

Abstract
Aspergillus fumigatus is the most prevalent airborne filamentous fungal pathogen in humans, causing severe and often fatal invasive infections in immunocompromised patients. Currently available antifungal drugs to treat invasive aspergillosis have limited modes of action, and few are safe and effective. To identify and prioritize antifungal drug targets, we have developed a conditional promoter replacement (CPR) strategy using the nitrogen-regulated A. fumigatus NiiA promoter (pNiiA). The gene essentiality for 35 A. fumigatus genes was directly demonstrated by this pNiiA-CPR strategy from a set of 54 genes representing broad biological functions whose orthologs are confirmed to be essential for growth in Candida albicans and Saccharomyces cerevisiae. Extending this approach, we show that the ERG11 gene family (ERG11A and ERG11B) is essential in A. fumigatus despite neither member being essential individually. In addition, we demonstrate the pNiiA-CPR strategy is suitable for in vivo phenotypic analyses, as a number of conditional mutants, including an ERG11 double mutant (erg11BΔ, pNiiA-ERG11A), failed to establish a terminal infection in an immunocompromised mouse model of systemic aspergillosis. Collectively, the pNiiA-CPR strategy enables a rapid and reliable means to directly identify, phenotypically characterize, and facilitate target-based whole cell assays to screen A. fumigatus essential genes for cognate antifungal inhibitors.
Author Summary
Aspergillus fumigatus is an opportunistic filamentous fungal pathogen of emerging clinical significance. Although virulence factors are seen as potential drug targets, neither genetic analyses nor genomic comparisons have identified genuine virulence factors in A. fumigatus. Essential genes required for fungal growth and viability also serve as potential drug targets, yet few have been described in this pathogen. To begin to catalog essential genes in A. fumigatus, we devised a genetic strategy for creating conditional mutants by promoter replacement of target genes using a nitrogen-regulated promoter. Applying this genetic approach to A. fumigatus genes orthologous to known essential genes of the nonpathogenic yeast, Saccharomyces cerevisiae and Candida albicans, we demonstrate a robust enrichment for identifying essential genes conserved within this pathogen. We show that A. fumigatus conditional mutants can be evaluated according to their terminal phenotypes (e.g., conidial germination, growth, morphology, and cidal versus static consequences) and pathogenesis in a murine model of systemic aspergillosis to prioritize essential genes as novel drug targets suitable for developing broad-spectrum antifungal agents.
Introduction
Aspergillus fumigatus is a ubiquitous soil-dwelling saprophytic fungus that propagates through the prolific production of air-borne conidia [1]. Large-scale genome comparisons have shown that no gene sets are shared exclusively by both Aspergillus fumigatus and any other human pathogenic fungi sequenced to date, such as Candida or Cryptococcus species [2]. Thus, it has been recently suggested that A. fumigatus pathogenesis is based on its saprophytic lifestyle in combination with the immunosuppressed state of the host, rather than from genuine fungal virulence factors [2]. Although A. fumigatus conidia are constantly inhaled and seldom cause serious medical conditions in healthy individuals, immunocompromised patients (e.g., those with HIV infection or AIDS, solid organ and bone marrow transplant recipients, and those receiving chemotherapy) are highly susceptible to invasive aspergillosis, a fatal systemic infection [1,3,4]. Current treatment options for invasive aspergillosis are limited to three classes of antifungal therapeutics: polyenes (amphotericin B and various liposomal formulations), azoles (e.g., fluconazole, voriconazole, itraconazole), and, more recently, semisynthetic echinocandins (e.g., caspofungin and anidulafungin) [4,5]. Despite current therapeutic options, mortality associated with invasive aspergillosis remains high (ranging from 60% to 90%) and more efficacious antifungal drugs with novel mechanisms of action are needed [4,5].
The identification of conserved essential genes required for the growth of fungal pathogens offers an ideal strategy for elucidating novel antifungal drug targets. A comprehensive determination of all the essential genes has been achieved in the nonpathogenic yeast Saccharomyces cerevisiae [6,7]. Extending similar genetic approaches to fungal pathogens has proved difficult due to limited available molecular technologies and to the asexual nature of most medically relevant fungi, preventing the use of classical genetics. Nonetheless, large-scale essential gene identification in Candida albicans has begun through a number of alternative approaches, including antisense-based gene inactivation [8], transposon-based heterozygote screens for hypomorphs [9], homozygote null mutants [10], and a promoter replacement strategy to construct conditional mutants [11].
Large-scale functional analysis and essential gene identification in A. fumigatus have proved more difficult. Although gene disruption methodologies have been adapted to A. fumigatus, they are limited due to the organism's poor efficiency of homologous recombination as well as the inherent inability to study essential genes by such means. A. fumigatus essential genes have been defined using parasexual genetics in which gene essentiality is inferred from the failure to recover haploid segregants carrying a gene knock-out [12,13]. Nevertheless, such an approach is likely unsuitable to systematically identify all possible essential genes due to irregularities in the parasexual cycle of A. fumigatus. Direct demonstration of A. fumigatus gene essentiality and phenotypic analyses, however, may be achieved using molecular genetics strategies including RNA interference or promoter replacement strategies [14,15]. Mouyna et al. (2004) have used RNA interference to produce the predicted phenotypes associated with both a nonessential gene involved in melanin biosynthesis (alb1) and an essential glucan synthase, FKS1 [14]. (Note: In this report, we maintain gene nomenclature for A. fumigatus genes as previously described [e.g., alb1 or FKS1]; in cases where previously uncharacterized A. fumigatus genes are described, standard S. cerevisiae gene nomenclature and provisional gene names are adopted according to their yeast ortholog.) To date, conditional promoter replacement (CPR) strategies applied to A. fumigatus have been restricted to heterologous promoters including the Aspergillus nidulans alcA promoter and Escherichia coli tetracycline-regulated promoter [15,16].
Here we report a CPR strategy to identify A. fumigatus essential genes and to prioritize potential antifungal drug targets. This strategy uses the A. fumigatus NiiA nitrogen-regulatable promoter (pNiiA) to delete and replace the endogenous promoter of selected genes. By applying this strategy to 54 A. fumigatus genes of diverse biological functions whose orthologs are known to be essential for growth in S. cerevisiae and C. albicans, we have identified 35 genes essential for mycelial growth, with many displaying a cidal terminal phenotype. We also demonstrate that the ERG11 gene family is essential in A. fumigatus and that the resulting pNiiA-CPR strains may be used as cell-based whole cell assays to examine target-specific chemical hypersensitivity. Finally, we show that a number of pNiiA-CPR mutants of essential genes fail to establish a terminal aspergillosis infection in a mouse model system. Therefore, both in vitro and in vivo phenotypic analysis of essential genes may be performed. This initial gene set comprises an experimentally validated drug target set that is broadly conserved within fungal pathogens and therefore of significant relevance to antifungal drug discovery.
Results
Construction of A. fumigatus Conditional Mutant by Promoter Replacement
A. fumigatus conditional mutants were constructed by CPR using pNiiA. pNiiA was chosen based on previous work in both A. fumigatus and A. nidulans demonstrating its tight nitrogen-dependent transcriptional regulation [17,18]. Two operationally independent signals control pNiiA expression: an inducer (e.g., nitrate or other secondary nitrogen source) and a repressor (e.g., ammonium or other primary nitrogen source). Expression is achieved solely in the absence of ammonium and the presence of nitrate; however, repression is achieved by the presence of ammonium regardless of the presence of other nitrogen [17,18]. pNiiA-CPR mutants were constructed by homologous recombination-mediated deletion of the endogenous promoter (approximately 250 base pairs of promoter sequence immediately preceding the start codon of the gene) and replacement with an NiiA promoter cassette marked with the pyrG selectable marker (Figure 1A and 1B; see Materials and Methods for details). To test the pNiiA-CPR strategy, TUB1 and MET2 were first selected. TUB1 encodes the broadly conserved and essential protein α-tubulin, and MET2 encodes a homoserine O-acetyltransferase involved in methionine biosynthesis, mutations of which cause methionine auxotrophy. TUB1 and MET2 pNiiA-CPR mutants displayed wild-type mycelial growth on inducing medium (Aspergillus minimal medium [AMM] plus nitrate), but neither mutant was able to grow on repressing medium (AMM plus ammonium, Figure 1C). Furthermore, the A. fumigatus MET2 conditional mutant displayed a tight methionine auxotrophy that was fully suppressed by methionine added to the medium (Figure 2A). pNiiA-CPR conditional mutants of GFA1 and ALR1, whose orthologs are essential in both S. cerevisiae and C. albicans, were also examined (Figure 1C). GFA1, a glutamine-fructose-6-phosphate aminotransferase catalyzing the first step in the chitin biosynthesis pathway, was shown to be essential in A. fumigatus. However, CPR mutants of ALR1 (a metal cation transporter belonging to a gene family in A. fumigatus) failed to display any growth defect under repressing conditions, revealing that ALR1 is nonessential in A. fumigatus (see below).
Figure 1. Outline of the A. fumigatus pNiiA-CPR Strategy.

(A) A cloning vector, pPyrG-pNiiA, was created specifically for the construction of CPR cassettes. This plasmid contains an A. niger pyrG (encoding orotidine-5′-phosphate decarboxylase and required for uridine-uracil prototrophy) as a selection marker [41], as well as an Amp+ selection marker and the A. fumigatus pNiiA conditional promoter. Unique restriction sites have been engineered at either side of the pyrG-pNiiA cassette to facilitate subcloning of flanking sequences.
(B) Schematic overview of the pNiiA-CPR strategy and strain confirmation by PCR genotyping. Promoter replacement cassettes were constructed by inserting approximately 1.5 kb of homologous flanking sequences of the target gene (L-arm and R-arm) into the Not1/Mlu1 and Asc1/Pac1 restriction sites, respectively. After Not1 and Pac1 double digestion, the linearized pNiiA-CPR cassette was introduced into CEA17 strain (pyrG−) by protoplast transformation. As indicated, three sets of primers were used to perform genotypic PCRs to map the expected promoter replacement junctions (L1/L2 for left-arm junction, R1/R2 for right-arm junction) and to confirm the deletion of the native promoter (L1/P).
(C) Phenotype of transformants obtained by the pNiiA-CPR strategy for MET2, TUB1, GFA1, and ALR1. CPR mutants were examined under pNiiA-inducing (AMM plus nitrate) and repressing (AMM plus ammonium) conditions after 36 h at 30 °C.
(D) Growth phenotypes of GFA1, PFS2, ALG7, CDC24, and ALR1 pNiiA-CPR mutants under these standard repressing conditions are scored qualitatively as the following: 4+, essential for cell viability, no growth under repressing conditions; 3.5+ or 3+, showing very strong or strong growth defect; 2+ or 1+, mild to minor growth defect; and 0+, no growth phenotype observed.
Figure 2. Genetic Evaluation of the pNiiA-CPR Strategy.

(A) Phenotypes of pNiiA-CPR mutants for HIS3, TRP5, MET16, MET2, LYS9, and LYS4. Under standard repressing conditions (Re, AMM plus ammonium), all strains lacked detectable growth (4+ phenotype). Growth was unimpaired under inducing conditions (In; AMM plus nitrate). Growth phenotypes of pNiiA-CPR mutants under repressing conditions were specifically suppressed if the cognate amino acid was provided to the growth media (His, histidine; Trp, tryptophan; Met, methionine; or Lys, lysine).
(B) Growth phenotypes of pNiiA-CPR mutants for the previously reported A. fumigatus essential genes, FKS1, GUS1, SPE2, and HEM15 [12,13]. Reproducible 4+ essential growth phenotypes are observed for each pNiiA-CPR mutant under repressing conditions with the exception of FKS1, which produced a 3.5+ growth phenotype.
(C) arp2 and ayg1 conidial color phenotypes by the pNiiA-CPR strategy. Wild-type (WT) strain CEA10, pNiiA-arp2, and pNiiA-ayg1 mutants display normal dark-green conidia color under inducing conditions. Gene-specific conidia color phenotypes characteristic of their known null phenotype [19] are specifically detected under repressing conditions.
(D) nudC growth phenotype by the pNiiA-CPR strategy. Highly reproducible growth and morphological phenotypes associated with nudC mutants are observed (see Figure S1) as similarly determined using an alcA heterologous conditional promoter [15].
According to the severity of the growth phenotype displayed by CPR mutants in repressing conditions, qualitative scores were assigned to each of the CPR mutants to classify the terminal phenotype (Figure 1D). A 4+ shutoff phenotype was assigned to those strains (e.g., GFA1) that completely fail to grow on repressing medium after 48 h at 30 °C. Similarly, a 3.5+ or 3+ shutoff phenotype was used to score those strains that showed a very severe (e.g., PFS2) or a severe (e.g., ALG7) growth phenotype, respectively. Other growth defects were scored as 2+ (mild), 1+ (minor; e.g., CDC24), or 0+ (no defect; e.g., ALR1). All essential genes described here showed either a complete absence of growth (4+) or a dramatic growth defect (3.5+ and 3+) under repressing conditions.
Evaluating pNiiA-CPR Reliability by Genetic Approaches
To test its reliability, the pNiiA-CPR strategy was further applied to multiple A. fumigatus genes whose null phenotype was either known or independently verified by other means. First, pNiiA-CPR mutants were constructed for five genes (HIS3, TRP5, MET16, LYS9, and LYS4) involved in amino acid biosynthesis. Growth was completely impaired for all strains under repressing conditions lacking amino acids but fully restored when the cognate amino acid was provided (Figure 2A). Second, pNiiA-CPR mutants were constructed for four genes (FKS1, GUS1, SPE2, and HEM1) whose essentiality in A. fumigatus has been demonstrated by a parasexual genetic strategy [12,13]. In each instance, the conditional mutant displayed an essential growth phenotype (Figure 2B). Third, we constructed pNiiA-CPR mutants of two nonessential genes (ayg1 and arp2) involved in conidial pigmentation whose null phenotypes result in the production of yellow-green and pink-red conidiospores, respectively [19]. Hence, conidial pigmentation phenotypes of ayg1 and arp2 mutants serve as whole cell reporters to monitor the level of repression achieved by the pNiiA-CPR system. Indeed, pNiiA-ayg1 and pNiiA-arp2 conidia color shifted from dark-green (wild-type conidia color) under inducing conditions to yellow-green (ayg1) or red-pink (arp2) under repressing conditions (Figure 2C). Finally, we applied this pNiiA-CPR strategy to A. fumigatus nudC, a gene shown to be essential by promoter replacement with A. nidulans alcA promoter [15]. Consistent with a previous report [15], the pNiiA-nudC mutant displayed (1) an essential growth phenotype (Figure 2D), (2) a germination defect of conidia with approximately 70% of conidia failing to germinate and approximately 30% of conidia forming short growth-arrested germ tubes (Figure S1), and (3) an nudC nuclear distribution terminal phenotype characterized by multinucleate conidia (Figure S1).
An advantage to the use of large homologous flanking sequences to pNiiA-CPR cassettes is that multiple independent promoter replacement mutants could be recovered for a given gene. Thus, gene essentiality can be independently validated from multiple pNiiA-CPR mutants. Indeed, multiple independent pNiiA-CPR mutants (n > 2) were recovered for 18 of the 19 genes mentioned above, and in all cases, their independent pNiiA-CPR mutants displayed reproducible growth phenotypes under repressing conditions. Although the essential growth phenotype of GUS1 was based on the single pNiiA-GUS1 mutant recovered, its essentiality is consistent with previous work and with its predicted function as a glutamate-tRNA sythetase [13].
Direct Comparison between pNiiA-CPR and Gene Disruption Terminal Phenotypes
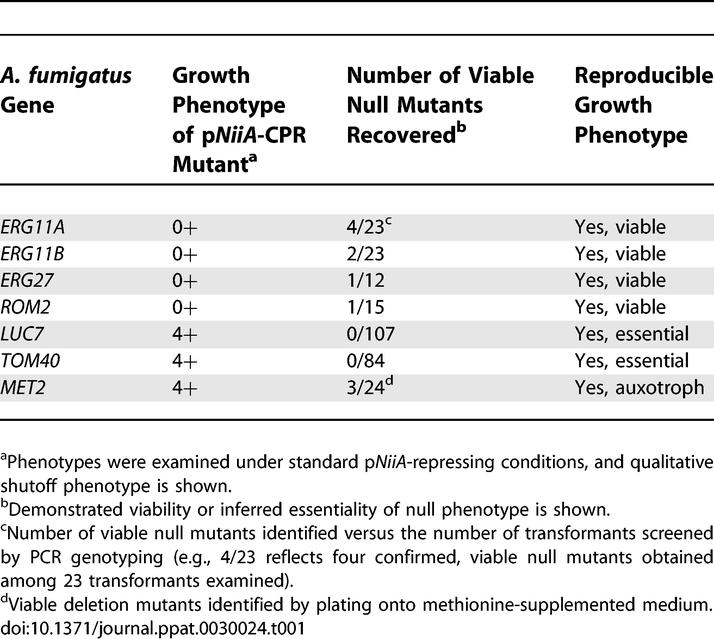
Reliability of the pNiiA-CPR system was also assessed by pyrG-based gene disruption of multiple genes shown to be essential by the pNiiA-CPR approach (Table 1). In fact, viable null mutants of ERG11A, ERG11B, ERG27, ROM2, and MET2 (when exogenous methionine was provided) were recovered, consistent with their nonessential or conditional essential phenotypes determined by pNiiA-CPR mutants. Moreover, no viable null mutants were recovered for either TOM40 or LUC7 by gene disruption (two essential genes identified by the pNiiA-CPR approach) despite a significantly large number of transformants examined (84 for TOM40 and 107 for LUC7, respectively) (Table 1). Thus, the essentiality of TOM40 and LUC7 is shown by both the pNiiA-CPR strategy and gene deletion analysis. Collectively, these data demonstrate that robust genetic conclusions spanning an extensive set of “phenotypic reporter genes” are reliably reproduced by the pNiiA-CPR strategy.
Table 1.
Comparison of Growth Phenotypes Observed by pNiiA-CPR Methods versus Gene Knockout
Quantitative Analysis of pNiiA-CPR Transcriptional Regulation
Reverse transcription (RT)-PCR was first performed to monitor pNiiA-CPR gene expression levels for all previously described genes in Table 1. As pNiiA-MET2 mutants grow normally in repressing media supplemented with methionine, MET2 mRNA expression could be assessed in both inducing and pNiiA-repressing conditions by RT-PCR. MET2 mRNA was detected in both the pNiiA-MET2 mutant and wild-type CEA10 on inducing medium; however, no MET2 transcript was detected in the pNiiA-MET2 mutant on repressing medium supplemented with methionine (Figure 3A). Similarly, mRNA transcripts were detected by RT-PCR in CEA10 and pNiiA-CPR mutants for ERG11A, ERG11B, ERG27, ALR1, and ROM2 under inducing conditions but not from pNiiA-CPR mutants maintained under repressing conditions (Figure S2). In addition, Northern blot analysis also confirmed a significant depletion of ALR1 mRNA levels under repressing conditions (despite the lack of a clear growth phenotype) versus its endogenous level of expression in wild-type CEA10 (Figure 3B). Although the ALR1 mRNA levels of the ALR1 conditional mutant were clearly elevated compared to the wild-type, no deleterious growth phenotype was detected (Figure 1C).
Figure 3. Expressional Level Analysis of pNiiA-CPR Mutants.

(A) RT-PCR of pNiiA-MET2: (a) pNiiA-MET2 mutant under inducing conditions, (b) pNiiA-MET2 mutant under repressing conditions plus 100 μg/ml methionine, (c) pNiiA-MET2 mutant under inducing condition plus 100 μg/ml methionine, and (d) wild-type A. fumigatus strain (CEA10) under repressing conditions. To monitor and ensure even sample loading, RT-PCRs for the ACT1 transcript were also performed using identical samples. In addition, standard PCR was performed to confirm that there is no detectable genomic-DNA contamination.
(B) Northern blot analysis of pNiiA-ALR1 mutant expression levels. Northern blot was performed with RNA samples prepared from the nonessential ALR1 mutant and wild-type cells growing in standard inducing (In) or repressing (Re) conditions. ACT1 transcript levels served as sample loading controls.
(C) Real-time RT-PCR analysis of expression level of pNiiA-ALR1 and pNiiA-MET2. pNiiA-CPR mutants and wild-type strain (CEA10) were grown in inducing (In) or repressing (Re) medium at 37 °C for 20 h, and total RNA was extracted from identical time points. The relative expression level normalized to total input RNA [43] is displayed on the y-axis. Error bars represent SD. Compared to wild-type, the relative expression level for ALR1 and MET2 is 0.013 and 0.061 under repressing conditions and 2.49 and 23.58 under inducing conditions, respectively.
(D) pNiiA-TUB1 expression level under inducing (In) and repressing (Re) conditions versus wild-type level is displayed on the y-axis. Error bar represents SD. The relative expression level of pNiiA-TUB1 versus wild-type is 0.06 under repressing conditions and 6.35 under inducing conditions, respectively. Note: Since real-time RT-PCR was performed using primers detecting both pNiiA-TUB1 and wild-type allele, data shown were calculated by subtracting wild-type level from total inducing (In) and total repressing (Re) level, respectively.
Real-time RT-PCR was performed to further evaluate the achievable range of expression levels for three pNiiA-CPR mutants shown to yield either a nonessential (ALR1), a conditional essential (MET2), or an essential (TUB1) terminal phenotype (Figure 3C and 3D). Compared to wild-type, ALR1 and MET2 expression levels in CPR mutants were dramatically reduced by 75.9-fold and 16.3-fold under repressing conditions and elevated by 2.8-fold and 23.6-fold under inducing conditions, respectively (Figure 3C). To overcome the technical difficulty of performing real-time RT-PCR on an essential gene, a TUB1 tandem duplication mutant was constructed in which the pNiiA-TUB1 allele is balanced by a wild-type TUB1 copy under the control of its native promoter. Thus, expression of the pNiiA-regulated TUB1 allele could be monitored under repressing conditions (see Materials and Methods). Real-time RT-PCR performed with this strain revealed expression of the pNiiA-TUB1 allele to be approximately 0.06-fold when repressed and 6.3-fold under inducing conditions relative to normal TUB1 expression levels (Figure 3D). Collectively, these data demonstrate that the pNiiA-CPR strategy typically achieves tight regulatable expression and suggest that it could be reliably applied to large-scale analysis of gene essentiality in A. fumigatus.
Large-Scale Identification of Essential Genes
The genomic sequence of A. fumigatus strain AF293 [20], as well as a second clinical isolate used in this study, CEA10, where approximately 10× coverage has been obtained (N. Fedorova, V. Joardar, J. Crabtree, M. Anderson, R. Maiti, et al., unpublished data), provides an opportunity to systematically identify essential genes. Although global annotation of the A. fumigatus genome is ongoing, homologs to genes of interest were identified through search alignment tools and corresponding promoter replacement cassettes designed accordingly. In total, 54 A. fumigatus genes were selected for pNiiA-CPR analysis, of which 49 are known to be essential (or conditionally essential) for growth in S. cerevisiae with all but three of these being essential for normal growth in C. albicans (Table 2). In addition, genes were selected so as to represent a broad diversity of gene functions and cellular processes. Biasing this A. fumigatus gene set toward those essential in both S. cerevisiae and C. albicans builds on previous work demonstrating that at least 60% of C. albicans genes homologous with essential S. cerevisiae genes were also essential in C. albicans [11]. Therefore, we reasoned that a set of genes experimentally demonstrated as essential in two distinct hemiascomycetes would be highly enriched for genes sharing conserved essential functions within the euascomycetes.
Table 2.
Growth Phenotypes of A. fumigatus Gene Set Examined and Concordance of Gene Essentiality in C. albicans and S. cerevisiae

Analysis of this A. fumigatus gene set revealed 35 genes displaying a 3+ or greater (3.5+ or 4+) essential growth phenotype (Table 2). Excluding genes involved in amino acid biosynthesis, no significant differences in terminal growth phenotypes were observed between minimum medium (AMM) and rich medium (Aspergillus complete medium [ACM]). Essential genes comprise a spectrum of biological functions including lipid, ergosterol, cell wall, amino acid, protein, and heme biosynthesis, as well as glycosylation, secretion, RNA processing, and novel genes of unknown functions (Table 2). Moreover, among those genes displaying an essential phenotype in both S. cerevisiae and C. albicans and predicted to have only a single ortholog in A. fumigatus (n = 44), 32 genes (73%) are experimentally demonstrated as sharing a conserved growth phenotype of 3+ or greater in A. fumigatus.
Essential genes identified in this manner constitute a collection of possible antifungal drug targets, and microscopic examination of their terminal growth phenotypes may assist in their prioritization (Figure 4). For example, repression of GFA1 produced enlarged and highly disrupted nongerminating conidia that failed to undergo any polarized growth. As GFA1 is predicted to encode the sole glutamine-fructose-6-phosphate aminotransferase activity responsible for the first and rate-limiting step in chitin synthesis, this terminal phenotype likely reflects the normal interdependence between cell wall biosynthesis and polarized growth. Conidia of SEC31 and SLY1 incubated under repressing conditions were unable to germinate, suggesting that these genes are required for the initiation of germ tubes, whereas terminal phenotypes of TUB1, ERG10, and, most prominently, PRI1, resulted in growth-arrested germ tubes. Interesting, HEM15 displayed a more complex terminal phenotype of dramatically swollen conidia with single or multiple germ tubes. Repression of pNiiA-FKS1 yielded stubby and highly branched micromycelial germlings that morphologically resemble the chemotype observed when A. fumigatus is treated with Fks1p-specific inhibitors such as caspofungin and related echinocandins [21]. Thus, distinct terminal phenotypes are identified among pNiiA-CPR mutants, and in some instances, they appear more severe than that caused by either genetic or chemical inactivation of FKS1.
Figure 4. Analysis of pNiiA-CPR Associated Morphological Terminal Phenotypes.

Terminal growth phenotypes of pNiiA-CPR mutants were observed under a microscope (×160) with conidia grown for 36 to 40 h at 30 °C under standard repressing conditions. A continuum of conidia germination phenotypes of high penetrance was observed; ranging from those completely failing to undergo polarized growth (SEC31, SLY1) or swollen and highly disorganized condidia (GFA1), to those displaying stunted (TUB1, ERG10) or nonbranching germlings with swollen conidia (HEM15) with only rudimentary polarized growth. Micromycelial colonies were observed for a pNiiA-FKS1 mutant and resembling the morphology of wild-type A. fumigatus when grown in the presence of minimum effective concentration (MEC) of the FKS1p inhibitor, caspofungin [21]. Growth phenotypes under inducing conditions are shown in Figure S3.
Essential genes with a cidal terminal phenotype are commonly preferred since genetic evidence predicts that chemical inhibitors of such targets may display similar chemotypes. An assay to evaluate cidal or static terminal phenotypes was applied to essential genes displaying a 3.5+ or 4+ growth phenotype (Table 2) by incubating conidia in pNiiA-repressing medium for various durations of time before washing and plating conidia onto pNiiA-inducing medium where viability was scored by counting colony-forming units (CFU) (see Materials and Methods and Figure 5). Of 28 genes tested, six genes (approximately 21%), including GFA1 (Figure 5), displayed a cidal terminal phenotype, as scored by a significant reduction (greater than 90%) of CFU after 24-h incubation in repressing medium. Additional genes demonstrating a cidal terminal phenotype include KRR1, SEC31, SLY1, TUB1, and GUS1 (Table 2). Additional genes displayed a slow cidal phenotype, as scored by significant reduction (greater than 90%) in CFU after 48- or 72-h incubation in repressing medium, or a static terminal phenotype after 48-h incubation in repressing conditions as shown with TRR1 (Figure 5, Table 2).
Figure 5. Determination of Cidal or Static Terminal Phenotypes.

A representative example of cidal and static terminal phenotypes for GFA1 and TRR1 pNiiA-CPR mutants is shown.
(A) pNiiA-GFA1 displayed a cidal terminal phenotype as a dramatic (greater than 90%) reduction in CFU was observed after incubation in pNiiA-repressing conditions for 24 or 48 h.
(B) pNiiA-TRR1 revealed a static terminal phenotype as no significant reduction in CFU counts was detected after 48-h incubation under repressing conditions. A summary of all additional cidal/static terminal phenotypes for A. fumigatus genes displaying 4+ qualitative growth phenotypes is provided (see Table 2).
The ERG11 Gene Family Is Essential in A. fumigatus
Identifying gene families that are essential for the growth and viability of A. fumigatus can be missed by our approach and thus require additional considerations. For example, duplicate ERG11 genes (encoding 14α-demethylase, the known target of azole-based antifungal agents) exist in A. fumigatus [22,23]. While disruption of ERG11A displays no effect on growth or ergosterol levels [23], it remained unknown whether ERG11B alone or the gene pair together is essential in A. fumigatus. To address this, individual pNiiA-CPR mutants of ERG11A and ERG11B were constructed, with neither producing a noticeable growth defect under repressing conditions (Figure 6, Table 1). Further, viable erg11AΔ and erg11BΔ null mutants displaying robust growth were readily recovered (Figure 6, Table 1). Thus, these data demonstrate that neither member of the ERG11 gene family is essential individually in A. fumigatus. To examine whether the ERG11 gene family is essential, a double mutant was constructed by creating a pNiiA-ERG11A allele within an ERG11B null mutant background (see Materials and Methods). The resulting ERG11 double mutant (erg11BΔ, pNiiA-ERG11A) displays robust growth under the inducing condition, with no growth evident under repressing conditions (Figure 6). Therefore, these data suggest that either of the ERG11 gene pair can functionally compensate for loss of the other, and the genetic inactivation of both ERG11A and ERG11B indicates their essentiality in A. fumigatus.
Figure 6. Phenotypic Analyses of ERG11 Gene Family.

Growth phenotypes of ERG11 gene family were observed with pNiiA-CPR mutants, null deletion mutants, and double mutants (erg11BΔ, pNiiA-ERG11A). Strains were grown on either inducing medium (AMM plus nitrate) or repressing medium (AMM plus ammonium) at 30 °C for 40 h.
In Vivo Analysis of pNiiA-CPR Mutants in a Murine Model of Systemic Aspergillosis
Animal models of fungal pathogenesis provide important experimental verification whether genetic mutations abrogate (or attenuate) the virulence of the pathogen. As mouse serum contains approximately 200 μM ammonium [24] and such levels of ammonium are theoretically sufficient to repress the pNiiA promoter, we tested this by streaking pNiiA-CPR mutants onto AMM plates containing 20% mouse serum (Figure 7A). In each case, TUB1, SEC31, GCD6, GFA1, MET2, AUR1, and pNiiA-ERG11A, erg11BΔ CPR mutants reproduced their terminal growth phenotypes as observed on either AMM or rich medium (Table 2), demonstrating that mouse serum levels of ammonium (or other primary nitrogen source) are sufficient to repress the NiiA promoter. Indeed, as even higher ammonium levels (approximately 1 to 4 mM) of repressor exist in mouse tissues (e.g., liver, kidney, brain, muscle) [24], we reasoned that the virulence of pNiiA-CPR mutants could be examined in a murine model of aspergillosis. To test this, an animal model was first established using mice that were immunocompromised by cyclophosphamide and infected with 100,000 conidia by tail vein injection (see Materials and Methods). Control experiments demonstrate that all mice (n = 5) infected with the wild-type virulent strain, CEA10, died from infection within 8 d, while 100% survival was observed for mice (n = 5) infected with a known avirulent strain, CEA17 [25]. Virulence of CPR mutants of TUB1, SEC31, GCD6, GFA1, AUR1, and MET2 was similarly examined (Figure 7B). No mortality or morbidity was observed for mice infected with pNiiA-TUB1 over the 12-d period, as predicted for a core essential gene that is significantly repressed and for which sufficient in vivo ammonium levels exist. Similarly, pNiiA-based repression was sufficient to fully abrogate virulence of SEC31, GFA1, and GCD6 (Figure 7B). Further, whereas both erg11AΔ and erg11BΔ mutants were similarly virulent as the wild-type CEA10, an ERG11 double mutant (erg11BΔ, pNiiA-ERG11A) was fully avirulent over an extended 22-d period (Figure 7C). On the other hand, attempted repression of MET2 and AUR1 had no effect on A. fumigatus virulence (Figure 7B). As pNiiA-MET2 growth was unaffected by serum, and a C. albicans MET2 tetracycline-regulatable conditional mutant is similarly virulent [10], this likely reflects that sufficient levels of methionine are scavenged to support growth both in vitro and in vivo. Although the observed virulence phenotype of pNiiA-AUR1 remains unclear (see Discussion), we demonstrate that pNiiA-CPR mutants generally gave in vivo phenotypes consistent with reliable repression being achieved.
Figure 7. In Vivo Validation of A. fumigatus pNiiA-CPR Mutants in an Immunocompromised Murine Model of Systemic Infection.

(A) pNiiA-CPR mutants growing on AMM plus 20% mouse serum (Sigma) reproduced their terminal growth phenotypes as observed on either AMM plus ammonium or rich medium (Table 2).
(B and C) In vivo validation of pNiiA-CPR mutants. (B) ICR male mice were immunocompromised by administration of cyclophosphamide at 150 mg/kg twice prior to infection and 100 mg/kg twice a week after infection. Approximately 105 viable conidia from individual pNiiA-CPR mutants (TUB1, SEC31, GCD6, GFA1, MET2, and AUR1) were injected into the tail vein of immunocompromised mice (five mice per group). Signs of infection were monitored for up to 12 d following infection. Wild-type strain CEA10 and the starting strain CEA17 (a pyrG − auxotroph of CEA10) [25,39] were included as positive controls for virulence and avirulence, respectively.
(C) Genetic inactivation of the ERG11 gene family promotes avirulence in an immunocompromised murine model of systemic infection. Pathogenesis of erg11AΔ, erg11BΔ, and an ERG11 double mutant (erg11BΔ, pNiiA-ERG11A) was similarly analyzed (as described above) but over a longer postinfection period (22 d), and animal survival was compared to CEA10 and CEA17 control strains.
Chemical Hypersensitivity of pNiiA-CPR Mutants
In principle, partially depleting the activity of a particular gene product sensitizes cells to compounds that act through that target. Modulating gene dosage may be achieved by deleting one copy in a diploid organism [6,26], antisense interference [8], or reduced gene expression through titratable repression of a regulatable promoter [11]. To test whether intermediate expression levels of pNiiA-CPR mutants could be achieved to produce chemical hypersensitivity, we first examined the chemical sensitivity of a pNiiA-ALG7 mutant to its cognate inhibitor, tunicamycin. A partial depletion of Alg7p was inferred by growing pNiiA-ALG7 in the presence of an ammonium/nitrate ratio sufficient to reduce growth rate by approximately 50%. Under such conditions, depletion of Alg7p resulted in an 8- to 16-fold shift in minimum inhibitory concentrations (MICs) to tunicamycin without any significant change in sensitivity to either fluconazole or itraconazole (Table 3). Moreover, no significant change in tunicamycin MIC was observed across a panel of similarly sensitized pNiiA-CPR mutants (Table 3). Therefore, partial depletion of the A. fumigatus Alg7p resulted in a specific hypersensitivity to tunicamycin.
Table 3.
MIC of Antifungal Drugs to pNiiA-CPR Strains
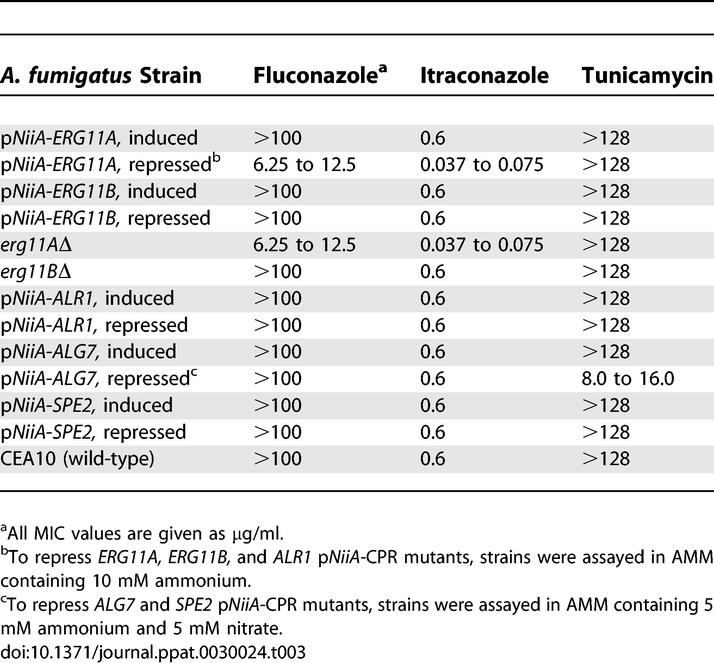
A. fumigatus exhibits an extremely low susceptibility to fluconazole (MIC > 256 μg/ml), a feature attributed to the genetic redundancy of ERG11 in this pathogen [22,23]. Using the conditional mutants, we tested whether inactivation of either member of the ERG11 gene family may sensitize A. fumigatus to ERG11-specific inhibitors (Table 3). As neither ERG11 gene displays growth defects under full repressing conditions, pNiiA-CPR mutants of ERG11A and ERG11B serve as whole cell assays by growing each mutant under full repressing conditions and determining their respective MIC to fluconazole. Indeed, the pNiiA-ERG11A mutant displayed an 8- to 16-fold lower MIC to fluconazole; no detectable drug hypersensitivity was observed for either pNiiA-ERG11B or control strains tested (Table 3). MIC determinations using null mutants of erg11AΔ and erg11BΔ independently confirmed that depletion of ERG11A (rather than ERG11B) similarly sensitizes A. fumigatus 8- to 16-fold to fluconazole. Moreover, only depletion of ERG11A, as either a pNiiA-CPR mutant or null mutant, produced hypersensitivity to itraconazole, another triazole that, unlike fluconazole, displays strong activity against wild-type A. fumigatus (Table 3). Neither pNiiA-CPR mutants nor null mutants of ERG11A or ERG11B displayed a change in MIC to tunicamycin. This preferential drug sensitivity of mutants depleted of A. fumigatus ERG11A further supports the feasibility of target-based sensitized whole cell assays using pNiiA-CPR mutants. These data also suggest ERG11A likely encodes the major 14α-demethylase activity and that ERG11A, rather than ERG11B, largely contributes to the poor susceptibility of A. fumigatus to fluconazole (see Discussion).
Discussion
In this report, we describe a pNiiA-CPR strategy to construct A. fumigatus conditional mutants and to directly identify and evaluate gene essentiality in vitro and in vivo. Replacement of the endogenous gene promoter with pNiiA typically enables a robust shutoff of gene expression to assess terminal growth phenotypes including intracellular, morphological, and cidal growth consequences. Collectively, the completion of the A. fumigatus genomic sequence, an understanding of essential genes in other yeasts, and the pNiiA-CPR strategy enable a rational approach to determine the essentiality of orthologous genes in A. fumigatus. Indeed, genomic comparisons between A. fumigatus and other eukaryotes reveals that, in all but one of 131 largely essential eukaryote orthologous groups, an A. fumigatus ortholog is identified [27]. Here we demonstrate that at least 73% of the predicted essential genes we examined were essential for growth. Hence, a broad spectrum of antifungal targets conserved within major human fungal pathogens was efficiently identified. We also demonstrate how this approach can be extended to gene families (e.g., ERG11) and that the resulting pNiiA-CPR mutants serve as target-based whole cell assays suitable for drug screening.
To evaluate the utility of the NiiA conditional promoter for studying gene essentiality, an extensive analysis of its regulatable expression was performed. First, its reliability was examined by genetic means. Conditional mutants of multiple genes encoding functions involved in amino acid biosynthesis were constructed, and all showed 4+ shutoff growth phenotypes that could be reversed by adding back appropriate amino acids to the medium (Figure 2A, Table 2). In addition, A. fumigatus genes examined by the pNiiA-CPR strategy and independently examined by a parasexual genetics strategy [12,13] yielded reproducible essential growth phenotypes (Figure 2B). pNiiA-CPR mutants of genes in the melanin biosynthetic pathway also gave the predicted terminal conidia color change characteristic to their known null phenotype (Figure 2C) [19]. Thus, genetic data strongly suggest that sufficiently achievable repression levels are obtained by the pNiiA promoter independent of the gene to which it is linked or its physical location within the genome. We also directly compared terminal phenotypes of A. fumigatus nudC genes achieved by pNiiA-CPR strategy to that achieved by the alcA conditional promoter system [15] and obtained highly comparable nudC terminal phenotypes resembling its reported null phenotype (Figures 2D and S1). Finally, a comparative analysis was performed of terminal phenotypes for seven genes evaluated by both pNiiA-CPR and gene disruption strategies. By both methods, three genes (LUC7, TOM40, and MET2) were shown to be essential, while four genes (ERG11A, ERG11B, ERG27, and ROM2) were found to be nonessential (Table 1).
Genetic interrogation of the pNiiA-CPR strategy supports a general reliability of this method, but quantitative metrics for expression levels achieved by the NiiA promoter were also required. Multiple genes monitored by RT-PCR displayed repression levels noticeably below wild-type expression levels (Figure 3A; also see Figure S2). Further, real-time RT-PCR analysis of expression levels among pNiiA-CPR mutants for ALR1, MET2, and TUB1 demonstrates achievable repression levels in the range of approximately 16- to 76-fold below wild-type (Figure 3C and 3D). In several cases, overexpression was detected by RT-PCR and/or real-time RT-PCR under inducing conditions. Although no deleterious growth consequences were observed, intrinsic overexpression may complicate pNiiA-CPR mutant construction and/or analysis in some instances.
Despite a high concordance in A. fumigatus gene essentiality to known essential S. cerevisiae and C. albicans orthologs, several exceptions were observed. In some instances, this is due to gene redundancy, as we have demonstrated by our genetic analysis of the ERG11 gene family and may similarly relate to ALR1. However, other nonessential phenotypes determined for this gene set were more surprising. For example, ERG27, although essential in both S. cerevisiae and C. albicans, lacked a clear growth phenotype in A. fumigatus. This phenotype cannot be attributed to the failure of the pNiiA promoter to sufficiently repress ERG27 expression since we could construct a viable ERG27 deletion mutant by gene disruption (Table 1). As ERG27 defines the last “essential” step in the S. cerevisiae ergosterol biosynthetic pathway and ergosterol intermediates that accumulate in ERG mutants are implicated as causing the phenotypes/chemotypes resulting from disrupting specific steps in the pathway [28], one possibility is that the presumed ergosterol intermediate (possibly 4-methylzymosterol or 4,4-dimethylzymosterol) is not deleterious for A. fumigatus growth. Indeed, an extended pNiiA-CPR analysis of the ergosterol pathway reveals that additional genes defining the last essential steps in this pathway in S. cerevisiae (e.g., ERG26) are not essential in A. fumigatus (W. Hu, H. Wang, S. Sillaots, S. Kauffman, J. Becker, et al., unpublished data). Other interesting examples include the GDP-GTP exchange factors (GEF) ROM2 and CDC24 that regulate Rho1p and Cdc42p, respectively, in S. cerevisiae [29,30]. Again, ROM2 was independently confirmed as nonessential in A. fumigatus as a viable knockout strain was constructed (Table 1) despite lacking a second clear homolog in A. fumigatus and sharing high homology (1e-155) to an essential C. albicans ortholog. In contrast, the A. fumigatus gene identified as sharing the greatest homology to CDC24 lacks striking homology (7e-30) to S. cerevisiae or C. albicans CDC24 and thus may be functionally distinct from CDC24. Regardless, it is unexpected that two GEF proteins critical to the process of polarized growth in yeast are either nonessential or absent from the A. fumigatus mycelial growth process, as is similarly the case for multiple polarized growth positional markers (e.g., BUD3, BUD8, and BUD9) [27]. Indeed, these examples stress the requirement for performing such analyses directly in A. fumigatus.
Our genetic analysis of the A. fumigatus ERG11 gene family begins to address the relative role of each ERG11 gene member during mycelial growth, as well as issues relating to differences in A. fumigatus activity among azole-based drugs. We demonstrate by genetic inactivation of either homolog that each enzyme can compensate for the lack of the other but that inactivation of both ERG11A and ERG11B is inviable in A. fumigatus. Therefore, azole-based antifungal drugs must inhibit both members of the ERG11 gene family in order to display potent inhibitory activity against A. fumigatus. This may (in part) reflect the highly variable A. fumigatus MIC levels between azole-based drugs. In addition, we demonstrate that ERG11A mutants (rather than ERG11B mutants) are specifically hypersensitive to fluconazole, suggesting that ERG11A encodes the major 14α-demethylase activity contributing to the poor susceptibility of A. fumigatus to this drug. As ERG11A mutant also displays hypersensitivity to itraconazole and additional azoles [23], ERG11A may encode the major 14α-demethylase activity required for mycelial growth. Consistent with this view, multiple missense mutations of ERG11A (rather than ERG11B) have been linked to itraconazole and posoconazole resistance by UV mutagenesis studies and analysis of azole-resistant clinical isolates of A. fumigatus [31–33]. ERG11Ap may also possess an inherently greater resistance to azoles, although biochemical studies are required to address this possibility. Additional studies are also required to address the functional role of ERG11B. Although we demonstrate that it is not solely required for A. fumigatus pathogenesis, it is unclear whether ERG11B serves exclusively a redundant role with ERG11A or a more specialized function under particular conditions.
A variety of alternative strategies (see Introduction) to genetically investigate gene function in A. fumigatus are currently being pursued, and where possible, we have compared pNiiA-CPR terminal phenotypes to those reported [12–15]. Recently, a doxycycline-regulated gene expression system in A. fumigatus was reported, although it remains to be tested using an endogenous A. fumigatus gene [16]. Unlike previous reports, our work employs a directed strategy to systematically evaluate A. fumigatus essential genes on a relatively large scale and distinguish between cidal or static growth-arrested terminal phenotypes. Importantly, the regulatable promoter approach circumvents the need to complement mutants with their corresponding wild-type gene; a difficult and time-consuming process in A. fumigatus. The ability to recover multiple, independently generated, promoter replacement mutants by this method also serves to reinforce the gene-specific phenotypes observed. Unlike alternative strategies, we demonstrate that pNiiA-CPR mutants can be used to validate the gene essentiality in an immunocompromised murine model of infection. Pathogenesis studies assist prioritization of essential genes for drug screening and provide a genetic prediction that cognate inhibitors would display similar efficacy in an animal model of infection. Finally, unlike a genetic screen, this approach allows an up-front systematic examination of specific A. fumigatus genes that meet bioinformatic spectrum criteria and/or essential phenotypes in other fungal organisms and, hence, enables a rapid and focused survey of potential broad-spectrum antifungal drug targets. In this way, we demonstrate a significant enrichment in the identification and validation of essential genes in this pathogen and envision that its utility could be expanded.
The principal limitation to the pNiiA-CPR strategy is that, as with any other promoter replacement strategy, it will fail to correctly identify gene essentiality in cases where endogenous gene expression levels are naturally lower than the residual level achieved by repression of the NiiA promoter. Although our data demonstrate that such limitations are not common, we can neither rule out this complication nor predict its occurrence among individual genes tested. In cases where a more rigorous analysis of a single (or relatively small number of genes) is of particular interest, an independent verification of the null phenotype(s) should be performed by gene disruption, as performed in Table 1. An advantage of this approach is that the pNiiA-CPR strategy offers a means to identify the majority of essential genes in this pathogen. Minor technical limitations of this approach relate to the requirement to predict the gene's correct start codon, which can be particularly difficult for those A. fumigatus genes that contain extremely short 5′ exons. In addition, construction of pNiiA-CPR cassettes involves step-by-step subcloning to introduce sufficient flanking homology to each given gene. Greater throughput of cassette constructions could be achieved using gap repair methods amenable in S. cerevisiae [34]. Although relatively large regions of flanking homology are required to correctly target promoter replacement cassettes, shorter flanking regions could be used in conjunction with a recipient strain deleted of Ku70 or Ku80 [35,36].
Limitations to the murine model described also exist. Again, residual levels of expression of pNiiA-CPR mutants in vivo may not always be sufficient to produce an avirulent phenotype. This depends not only quantitatively on the differential level of repression achieved versus wild-type expression but also on the qualitative level of expression necessary to support function, which also can vary between genes and cannot be predicted. For example, we cannot rule out the possibility that the observed pNiiA-AUR1 virulence (despite this gene displaying a 4+ growth phenotype) may reflect an insufficient shutoff within the host. Alternatively, the pNiiA-AUR1 mutant may be suppressed in vivo by the uptake of host sphingolipids in a manner analogous to that reported of Candida glabrata ERG11 and ERG9 mutants (see below) [37,38]. Notwithstanding this limitation, multiple (n = 5) individual pNiiA-CPR mutants could unambiguously be confirmed as avirulent by this murine model, strongly suggesting its general utility (Figure 7B and 7C). Indeed, although potential drug targets may be missed, others are definitively identified, such as GFA1, which is directly confirmed as (1) essential for growth, (2) displaying a cidal terminal phenotype, and (3) avirulent in a murine model of pathogenesis. Although the murine model does not reflect a true pulmonary aspergillosis infection model (because mice were infected through tail vein injection rather than the normal route of infection, namely inhalation), such improvements can be envisioned. Also, unlike that of a tetracycline-regulatable promoter system [11], virulence of pNiiA-CPR mutants can only be evaluated from the perspective of prophylactic antifungal drug targets because repression is constitutive rather than controlled. Nonetheless, pNiiA-CPR mutants provide a unique advantage over alternative methods [12,13,15] to validate A. fumigatus genes in pathogenesis studies.
Formal demonstration that gene essentiality confirmed in vitro indeed confers an avirulent phenotype is necessary, as exemplified by phenotypic analysis of ERG11 across fungal pathogens. Surprisingly, neither ERG11 nor ERG9 is essential in a murine model of C. glabrata infection, despite both genes being confirmed as essential in vitro [37,38]. Instead, C. glabrata ERG11 and ERG9 mutants are virulent in a host environment, and it is demonstrated that they scavenge host cholesterol to suppress ergosterol biosynthetic defects [37,38]. Direct experimentation was required to rule out that this phenotype extends to A. fumigatus. Although neither ERG11A nor ERG11B mutants were avirulent, the ERG11 double mutant (erg11BΔ, pNiiA-ERG11A) is completely avirulent. In the case of novel antifungal targets, one can neither predict the consequences of their inactivation in an animal model nor neglect their direct experimental confirmation. Moreover, as A. fumigatus pathogenesis likely reflects its saprophytic lifestyle and virulence factors remain to be identified, a pNiiA-CPR strategy demonstrating avirulence of essential genes offers a broad set of potential drug targets.
In conclusion, the pNiiA-CPR strategy overcomes a number of historical challenges in applying molecular genetics to A. fumigatus and is highly amenable to genetically investigate A. fumigatus biology as well as to validate essential genes in this important human fungal pathogen. This approach facilitates their phenotypic analysis both in vitro and in vivo, and the resulting conditional mutants serve as target-based whole cell assays, thereby enabling the discovery, prioritization, and screening of novel antifungal targets directly in A. fumigatus. Indeed, promoter replacement strategies in C. albicans [11] and here in A. fumigatus encourage its broader application.
Materials and Methods
Strains, media, and cultural conditions.
A. fumigatus wild-type strain CEA10 and its uracil and uridine auxotroph, CEA17, were used in this study [39]. ACM and AMM were prepared as reported [40]. ACM supplemented with uracil and uridine was used to obtain CEA17 conidia suspensions and to produce mycelia for protoplast preparation. For protoplast transformation, a modified AMM was used that contained 10 mM Mg(NO3)2 as the sole nitrogen source and 1% (w/v) glucose as the sole carbon source (AMM plus nitrate). In addition, AMM (AMM plus ammonium) that contained 10 mM ammonium tartrate (C4H12N2O6; Sigma, http://www.sigmaaldrich.com) as the sole nitrogen source was used to repress the pNiiA for phenotypic analysis of pNiiA-CPR mutant. A. fumigatus was grown at 37 °C except where otherwise mentioned.
Construction of plasmid pPyrG-pNiiA.
Plasmid pPyrG-pNiiA was constructed as follows (Figure 1A). Briefly, the A. niger pyrG selectable marker and the pNiiA sequence were amplified by PCR from plasmid pAB4-ARp [41] (kindly provided by Christophe d'Enfert, Institut Pasteur), and A. fumigatus genomic DNA, respectively, using gene-specific primers containing multiple cloning sites (Table S1). The above PCR products of pyrG and pNiiA were first digested with Not1/Nde1 and Nde1/Xho1, respectively, and then subcloned into pBluescript (SK; Stratagene, http://www.stratagene.com) at the Not1/Xho1 sites. The resulting plasmid is pPyrG-pNiiA, which contains pyrG-pNiiA cassette flanked on either side with a couple of cloning sites (Figure 1A).
Construction of pNiiA-CPR cassette.
Through a subcloning strategy, CPR cassette was constructed that included the A. niger PyrG selectable marker and the pNiiA sequence flanked on either side with approximately 1.5 kb of gene-specific flanking sequence (Figure 1B). Briefly, the CPR cassette was constructed in two steps. In step 1, approximately 1.5-kb 5′-flanking sequence (left arm) that was usually 250 bp upstream of the ATG start codon of the target gene was amplified by PCR from A. fumigatus CEA10 genomic DNA with two gene-specific primers and then subcloned into suitable cloning sites (usually at Not1 and Mlu1) of the pyrG side of plasmid pPyrG-pNiiA. In step 2, approximately 1.5-kb downstream (3′-) genomic flanking sequence (right arm) that starts at the ATG start codon was amplified from A. fumigatus CEA10 genomic DNA with two gene-specific primers and then subcloned into the pNiiA side of plasmid pPyrG-pNiiA (usually at Asc1 and Pac1 sites). The resulting pNiiA-CPR cassette was excised from the plasmid by digesting with restriction enzymes at either end of the cassette (usually Not1 and Pac1) and used to transform A. fumigatus strain CEA17 (see below).
Protoplast preparation and transformation.
Promoter replacement cassette (pNiiA-CPR cassette) DNA was introduced into A. fumigatus strain CEA17 (pyrG−) by protoplast transformation according to established procedures with minor modifications [42]. Briefly, protoplasts were prepared with freshly grown mycelium using lysing enzyme (Sigma) (0.5 g for 50 ml digestion). From 5 to 10 μg of excised CPR-cassette DNA was used for each transformation, and the transformants were grown at 37 °C for 3 d on AMM (AMM plus 10 mM nitrate) containing 1.2 M sorbitol as osmotic stabilizer but lacking uridine and uracil. Three days post-transformation, transformants were transferred to fresh AMM plus nitrate plates and then subjected to genotypic and phenotypic analysis.
Genotypic analysis.
A PCR strategy was used to confirm double-crossover integrated promoter replacements in mutants in which the endogenous promoter of the target gene has been precisely replaced with pNiiA (Figure 1B). Genomic DNA was prepared using the DNeasy plant kit (Qiagen, http://www.qiagen.com) from transformants grown on AMM plus nitrate and then subjected to genotypic analysis by PCR. Three PCRs were performed, of which two mapped the promoter replacement junctions by using two sets of primers (L1 and L2 for left-arm junction, R1 and R2 for right-arm junction), with the third PCR using primer set L1/P to confirm the presence of the endogenous promoter in wild-type CEA10 but not in the PR mutants (Figure 1B). pNiiA-CPR mutants that have been confirmed by genotypic analysis were subjected to monoconidial purification, and the resulting monoconidial isolates were reconfirmed by PCR as described above.
Phenotypic analysis of CPR mutants.
The phenotypes of all CPR mutants were evaluated by comparing the growth phenotype on inducing (AMM plus 10 mM nitrate) and repressing (AMM plus 10 mM ammonium) conditions. Typically, the terminal growth phenotype was observed after 36 to 40 h at 30 °C by streaking conidiospores onto inducing and repressing media. Qualitative scores were assigned to each of the CPR mutants to classify the severity of the terminal growth phenotype observed. A 4+ phenotype was assigned to CPR mutants that failed to grow in repressed media (AMM plus ammonium). Other growth phenotypes were scored as 3.5+ (very strong), 3+ (strong), 2+ (mild), 1+ (minor), or 0+ (no growth phenotype) (Figure 1C).
Cidal/static phenotypic analysis.
To determine the terminal cidal/static phenotypes of A. fumigatus essential genes, 1 × 103 to 1 × 104 conidiospores were incubated in broth AMM plus 10 mM ammonium tartrate (repressing condition) for 0 (time zero control), 24, 48, and 72 h; washed with PBS twice; and then plated onto inducing media plate (AMM plus 10 mM nitrate) for CFU counting. A cidal phenotype was assigned to those genes whose conditional mutant displayed a significant CFU reduction (greater than 90%) after 24 h under repressing conditions. Slow cidal targets are those that displayed reduced cell viability after 48 or 72 h, respectively.
Immunocompromised murine model of systemic infection.
A. fumigatus strains were tested for virulence in an immunocompromised murine model for systemic infection using CD-1 male mice (22 to 25 g; Charles River Laboratories, http://www.criver.com). Mice were housed five per cage with food and water supplied ad libitum. All experiments were performed according to the National Institutes of Health guidelines for the ethical treatment of animals. Mice were rendered immunocompromised by treatment with cyclophosphamide at 150 mg/kg 4 d and 1 d prior to infection and 100 mg/kg twice a week after infection. A. fumigatus conidial suspensions were prepared from 4- to 5-d-old cultures grown on solid medium by harvesting the conidia with gentle shaking by hand using 5 ml of PBS, 0.2% Tween-20, and 3 ml of glass beads. The conidial suspension was centrifuged (3,000g, 5 min), the pellet was resuspended in 0.01% Tween-20, and the conidia were counted on a hemacytometer. Immunocompromised mice were infected by tail vein injection of 0.1 ml of a conidial suspension of 1 × 106 conidia/ml. After injection, the conidia suspensions were diluted and plated to determine the viable count of the inoculum. Mice were monitored three times daily for signs of infection, up to 12 to 22 d. Animals were considered moribund when they could no longer reach food or water. Moribund mice, along with those surviving to the end of the experiment, were killed by anesthetization followed by cervical dislocation. All tested CPR mutants were coded so that laboratory workers did not know the identity of the strains. For each strain, five mice were infected.
Essential gene validation by ORF disruption.
To disrupt the complete ORF of a target gene, a disruption cassette was constructed. Briefly, 5′-flanking sequence (approximately 1.5 kb, upstream of the ATG start codon) of the target gene was amplified by PCR using two gene-specific primers and then subcloned into the Not1/Mlu1 sites of plasmid pPyrG-pNiiA (Figure 1A). Similarly, the 3′-flanking sequence (approximately 1.5 kb, downstream of the stop codon) was amplified by PCR and subsequently subcloned into the Nde1/Pac sites of plasmid pPyrG-pNiiA. The resulting plasmid was digested using Not1/Pac1 and then introduced into CEA17 strain by protoplast transformation (see above). Transformants were incubated on ACM without uracil and uridine and then subjected to genotypic analysis by PCR using primer sets to verify the disruption junctions and ORF disruption. Gene essentiality was inferred if no genotype-confirmed transformants could be recovered from a large population of transformants (n > 80). Otherwise, if genotype-confirmed transformants were recovered, the target gene was defined as nonessential.
ERG11 double knockout.
To validate the gene essentiality of the ERG11A/ERG11B gene family, a double knockout mutant was constructed as follows. In step 1, an ERG11B null mutant (erg11BΔ) was constructed by disruption of the whole ERG11B ORF using a pyrG-based gene disruption cassette as described above. In step 2, the pyrG selection marker was recycled from erg11BΔ to facilitate the promoter replacement of ERG11A. To achieve this, the ERG11B gene disruption plasmid was digested with Mlu1 and Asc1 to remove the PyrG-pNiiA insert from the cassette and religated (Mlu1 and Asc1 provide compatible 5′ overhangs) prior to subsequent transformation with Escherichia coli. The resulting plasmid was then purified and digested with Not1 and Pac1 to excise the ERG11B insert (comprising fused 5′ and 3′ flanking sequences and no intervening pyrG-pNiiA insert) and used to transform erg11BΔ. Transformants were grown at 30 °C for 48 h on ACM containing 5 mM uridine and uricine, respectively. The conidiospores of resulting transformants were collected and then plated onto AMM plates containing 10 mM nitrate and 1 mg/ml 5-FOA to counterselect pyrG − strains. The resulting pyrG − strain was then genotyped by PCR to confirm that the pyrG − selectable marker has been removed from erg11BΔ. In step 3, the resulting pyrG − , erg11BΔ strain was then used as the starting strain to perform promoter replacement of ERG11A as described above.
Construction of TUB1 tandem duplicate mutant.
To construct TUB1 tandem duplicate mutants, TUB1 ORF plus approximately 1 kb of the DNA sequence directly downstream of the stop codon was amplified by PCR and then subcloned into the Asc1 and Pac1 sites of plasmid pPyrG-pNiiA (Figure 1A). The resulting plasmid was used to transform A. fumigatus CEA17 by protoplast transformation as described above. Transformants were incubated on AMM plus nitrate without uracil and uridine and then subjected to genotypic analysis by PCR using primer sets to verify single crossover event.
RT-PCR and real-time RT-PCR.
To perform RT-PCR, pNiiA-CPR mutants and wild-type were grown either in AMM plus nitrate (inducing) or in AMM plus ammonium (repressing) at 37 °C for 20 h. Total RNA was extracted using the RNeasy plant mini kit (Qiagen) according to the manufacturer's instructions. Total RNA samples were treated with Turbo-DNase I at 37 °C for 30 to 40 min according to the manufacturer's manual (Ambion, http://www.ambion.com). RNA samples obtained using these procedures lacked detectable genomic DNA as assayed by control PCR tests. The first-strand cDNA was synthesized by using ThermoScript RT-PCR system and oligo(dT) primers (Invitrogen, http://www.invitrogen.com) according to the manufacturer's instructions. Double-strand cDNAs were then amplified from the above first-strand cDNA by PCR using a set of gene-specific forward and reverse primers (Table S1). A typical PCR program is as the following: 94 °C for 2 min, 1 cycle; 94 °C for 30 s, 60 °C for 30 s and 72 °C for 2 min, 16 to 20 cycles; and then extension at 72 °C for 10 min. PCR products were then subject to 1% agarose-gel analysis.
To perform real-time RT-PCR, total RNA samples was extracted and treated with Turbo-DNase (Ambion), and the first-strand cDNA was synthesized as described above. Real-time PCR was then performed using the ABI 7500 thermocycler (Applied Biosystems, http://www.appliedbiosystems.com). Reactions containing cDNA, forward and reverse primers, and a fluorescent probe (integrated DNA technologies, invasive aspergillosis, and applied biosystems; see Table S2) and TaqMan Master Mix were run in triplicate using the standard conditions suggested by the manufacturer. When possible, primers and probes were specifically designed to cross adjacent exons to avoid amplification of trace genomic DNA. The mean threshold cycle value (Ct) for each sample was used to calculate cDNA abundance under inducing or repressing growth conditions, and samples were normalized based on total input RNA as previously described [43]. Changes in expression were calculated relative to the Ct for each gene expressed by a wild-type strain.
Northern blot analysis.
Northern blot analysis was carried out according to established procedures [14,15]. Briefly, total fungal RNA was prepared using the RNeasy plant mini kit (Qiagen) according to the manufacturer's instructions. From 5 to 10 μg of total RNA was used for each sample. P32-labeled ALR1 and ACT1 probes were made from PCR products amplified using gene-specific primers (Table S1).
Drug susceptibility testing.
MIC testing of antifungal drugs against A. fumigatus wild-type strain CEA10 and conditional mutants constructed from promoter replacement was performed by broth microdilution as described by the National Committee for Clinical Laboratory Standards with minor modifications [44]. The A. fumigatus inoculum was adjusted to concentrations of approximately 2 × 103 conidiospores/ml in AMM, and an aliquot of 0.1 ml was added to microtiter wells containing 0.1 ml of antifungal drug solutions in AMM. For inducing conditions, AMM plus 10 mM Mg(NO3)2 was used. To sensitize the CPR mutants, ammonium tartrate was added to AMM plus 10 mM nitrate at a final concentration of 10 to 160 μM. The microtiter plate was incubated at 37 °C for 48 h, and the MICs were determined as the lowest concentration that completely inhibited mycelia growth.
Supporting Information
(A) Phase contrast microscopy (×160) demonstrates normal hyphal growth of pNiiA-nudC mutants incubated under inducing conditions (16 h at 30 °C).
(B) Phase contrast microscopy demonstrates pNiiA-nudC conidia typically failed to germinate (greater than 70%) and a minority (less than 30%) formed short growth-arrested germ tubes when incubated under repressing conditions (16 h at 30 °C).
(C and D) Higher magnification (×400) phase contrast microscopy and DAPI staining of pNiiA-nudC mutant incubated under the same conditions as above.
(E and F) DAPI staining demonstrates a nuclear distribution phenotype of the pNiiA-nudC mutant as multiple nuclei remain inside the ungerminated conidia and short growth-arrested germ tubes under repressing conditions (F) but not in mycelia under inducing conditions (E).
(5.3 MB PPT)
pNiiA-CPR mutants were grown in AMM plus nitrate (In) or AMM plus ammonium (Re) at 37 °C for 20 h, and total RNA was extracted and normalized. To monitor and ensure even sample loading, RT-PCRs for the ACT1 transcript were also performed using identical samples (unpublished data). Standard PCR was also performed to confirm that there is no detectable genomic-DNA contamination (unpublished data).
(197 KB PPT)
To compare terminal phenotypes characterized in Figure 4, pNiiA-CPR mutants of GFA1, TUB1, SEC31, SLY1, ERG10, HEM15, PRI1, and FKS1 were grown on AMM plus nitrate (inducing conditions) at 30 °C for 2 d. Micrographs were taken under a microscope at a magnification of approximately ×80.
(299 KB PPT)
(35 KB DOC)
(26 KB DOC)
Acknowledgments
We would like to especially thank Dr. Christophe d'Enfert (Institut Pasteur) for kindly providing the pyrG-containing plasmid and corresponding strains (CEA10 and CEA17) and Dr. Howard Bussey for helpful advice throughout this project. We thank Dr. Cameron Douglas for his technical help in real-time RT-PCR and for critical review of and helpful suggestions for the manuscript. We also thank Drs. William Nierman, Malcolm Whiteway, Deming Xu, and Roberto Rodriguez-Suarez for their critical review of and helpful suggestions for the manuscript.
Abbreviations
- ACM
Aspergillus complete medium
- AMM
Aspergillus minimal medium
- CFU
colony-forming units
- CPR
conditional promoter replacement
- MIC
minimum inhibitory concentration
- pNiiA
A. fumigatus NiiA nitrogen-regulatable promoter
- RT-PCR
reverse transcription PCR
Footnotes
¤ Current address: Institute of Research in Immunology and Cancer, Université de Montréal, Montreal, Quebec, Canada
Competing interests. W. Hu, S. Sillaots, S. Lemieux, J. Davison, A. Breton, A. Linteau, C. Xin, J. Bowman, B. Jiang, and T. Roemer are current or previous employees of Merck & Co., Inc. and Elitra Phamarceuticals Inc. S. Kauffman and J. Becker are consultants to Elitra Phamarceuticals Inc.
Author contributions. W. Hu and T. Roemer conceived and designed the experiments and wrote the paper. W. Hu, S. Sillaots, J. Davison, S. Kauffman, A. Breton, A. Linteau, C. Xin, and J. Bowman performed the experiments. W. Hu, S. Lemieux, S. Kauffman, J. Bowman, J. Becker, B. Jiang, and T. Roemer analyzed the data. S. Lemieux, J. Becker, and B. Jiang contributed reagents/materials/analysis tools.
Funding. This study was supported in part by a grant from Genome Canada and Genome Quebec.
References
- Latge JP. The pathobiology of Aspergillus fumigatus . Trends Microbiol. 2001;9:382–389. doi: 10.1016/s0966-842x(01)02104-7. [DOI] [PubMed] [Google Scholar]
- Tekaia F, Latge JP. Aspergillus fumigatus: Saprophyte or pathogen? Curr Opin Microbiol. 2005;8:385–392. doi: 10.1016/j.mib.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Singh N, Paterson DL. Aspergillus infections in transplant recipients. Clin Microbiol Rev. 2005;18:44–69. doi: 10.1128/CMR.18.1.44-69.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latge JP. Aspergillus fumigatus and aspergillosis. Clin Microbiol Rev. 1999;12:310–350. doi: 10.1128/cmr.12.2.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach WS, Latgé JP, Stevens DA. Advances against aspergillosis. Med Mycol. 2005;43:S1. [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, et al. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet. 1999;21:278–283. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- Ross-Macdonald P, Coelho PS, Roemer T, Agarwal S, Kumar A, et al. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature. 1999;402:413–418. doi: 10.1038/46558. [DOI] [PubMed] [Google Scholar]
- De Backer MD, Nelissen B, Logghe M, Viaene J, Loonen I, et al. An antisense-based functional genomics approach for identification of genes critical for growth of Candida albicans . Nat Biotechnol. 2001;19:235–241. doi: 10.1038/85677. [DOI] [PubMed] [Google Scholar]
- Uhl MA, Biery M, Craig N, Johnson AD. Haploinsufficiency-based large-scale forward genetic analysis of filamentous growth in the diploid human fungal pathogen C. albicans . EMBO J. 2003;22:2668–2678. doi: 10.1093/emboj/cdg256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno VM, Kalachikov S, Subaran R, Nobile CJ, Kyratsous C, et al. Control of the C. albicans cell wall damage response by transcriptional regulator Cas5. PLoS Pathog. 2006;2:e21. doi: 10.1371/journal.ppat.0020021. doi: 10.1371/journal.ppat.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer T, Jiang B, Davison J, Ketela T, Veillette K, et al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol Microbiol. 2003;50:167–181. doi: 10.1046/j.1365-2958.2003.03697.x. [DOI] [PubMed] [Google Scholar]
- Firon A, Beauvais A, Latge JP, Couve E, Grosjean-Cournoyer MC, et al. Characterization of essential genes by parasexual genetics in the human fungal pathogen Aspergillus fumigatus: Impact of genomic rearrangements associated with electroporation of DNA. Genetics. 2002;161:1077–1087. doi: 10.1093/genetics/161.3.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firon A, Villalba F, Beffa R, D'Enfert C. Identification of essential genes in the human fungal pathogen Aspergillus fumigatus by transposon mutagenesis. Eukaryot Cell. 2003;2:247–255. doi: 10.1128/EC.2.2.247-255.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouyna I, Henry C, Doering TL, Latge JP. Gene silencing with RNA interference in the human pathogenic fungus Aspergillus fumigatus . FEMS Microbiol Lett. 2004;237:317–324. doi: 10.1016/j.femsle.2004.06.048. [DOI] [PubMed] [Google Scholar]
- Romero B, Turner G, Olivas I, Laborda F, De Lucas JR. The Aspergillus nidulans alcA promoter drives tightly regulated conditional gene expression in Aspergillus fumigatus permitting validation of essential genes in this human pathogen. Fungal Genet Biol. 2003;40:103–114. doi: 10.1016/s1087-1845(03)00090-2. [DOI] [PubMed] [Google Scholar]
- Vogt K, Bhabhra R, Rhodes JC, Askew DS. Doxycycline-regulated gene expression in the opportunistic fungal pathogen Aspergillus fumigatus . BMC Microbiol. 2005;5:1–11. doi: 10.1186/1471-2180-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaar YG, Moore MM. Mapping of the nitrate-assimilation gene cluster (crnA-niiA-niaD) and characterization of the nitrite reductase gene (niiA) in the opportunistic fungal pathogen Aspergillus fumigatus . Curr Genet. 1998;33:206–215. doi: 10.1007/s002940050328. [DOI] [PubMed] [Google Scholar]
- Muro-Pastor MI, Gonzalez R, Strauss J, Narendja F, Scazzocchio C. The GATA factor AreA is essential for chromatin remodelling in a eukaryotic bidirectional promoter. EMBO J. 1999;18:1584–1597. doi: 10.1093/emboj/18.6.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HF, Wheeler MH, Chang YC, Kwon-Chung KJ. A developmentally regulated gene cluster involved in conidial pigment biosynthesis in Aspergillus fumigatus . J Bacteriol. 1999;181:6469–6477. doi: 10.1128/jb.181.20.6469-6477.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus . Nature. 2005;438:1151–1156. doi: 10.1038/nature04332. [DOI] [PubMed] [Google Scholar]
- Kurtz MB, Heath IB, Marrinan J, Dreikorn S, Onishi J, et al. Morphological effects of lipopeptides against Aspergillus fumigatus correlate with activities against (1,3)-beta-D-glucan synthase. Antimicrob Agents Chemother. 1994;38:1480–1489. doi: 10.1128/aac.38.7.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellado E, Diaz-Guerra TM, Cuenca-Estrella M, Rodriguez-Tudela JL. Identification of two different 14-alpha sterol demethylase-related genes (cyp51A and cyp51B) in Aspergillus fumigatus and other Aspergillus species. J Clin Microbiol. 2001;39:2431–2438. doi: 10.1128/JCM.39.7.2431-2438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellado E, Garcia-Effron G, Buitrago MJ, Alcazar-Fuoli L, Cuenca-Estrella M, et al. Targeted gene disruption of the 14-alpha sterol demethylase (cyp51A) in Aspergillus fumigatus and its role in azole drug susceptibility. Antimicrob Agents Chemother. 2005;49:2536–2538. doi: 10.1128/AAC.49.6.2536-2538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Zhao D, Solenov E, Verkman AS. Evidence from knockout mice against physiologically significant aquaporin 8-facilitated ammonia transport. Am J Physiol Cell Physiol. 2006;291:C417–C423. doi: 10.1152/ajpcell.00057.2006. [DOI] [PubMed] [Google Scholar]
- D'Enfert C, Diaquin M, Delit A, Wuscher N, Debeaupuis JP, et al. Attenuated virulence of uridine-uracil auxotrophs of Aspergillus fumigatus . Infect Immun. 1996;64:4401–4405. doi: 10.1128/iai.64.10.4401-4405.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum PY, Armour CD, Stepaniants SB, Cavet G, Wolf MK, et al. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell. 2004;116:121–137. doi: 10.1016/s0092-8674(03)01035-3. [DOI] [PubMed] [Google Scholar]
- Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae . Nature. 2005;438:1105–1115. doi: 10.1038/nature04341. [DOI] [PubMed] [Google Scholar]
- Beauvais A, Latge JP. Membrane and cell wall targets in Aspergillus fumigatus . Drug Resist Update. 2001;4:38–49. doi: 10.1054/drup.2001.0185. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Cerione R, Bender A. Control of the yeast bud-site assembly GTPase Cdc42. Catalysis of guanine nucleotide exchange by Cdc24 and stimulation of GTPase activity by Bem3. J Biol Chem. 1994;26:2369–2372. [PubMed] [Google Scholar]
- Ozaki K, Tanaka K, Imamura H, Hihara T, Kameyama T, et al. Rom1p and ROM2p are GDP/GTP exchange proteins (GEPs) for the Rho1p small GTP binding protein in Saccharomyces cerevisiae . EMBO J. 1996;15:2196–2207. [PMC free article] [PubMed] [Google Scholar]
- Diaz-Guerra TM, Mellado E, Cuenca-Estrella M, Rodriguez-Tudela JL. A point mutation in the 14alpha-sterol demethylase gene cyp51A contributes to itraconazole resistance in Aspergillus fumigatus . Antimicrob Agents Chemother. 2003;47:1120–1124. doi: 10.1128/AAC.47.3.1120-1124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann PA, Parmegiani RM, Wei SQ, Mendrick CA, Li X, et al. Mutations in Aspergillus fumigatus resulting in reduced susceptibility to posaconazole appear to be restricted to a single amino acid in the cytochrome P450 14alpha-demethylase. Antimicrob Agents Chemother. 2003;47:577–581. doi: 10.1128/AAC.47.2.577-581.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento AM, Goldman GH, Park S, Marras SA, Delmas G, et al. Multiple resistance mechanisms among Aspergillus fumigatus mutants with high-level resistance to itraconazole. Antimicrob Agents Chemother. 2003;47:1719–1726. doi: 10.1128/AAC.47.5.1719-1726.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, et al. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci U S A. 2006;103:10352–10357. doi: 10.1073/pnas.0601456103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krappmann S, Sasse C, Braus GH. Gene targeting in Aspergillus fumigatus by homologous recombination is facilitated in a nonhomologous end-joining–deficient genetic background. Eukaryot Cell. 2006;5:212–215. doi: 10.1128/EC.5.1.212-215.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Ferreira ME, Kress MR, Savoldi M, Goldman MH, Hartl A, et al. The akuB(KU80) mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus . Eukaryot Cell. 2006;5:207–211. doi: 10.1128/EC.5.1.207-211.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Izuta M, Nakayama N, Arisawa M, Aoki Y. Depletion of the squalene synthase (ERG9) gene does not impair growth of Candida glabrata in mice. Antimicrob Agents Chemother. 2000;44:2411–2418. doi: 10.1128/aac.44.9.2411-2418.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Nakayama N, Arisawa M, Aoki Y. In vitro and in vivo effects of 14alpha-demethylase (ERG11) depletion in Candida glabrata . Antimicrob Agents Chemother. 2001;45:3037–3045. doi: 10.1128/AAC.45.11.3037-3045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Enfert C. Selection of multiple disruption events in Aspergillus fumigatus using the orotidine-5′-decarboxylase gene, pyrG, as a unique transformation marker. Curr Genet. 1996;30:76–82. doi: 10.1007/s002940050103. [DOI] [PubMed] [Google Scholar]
- Cove DJ. The induction and repression of nitrate reductase in the fungus Aspergillus nidulans . Biochim Biophys Acta. 1966;113:51–56. doi: 10.1016/s0926-6593(66)80120-0. [DOI] [PubMed] [Google Scholar]
- Verdoes JC, Punt PJ, van der Berg P, Debets F, Stouthamer AH, et al. Characterization of an efficient gene cloning strategy for Aspergillus niger based on an autonomously replicating plasmid: Cloning of the nicB gene of A. niger . Gene. 1994;146:159–165. doi: 10.1016/0378-1119(94)90288-7. [DOI] [PubMed] [Google Scholar]
- Tilburn J, Scazzocchio C, Taylor GG, Zabicky-Zissman JH, Lockington RA, et al. Transformation by integration in Aspergillus nidulans . Gene. 1983;26:205–221. doi: 10.1016/0378-1119(83)90191-9. [DOI] [PubMed] [Google Scholar]
- Bustin SA. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): Trends and problems. J Mol Endocrinol. 2002;29:23–39. doi: 10.1677/jme.0.0290023. [DOI] [PubMed] [Google Scholar]
- National Committee for Clinical Laboratory Standards. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi. Wayne (Pennsylvania): National Committee for Clinical Laboratory Standards; 2002. 7 Document M-38A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Phase contrast microscopy (×160) demonstrates normal hyphal growth of pNiiA-nudC mutants incubated under inducing conditions (16 h at 30 °C).
(B) Phase contrast microscopy demonstrates pNiiA-nudC conidia typically failed to germinate (greater than 70%) and a minority (less than 30%) formed short growth-arrested germ tubes when incubated under repressing conditions (16 h at 30 °C).
(C and D) Higher magnification (×400) phase contrast microscopy and DAPI staining of pNiiA-nudC mutant incubated under the same conditions as above.
(E and F) DAPI staining demonstrates a nuclear distribution phenotype of the pNiiA-nudC mutant as multiple nuclei remain inside the ungerminated conidia and short growth-arrested germ tubes under repressing conditions (F) but not in mycelia under inducing conditions (E).
(5.3 MB PPT)
pNiiA-CPR mutants were grown in AMM plus nitrate (In) or AMM plus ammonium (Re) at 37 °C for 20 h, and total RNA was extracted and normalized. To monitor and ensure even sample loading, RT-PCRs for the ACT1 transcript were also performed using identical samples (unpublished data). Standard PCR was also performed to confirm that there is no detectable genomic-DNA contamination (unpublished data).
(197 KB PPT)
To compare terminal phenotypes characterized in Figure 4, pNiiA-CPR mutants of GFA1, TUB1, SEC31, SLY1, ERG10, HEM15, PRI1, and FKS1 were grown on AMM plus nitrate (inducing conditions) at 30 °C for 2 d. Micrographs were taken under a microscope at a magnification of approximately ×80.
(299 KB PPT)
(35 KB DOC)
(26 KB DOC)