (d) and (h). Scale bar: 0.2 μm. Desmosome/hemidesmosome and keratin filaments are indicated by arrows and arrowheads, respectively.
Cell–cell adhesions were further characterized by electron microscopy (Figure 4B). In the mutant epidermis, the number of desmosomes was significantly reduced, and the intercellular spaces were expanded (Figure 4Be and Supplementary Figure 3). A higher magnification examination of the mutant epidermis revealed that desmosomes and hemidesmosomes as well as keratin filaments that support these adhesion structures did not develop fully (Figure 4Bf and g). These observations demonstrate that formation of not only adherence junctions but also desmosomal junctions is attenuated in the mutant epidermis. In the epidermis of wild-type newborn mice (P5), zipper-lock structures or cellular interdigitation structures were formed during the initial stages of cell–cell adhesion (Figure 4Bd). However, the mutant mice did not form such interdigitated structures, and instead extended protruding lamellipodia-like structures (Figure 4Bh), suggesting that development of intrinsic cell–cell adhesion in the epidermis is attenuated by Csk inactivation.
Defective cell–cell adhesion in K5 csk-KO keratinocytes
To characterize the defects caused by Csk inactivation in vitro, keratinocytes from newborn mouse skin were cultured. Immunostaining for K1 and Ki67 revealed that Csk inactivation did not appear to influence the ability of differentiation or the cell-autonomous proliferation potential (data not shown). However, the mutant keratinocytes exhibited apparent defects in cell–cell adhesion. Immunostaining for E-cadherin during differentiation showed that wild-type cells accumulated E-cadherin at cell–cell contacts within 2 h after Ca2+ stimulation, whereas mutant cells required more than 8 h of stimulation for E-cadherin accumulation (Figure 5A). In the mutant cells, staining for plakoglobin and K5 also showed that the formation of desmosomes was attenuated and radial keratin fibers did not fully develop to support the desmosomes (Figure 5B). During these differentiation processes, however, the mutant cells did not show significant changes in either the levels of E-cadherin, β-catenin, and plakoglobin, or their tyrosine phosphorylation (Figure 5C and D), suggesting that degradation or phosphorylation of these cell adhesion proteins is not associated with the cell–cell adhesion defects observed in the mutant cells.
Figure 5.
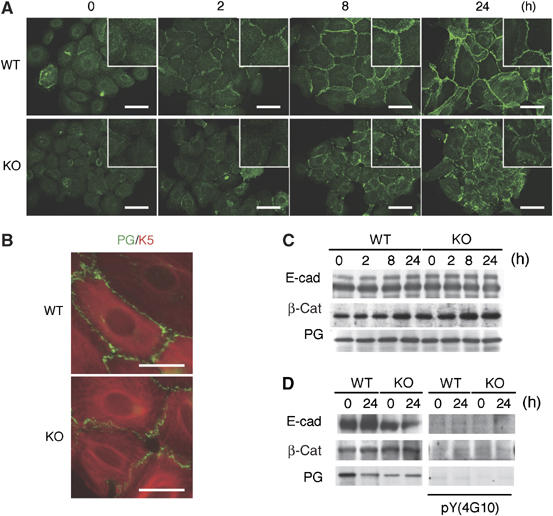
Characterization of primary cultured K5 csk-KO keratinocytes. (A) Wild type (WT) and K5 csk-KO (KO) keratinocytes were treated with 1 mM Ca2+ for the indicated periods and then immunostained with anti-E-cadherin (green). Insets are higher magnification views. Scale bars: 10 μm. (B) WT and KO keratinocytes treated with 1 mM Ca2+ for 8 h were stained with anti-plakoglobin (PG; green) and anti-K5 (red). Merged images are shown. Scale bars: 5 μm. (C) The levels of E-cadherin (E-cad), β-catenin (β-cat), and plakoglobin (PG) during keratinocyte differentiation were examined by immunoblotting. (D) E-cadherin, β-catenin, and plakoglobin were immunoprecipitated from cell lysates prepared before or after 24 h of differentiation (left panels), and tyrosine phosphorylation was detected by immunoblotting with the 4G10 antibody (right panels).
Tyrosine phosphorylation and actin cytoskeletal organization in K5 csk-KO keratinocytes
Tyrosine phosphorylation of cellular proteins in wild-type keratinocytes gradually increased during differentiation (Figure 6A), concomitant with the activation of SFK (Figure 6B, upper panel). In contrast, mutant keratinocytes had high levels of phosphorylation even before differentiation. Phosphorylation of FAK at Y397, an autophosphorylation site, and Y576, an Src phosphorylation site, was constitutively high in the mutant cells (Figure 6B). Phosphorylation of paxillin, another critical component of focal adhesions, was also increased in mutant cells (data not shown), suggesting that focal adhesion formation is activated in the mutant cells. Indeed, immunocytochemical analysis showed that the number and size of focal adhesions detected by staining for tyrosine-phosphorylated proteins were greatly increased in the mutant cells (Figure 6C, upper panels). In addition, accumulation of tyrosine-phosphorylated proteins at podosome-like punctate structures was observed in these cells. Phalloidin staining showed that differentiated wild-type cells had concentrated cortical actin along the cell–cell contacts and radial actin fibers emanating from the adherence junctions (Figure 6C, middle panels). In the mutant cells, however, the actin cytoskeleton appeared dramatically disorganized: actin fibers were nonpolarized, fragmented, and multiply branched, and some fibers terminated at focal adhesions as well as at podosome-like structures.
Rac1-mediated cytoskeletal remodeling in K5 csk-KO keratinocytes
To address the mechanism of cytoskeletal remodeling induced by Csk inactivation, we assessed the activity of the small GTPases, Rac1, Cdc42, and RhoA, which are key regulators of actin cytoskeletal organization (Takai et al, 2001). Pull-down assays revealed that the activity of Rac1 was constitutively elevated, whereas the activity of RhoA was downregulated in the mutant cells (Figure 7A and B).
Figure 7.
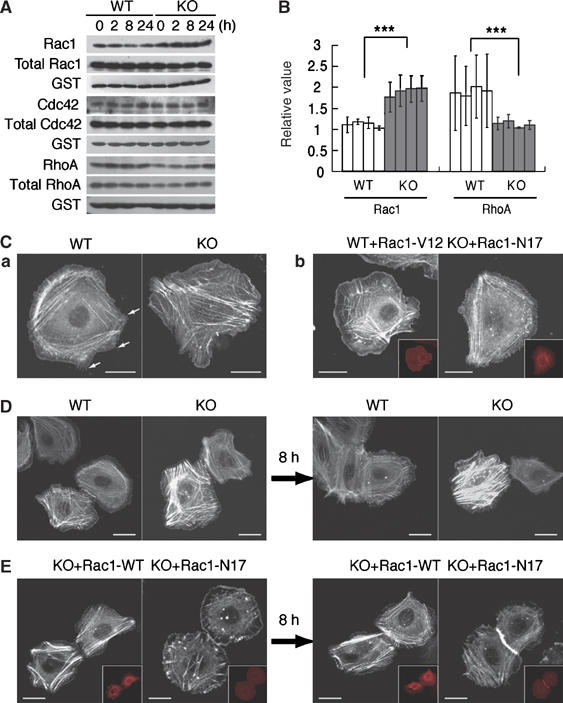
Rac1-mediated cytoskeletal remodeling in K5 csk-KO keratinocytes. (A) Cell lysates prepared from wild-type (WT) and KO keratinocytes at the indicated differentiation stages were incubated with GST-PAK-CRIB, and activated Rac1 was pulled down with glutathione–Sepharose beads. Bound, activated Rac1 (Rac1) and Rac1 in the cell lysates (total Rac1) were detected by immunoblotting with anti-Rac1. GST-PAK-CRIB was detected with anti-GST (GST). Similar assays were performed to detect Cdc42 activity (middle panels). RhoA activity was detected using GST-Rhotekin (lower panels). (B) Signal intensities in the blots for Rac1 (A) were quantified using Image J (NIH), and the relative ratios of activated Rac1 to total Rac1 and activated RhoA to total RhoA were plotted. Data are means±s.e. of three independent assays (t-test; ***P<0.001). (C) WT and KO keratinocytes expressing GFP-actin were monitored for 1 h by time-lapse confocal microscopy. Fluorescent signals for GFP-actin are shown in white (a; Supplementary Movie). Small fillopodia-like structures are shown by arrows (a). WT keratinocytes coexpressing GFP-actin and constitutively active Rac1 (Rac1-V12) and KO keratinocytes coexpressing GFP-actin and dominant negative Rac1 (Rac1-N17) were monitored (b; Supplementary Movie). Fluorescent signals for mRFP-Rac1 are shown in red (insets). (D) WT and KO keratinocytes expressing GFP-actin were treated with 1 mM Ca2+ and monitored for 8 h (Supplementary Movie). Images before (a) and after (b) treatment with Ca2+ are shown. (E) KO keratinocytes expressing WT Rac1 (Rac1-WT) or Rac1-N17 were treated with 1 mM Ca2+ and monitored for 8 h (Supplementary Movie). Images before (a) and after (b) treatment with Ca2+ are shown. Fluorescent signals for mRFP-Rac1 are shown in red (insets). Scale bars: 5 μm.
The contribution of Rac1 was confirmed by time-lapse analysis of keratinocytes expressing GFP-actin. As it is known that other Rac family members (Rac2 and Rac3) are expressed at quite low levels in mouse keratinocytes (Chrostek et al, 2006), we focused on the function of Rac1 in this study. Wild-type keratinocytes had cortically extended actin stress fibers, whereas mutant keratinocytes formed multiply branched and nonpolarized actin fibers and displayed a dynamic formation of lamellipodia as well as ruffled membranes in a wide area of the plasma membrane (Figure 7Ca and Supplementary Movie). When a constitutively active form of Rac1 (Rac1-V12) was expressed in wild-type cells, they gained the ability to extend lamellipodia and exhibited disordered actin fibers similar to the mutant cells. Conversely, expression of a dominant negative form of Rac1 (Rac1-N17) in mutant cells suppressed the extension of lamellipodia and induced the formation of well-organized actin stress fibers, resulting in a phenotype similar to wild-type cells (Figure 7Cb and Supplementary Movie). These observations suggest that Rac1 is one of the key regulators of actin cytoskeletal remodeling by Csk inactivation. When differentiating wild-type cells contacted other cells, they rapidly formed stable cell–cell adhesions (Figure 7Da and Supplementary Movie). In contrast, mutant cells continued formation and destruction of cell–cell contacts and failed to form stable adhesions (Figure 7Db, Supplementary Movie). The expression of Rac1-N17, however, conferred the ability to form stable cell–cell adhesions (Figure 7Eb, Supplementary Movie). The expression of dominant negative RhoA in wild-type cells could also be suppressive for cell–cell adhesion formation (data not shown). These results suggest that activation of Rac1 and inactivation of RhoA are cooperatively involved in the actin cytoskeletal remodeling that leads to destabilization of cell–cell adhesion.
Hyperplasia in K5 csk-KO epithelia is associated with inflammatory events
Finally, we examined the causal link between the in vivo defects, hyperplasia, and inflammation, in K5 csk-KO mice. As the ability of mutant keratinocytes to proliferate in vitro was not significantly affected, we hypothesized that the hyperplasia might be linked to an environmental cue such as inflammation. To examine the effect of Csk inactivation on phenotypic conversion of keratinocytes, we assessed changes in gene expression of secretory factors involved in the onset of inflammation and of the EMT marker proteins, snail and twist (Martin et al, 2005) (Figure 8A). Mutant keratinocytes displayed enhanced expression of MMP2 and MMP9, which have been implicated in both inflammation and EMT (Tester et al, 2000), as well as cancer progression (Ito et al, 2003; Chin et al, 2005). Furthermore, expression of TNF-α, a critical inducer of inflammation (Bates and Mercurio, 2003), was significantly enhanced in the mutant cells. To examine the role of activation of TNF-α and MMP9 expression, we used the anti-inflammatory reagent FK506 (Dumont, 2000), which has been reported to inhibit expression of TNF-α (Lan et al, 2005) and MMP9 (Migita et al, 2006). Indeed, FK506 treatment reduced TNF-α and MMP9 expression to nearly basal levels in the mutant cells (Figure 8A). A gelatin zymography assay further confirmed the FK506-mediated inhibition of MMP9 in the mutant cells (Figure 8B). FK506 treatment, however, did not significantly influence tyrosine phosphorylation of major cellular proteins, such as FAK and paxillin, or actin cytoskeletal organization (data not shown), suggesting that FK506 preferentially acts on a pathway leading to gene expression of TNF-α and MMP9.
Figure 8.
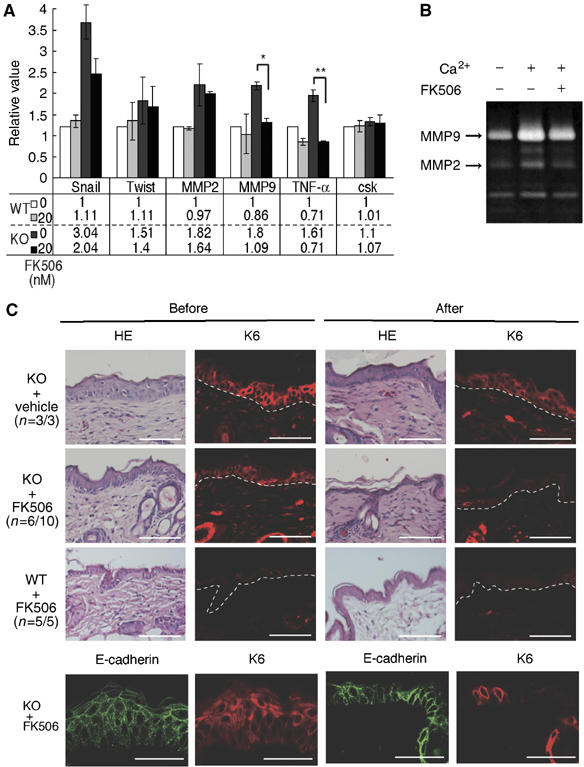
Effects of FK506 treatment on keratinocytes and epidermis in K5 csk-KO mice. (A) Expression of proinflammatory factors and EMT markers in K5 csk-KO keratinocytes. Expression of the indicated genes in wild-type (WT) and K5 csk-KO (KO) keratinocytes treated in the presence or absence of FK506 (20 nM) was quantified by real-time PCR. Values relative to nontreated WT controls are shown in the upper graph (mean±s.e. of five independent experiments, t-test; **P<0.01, *P<0.05). Actual mean values are shown in the lower table. (B) Zymogram of MMP activity. Conditioned medium was prepared from K5 csk-KO keratinocytes treated with the indicated reagents and then subjected to zymography in a gelatin-containing gel. Positions corresponding to MMP9 and MMP2 are indicated by the arrows. (C) The skin of K5 csk-KO mice (KO) was treated with FK506 ointment or control vehicle (petroleum jelly). The skin of WT mice was treated with FK506 as a control. Dorsal skin sections stained with HE and anti-K6 before (left) and after (right) treatment are shown. Frequencies (n=affected individuals/total individuals) are shown in parentheses. The skin of K5 csk-KO mice (KO) was further subjected to staining for E-cadherin before and after treatment with FK506 (lower panels). The dotted white line represents the epidermal–dermal border. Scale bars: 10 μm.
The in vivo effect of FK506 was then evaluated by applying FK506 ointment to areas of mutant mouse skin exhibiting chronic dermatitis. In a significant number of cases, FK506 treatment reduced the thickness of the mutant epidermal layers to the normal size and substantially decreased the number of K6-positive proliferating cells (Figure 8C). Accumulation of E-cadherin at cell–cell contacts was also improved in FK506-treated mutant epidermis (Figure 8C). Treatment with petroleum jelly as a control did not affect hyperplasia in any of the mutant mice. In addition, FK506 treatment of wild-type skin did not induce any significant effects, confirming that FK506 has no harmful effect on normal skin. These observations indicate that epidermal hyperplasia could be cured by FK506 treatment, suggesting that hyperplasia in the mutant skin is linked to inflammatory events induced by Csk inactivation.
Discussion
To elucidate the in vivo roles of Csk in epithelial tissues, we developed mutant mice in which Csk was conditionally inactivated in the basal layer of squamous epithelia. The mutant mice developed apparent defects in skin, esophagus, and forestomach, and displayed epithelial hyperplasia and chronic inflammation. Analysis of the mutant epidermis and cultured keratinocytes revealed that attenuation of cell–cell adhesion is an intrinsic defect caused by Csk inactivation. Previous studies have shown that activated Src directly induces downregulation of cell adhesion molecules (Frame, 2002; Fujita et al, 2002), but neither the extent of tyrosine phosphorylation nor the level of these proteins was significantly changed in the mutant keratinocytes. However, a dramatic remodeling of the cytoskeleton was observed in these cells. There was a substantial reduction in radial actin stress fibers and keratin filaments supporting cell–cell adhesion, whereas formation of depolarized actin fibers, podosomes, ruffled membranes, and lamellipodia was greatly activated. Thus, it appears that the cytoskeletal remodeling induced by Csk inactivation accounts for defective cell–cell adhesion in the mutant epidermis.
To address the mechanism underlying cytoskeletal remodeling, we examined the activity of small GTPases and found that Rac1 was constitutively activated in K5 csk-KO keratinocytes. Introduction of dominant negative Rac1 into the mutant cells suppressed the extension of lamellipodia and formation of ruffled membranes, and, instead, promoted the formation of radial actin fibers. Consequently, the cells successfully restored stable cell–cell adhesions. Furthermore, small fillopodia, which are needed for initial cell–cell contact, could be restored by the dominant negative Rac1. These results suggest that a constitutively activated Rac1 pathway is crucial for the defects induced by Csk inactivation. SFK-dependent phosphorylation and/or recruitment of adaptor proteins, such as FAK, Cas, and Crk (Brown and Cooper, 1996; Fukuyama et al, 2005), and activation of guanine nucleotide exchange factors, such as Vav and DOCK180 (Crespo et al, 1997), may mediate Rac1 activation in the mutant keratinocytes. Recently, it has been reported that Rac1-null epidermal cells show expanded intercellular spaces with concomitant loss of E-cadherin expression (Chrostek et al, 2006). In contrast, K5 csk-KO epidermis shows expanded intercellular spaces despite constitutive Rac1 activation. This inconsistency can be explained by the observation that upregulation of Rac1 activity could abrogate the function of E-cadherin by inducing dramatic remodeling of the cytoskeleton stabilizing the adherence junctions.
In K5 csk-KO keratinocytes, we also detected a significant reduction of RhoA activity, which might result from Rac1 activation (Nimnual et al, 2003). The downregulation of RhoA may account for the destruction of polarized actin stress fibers as well as the attenuated desmosomal adhesion (Waschke et al, 2006). RhoA and Rac1 work competitively in focal adhesion formation (Rottner et al, 1999) and they are involved in crosstalk among adhesive receptors in epithelial junctions (Braga and Yap, 2005). In this study, we showed that cell–cell adhesion and cell–substrate adhesion were reciprocally regulated by SFK/Csk: K5 csk-KO cells displayed increased cell–substrate adhesion and rather attenuated cell–cell adhesion. These observations suggest that the interplay between Rac1 and RhoA regulated by an SFK/Csk circuit would largely contribute to the maintenance of epithelial organization.
Another critical defect caused by Csk inactivation was epidermal hyperplasia. Epidermal hyperplasia was not observed in newborn mice, in which there was no significant sign of inflammation. During mouse development, inflammatory events were observed at relatively earlier stages (by P20), whereas epidermal hyperplasia became evident at approximately P50 and thereafter spread over the entire skin of the mutant mice. These observations suggest that epidermal hyperplasia is linked to the onset of inflammation. Inflammation induced by physical stimuli has been reported previously to cause epidermal hyperplasia, which could be suppressed by the anti-inflammatory agent FK506 (Hiroi et al, 1998; Fujii et al, 2002). Treatment of mutant mouse skin with FK506 could also cure hyperplasia in the mutant mouse, suggesting a functional link between epidermal hyperplasia and inflammation in K5 csk-KO mice.
We examined the cause of inflammation in K5 csk-KO skin. Infection by pathogens was unlikely, because the inflammation was chronic and widespread and there was no increase in neutrophils in the mutant skin. In keratinocyte cultures, we observed that Csk inactivation induced expression of the proinflammatory cytokine TNF-α and MMPs. It is, thus, likely that these proinflammatory factors would activate lymphocytes and/or stromal cells to trigger inflammation, resulting in the outflow of growth-promoting cytokines that induce epidermal hyperplasia. In support of this hypothesis, FK506-mediated inhibition of TNF-α and MMP9 expression cured the inflammation as well as the hyperplasia in K5 csk-KO mice. These findings suggest that the SFK/Csk circuit is involved in the regulation of inflammatory events and potentially innate immunity in the epithelial cells, as previously observed in granulocytes (Thomas et al, 2004) and macrophages (Aki et al, 2005). However, the mechanisms underlying the induction of proinflammatory factors downstream of SFK still remain unknown. Further analysis of SFK responsive cis and trans elements and potential crosstalk with the Rac1 pathway will be required.
Recently, another group generated transgenic mice that overexpress c-Src in epidermal basal cells under the control of the bovine keratin 5 (BK5) promoter (Matsumoto et al, 2002, 2003). BK5 c-src mice exhibited epidermal hyperplasia and hyperkeratosis, phenotypes that are quite similar to those observed in K5 csk-KO mice. Interestingly, some lines of BK5 c-src transgenic mice developed squamous cell carcinomas (SCCs) in the skin, although the K5 csk-KO mice do not produce carcinomas. This phenotypic difference is probably due to differences in the levels of kinase activities between overexpressed c-Src in BK5 c-src mice and endogenous SFKs in K5 csk-KO mice. As previously observed (Harris et al, 1999), there was a significant degradation of activated SFK proteins in the K5 csk-KO fibroblasts, which may also contribute to the reduction of total kinase activity in these cells. These observations suggest that the activation of endogenous SFK is not sufficient for cancer initiation. Nonetheless, we have shown that constitutive activation of endogenous SFK by Csk inactivation could induce a phenotypic conversion in epithelial cells; dramatic remodeling of cytoskeletal organization and upregulation of some cytokines and mesenchymal proteins such as snail and twist, as well as MMPs. These are characteristic events in the EMT that favor the invasive and metastatic potential of cancers. Furthermore, these phenotypic changes influence the extracellular environment to induce the inflammation and vascularization that further promote cancer progression (Coussens and Werb, 2002). Thus, it is likely that activation of endogenous SFK can produce an intra- and extracellular environment that promotes malignancy upon subsequent mutation and/or external insult. Given that the metastatic potential of human cancers is frequently associated with upregulation of endogenous SFK activity, molecules involved in Csk-mediated negative regulation of SFK could serve as potential therapeutic targets for the suppression of cancer progression.
Materials and methods
K5-Csk knockout mice
K5-Cre transgenic mice (gifts from J Takeda) were mated with cskflox/flox mice. The F2 offspring carrying the cskflox/flox locus and the K5-Cre transgene (K5-Cre-cskflox/flox), and their littermates carrying no K5-Cre transgene (cskflox/flox), were used as K5 csk-KO mice and control mice, respectively. Genotypes were confirmed by detection of K5-Cre and the cskflox locus by allele-specific PCR, as described previously (Tarutani et al, 1997; Schmedt et al, 1998). Mice were handled and maintained according to the Osaka University guidelines for animal experimentation.
Antibodies
Anti-Csk (C-20) and anti-β-catenin (H-102) were purchased from Santa Cruz Biotechnology. Anti-Src pY418, anti-Src pY529, anti-FAK pY576, and anti-FAK pY397 were purchased from Biosource. Anti-Thy1.2 and anti-CD11b were obtained from e-Bioscience. Anti-plakoglobin was purchased from PROGEN. Anti-keratin 1 (K1), anti-keratin 5 (K5), anti-keratin 6 (K6), and anti-involucrin were purchased from Berkeley Antibody. Anti-Ki67 was purchased from Novocastra. Anti-E-cadherin, anti-paxillin, anti-FAK, anti-Rac1, and anti-Cdc42 were obtained from BD Transduction Laboratories. Anti-phosphotyrosine (4G10) was purchased from Upstate Biotechnology.
Histological analysis
Dorsal skin was excised from mice deeply anesthetized with 10% Nembutal. The samples were fixed in paraformaldehyde (4% PFA in phosphate-buffered saline) and embedded in paraffin. Sections (2 μm) on glass slides were rehydrated and incubated with Histo-HV One solution (Nacalai tesque) for antigen reactivation. The specimens were incubated with blocking solution (50 mM Tris–HCl, pH 7.4, 0.9% NaCl, 0.25% gelatin, 0.5% (w/v) Triton X-100) or 5% normal goat serum/Tris-buffered saline containing 0.1% Tween 20 (TTBS), and then incubated with primary antibodies in blocking solution or TTBS overnight at 4°C, followed by incubation with labeled secondary antibodies. The labeled sections were coverslipped and examined by confocal laser-scanning microscopy (Olympus, FV-1000). Anti-Thy1.2, anti-β-catenin, and anti-CD11b were used at dilutions of 1:1000, 1:200, and 1:100, respectively. All other antibodies were used at a dilution of 1:500. For HE staining, rehydrated sections were stained with Mayer's hematoxylin (Waco chemical co.) and eosin. For periodic acid Schiff (PAS) staining, rehydrated paraffin sections were pretreated with 0.5% periodic acid solution (Wako chemical co.). After washing, samples were incubated with the Schiff reagent and washed three times with sodium pyrosulfite (0.5% hydrosulfite in 0.05 N HCl).
Preparation of keratinocytes
Skin from newborn mice was treated with 100 U/ml of dispase for 20 h at 4°C, and the epidermis was peeled away from the dermis and trypsinized for 10 min at 37°C. Keratinocytes were cultured on collagen type I-coated plates in keratinocyte growth medium (KGM, modified MCDB 153 medium, Kyokuto) supplemented with 5 μg/ml insulin, 0.5 μM hydrocortisone, and bovine pituitary extract. To induce differentiation, the medium was changed from normal KGM (<0.03 mM Ca2+) to Ca2+-rich KGM (1 or 2 mM CaCl2).
Immunocytochemistry
Keratinocytes cultured in 24-well plates were fixed with 4% PFA for 15 min at room temperature. For analysis of cell adhesion proteins, cells were fixed with cold methanol (precooled at −20°C) for 30 min at 4°C. After washing with TTBS, the samples were incubated with 5% BSA/TTBS, followed by incubation with primary antibodies in TTBS overnight at 4°C. After incubation with secondary antibodies for 45 min at room temperature, coverslips were mounted on glass slides. The specimens were examined by fluorescence microscopy (Olympus, BX60) or confocal laser-scanning microscopy (Olympus, FV-1000). Antibodies were used at the same dilutions as for immunohistochemistry.
Electron microscopy
Mice (37-week-old or P5) were deeply anesthetized with 10% Nembutal and perfused with 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Skin samples were removed and cut into small pieces that were again immersed in the same fixative at 4°C. After washing with 7.5% sucrose/0.1 M phosphate buffer, tissues were post-fixed with 1% OsO4 in the same buffer and then block-stained with an aqueous solution of uranyl acetate for 1 h. After dehydration, tissues were embedded in Poly/Bed 812. Ultrathin sections were stained with uranyl acetate and lead citrate. The specimens were examined with an HV-7100 electron microscope (Hitachi).
Immunoblotting
Whole-cell lysates were prepared from keratinocytes in RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% (w/v) Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 2 mM EGTA, 1 mM sodium orthovanadate, 0.1% leupeptin, 0.1% aprotinin, and 1 M PMSF) or ODG buffer (50 mM Tris–HCl, pH 7.4, 1 mM EDTA, 0.15 M NaCl, 5% (w/v) glycerol, 1% (w/v) NP-40, 20 mM NaF, 2% N-octyl-β-D-glucoside (ODG), 5 μM 2-mercaptoethanol, 1 mM sodium orthovanadate, 0.1% leupeptin, 0.1% aprotinin, and 1 mM PMSF), for immunoprecipitation and immunoblotting, respectively. Equal amounts of total protein were separated by SDS–PAGE and transferred onto nitrocellulose membranes. The membranes were blocked and incubated with primary antibodies, followed by incubation with HRP-conjugated secondary antibodies. Primary antibodies were used at dilutions of 1:5000 (for 4G10), 1:1000 (for Rac1, RhoA, and Cdc42), and 1:2000 (for all others). Signals from immunopositive bands were visualized on X-ray film using an enhanced chemiluminescence system (Amersham).
Assays of Rho family small G proteins
Whole-cell lysates were prepared from keratinocytes in ODG buffer. Lysates (100 μg protein) were incubated with purified GST-PAK-CRIB (a gift from M Matsuda) or GST-Rhotekin bound to glutathione-coupled Sepharose beads (PAK, 4°C, overnight; Rhotekin, 4°C, 1 h). Following pull-down, activated Rac1, Cdc42, and RhoA were detected by immunoblotting and quantified using the Image J software package. For time-lapse analysis, EGFP-actin (BD Bioscience) and/or mRFP-Rac1 (wild type, V12 and N17; gifts from M Matsuda) were transiently transfected into keratinocyte cultures using a retrovirus vector (pCX4bleo). The transfected keratinocytes, cultured in the presence or absence of 1 mM CaCl2, were monitored for 8 h using confocal laser-scanning microscopy (Olympus, FV-1000).
Real-time quantitative PCR
Total RNA was prepared from differentiated keratinocytes using Sepasol (Nacalai Tesque), and reverse transcribed using SuperScript II reverse transcriptase (Invitrogen). Gene-specific primers and probes were obtained from Applied Biosystems (TaqMan Gene Expression Assays). PCR was performed using an Applied Biosystems 7900HT Fast Real-Time PCR System and Sequence Detection System Software v2.2.1 according to the manufacturer's instructions.
FK506 treatment
Dorsal skin from 13-week-old K5 csk-KO mice that exhibited spontaneous dermatitis was used for analysis. K5 csk-KO mice were treated with 0.1% FK506 ointment (Astellas Pharma Inc.) or Petroleum jelly for 10 days. After treatment, dorsal skin from these mice was excised and observed by HE staining.
Gelatin zymography
Keratinocytes (0.5 × 105 cells) cultured for 3 days were further cultured for 24 h in MCDB153 medium in the presence or absence of 1 mM CaCl2 and 20 nM FK506. These media were electrophoresed under non-reducing conditions on 10% SDS–PAGE gels containing 1% gelatin (15 mA for 2.5 h, 4°C). Gels were washed with 2.5% (w/v) Triton X-100 for 30 min and incubated in a reaction buffer (50 mM Tris–HCl, pH 7.4, 0.2 M NaCl, 0.5 mM CaCl2, 1% (w/v) Triton X-100, and 0.02% NaN3) for 36 h at 37°C. Gels were then stained with 0.25% Coomassie brilliant blue R-250.
Supplementary Material
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Figures
Acknowledgments
We thank Alexander Tarakhovsky (Rockefeller University) for discussions and critical reading of the manuscript, Jyunji Takeda (Osaka University) for the kind gift of K5-Cre transgenic mouse, and Michiyuki Matsuda (Kyoto University) for the kind gift of Rac1 expression vectors. This work was supported by a Grant-in-Aid for Scientific Research on Priority Areas ‘Cancer' from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Aki D, Mashima R, Saeki K, Minoda Y, Yamauchi M, Yoshimura A (2005) Modulation of TLR signalling by the C-terminal Src kinase (Csk) in macrophages. Genes Cells 10: 357–368 [DOI] [PubMed] [Google Scholar]
- Avizienyte E, Frame MC (2005) Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr Opin Cell Biol 17: 542–547 [DOI] [PubMed] [Google Scholar]
- Bates RC, Mercurio AM (2003) Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell 14: 1790–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel MM, Birchmeier W (1993) Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol 120: 757–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga VM, Yap AS (2005) The challenges of abundance: epithelial junctions and small GTPase. Curr Opin Cell Biol 17: 466–474 [DOI] [PubMed] [Google Scholar]
- Brown MT, Cooper JA (1996) Regulation, substrates and functions of src. Biochim Biophys Acta 1287: 121–149 [DOI] [PubMed] [Google Scholar]
- Chin D, Boyle GM, Kane AJ, Theile DR, Hayward NK, Parson PG, Coman WB (2005) Invasion and metastasis markers in cancers. Br J Plast Surg 58: 466–474 [DOI] [PubMed] [Google Scholar]
- Chow LM, Fournel M, Davidson D, Veillette A (1993) Negative regulation of T-cell receptor signalling by tyrosine protein kinase p50csk. Nature 365: 156–160 [DOI] [PubMed] [Google Scholar]
- Chrostek A, Wu X, Quondamatteo F, Hu R, Sanecka A, Niemann C, Langbein L, Haase I, Brakebusch C (2006) Rac1 is crucial for hair follicle integrity but is not essential for maintenance of the epidermis. Mol Cell Biol 26: 6957–6970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420: 560–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo P, Schuebel K, Ostrom A, Gutkind J, Bustelo X (1997) Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature 385: 169–172 [DOI] [PubMed] [Google Scholar]
- Dumont FJ (2000) FK506, an immunosuppressant targeting calcineurin function. Curr Med Chem 7: 731–748 [DOI] [PubMed] [Google Scholar]
- Frame MC (2002) Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta 1602: 114–130 [DOI] [PubMed] [Google Scholar]
- Fujii Y, Takeuchi H, Tanaka K, Sakuma S, Ohkubo Y, Mutoh S (2002) Effects of FK506 (tacrolimus hydrate) on chronic oxazolone-induced dermatitis in rats. Eur J Pharmacol 456: 115–121 [DOI] [PubMed] [Google Scholar]
- Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W (2002) Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 4: 222–231 [DOI] [PubMed] [Google Scholar]
- Fukuyama T, Ogita H, Kawakatsu T, Fukuhara T, Yamada T, Sato T, Shimizu K, Nakamura T, Matsuda M, Takai Y (2005) Involvement of the c-Src–Crk–C3G–Rap1 signaling in the nectin-induced activation of Cdc42 and formation of adherens junctions. J Biol Chem 280: 815–825 [DOI] [PubMed] [Google Scholar]
- Hamaguchi M, Yamagata S, Thant AA, Xiao H, Iwata H, Mazaki T, Hanafusa H (1995) Augmentation of metalloproteinase (gelatinase) activity secreted from Rous sarcoma virus-infected cells correlates with transforming activity of src. Oncogene 10: 1037–1043 [PubMed] [Google Scholar]
- Harris KF, Shoji I, Cooper EM, Kumar S, Oda H, Howley PM (1999) Ubiquitin-mediated degradation of active Src tyrosine kinase. Proc Natl Acad Sci USA 96: 13738–13743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroi J, Sengoku T, Morita K, Kishi S, Sato S, Ogawa T, Tsudzuki M, Matsuda H, Wada A, Esaki K (1998) Effect of tacrolimus hydrate (FK506) ointment on spontaneous dermatitis in NC/Nga mice. Jpn J Pharmacol 76: 175–183 [DOI] [PubMed] [Google Scholar]
- Hsia DA, Mitra SK, Hauck CR, Streblow DN, Nelson JA, Ilic D, Huang S, Li E, Nemerow GR, Leng J, Spencer KS, Cheresh DA, Schlaepfer DD (2003) Differential regulation of cell motility and invasion by FAK. J Cell Biol 160: 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber MA, Kraut N, Beug H (2005) Molecular requirements for epithelial–mesenchymal transition during tumor progression. Curr Opin Cell Biol 17: 548–558 [DOI] [PubMed] [Google Scholar]
- Hunter T, Sefton BM (1980) Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci USA 77: 1311–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamoto A, Soriano P (1993) Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell 73: 1117–1124 [DOI] [PubMed] [Google Scholar]
- Irby RB, Yeatman TJ (2000) Role of Src expression and activation in human cancer. Oncogene 19: 5636–5642 [DOI] [PubMed] [Google Scholar]
- Ito H, Gardner-Thorpe J, Zinner MJ, Ashley SW, Whang EE (2003) Inhibition of tyrosine kinase Src suppresses pancreatic cancer invasiveness. Surgery 134: 221–226 [DOI] [PubMed] [Google Scholar]
- Jove R, Hanafusa H (1987) Cell transformation by the viral src oncogene. Annu Rev Cell Biol 3: 31–56 [DOI] [PubMed] [Google Scholar]
- Lan CC, Yu HS, Wu CS, Kuo HY, Chai CY, Chen GS (2005) FK506 inhibits tumour necrosis factor-alpha secretion in human keratinocytes via regulation of nuclear factor-kappaB. Br J Dermatol 153: 725–732 [DOI] [PubMed] [Google Scholar]
- Martin TA, Goyal A, Watkins G, Jiang WG (2005) Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol 12: 488–496 [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Jiang J, Kiguchi K, Carbajal S, Rho O, Gimenez-Conti I, Beltran L, DiGiovanni J (2002) Overexpression of a constitutively active form of c-src in skin epidermis increases sensitivity to tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Mol Carcinog 33: 146–155 [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Jiang J, Kiguchi K, Ruffino L, Carbajal S, Beltran L, Bol DK, Rosenberg MP, DiGiovanni J (2003) Targeted expression of c-Src in epidermal basal cells leads to enhanced skin tumor promotion, malignant progression, and metastasis. Cancer Res 63: 4819–4828 [PubMed] [Google Scholar]
- Matsuyoshi N, Hamaguchi M, Taniguchi S, Nagafuchi A, Tsukita S, Takeichi M (1992) Cadherin-mediated cell–cell adhesion is perturbed by v-src tyrosine phosphorylation in metastatic fibroblasts. J Cell Biol 118: 703–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migita K, Maeda Y, Abiru S, Nakamura M, Komori A, Yokoyama T, Takii Y, Mori T, Yatsuhashi H, Eguchi K, Ishibashi H (2006) Immunosuppressant FK506 inhibits matrix metalloproteinase-9 induction in TNF-alpha-stimulated human hepatic stellate cells. Life Sci 78: 2510–2515 [DOI] [PubMed] [Google Scholar]
- Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H (1991) Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature 351: 69–72 [DOI] [PubMed] [Google Scholar]
- Nada S, Yagi T, Takeda H, Tokunaga T, Nakagawa H, Ikawa Y, Okada M, Aizawa S (1993) Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell 73: 1125–1135 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Tanaka S, Suzuki H, Takayanagi H, Miyazaki T, Nakamura K, Tsuruo T (2000) Overexpression of the csk gene suppresses tumor metastasis in vivo. Int J Cancer 88: 384–391 [PubMed] [Google Scholar]
- Nimnual AS, Taylor LJ, Bar-Sagi D (2003) Redox-dependent downregulation of Rho by Rac. Nat Cell Biol 5: 236–241 [DOI] [PubMed] [Google Scholar]
- Okada M, Nada S, Yamanashi Y, Yamamoto T, Nakagawa H (1991) CSK: a protein-tyrosine kinase involved in regulation of src family kinases. J Biol Chem 266: 24249–24252 [PubMed] [Google Scholar]
- Rengifo-Cam W, Konishi A, Morishita N, Matsuoka H, Yamori T, Nada S, Okada M (2004) Csk defines the ability of integrin-mediated cell adhesion and migration in human colon cancer cells: implication for a potential role in cancer metastasis. Oncogene 23: 289–297 [DOI] [PubMed] [Google Scholar]
- Rottner K, Hall A, Small JV (1999) Interplay between Rac and Rho in the control of substrate contact dynamics. Curr Biol 9: 640–648 [DOI] [PubMed] [Google Scholar]
- Schmedt C, Saijo K, Niidome T, Kuhn R, Aizawa S, Tarakhovsky A (1998) Csk controls antigen receptor-mediated development and selection of T-lineage cells. Nature 394: 901–904 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Shoji S, Yamamoto K, Nada S, Okada M, Yamamoto T, Honda Z (1998) Essential roles of Lyn in fibronectin-mediated filamentous actin assembly and cell motility in mast cells. J Immunol 161: 3694–3701 [PubMed] [Google Scholar]
- Takai Y, Sasaki T, Matozaki T (2001) Small GTP-binding proteins. Physiol Rev 81: 153–208 [DOI] [PubMed] [Google Scholar]
- Talamonti MS, Roh MS, Curley SA, Gallick GE (1993) Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J Clin Invest 91: 53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarutani M, Itami S, Okabe M, Ikawa M, Tezuka T, Yoshikawa K, Kinoshita T, Takeda J (1997) Tissue-specific knockout of the mouse Pig-a gene reveals important roles for GPI-anchored proteins in skin development. Proc Natl Acad Sci USA 94: 7400–7405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester AM, Ruangpanit N, Anderson RL, Thompson EW (2000) MMP-9 secretion and MMP-2 activation distinguish invasive and metastatic sublines of a mouse mammary carcinoma system showing epithelial–mesenchymal transition traits. Clin Exp Metast 18: 553–560 [DOI] [PubMed] [Google Scholar]
- Thomas RM, Schmedt C, Novelli M, Choi BK, Skok J, Tarakhovsky A, Roes J (2004) C-terminal SRC kinase controls acute inflammation and granulocyte adhesion. Immunity 20: 181–191 [DOI] [PubMed] [Google Scholar]
- Waschke J, Spindler V, Bruggeman P, Zillikens D, Schmidt G, Drenckhahn D (2006) Inhibition of Rho A activity causes pemphigus skin blistering. J Cell Biol 175: 721–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ (1997) Three-dimensional structure of the tyrosine kinase c-Src. Nature 385: 595–602 [DOI] [PubMed] [Google Scholar]
- Yu H, Jove R (2004) The STATs of cancer—new molecular targets come of age. Nat Rev Cancer 4: 97–105 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Figures