

Abstract
Cytokines of the tumor necrosis factor (TNF) family regulate inflammation and immunity, and a subset of this family can also induce cell death in a context-dependent manner. Although TNFα is cytotoxic to certain tumor cell lines, it induces apoptosis in normal cells only when NFκB signaling is blocked. Here we show that the matricellular protein CCN1/CYR61 can unmask the cytotoxic potential of TNFα without perturbation of NFκB signaling or de novo protein synthesis, leading to rapid apoptosis in the otherwise resistant primary human fibroblasts. CCN1 acts through binding to integrins αvβ5, α6β1, and syndecan-4, triggering the generation of reactive oxygen species (ROS) through a Rac1-dependent mechanism via 5-lipoxygenase and the mitochondria, leading to the biphasic activation of JNK necessary for apoptosis. Mice with the genomic Ccn1 locus replaced with an apoptosis-defective Ccn1 allele are substantially resistant to TNFα-induced apoptosis in vivo. These results indicate that CCN1 may act as a physiologic regulator of TNFα cytotoxicity, providing the contextual cues from the extracellular matrix for TNFα-mediated cell death.
Keywords: CCN2, CCN3, CTGF, NADPH oxidase, wound healing
Introduction
The tumor necrosis factor (TNF) superfamily subsumes at least 19 cytokines that play critical roles in regulating the development and function of the immune system (Locksley et al, 2001; Aggarwal, 2003; Wajant et al, 2003; Hehlgans and Pfeffer, 2005). A subset of this family, notably TNFα and FasL, can also induce cell death. Although TNFα is cytotoxic to certain tumor cell lines, it does not trigger apoptosis in normal cells, but instead stimulates the proliferation of normal fibroblasts (Sugarman et al, 1985; Battegay et al, 1995). This dichotomy is in part due to the potency of TNFα as an activator of NFκB, a pro-inflammatory transcription factor that promotes cell survival through activation of prosurvival genes and suppression of proapoptosis genes. Thus, although TNFα also induces apoptotic signals, its cytotoxicity in normal cells in culture is completely dependent on the blockade of NFκB signaling or de novo protein synthesis (Karin and Lin, 2002; Varfolomeev and Ashkenazi, 2004). This dependence is also observed in vivo: targeted deletion of genes encoding the NFκB p65 subunit RelA or the NFκB-activating kinase IKKβ results in massive hepatocyte apoptosis and embryonic lethality, phenotypes that are eliminated by further genetic ablation of TNFα or its receptor (Doi et al, 1999; Rosenfeld et al, 2000; Senftleben et al, 2001). A critical signaling element in TNFα-induced apoptosis is the robust and prolonged activation of JNK, which occurs when NFκB is inhibited (Sakon et al, 2003; Kamata et al, 2005). These findings illuminate the mechanism by which TNFα can induce apoptosis and suggest that the cytotoxicity of TNFα is highly contextual; however, the specific signals that can unleash this cytotoxicity in a physiological context are not well understood.
The CCN family is comprised of six secreted matricellular proteins that regulate diverse cellular processes (Lau and Lam, 1999; Bornstein and Sage, 2002; Brigstock, 2003; Planque and Perbal, 2003). CCN1/CYR61, CCN2/CTGF, and CCN3/Nov are angiogenic inducers in vivo. Consistently, Ccn1-null mice suffer embryonic lethality due to cardiovascular defects (Mo et al, 2002; Mo and Lau, 2006), whereas Ccn2-deficient mice are perinatal lethal due to respiratory failure as a secondary consequence of severe skeletal malformations and impaired angiogenesis in the skeletal growth plates (Ivkovic et al, 2003). In keeping with their structural homology with conserved domains of extracellular matrix (ECM) proteins, CCNs bind to and function through integrin receptors (Supplementary Figure 1) (Lau and Lam, 2005). Thus, CCN1 supports cell adhesion and spreading in fibroblasts through integrin α6β1 and cell surface heparan sulfate proteoglycans (HSPGs), whereas αvβ3 mediates the proangiogenic activities of CCN1 in activated endothelial cells (Chen et al, 2000; Leu et al, 2002).
The expression of CCN proteins is associated with sites of angiogenesis and inflammation, such as in wound healing, arthritis, tumors, and vessels damaged by angioplasty or atherosclerosis (Lau and Lam, 2005). As these are also sites of TNFα expression, we hypothesize that CCNs and TNFα may cooperate to induce cellular responses, such as apoptosis. Remarkably, we found that CCNs can unmask the apoptotic activity of TNFα without perturbation of NFκB signaling or de novo protein synthesis, thus enabling TNFα to induce rapid apoptosis in the otherwise resistant normal human skin fibroblasts (HSFs). CCN1 accomplishes this effect through direct binding to integrins αvβ5, α6β1 and the HSPG syndecan-4 to induce a high level of reactive oxygen species (ROS) accumulation, resulting in the reactivation of JNK after the initial rapid and transient JNK activation induced by TNFα. This novel mechanism overrides the antiapoptotic effects of NFκB to achieve reactivation of JNK, which is critical for apoptosis. Furthermore, mice with the genomic Ccn1 locus replaced with an apoptosis-defective Ccn1 allele are significantly resistant to TNFα-induced apoptosis in vivo. These results show that the extracellular matrix milieu can profoundly regulate the cytotoxicity of TNFα. We propose that the dynamic expression of CCN proteins and TNFα during inflammatory responses allow their interaction at critical stages, resulting in apoptosis of selected cell types specified by the combinatorial activation of death receptors, integrins, and HSPGs.
Results
TNFα-induced fibroblast apoptosis is ECM dependent
To test whether the cytotoxicity of TNFα may be regulated by the ECM, HSFs were adhered on surfaces coated with various ECM proteins. CCNs are matricellular proteins that support cell adhesion and cell spreading, and induce adhesive signaling including activation of FAK, paxillin, and Rac (Chen et al, 2001a; Todorovic et al, 2005). About 20–40% of cells adhered to CCN1, CCN2, or CCN3 were apoptotic after incubation with TNFα (10 ng/ml) for 4 h as judged by TUNEL assay and DAPI staining (Figure 1A and B). By contrast, <3% cell death occurred in cells adhered to fibronectin (FN), laminin (LN), vitronectin (VN), collagen I (Col. I), or fibrinogen (FBN) with or without TNFα. Thus, CCNs provide a unique matrix environment that enables the cytotoxicity of TNFα in fibroblasts, a cell type in which TNFα alone stimulates proliferation rather than cell death (Sugarman et al, 1985). As cell adhesion to ECM proteins is known to induce prosurvival signals, we tested whether this process can protect against CCN-dependent apoptosis. Soluble CCN1 and TNFα were added alone or in combination to cells adhered to Col. I, FBN, FN, LN, or VN. In all cases, CCN1 and TNFα together induced >20% apoptosis, whereas minimal cell death occurred with either factor alone (Figure 1C). Thus, CCN1 and TNFα prevailed over the prosurvival signals resulting from cell adhesion to these ECM proteins. Further, in cells cultured in 10% serum for 2 days to allow deposition of endogenous matrix before serum deprivation, CCN1 cooperated with TNFα to induce cell death in a dose-dependent manner, with 20% cell death observed at 2 μg/ml CCN1 (Figure 1D).
Figure 1.
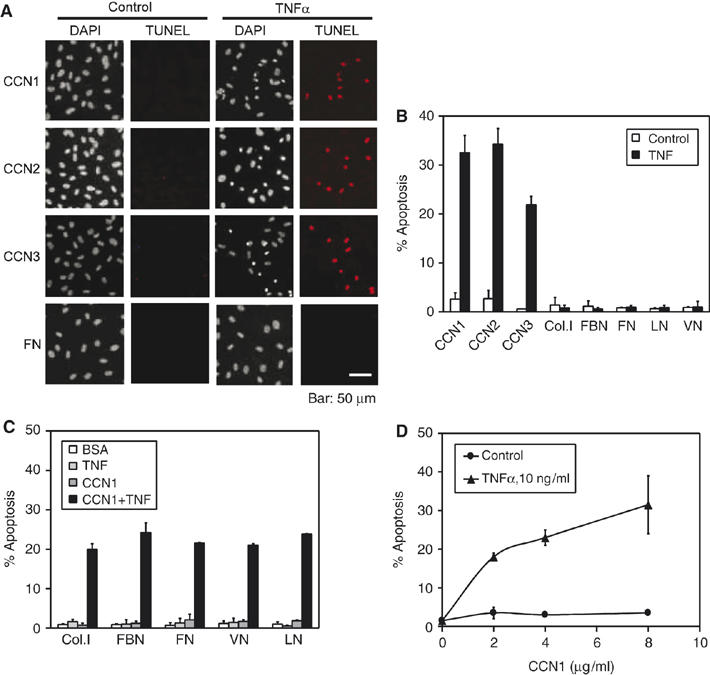
Fibroblast adhesion to CCN proteins enables TNFα to induce apoptosis. Primary HSFs were treated as described and exposed to CCN1 and/or TNFα for 4.5 h where indicated. Apoptotic cells were scored as condensed cell nuclei. (A) HSFs were adhered on glass cover slips coated with purified CCN1, CCN2, CCN3, or FN in serum-free medium, and treated with TNFα or vehicle (control). Cells were subjected to TUNEL assay and counterstained with DAPI to assess apoptosis. (B) Percents of apoptotic nuclei in cells treated as in (A) were quantified, including those adhered on FN, LN, VN, Col. I, and FBN. (C) Cells adhered to matrix substrates as indicated were treated with soluble CCN1 and/or TNFα and scored for apoptosis. (D) HSFs were cultured for 2 days in medium with 10% FBS to allow deposition of endogenous ECM. After serum starvation, cells were treated with various concentrations of CCN1 as indicated with or without TNFα. In this entire study, all bar graphs show standard deviation of the mean of triplicate determinations. Each experiment in this study was repeated at least three times with similar results; one representative experiment is shown.
Ligation of TNFα to its receptor TNFRI is known to activate caspase-8 and -10, initiator caspases with similar activities that mobilize the extrinsic apoptotic pathway through direct activation of caspase-3 (Fischer et al, 2005). Depending on the cell type, mitochondrial amplification of caspase activation through cytochrome c release and activation of caspase-9 may be necessary for cell death (Wajant et al, 2003). Caspase-3, -8, -9, and -10 were all activated in CCN1/TNFα-treated cells, and inhibitors of caspase-3, -9, or -10 effectively blocked apoptosis (Supplementary Figures 2 and 3A). These results confirmed the apoptotic nature of cell death and suggested that receptor-mediated death signals were amplified through cytochrome c release, a process mediated by proapoptotic Bcl2 family proteins such as Bax (Cory and Adams, 2002). Indeed, apoptosis requires activation of an initiator caspase, and Bax activation and cytochrome c release were observed in CCN1/TNFα-treated cells (Supplementary Figure 3B). Whereas caspase-10 inhibitor blocked apoptosis in human fibroblasts, caspase-8 inhibitor or knockdown of caspase-8 by siRNA did not have any effect; however, caspase-8 inhibitor blocked CCN1/TNFα-induced apoptosis in mouse fibroblasts, which lack caspase-10 (Supplementary Figure 3C and D).
Receptors mediating CCN1/TNFα-induced apoptosis
TNFα binds to TNFα type 1 (TNFR1) and type 2 receptors (TNFR2). TNFR1 contains in its cytoplasmic tail a death domain (DD), which recruits the adaptors TRADD and FADD to activate procaspases-8 and -10, whereas TNFR2 lacks DD and its role in apoptosis appears auxiliary (Wajant et al, 2003). Consistently, pre-incubation of cells with a monoclonal antibody (mAb) against TNFR1 effectively blocked apoptosis (Figure 2A). TNFR2 is preferentially activated by the membrane-bound form of TNFα and does not play a role under the current experimental condition (Figure 2A) (Grell et al, 1995).
Figure 2.
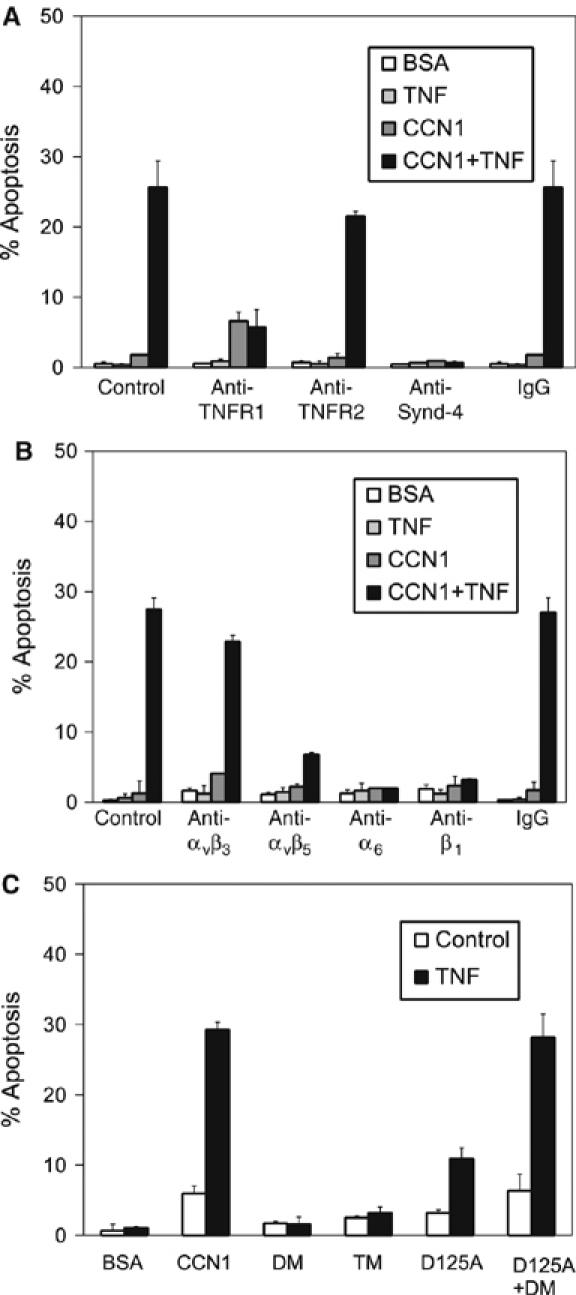
Receptors required for CCN1/TNFα-induced apoptosis. Primary HSFs were subjected to various treatments as described below, and then treated with soluble CCN1, TNFα, or both for 4.5 h before scoring for apoptosis. (A) HSFs were pre-incubated with 100 μg/ml mAbs that neutralize TNFRI, TNFRII, or with anti-syndecan-4 polyclonal antibodies (100 μg/ml), or normal rabbit IgG for 30 min before treatment with CCN1 and TNFα. (B) Cells were incubated with various inhibitory mAbs (50 μg/ml) before treatment with CCN1 and/or TNFα. Antibodies used included GoH3 (anti-integrin α6), P5D2 (anti-β1), LM609 (anti-αvβ3), P1F6 (anti-αvβ5), and normal mouse IgG. (C) HSFs were treated with CCN1 or the mutants DM, TM, or D125A (4 μg/ml each), or a combination of DM and D125A, either with or without TNFα.
CCN proteins are ligands of integrins, which function as receptors for ECM proteins and regulate diverse cellular processes (Hynes, 2002). In fibroblasts, CCN1 binds to and acts through α6β1-HSPGs, αvβ5, and αvβ3, which mediate the promotion of cell adhesion, migration, and proliferation, respectively (Grzeszkiewicz et al, 2001). Inhibitory mAb against αvβ5 (P1F6) blocked apoptosis substantially, whereas anti-αvβ3 mAb (LM609) had minimal effect (Figure 2B). Thus, αvβ5 is critical for CCN1/TNFα-mediated apoptosis, and αvβ3 plays a relatively minor role. Furthermore, mAbs against α6 (GoH3) or β1 (P5D2) both abrogated CCN1/TNFα-induced apoptosis (Figure 2B), indicating the involvement of α6β1. The presence of soluble heparin or treatment of cells with heparinase also obliterated apoptosis (data not shown), consistent with requirement of cell surface HSPGs. Among HSPGs expressed in fibroblasts, syndecan-4 is uniquely co-localized with integrins in focal adhesions and promotes cell adhesion and spreading (Woods and Couchman, 2001). Pre-incubation of fibroblasts with anti-syndecan-4 antibodies obliterated apoptosis, whereas control IgG had no effect (Figure 2A), implicating syndecan-4 as the cell surface HSPG critical for CCN1/TNFα-induced cytotoxicity. These results indicate the involvement of integrins αvβ5, α6β1, and syndecan-4 in CCN1/TNFα-induced apoptosis.
We have previously identified the CCN1-binding sites for integrins αv, α6β1, and HSPGs, and created full-length mutant proteins disrupted in these sites (Supplementary Figure 1). These include DM (disrupted two α6β1-HSPG-binding sites in domain IV) and TM (combined mutations in DM and the T1-binding site for α6β1 in domain III), which are defective in α6β1-HSPG-dependent activities but are fully active in αvβ3-mediated functions (Leu et al, 2004). DM and TM were completely unable to induce apoptosis with TNFα, reinforcing the conclusion that direct binding of CCN1 to α6β1-HSPGs is crucial for this activity (Figure 2C). D125A, a single a.a. substitution CCN1 mutant that is disrupted in the αv-binding site but retains all α6β1-HSPG-mediated functions (Chen et al, 2004), was also substantially impaired in apoptotic activity (Figure 2C). Interestingly, a combination of DM and D125A reconstituted full apoptotic activity. These results show that direct interaction of CCN1 with both αv integrins and α6β1-HSPG is essential for induction of apoptosis with TNFα, but engagement of these two receptor systems need not occur through the same CCN1 molecule.
CCN1/TNFα induce apoptosis independent of de novo protein synthesis or NFκB signaling
TNFα activates NFκB, which induces transcription of genes encoding antiapoptotic factors such as antioxidant proteins, caspase inhibitors, and antiapoptotic members of the Bcl2 family. Thus, blockade of de novo protein synthesis or NFκB signaling is necessary for TNFα to induce apoptosis (Karin and Lin, 2002). However, CCN1 did not affect the rate of protein synthesis, either alone or in combination with TNFα (Figure 3A), and treatment of cells with cycloheximide (CHX) did not diminish the apoptotic effects of CCN1 with TNFα (Figure 3B). Furthermore, a combination of CCN1 with TNFα induced >2-fold higher apoptotic index (>25%) than achieved with CHX and TNFα (∼12%). Thus, CCN1 enables TNFα to induce a greater degree of cell death than CHX, without requiring de novo protein synthesis. To test whether CCN1 modulates NFκB signaling, we monitored the phosphorylation of p65 NFκB and NFκB-dependent transcription in CCN1-treated fibroblasts. As expected, TNFα induced rapid and pronounced p65 phosphorylation within 15 min (Figure 3C), and enhanced NFκB-dependent transcription by ∼6-fold as judged by luciferase activity in cells transiently transfected with a NFκB-luciferase reporter construct (Figure 3D). CCN1 had no effect on p65 phosphorylation or NFκB-dependent transcription either alone or in combination with TNFα. Together, these results show that CCN1 does not promote TNFα-induced apoptosis through the established paradigm of blocking de novo protein synthesis or NFκB signaling, indicating involvement of a distinct pathway.
Figure 3.
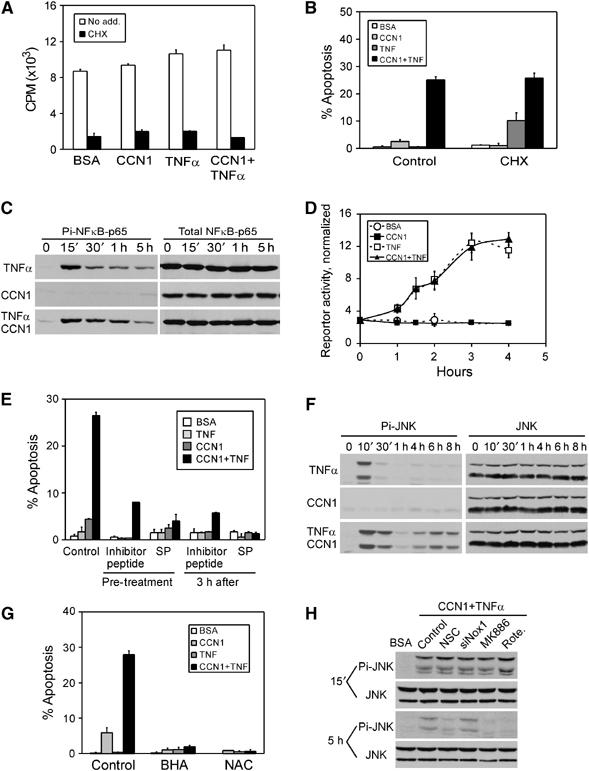
CCN1/TNFα-induced apoptosis is independent of NFκB signaling but completely dependent on ROS accumulation and the consequent biphasic JNK1/2 activation. HSFs were subjected to various treatments as described, and incubated with CCN1, TNFα, CHX, or a combination as indicated. (A) Fibroblasts were incubated in serum-free medium containing 1 μCi/ml of 35S-methionine together with CCN1 and/or TNFα for 4 h, and incorporated radioactivity was measured as acid precipitable counts. CHX was included as control. (B) HSFs were pre-incubated with CHX for 30 min, and then treated for 4 h with CCN1, with or without TNFα. Apoptotic cells were counted after DAPI staining. (C) HSFs were incubated with CCN1 and/or TNFα for various times as indicated, and cell lysates were resolved on SDS–PAGE followed by immunoblotting with phospho-specific antibodies against S536 of NFκB-p65. Blots were stripped and re-probed with antibodies against total NFκB-p65. (D) NFκB-dependent transcription was assessed by transient transfection with a luciferase reporter construct driven by an NFκB-responsive sequence (Takada et al, 2004). Transfected cells were treated with CCN1 and/or TNFα for the indicated times, and luciferase activity in the cell lysates determined. The activities were normalized against transfection efficiency controls. (E) HSFs were pre-incubated for 30 min with either a cell-permeable JNK-inhibitory peptide (50 μM) or SP600125 (25 μM), and then treated with CCN1 and/or TNFα for 4 h before scoring for apoptosis. Where indicated, JNK inhibitors were added 3 h after CCN1/TNFα treatment. (F) HSFs were treated with CCN1 and/or TNFα for various times (10 min to 8 h) as indicated, and cell lysates were electrophoresed and immunoblotted with antibodies against dually phosphorylated JNK1/2 (T183/Y185). Blots were stripped and re-probed with antibodies recognizing total JNK1/2. (G) Cells were pre-incubated for 30 min with 10 mM NAC or 0.4 mM BHA and then treated with CCN1 and/or TNFα for 4 h before scoring for apoptosis. (H) Cells were either transfected with Nox1 siRNA for 48 h to downregulate Nox1, or pre-incubated for 30 min with Rac1 inhibitor NSC23766 (0.4 mM), MK886 (10 μM), or rotenone (10 μM), before treatment with CCN1/TNFα for 15 min or 5 h (see Figure 5 for further details). Cell lysates were resolved on SDS–PAGE and immunoblotted with antibodies against dually phosphorylated and total JNK1/2.
ROS-dependent biphasic JNK activation is required for CCN1/TNFα-induced apoptosis
JNK activation by TNFα is normally modest and transient, which is insufficient for apoptosis, due to the action of MAPK phosphatases (MKPs) that inactivate JNK. When NFκB-dependent signaling is inhibited and thus antioxidant proteins are not induced, TNFα-induced ROS is maintained at a high level. This high level of ROS, in turn, inactivates MKPs by cysteine oxidation at their active sites and leads to prolonged JNK activation, resulting in cell death (Sakon et al, 2003; Kamata et al, 2005). Thus, we tested whether ROS accumulation and JNK activation are necessary for CCN1/TNFα-induced apoptosis. Pretreatment of HSFs with either a cell-permeable JNK inhibitory peptide derived from JIP-1, or SP600125, which competitively inhibits ATP-JNK binding, effectively blocked CCN1/TNFα-induced apoptosis (Figure 3E), indicating that JNK activity is required for this process. As expected, TNFα rapidly induced maximal JNK phosphorylation at T183 and Y185 within 10 min, with phosphorylation declining to background undetectable level 1 h after treatment (Figure 3F). By contrast, CCN1 alone did not activate JNK, but leads to a second phase of JNK activation in the presence of TNFα 4–8 h after stimulation, concomitant with cell death. Remarkably, both the JNK inhibitory peptide and SP600125 effectively blocked apoptosis even when added 3 h after CCN1/TNFα addition to cells (Figure 3E). This result indicates that the first wave of JNK activation, which peaked and declined to undetectable level within 1 h, is not sufficient for CCN1/TNFα-induced apoptosis. Instead, the second wave of JNK activation induced by the combination of CCN1 and TNFα is necessary for cell death. The ROS scavengers butylated hydroxyanisole (BHA) and N-acetyl-cysteine (NAC) both blocked apoptosis completely (Figure 3G), suggesting that ROS may be critical for the JNK activation essential for apoptosis. Furthermore, both BHA and NAC (data not shown), as well as inhibitors of specific cellular sources of ROS critical for apoptosis (see below), blocked the second wave of JNK activation but not the first, which is CCN1 independent (Figure 3H). Thus, the second phase of JNK activation occurring >4 h after CCN1/TNFα stimulation is both required for apoptosis and dependent on ROS.
CCN1 induces ROS accumulation through a Rac1- and 5-lipoxygenase-dependent mechanism
TNFα is known to induce ROS accumulation, which is critical for its cytotoxicity (Sakon et al, 2003; Shakibaei et al, 2005). Surprisingly, we found that CCN1 alone can also induce a dramatic increase in intracellular level of H2O2, substantially higher than that induced by TNFα alone (Figure 4A). Although TNFα signaling attenuated the level of ROS induced by CCN1, apparently through NFκB-induced antioxidant proteins (Sakon et al, 2003; Pham et al, 2004), CCN1 and TNFα together induced significantly higher ROS levels than TNFα alone, particularly at 4 h post-treatment when apoptosis was notable. This level of ROS induced by CCN1 even in the presence of NFκB signaling is apparently sufficient for the ROS-dependent reactivation of JNK necessary for apoptosis (Figure 3E–H). Antibodies against integrin αvβ5, α6, or syndecan-4 all blocked CCN1-induced ROS accumulation completely, whereas antibodies against αvβ3 had little effect (Figure 4B), indicating that combinatorial engagement of all three receptors of CCN1 (αvβ5, α6β1, and syndecan-4) necessary for apoptosis is required to induce ROS. Consistently, CCN1 mutants defective for binding α6β1-HSPGs (DM and TM) or αv integrins (D125A) were unable to induce ROS or apoptosis, and the combination of DM and D125A reconstituted both ROS induction and apoptotic activity (Figures 2C and 4C).
Figure 4.
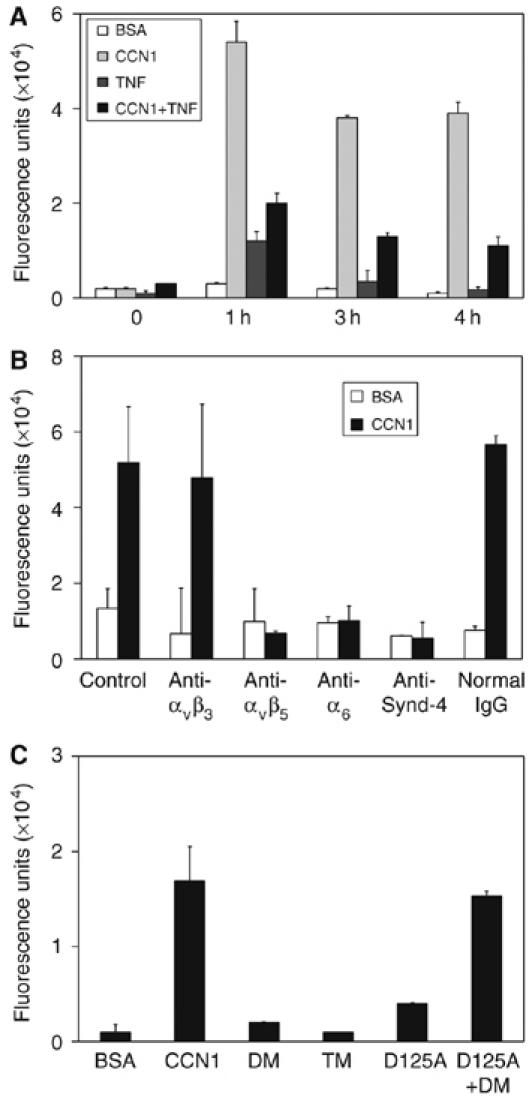
CCN1 induces ROS through its apoptotic receptors. HSFs were cultured on glass cover slips and incubated with CCN1 and/or TNFα before ROS detection by fluorescent microscopy after loading with H2DCF-DA (5 μM in PBS). Ten randomly selected high-power fields were photographed for each sample, and the average fluorescence intensity per cell is presented in arbitrary units. (A) Cells were treated with CCN1 and/or TNFα for various times indicated. (B) Cells were pre-incubated (30 min) with 50 μg/ml each of the following antibodies before being treated with CCN1 for 2 h and assayed for ROS accumulation as above: P1F6 (anti-αvβ5), LM609 (anti-αvβ3), GoH3 (anti-α6), rabbit polyclonal anti-syndecan-4 antibodies, and normal IgG as control. (C) Cells were treated with wild-type CCN1, DM, TM, D125A, or a combination of DM and D125A for 2 h.
Major cellular sources of ROS associated with apoptosis include NADPH oxidase (Nox), 5-lipoxygenase, and the mitochondria. The small GTPase Rac1, which is activated in HSFs upon integrin-mediated cell adhesion to CCN1 (Chen et al, 2001a), has been linked to activation of Nox and 5-lipoxygenase, as well as mitochondrial ROS production in fibroblasts (Werner and Werb, 2002; Chiarugi et al, 2003; Hordijk, 2006). Silencing of Rac1 by siRNA abrogated both CCN1- and TNFα-induced ROS accumulation and blocked apoptosis induced by either CCN1/TNFα or TNFα in the presence of CHX (Figure 5A and B). Likewise, the Rac1 inhibitor NSC23766, which blocks Rac1 interaction with its guanine exchange factors, also annihilated CCN1-dependent ROS accumulation and apoptosis, confirming the requirement of Rac1 for both CCN1/TNFα and TNFα/CHX-induced apoptosis (Supplementary Figure 4A,C). Nox1, a homolog of the gp91phox/Nox2 found in neutrophils, is expressed in smooth muscle cells and fibroblasts (Hordijk, 2006) (Figure 5F). Interestingly, knockdown of Nox1 by siRNA only partially inhibited CCN1-induced ROS accumulation and did not block CCN1/TNFα-induced apoptosis, but efficiently blocked TNFα-induced ROS generation and TNFα/CHX-induced apoptosis (Figure 5A and B). Similar results were also observed with the Nox inhibitor apocynin, which prevents assembly of the Nox enzyme complex (Supplementary Figure 4). Thus, Nox is dispensable for CCN1-induced ROS accumulation and CCN1/TNFα-induced apoptosis, but is required for TNFα/CHX-induced ROS and apoptosis. By contrast, MK886, which blocks the arachidonic acid transfer protein FLAP from delivering substrate to 5-lipoxygenase, decimated CCN1-dependent ROS accumulation and CCN1/TNFα-induced apoptosis but not TNFα/CHX-induced ROS or apoptosis (Figure 5C and D). The 5-lipoxygenase inhibitor NDGA also blocked CCN1-dependent ROS generation and apoptosis, further supporting a role for 5-lipoxygenase in these processes (Supplementary Figure 4A and C). Another important source of ROS is the mitochondrion. Rotenone, a cell-permeable toxin that blocks electron transport in complex I of the mitochondrial respiratory chain, inhibited CCN1- and TNFα-dependent ROS accumulation and apoptosis induced by CCN1/TNFα and TNFα/CHX (Figure 5C and D). Mitochondrial complex III inhibitors, including stigmatellin and myxothiazol, also blocked CCN1/TNFα and TNFα/CHX-induced apoptosis (Supplementary Figure 4C and D). CCN1/TNFα-induced ROS is required for the second phase of JNK activation necessary for cell death, as inhibitors of Rac1 (NSC23766), 5-lipoxygenase (MK886), or mitochondrial complex I (rotenone) each blocked the second phase of JNK activation but not the first, whereas Nox1 siRNA had no effect (Figure 3H). Together, these results show that CCN1 induces ROS accumulation, second phase JNK activation, and the consequent apoptosis in the presence of TNFα through a Rac1-dependent mechanism via 5-lipoxygenase and the mitochondria, whereas Nox1 is dispensable. By contrast, TNFα/CHX-induced apoptosis requires ROS generated through Nox1 and the mitochondria, but 5-lipoxygenase is not required in this system.
Figure 5.
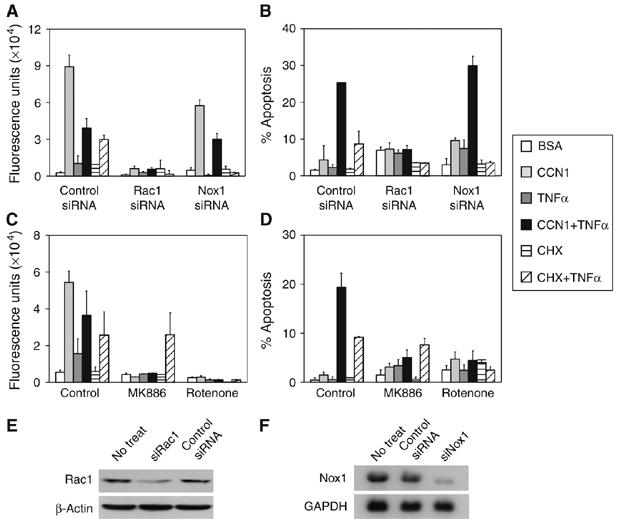
Induction of ROS accumulation by CCN1 and TNFα through distinct cellular sources and the requirement of ROS for apoptosis. HSFs were transfected with control siRNA (mixture of four irrelevant sequences), Rac1 siRNA, or Nox1 siRNA, or pretreated with the 5-lipoxygenase inhibitor MK886 (10 μM) or the mitochondrial complex I inhibitor rotenone (10 μM). Cells were then incubated with CCN1, CHX, and/or TNFα for 1 h before ROS detection or 4.5 h before apoptosis assays. The effects of siRNAs and inhibitors on ROS accumulation (A, C), and apoptosis (B, D) are shown. The efficacies of Rac1 and Nox1 siRNAs in transfected cells are shown by immunoblotting total cell lysates with anti-Rac1 and β-actin antibodies (E) or by RNA blotting probed with 32P-labeled human Nox1 (revealing the 2.0 kb Nox1 mRNA) and GAPDH cDNA (F).
Allelic replacement of Ccn1 with an apoptosis-defective mutant blunts TNFα-mediated apoptosis in vivo
To determine whether the apoptotic synergism between CCN1 and TNFα occurs in a physiological context, we constructed mutant mice with the endogenous Ccn1 genomic locus replaced with a mutant allele that encodes DM (Leu et al, 2004), a CCN1 mutant completely defective in apoptotic synergism with TNFα (Figure 2C). Knock in of the mutant allele was confirmed by Southern blotting, PCR analysis, and the presence of a diagnostic SphI site (Figure 6A-C). As DM is defective for binding α6β1-HSPGs (Chen et al, 2000), the mutant CCN1 produced in MEFs from Ccn1dm/dm mice was unable to bind heparin, whereas CCN1 from the wild-type littermate bound heparin with high affinity (Figure 6D). In contrast to the embryonic lethality of Ccn1-null mice (Mo et al, 2002), Ccn1dm/dm mice are viable, fertile, and exhibit no apparent abnormalities, indicating that the Ccn1dm allele is biologically active and does not significantly impair CCN1 function in development.
Figure 6.
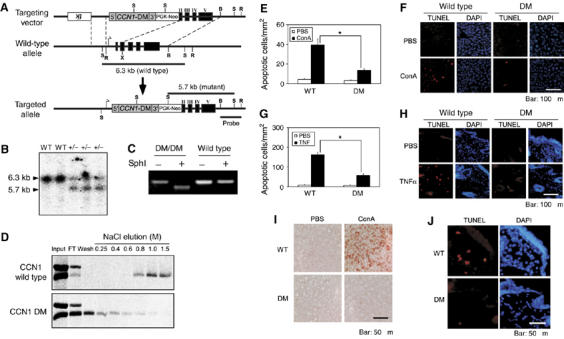
Generation of Ccn1dm/dm mice and blunted TNFα-mediated apoptosis in vivo. (A) A gene targeting construct replaced the EcoRI–XmaI fragment of Ccn1 with a cDNA encoding Ccn1dm. The recombined allele maintained the Ccn1 promoter and transcription start site and expresses the Ccn1dm cDNA that preserved the 5′ and 3′ untranslated sequences. Thymidine kinase (tk) and PGK-neo were used as selectable markers in homologous recombination in ES cells. B, BamHI; X, XmaI; R, EcoRI; S, SphI. (B) DNA samples isolated from wild-type (WT) or Ccn1dm/Ccn1 mice (+/−) were digested with SphI and probed with a BamHI/EcoRI fragment, yielding 6.3 (wild-type) and 5.7 (targeted) kb bands illustrated in (A). (C) Total RNA was isolated from MEFs, reverse-transcribed and the Ccn1 sequence amplified by PCR. The Ccn1dm cDNA contains an engineered diagnostic Sph1 site, which is absent in the wild-type sequence. (D) MEFs from Ccn1dm/dm mice and their wild-type littermates were serum-stimulated to induce synthesis of CCN1 while being labeled with 35S-cysteine. Total cell lysates were passed through a heparin-sepharose column, and eluted with buffer containing varying concentrations of NaCl as indicated. Eluted proteins were immunoprecipitated with anti-CCN1 antibodies, resolved on SDS–PAGE and exposed to X-ray film. (E) ConA (20 mg/kg body weight) was delivered by tail-vein injection, and liver apoptosis analyzed 8 h thereafter by TUNEL assay (labeled with rhodamine). Numbers of apoptotic cells were counted in three randomly chosen fields and expressed as cells/1 mm2 of tissue. (WT, n=5; Ccn1dm/dm, n=8; *P<0.001). Histological sections are shown in (F). (G) Apoptosis was induced by subcutaneous injection of TNFα (400 ng in 50 μl). After 8 h, skin tissue from injection sites was collected, processed, and subjected to TUNEL assay. Numbers of apoptotic cells were counted as above. (WT, n=4; Ccn1dm/dm, n=7; *P<0.001). Histological sections are shown in (H), with a higher magnification view shown in (J). (I) Liver tissue of ConA or PBS-treated WT or Ccn1dm/dm mice were stained with anti-8-OHdG antibodies.
A widely used and well-documented model of TNFα-mediated apoptosis in vivo is the intravenous administration of concanavalin A (ConA), which causes pan-T-cell activation in the liver and natural killer T cells-dependent synthesis of TNFα, resulting in hepatitis and TNFα-dependent hepatocyte apoptosis that can be obliterated by treatment with anti-TNFα antibodies or genetic ablation of TNFR1 and TNFR2 (Trautwein et al, 1998; Wolf et al, 2001). Remarkably, ConA-induced apoptosis was suppressed by >60% in Ccn1dm/dm mice compared to wild-type mice, showing that CCN1 is important for ConA-induced apoptosis in vivo (Figure 6E and F). The numbers of infiltrated CD3+ T lymphocytes in ConA-treated wild-type and mutant mice were similar, indicating a similar T-cell response (data not shown). Antibodies specific to 8-hydroxy-2′-deoxyguanosine (8-OHdG), a marker of oxidative DNA damage (Maeda et al, 2005), detected a much higher level of reactivity in livers of ConA-induced WT mice than those of Ccn1dm/dm mice (Figure 6I). These results indicate that CCN1 is an important contributor to oxidative DNA damage in ConA-treated livers, and suggest that reduced apoptosis in ConA-treated Ccn1dm/dm mice is correlated with impaired ROS generation. To confirm further that CCN1 can synergize with TNFα in vivo, skin fibroblast apoptosis following subcutaneous injection of purified soluble TNFα was examined (Figure 6G, H and J). Apoptosis in Ccn1dm/dm mice was also reduced by >60% compared to wild-type mice. Together, these results show that TNFα-mediated apoptosis is severely blunted in mice expressing an apoptosis-defective form of CCN1, thus establishing CCN1/TNFα synergism as an important apoptotic pathway in vivo.
Discussion
The present study uncovers a novel and unexpected proapoptotic synergism between TNFα and the CCN family of matricellular proteins. TNFα is an important regulator of inflammation and immunity, whereas CCN proteins modulate angiogenesis, matrix remodeling, and injury repair. As the cytotoxicity of TNFα is contextual and requires blockade of NFκB signaling in vitro, how it occurs in vivo is not well understood. Remarkably, CCNs can unmask the cytotoxicity of TNFα and convert it from a cytokine that normally promotes cell proliferation in fibroblasts into one that induces rapid apoptosis. These findings indicate that the cytotoxicity of TNFα may be regulated by the ECM microenvironment, and identify CCNs as important modulators of TNFα cytotoxicity.
A striking finding of this study is that mice with allelic replacement of Ccn1 with an apoptosis-defective mutant (Ccn1dm) are significantly resistance to TNFα-mediated apoptosis, thus establishing the physiological significance of CCN1/TNFα synergism in apoptosis. Both ConA-induced hepatocyte apoptosis and skin fibroblast cell death following subcutaneous injection of TNFα were greatly diminished in Ccn1dm/dm mice (Figure 6). IFN-γ has been shown to facilitate TNFα-induced apoptosis in certain cell lines by inhibiting NFκB-dependent transcription (Suk et al, 2001). Our results present a distinct paradigm whereby the matrix protein CCN1 controls TNFα-mediated apoptosis through integrin signaling, overriding the antiapoptotic effects of NFκB without inhibiting its transcriptional activity. As CCN proteins and TNFα are dynamically regulated during tissue injury and inflammation (Chen et al, 2001b; Mori et al, 2002), their coexpression at critical stages may provide the environmental context that dictates whether TNFα acts as a prosurvival or prodeath cytokine in a cell type-dependent manner.
Mechanistically, CCN1 initiates this apoptotic pathway by concomitant engagement of αvβ5, α6β1, and syndecan-4, leading to a high level of ROS accumulation that far exceeds that induced by TNFα alone (Figure 4A). This high level of CCN1-induced ROS overrides the effects of NFκB, leading to JNK reactivation and apoptosis (Figures 3 and 5). However, ROS accumulation per se is not sufficient for apoptosis, as H2O2 added exogenously in the presence of TNFα induces necrotic cell death rather than apoptosis (data not shown), suggesting that the nature, quality, and quantity of ROS may influence the biological outcome. Although TNFα can induce cell death through apoptosis and necrosis (Aggarwal, 2003; Wajant et al, 2003), CCN1/TNFα-induced cell death occurs through apoptotic mechanisms, and we have detected only minimal background level of necrosis in HSFs exposed to CCN1/TNFα for up to 24 h by flow cytometry with annexin-V and propidium iodide staining (data not shown). Both CCN1 and TNFα induce ROS accumulation through a Rac1-dependent mechanism involving the mitochondria (Werner and Werb, 2002) (Figure 5). However, CCN1/TNFα-induced ROS accumulation requires 5-lipoxygenase but Nox is not essential, whereas CHX/TNFα-induced ROS accumulation requires Nox but not 5-lipoxygenase, underscoring a distinct mechanism of ROS generation induced by CCN1 and TNFα (Figure 5). As CCN1/TNFα induced a comparable amount of ROS as compared to TNFα/CHX but a higher level of cell death (Figure 5), this difference in cellular sources of ROS may potentially contribute to the relative efficacy of apoptosis induced by CCN1/TNFα. As Nox and 5-lipoxygenase are localized in the plasma and nuclear membranes, respectively, the subcellular localization of ROS they generate may influence their roles in apoptosis (Ushio-Fukai, 2006).
The specific role of ROS in CCN1/TNFα-induced apoptosis appears to be the reactivation of JNK (Figure 3E-H), which is required for the cytotoxicity of TNFα (Varfolomeev and Ashkenazi, 2004) (Figure 7). It has been observed that transient and modest JNK activation promotes cell proliferation, whereas prolonged and robust JNK activation promotes cell death (Kamata et al, 2005). JNK has been shown to mediate apoptosis either by inducing the cleavage and activation of the BH3-only protein Bid (Deng et al, 2003), or by promoting the degradation of FLIP, an inhibitor of caspase-8/10 activation, through phosphorylation of the ubiquitin ligase ITCH (Chang et al, 2006) (Figure 7). Prolonged JNK activation is needed to degrade FLIP from the pre-existing intracellular pool and from de novo synthesis induced by NFκB. However, TNFα induces transient JNK activation that is insufficient for apoptosis (Figure 3F), as JNK is rapidly inactivated by MKPs, some of which are induced by NFκB. Inhibition of NFκB results in elevated ROS accumulation and sustained JNK activation, as NFκB induces MKPs as well as antioxidant proteins such as Mn2+ superoxide dismutase and ferritin heavy chain. Sustained ROS accumulation has been shown to inactivate MKPs by cysteine oxidation at their active sites, allowing JNK activation to be prolonged (Kamata et al, 2005). The high level of ROS induced by CCN1 triggers a second wave of JNK activation (Figure 3H), thus bypassing the need to inhibit NFκB signaling to achieve sustained JNK activation necessary for apoptosis. It is interesting to note that JNK activation in TNFα-stimulated MEFs deficient for NFκB p65 also appears biphasic (Sakon et al, 2003), although the two waves of JNK activation occurs with more compressed kinetics.
Figure 7.
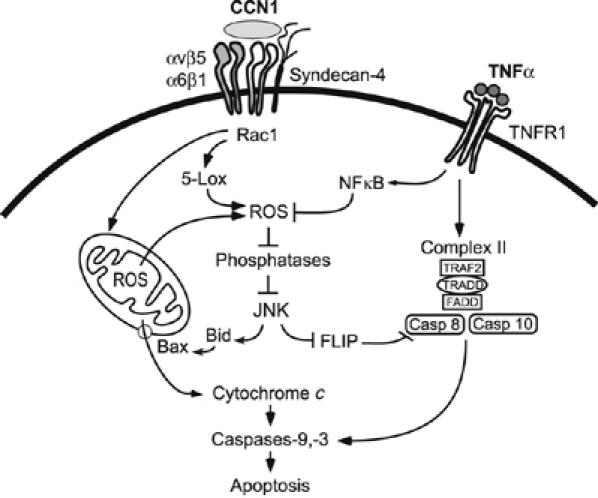
Model for CCN1/TNFα synergism. Matrix signaling through CCN1 can override the antiapoptotic effects of NFκB, allowing TNFα to induce apoptosis (Figures 1, 2 and 3). Data presented in this paper support a model in which CCN1 binding to its receptors (integrins αvβ5, α6β1, and syndecan-4) results in elevated and prolonged ROS accumulation that is dependent on Rac1, 5-lipoxygenase, and mitochondria, leading to the biphasic activation of JNK in the presence of TNFα (Figures 3, 4 and 5). Others have shown that ROS inactivates phosphatases (Kamata et al, 2005), leading to sustained activation of JNK, which in turn triggers the degradation of c-FLIP (Chang et al, 2006) to allow the activation of caspases-8 by the TNFα-dependent cytoplasmic complex II (Micheau and Tschopp, 2003). Signals of caspases-8/10 are amplified through the mitochondria via Bax-mediated cytochrome c release and activation of caspase-9 and -3, leading to apoptosis.
CCNs appear unique among matrix proteins in their apoptotic synergism with TNFα (Figure 1). While ROS is also generated in a Rac1-dependent manner upon integrin-mediated cell adhesion to FN, there is no mitochondrial involvement in this instance (Chiarugi et al, 2003). Thus, CCN1 induces ROS generation differently from FN. The requirement of multiple CCN1 receptors (αvβ5, α6β1, and syndecan-4) for CCN1/TNFα-induced ROS accumulation and apoptosis may serve to specify the target cells with relative precision, identifying the specific cell types for elimination. For example, CCN1 is prosurvival in endothelial cells and does not induce apoptosis even in the presence of TNFα (data not shown). Of note, CCN1 and CCN2 on their own have either prosurvival or proapoptotic effects in specific cell types, and CCN1 can induce apoptosis in the p21-defective Rat1a fibroblasts through a p53-dependent mechanism while promoting survival in activated endothelial cells (Todorovic et al, 2005).
Our current understanding of how CCN1 synergizes with TNFα is summarized in Figure 7. Where examined, CCN2 also acts through ROS, JNK, caspases, and the same receptors as CCN1, suggesting a similar mechanism of action (Supplementary Figure 5). The apoptotic synergism observed between CCNs and TNFα raises the intriguing possibility that other members of these two multifunctional protein families may also cooperate, and their synergism may extend beyond apoptosis. The notion that CCN matricellular proteins can provide the environmental context that dictates TNFα cytotoxicity, and the possibility that the matrix environment may profoundly affect the diverse actions of the larger family of TNF cytokines, clearly merit further investigation.
Materials and methods
Cell culture
Primary normal HSFs from newborns were from American Type Culture Collection, maintained in Iscove's modified Dulbecco's Medium (IMDM; Invitrogen) containing 10% fetal bovine serum (FBS; Hyclone) and used before passage 6. Cells were serum-starved overnight before experiments in IMDM containing 0.1% BSA. Primary synovial fibroblasts from joints of normal adult mice were a generous gift from Dr John Varga (Northwestern University) and maintained in Eagle's minimum essential medium (EMEM; Cambrex) supplemented with Earl's balanced salt solution, 2 mM glutamine, 1 mM pyruvate, 0.1 mM non-essential amino acids, and 10% FBS. Cells were used before passage 5.
Proteins, reagents, antibodies
Recombinant CCN1, CCN2, and CCN3 were produced using a baculovirus expression system in insect cells and purified by ion-exchange (Chen et al, 2001b) or immuno-affinity (Leu et al, 2004) chromatography. DM and TM were purified using anti-Flag immuno-affinity chromatography (Leu et al, 2004), whereas D125A was purified using Ni2+columns via its histidine tag (Chen et al, 2004). Vendors of reagents are listed in Supplementary Materials and methods. In all experiments, CCN1 was used at 4 μg/ml, TNFα at 10 ng/ml, and CHX at 10 μg/ml unless otherwise indicated.
Apoptosis assays
For DAPI staining, fibroblasts were cultured in 24-well plates (105 cells per well), serum-starved and treated with apoptosis-inducing factors, then fixed with 10% formalin at room temperature (RT) overnight. After washing, cells were incubated with DAPI at 1 μg/ml in PBS for 5 min, and 2 drops of Fluoromount-G were added per well. Using a fluorescent microscope, 10 randomly selected high-power fields (∼60 cells per field) per well were counted for both apoptotic (condensed nuclei) and nonapoptotic cells. TUNEL assay was performed using the ApopTag Red detection kit (Chemicon, Inc.) following manufacturer's protocol. Samples were counterstained with DAPI before mounting.
NFκB-dependent transcription activity assay
Reporter constructs for NFκB activity (pNFκB-Luc) and transfection efficiency (pRL-CMV) were from Promega (Madison, WI), and transfected using Lipofectamine-2000 (Invitrogen Corp., Carlsbad, CA) as described by the vendor. Luciferase activity was assayed using a dual-luciferase reporter system (Promega).
Measurement of ROS
Intracellular peroxide was measured using the cell-permeable dye, H2DCF-DA, which becomes fluorescent upon oxidation by intracellular peroxide/hydroperoxides (Ohba et al, 1994). Normal HSFs were plated (105 cells per well) on glass cover slips in 24-well plates and cultured in FBS-containing media. Cells were then serum-starved in phenol red-free medium overnight before apoptotic treatment as described above. For experiments using blocking antibodies, cells were incubated with 50 μg/ml of antibodies or control normal IgG for 30 min before factor treatment. Media were replaced with PBS containing 5 μM H2DCF-DA at the indicated times after stimulation and incubated for 10 min at 37°C. Cover slips were then mounted on slides and imaged by fluorescence microscopy. Ten randomly selected high-power fields per condition were photographed and the integrated densities (60–100 cells per condition) were measured using ImageJ software from NIH.
siRNA
Gene silencing by siRNA against caspase-8, Rac1, and Nox1 employed established sequences and protocols, as detailed in Supplementary data.
Generation of Ccn1dm/dm mice and apoptosis in vivo
Mice with the Ccn1dm mutation (Chen et al, 2000) were created by replacement of the EcoRI–XmaI fragment of the endogenous Ccn1 genomic locus, which contains the transcription start site, exon I and half of exon II, with a DNA cassette containing the Ccn1 promoter, a cDNA encoding Ccn1dm and the selection marker PGK-Neo (Figure 6A). The 5′ and 3′ untranslated regions of Ccn1 were maintained in the cDNA. Recombinants with the allelic replacement were selected in svJ129 ES cells, and implanted into blastocysts of C57/BL6 mice as described (Mo et al, 2002). Germline chimeras were identified by genomic blot and PCR analysis, and intercrossed to produce Ccn1dm/dm mice (Figure 6A–D). Mice were treated with ConA (20 mg/kg body weight) through tail vein injections and killed 8 h thereafter. Liver was fixed with 3.0% buffered paraformaldehyde and paraffin sections were subjected to TUNEL assay and counterstained with DAPI. For TNFα-induced skin fibroblast apoptosis (Alikhani et al, 2004), each mouse received 0.4 μg soluble TNFα in 50 μl by subcutaneous injection. After 8 h, tissue was collected, processed, and TUNEL assay performed as above.
Supplementary Material
Supplementary Materials and methods
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
We thank John Varga for a gift of mouse synovial fibroblasts, and GR Scott Budinger, V Juric, and N Hay for helpful discussions. JLY was supported by postdoctoral fellowships from the NIH (T32 HL07829) and the American Heart Association. VT was supported by a predoctoral fellowship from the American Heart Association. This work was supported by a grant from the National Cancer Institute (CA46565) to LFL.
References
- Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3: 745–756 [DOI] [PubMed] [Google Scholar]
- Alikhani M, Alikhani Z, Raptis M, Graves DT (2004) TNF-alpha in vivo stimulates apoptosis in fibroblasts through caspase-8 activation and modulates the expression of pro-apoptotic genes. J Cell Physiol 201: 341–348 [DOI] [PubMed] [Google Scholar]
- Battegay EJ, Raines EW, Colbert T, Ross R (1995) TNF-alpha stimulation of fibroblast proliferation. Dependence on platelet-derived growth factor (PDGF) secretion and alteration of PDGF receptor expression. J Immunol 154: 6040–6047 [PubMed] [Google Scholar]
- Bornstein P, Sage EH (2002) Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 14: 608–616 [DOI] [PubMed] [Google Scholar]
- Brigstock DR (2003) The CCN family: a new stimulus package. J Endocrinol 178: 169–175 [DOI] [PubMed] [Google Scholar]
- Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M (2006) The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 124: 601–613 [DOI] [PubMed] [Google Scholar]
- Chen C-C, Chen N, Lau LF (2001a) The angiogenic factors Cyr61 and CTGF induce adhesive signaling in primary human skin fibroblasts. J Biol Chem 276: 10443–10452 [DOI] [PubMed] [Google Scholar]
- Chen C-C, Mo FE, Lau LF (2001b) The angiogenic inducer Cyr61 induces a genetic program for wound healing in human skin fibroblasts. J Biol Chem 276: 47329–47337 [DOI] [PubMed] [Google Scholar]
- Chen N, Chen CC, Lau LF (2000) Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin a6b1 and cell surface heparan sulfate proteoglycans. J Biol Chem 275: 24953–24961 [DOI] [PubMed] [Google Scholar]
- Chen N, Leu S-J, Todorovic V, Lam SCT, Lau LF (2004) Identification of a novel integrin avb3 binding site in CCN1 (CYR61) critical for pro-angiogenic activities in vascular endothelial cells. J Biol Chem 279: 44166–44176 [DOI] [PubMed] [Google Scholar]
- Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, Ramponi G (2003) Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol 161: 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory S, Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2: 647–656 [DOI] [PubMed] [Google Scholar]
- Deng Y, Ren X, Yang L, Lin Y, Wu X (2003) A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 115: 61–70 [DOI] [PubMed] [Google Scholar]
- Doi TS, Marino MW, Takahashi T, Yoshida T, Sakakura T, Old LJ, Obata Y (1999) Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc Natl Acad Sci USA 96: 2994–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Stroh C, Schulze-Osthoff K (2005) Unique and overlapping substrate specificities of caspase-8 and caspase-10. Oncogene 25: 152–159 [DOI] [PubMed] [Google Scholar]
- Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P (1995) The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83: 793–802 [DOI] [PubMed] [Google Scholar]
- Grzeszkiewicz TM, Kirschling DJ, Chen N, Lau LF (2001) CYR61 stimulates human skin fibroblasts migration through integrin avb5 and enhances mitogenesis through integrin avb3, independent of its carboxyl-terminal domain. J Biol Chem 276: 21943–21950 [DOI] [PubMed] [Google Scholar]
- Hehlgans T, Pfeffer K (2005) The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology 115: 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hordijk PL (2006) Regulation of NADPH oxidases: the role of Rac proteins. Circ Res 98: 453–462 [DOI] [PubMed] [Google Scholar]
- Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–687 [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Popoff SN, Safadi FF, Zhao M, Stephenson RC, Yoon BS, Daluiski A, Segarini P, Lyons KM (2003) Connective tissue growth factor is an essential regulator of skeletal development. Development 130: 2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M (2005) Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120: 649–661 [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A (2002) NF-kappaB at the crossroads of life and death. Nat Immunol 3: 221–227 [DOI] [PubMed] [Google Scholar]
- Lau LF, Lam SC (1999) The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res 248: 44–57 [DOI] [PubMed] [Google Scholar]
- Lau LF, Lam SCT (2005) Integrin-mediated CCN functions. In CCN Proteins: a New Family of Cell Growth and Differentiation Regulators, Perbal B, Takigawa M (eds), pp 61–79. London: Imperial College Press [Google Scholar]
- Leu S-J, Chen N, Chen C-C, Todorovic V, Bai T, Juric V, Liu Y, Yan G, Lam SCT, Lau LF (2004) Targeted mutagenesis of the matricellular protein CCN1 (CYR61): selective inactivation of integrin a6b1-heparan sulfate proteoglycan coreceptor-mediated cellular activities. J Biol Chem 279: 44177–44187 [DOI] [PubMed] [Google Scholar]
- Leu S-J, Lam SCT, Lau LF (2002) Proangiogenic activities of CYR61 (CCN1) mediated through integrins avb3 and a6b1 in human umbilical vein endothelial cells. J Biol Chem 277: 46248–46255 [DOI] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104: 487–501 [DOI] [PubMed] [Google Scholar]
- Maeda S, Kamata H, Luo JL, Leffert H, Karin M (2005) IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 121: 977–990 [DOI] [PubMed] [Google Scholar]
- Micheau O, Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190 [DOI] [PubMed] [Google Scholar]
- Mo FE, Lau LF (2006) The matricellular protein CCN1 is essential for cardiac development. Circulat Res 99: 961–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF (2002) CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol 22: 8709–8720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori R, Kondo T, Ohshima T, Ishida Y, Mukaida N (2002) Accelerated wound healing in tumor necrosis factor receptor p55-deficient mice with reduced leukocyte infiltration. FASEB J 16: 963–974 [DOI] [PubMed] [Google Scholar]
- Ohba M, Shibanuma M, Kuroki T, Nose K (1994) Production of hydrogen peroxide by transforming growth factor-beta 1 and its involvement in induction of egr-1 in mouse osteoblastic cells. J Cell Biol 126: 1079–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, Franzoso G (2004) Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 119: 529–542 [DOI] [PubMed] [Google Scholar]
- Planque N, Perbal B (2003) A structural approach to the role of CCN (CYR61/CTGF/NOV) proteins in tumorigenesis. Cancer Cell Int 3: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld ME, Prichard L, Shiojiri N, Fausto N (2000) Prevention of hepatic apoptosis and embryonic lethality in RelA/TNFR-1 double knockout mice. Am J Pathol 156: 997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H (2003) NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J 22: 3898–3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senftleben U, Li ZW, Baud V, Karin M (2001) IKKbeta is essential for protecting T cells from TNFalpha-induced apoptosis. Immunity 14: 217–230 [DOI] [PubMed] [Google Scholar]
- Shakibaei M, Schulze-Tanzil G, Takada Y, Aggarwal BB (2005) Redox regulation of apoptosis by members of the TNF superfamily. Antioxid Redox Signal 7: 482–496 [DOI] [PubMed] [Google Scholar]
- Sugarman BJ, Aggarwal BB, Hass PE, Figari IS, Palladino MA Jr, Shepard HM (1985) Recombinant human tumor necrosis factor-alpha: effects on proliferation of normal and transformed cells in vitro. Science 230: 943–945 [DOI] [PubMed] [Google Scholar]
- Suk K, Chang I, Kim YH, Kim S, Kim JY, Kim H, Lee MS (2001) Interferon gamma (IFNgamma) and tumor necrosis factor alpha synergism in ME-180 cervical cancer cell apoptosis and necrosis. IFNgamma inhibits cytoprotective NF-kappa B through STAT1/IRF-1 pathways. J Biol Chem 276: 13153–13159 [DOI] [PubMed] [Google Scholar]
- Takada Y, Fang X, Jamaluddin MS, Boyd DD, Aggarwal BB (2004) Genetic deletion of glycogen synthase kinase-3beta abrogates activation of IkappaBalpha kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J Biol Chem 279: 39541–39554 [DOI] [PubMed] [Google Scholar]
- Todorovic V, Chen C-C, Hay N, Lau LF (2005) The matrix protein CCN1 (CYR61) induces apoptosis in fibroblasts. J Cell Biol 171: 559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein C, Rakemann T, Brenner DA, Streetz K, Licato L, Manns MP, Tiegs G (1998) Concanavalin A-induced liver cell damage: activation of intracellular pathways triggered by tumor necrosis factor in mice. Gastroenterology 114: 1035–1045 [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M (2006) Localizing NADPH oxidase-derived ROS. Sci STKE 2006: re8. [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Ashkenazi A (2004) Tumor necrosis factor: an apoptosis JuNKie? Cell 116: 491–497 [DOI] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P (2003) Tumor necrosis factor signaling. Cell Death Differ 10: 45–65 [DOI] [PubMed] [Google Scholar]
- Werner E, Werb Z (2002) Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J Cell Biol 158: 357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, Hallmann R, Sass G, Sixt M, Kusters S, Fregien B, Trautwein C, Tiegs G (2001) TNF-alpha-induced expression of adhesion molecules in the liver is under the control of TNFR1 relevance for concanavalin A-induced hepatitis. J Immunol 166: 1300–1307 [DOI] [PubMed] [Google Scholar]
- Woods A, Couchman JR (2001) Syndecan-4 and focal adhesion function. Curr Opin Cell Biol 13: 578–583 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials and methods
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5