

Abstract
Sphingolipids are ubiquitous and essential components of eukaryotic membranes, particularly the plasma membrane. The biosynthetic pathway for the formation of these lipid species is conserved up to the formation of sphinganine. However, a divergence is apparent in the synthesis of complex sphingolipids. In animal cells, ceramide is a substrate for sphingomyelin (SM) production via the enzyme SM synthase. In contrast, fungi utilise phytoceramide in the synthesis of inositol phosphorylceramide (IPC) catalysed by IPC synthase. Due to the absence of a mammalian equivalent, this essential enzyme represents an attractive target for anti-fungal compounds. In common with the fungi, the kinetoplastid protozoa (and higher plants) synthesize IPC rather than SM. However, orthologues of the gene believed to encode the fungal IPC synthase (AUR1) are not readily identified in the complete genome databases of these species. By utilising bioinformatic and functional genetic approaches, we have isolated a functional orthologue of AUR1 in the kinetoplastids, causative agents of a range of important human diseases. Expression of this gene in a mammalian cell line led to the synthesis of an IPC-like species strongly indicating that IPC synthase activity is reconstituted. Furthermore the gene product can be specifically inhibited by an anti-fungal targeting IPC synthase. We propose that the kinetoplastid AUR1 functional orthologue encodes an enzyme which defines a new class of protozoan sphingolipid synthase. The identification and characterisation of the protozoan IPC synthase, an enzyme with no mammalian equivalent, will raise the possibility of developing anti-protozoal drugs with minimal toxic side-affects.
INTRODUCTION
The evolutionarily divergent, insect vector borne protozoan parasites of the order Kinetoplastidae cause a range of human diseases, including the leishmaniases (a broad spectrum of infections caused by Leishmania species), Chagas’ disease (Trypanosoma cruzi) and African sleeping sickness (T. brucei). These infections are of increasing prevalence in developing countries where mortality is often affected by poor access to health care. Many of the drugs available to treat these diseases are expensive, difficult to administer or toxic and no effective vaccines are available (1,2). These facts make the discovery of new anti-kinetoplastid therapeutic targets and compounds of paramount importance (http://www.who.int/tdr).
Sphingolipids are amphipathic lipids that have a sphingoside backbone with a long-chain fatty acid and a polar alcohol as attachments. Ceramide is an unmodified sphingolipid that functions as a key player in cell signalling and as a precurser of complex sphingolipids which are major components of the outer leaflet of eukaryotic plasma membranes thought to be involved, together with sterols, in the formation of micro-domains known as lipid rafts (3). These rafts have been proposed to function in a diverse array of processes from the polarised trafficking of lipid-modified proteins (4), to the assembly and activation of signal transduction complexes (5,6). Unlike mammalian cells, which synthesize sphingomyelin (SM), the primary complex sphingolipid species in Leishmania spp. and Trypanosoma spp. is inositol phosphorylceramide (IPC) (7). Similarly, in fungi and plants, IPC and its glycosylated derivatives are abundant complex sphingolipid species (7,8) (see figure 1). IPC is produced by the transfer of a phosphorylinositol head group from phosphatidylinositol (PI) to ceramide, a process catalysed by IPC synthase (9). In the fungi, IPC synthase or a subunit of this enzyme is believed to be encoded by the AUR1 gene that was first identified in Saccharomyces cerevisiae and subsequently in several pathogenic species (10). As in the protozoan parasites, the search for specific drug targets in the pathogenic fungi is hampered by the conservation of biochemical processes between the eukaryotic microbe and its eukaryotic host. Consequently, as for anti-protozoan therapies, many of the available anti-fungals exhibit toxic side-effects. IPC synthase has no functional equivalent in mammals. As such this enzyme activity has been established as a target for the development of anti-fungal compounds (11), a number of which have been characterized (12).
Figure 1.
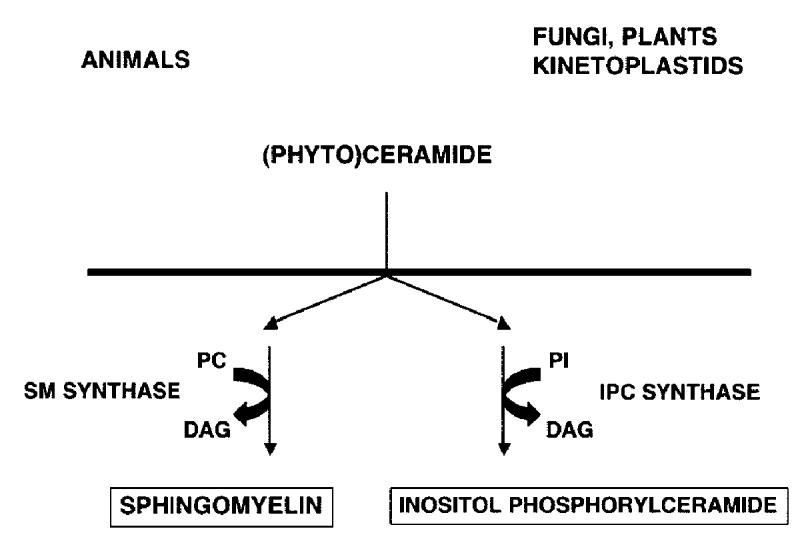
Schematic illustrating the dichotomy of complex sphingolipid biosynthesis: mammals (and other animals) producing sphingomyelin (SM) via SM synthase; and fungi, plants and kinetoplastids synthesizing inositol phosphorylceramide (IPC) utilising IPC synthase. Adapted from Denny and Smith, 2004 (44).
No homologue of the fungal AUR1 gene has been found in either the complete protozoan or plant genome sequence databases. However, given the efficacy of known yeast inhibitors against the plant IPC synthase activity, it appears likely that the enzyme is conserved in plants at least (13). The biosynthesis of sphingoid base and ceramide, precursors of complex sphingolipids, are non-essential for Leishmania pathogenesis in an animal model system (14). However, it is known that intra-macrophage amastigotes possess an active IPC synthase presumably utilising precursors from alternative sources (15). Indeed it has been shown that L. donovani stimulates host macrophages to up-regulate the production of ceramide, a precursor of IPC and a substrate of IPC synthase (see figure 1) (16). Moreover, sphingolipid synthesis is essential for replication of pathogenic bloodstream form T. brucei (Denny et al. in preparation). IPC synthase activity has been characterised in T. cruzi (17) where it has been suggested that IPC synthesis is important for infectivity (18). In this context, it should be noted that IPC synthase is believed to play a pivotal role in the pathogenesis of the fungus Cryptococcus neoformans which, like Leishmania spp. and T. cruzi, resides in an acidic, macrophage phagolysosome. Genetically induced down-regulation of the C. neoformans AUR1 confers an in vivo growth defect through a pH-dependent mechanism (19).
Here we describe the identification and characterisation of an AUR1 functional orthologue in Leishmania major, the putative protozoan IPC synthase (LmIPCS). It is proposed that this protein defines a new class of eukaryotic sphingolipid synthases. In addition, the development of an ex vivo assay system has allowed the identification of an inhibitor with specific activity against LmIPCS. This work will facilitate the rational development of inhibitors against a protozoan enzyme with no mammalian equivalent, leading to the prospect of anti-protozoal compounds with minimal toxic side effects.
EXPERIMENTAL PROCEDURES
Selection and cloning of a candidate AUR1 orthologue
Candidate genes were identified in a Motif Search of the genome sequence databases of L. major, T. brucei and T. cruzi using the sequence motif H-[YFWH]-X2-D-[VLI]-X2-[GA]-X3-[GSTA] (Sanger GeneDB). Candidates were then further selected using the additional criteria described in the Results.
The Leishmania candidate, CDS - LmjF35.4990, was amplified from genomic DNA (14) using Pfu polymerase (Stratagene) and the primer pairs: 5’LmIPCSBamHI (5’cgc gga tcc ATG ACG AGT CAC GTG ACA GC) and 3’LmIPCS*HindIII (ccc aag ctt TTA GTG CTC AGG CAA AGC CGC CG); and 5’LmIPCSBamHI and 3’LmIPCSHindIII (ccc aag ctt GTG CTC AGG CAA AGC CGC CG). The products were cloned into the yeast and mammalian expression vectors pRS426MET25 and pcDNA3.1/Myc-HisA (Invitrogen), creating pRS426 LmIPCS and pcDNA3.1A LmIPCSMyc-His respectively. The S. cerevisiae AUR1 gene was similarly amplified from genomic DNA (Invitrogen) using the primer pair AURFXbaI (cat aga tct aga ATG GCA AAC CCT TT TTC GAG ATG G) and 3’ScIPCS*HindIII (ccc aag ctt TTA AGC CCT CTT TAC ACC TAG TGA CG), and the product cloned into pRS426MET25 to create pRS426 ScAUR1. Cloning sites are shown in lower case, with Leishmania and S. cerevisiae sequence in upper case.
Construction of YPH499-HIS-GAL-AUR1 auxotrophic S. cerevisiae strain
YPH499-HIS-GAL-AUR1 S. cerevisiae strain was constructed in YPH499 [Mat a; ura3-52; lys2-801amber; ade2-101ochre; trp1-63; his3-200; leu2-1] (Stratagene) by bringing the expression of the yeast AUR1 gene under the control of the stringently regulated GAL1 promoter that is repressed in the presence of glucose (20). The AUR1 promoter in the yeast genome was exchanged by a selection marker/promoter HIS/GAL1-cassette using previously described methodology (21). The primer sequences for amplification of the HIS-GAL cassette were chosen as follows: (a) sequence for integration upstream of the coding region (nucleotides -200 to - 150) - AuriHISGalS 5′GGT AGT TGG TTA GTC CGA TCG CTC ACT TTT GGT TGT TGT TAA GTA CTT CAG GGC GAA TTG GAG CTC CAC3′; (b) sequence for integration at the initiation codon (nucleotides +1 to +50) - AuriHISGalAS 5′CAG TTT GGA GGT CTC TCT GAT AGA AAC CAT CTC GAA AAA GGG TTT GCC ATG GGG ATC CAC TAG TTC TAG3′. The numbers indicate the nucleotide positions in the S. cerevisiae DNA sequence, with the adenosine of the ATG initiation codon being defined as position +1. The 19 bp sequences at the 3′ ends of these oligonucleotides that are homologous to the sequences of the vector pGAL/HIS3 (22) and serve as a template for amplification of the GAL1/HIS3-cassette are underlined. Transformation into the haploid YPH499 strain, selection on minimal medium lacking histidine but containing galactose and confirmation of the insertion of the HIS-GAL fragment were performed as previously described (23). YPH499-HIS-GAL-AUR1 was maintained in SGR medium (4% galactose, 2% raffinose, 0.17% Bacto yeast nitrogen base, 0.5% ammonium sulphate) with galactose/raffinose rather than non-permissive dextrose as the carbohydrate source. For rapid cultivation of the mutant, YPGR medium (4% galactose, 2% raffinose, 1% yeast extract, 2% peptone) was routinely used.
Identification of Leishmania AUR1 orthologue
The YPH499-HIS-GAL-AUR1 S. cerevisiae strain was transformed with pRS426 ScAUR1 or pRS426 LmIPCS and functionally complemented transformants selected on non-permissive SD medium (0.17% Bacto yeast nitrogen base, 0.5% ammonium sulphate and 2% dextrose) containing the nutritional supplements necessary to allow selection of transformants.
Sequence analyses
Based on primary sequence, orthologues of LmIPCS (CDS - LmjF35.4990) were found by BLAST search of the T. brucei and T. cruzi databases (Sanger GeneDB) using WU-BLASTp (Gish, W. (1996-2001) http://blast.wustl.edu). Sequence alignments were made using CLUSTALW (24). Phylogenetic analyses were performed on the edited alignments using maximum parsimony (Protpars, Felsenstein, J. 1993. PHYLIP (Phylogeny Inference Package version 3.5c); distributed by the author, Department of Genetics, University of Washington, Seattle, USA). Topology predictions were performing using the PHD package (25).
Expression of LmIPCS in a mammalian cell line
Human ARF1 was amplified from lymph node cDNA (Clontech) using the primers hARF1F (5′CCT GTC CAC AAG CAT GGG GAA CAT3′ and hARF1R, 5′CCT TCT GGT TCC GGA GCT GAT TGG3′. The fragment was cloned into pcDNA3.1/CT-GFP-TOPO (Invitrogen) to produce the construct pcDNA3.1 hARF1GFP.
The cell line HEK293 was grown in Dulbecco′s modified eagle medium (DMEM), containing 10% FCS, 100 U/ml penicillin and 100 U/ml streptomycin at 37°C in a humidified atmosphere with 5% CO2.
For transient transfection, cells were seeded in 6-well plates +/- glass coverslips at a cell density of 2 × 105 cells per well, and allowed to grow for 24 hours. Cells were then transfected with pcDNA3.1A LmIPCSMyc-His, pcDNA3.1 hARF1GFP, pcDNA3.1/Myc/His/lacZ or pcDNA3.1A vector alone, using Fugene 6 Transfection Reagent (Roche). For each well, 3 μl of Fugene reagent was combined with 2 μg of plasmid DNA. For co-transfections, 6 μl of Fugene was used per well, with 2 μg of each construct.
Immunofluorescence
36 hours post-transfection HEK293cells were fixed with 4% paraformaldehyde (w/v) for 45 minutes at RT. Expressed LmIPCS was detected by indirect immunofluorescence. Cells were washed in PBS, permeabilised in 0.5% Triton X-100/PBS (v/v) for 10 mins and then blocked in 10% FCS/PBS (v/v) for 1 hour. Samples were incubated with rabbit polyclonal anti-myc (Abcam, 1:200 dilution) in blocking solution for 1 hour. After washing in PBS, cells were incubated in Alexa Fluor 633 goat anti-rabbit IgG (Invitrogen, 1:250) in blocking solution for 1 hour. After washing in PBS, coverslips were mounted on slides using Vectashield with DAPI (Vector Laboratories, UK). Samples were visualized by confocal microscopy using a Zeiss LSM 510 meta with a Plan-Apochromat 63x/1.4 Oil DIC I objective lens. Images were acquired using LSM 510 version 3.2 software (Zeiss).
Metabolic labelling and analyses
HEK293 cells, 36 hours post-transfection, were labelled for 16 hours in DMEM supplemented with 10% foetal calf serum and 20 μCi/ml of myo-[3H] inositol (102 Ci/mmol) (Amersham). These cells were also labelled with NBD C6 –ceramide complexed with BSA (Molecular Probes) for 1 hour at 37°C in serum-free media as previously described (26). L. major (MHOM/IL/81/Friedlin; FV1 strain) maintained as previously described (14) were similarly labelled with NBD ceramide but at 26°C for 2 hours. Lipids were extracted and analysed as previously described (27,28), although the NBD ceramide labelled samples were, following fractionation by thin layer chromatography, scanned using a FLA3000 (Fuji).
Logarithmic phase yeast (unless stated otherwise) were harvested and incubated at 10 OD600/ml in 0.25 ml inositol-free SD medium at 30°C for 20 minutes. 10 μCi [3H]inositol were added and the cells incubated for 40 minutes. 0.75 ml of fresh medium was added and the incubation continued for 80 minutes. Yeast were also labelled with C6 -ceramide complexed with BSA as previously described (29). Cells were harvested by centrifugation and washed twice with 1 M sorbitol. Chloroform/methanol (0.4 ml; 1:1 v/v) was added and cells were disintegrated with glass beads. The pellet was re-extracted several times with chloroform/methanol/water (10:10:3, per vol.), the combined supernatants dried and the lipids prepared and analysed as previously described (27,28). Again NBD ceramide labelled samples were imaged using a FLA3000 (Fuji).
Agar diffusion assay
Wild type YPH499 yeast and the transgenic strain YPH499-HIS-GAL-AUR1 complemented with ScAUR1 or LmIPCS were assayed for susceptibility to Aureobasidin A (Takara), myriocin (Sigma) and cycloheximide (Sigma) as previously described (30). Briefly, 2.4×107 logarithmically dividing cells were embedded in 15 ml of YPD agarose (2% dextrose, 1% yeast extract, 2% peptone, 0.8% agarose) on 100mm2 square Petri dishes (Sarstedt). Inhibitors were applied in DMSO at the concentrations described below and the dishes incubated at 30°C.
RESULTS
Functional cloning of the Leishmania AUR1 orthologue
Candidate genes encoding the putative protozoan protein were selected using a bioinformatic approach based on that used in the identification of the metazoan enzyme sphingomyelin (SM) synthase (29). Briefly, a conserved motif shared by the lipid phosphate phosphatase (LPP) family, fungal AUR1p proteins and animal sphingolipid synthases (H-[YFWH]-X2-D-[VLI]-X2-[GA]-X3-[GSTA]) was used to screen the genome sequence databases of the kinetoplastids (www.genedb.org/). Of the 8 candidates identified, one was of unknown function, had sequence orthologues in both Trypanosoma and Leishmania spp., had transmembrane domains (TMDs) consistent with a Golgi localised IPC synthase and had no orthologue in mammalian cells (which do not possess an IPC activity). The gene encoding this putative protozoan IPC synthase is present as a single copy in L. major. In contrast, the trypanosomes possess multiple non-identical loci, 2 in T. cruzi, and 4 in T. brucei. The Leishmania candidate was amplified and cloned into a URA3 selectable yeast expression vector to create pRS426 LmIPCS. In the auxotrophic yeast strain YPH499-HIS-GAL-AUR1 (see Experimental Procedures), the essential AUR1 gene, believed to encode at least part of an IPC synthase (31), is under the control of the GAL1 promoter and so is repressed in the presence of glucose. In non-permissive glucose-containing SD media, transformation with pRS426 LmIPCS restored the growth of YPH499-HIS-GAL-AUR1, as did the ectopic expression of S. cerevisiae AUR1p (ScAUR1) (figure 2A). The wild type (YPH499) cannot grow without uracil.
Figure 2.

-
Rescue of YPH499-HIS-GAL-AUR1 by complementation with either the yeast AUR1 (ScAUR1) or a predicted Leishmania orthologue identified using bioinformatics (LmIPCS). YPH499 - wild type; pRS426 - YPH499-HIS-GAL-AUR1 pRS426MET25; ScAUR1 - YPH499-HIS-GAL-AUR1 pRS426 ScAUR1; LmIPCS - YPH499-HIS-GAL-AUR1 pRS426 LmIPCS.SD - synthetic minimal media supplemented (or not) with histidine (HIS) and uracil (URA) for selection of the pRS426MET25 plasmid.
-
YPH499-HIS-GAL-AUR1 is deficient in inositol sphingolipid biosynthesis when incubated in non-permissive, glucose-containing SD media, however complementation with either yeast AUR1 (ScAUR1) or the protozoan orthologue (LmIPCS) open reading frames facilitates synthesis of a base-resistant inositol sphingolipid profile identical to that of wild type yeast. Material extracted from wild type YPH499, IPT1 mutant RCD113 (45) and YPH499-HIS-GAL-AUR1 transformed with pRS426MET25, pRS426 ScAUR1 or pRS426 LmIPCS, maintained in media indicated and inositol labelled as described in Experimental Procedures. The base resistant lipids (BASE +, indicated left) which co-migrated with base sensitive PI species (BASE -, indicated right) are designated as IPC and are likely to represent species with varying levels of hydroxylation of the sphinganine and fatty acid moieties (7,46). Unlike the other samples YPH499-HIS-GAL-AUR1 pRS426 was labelled, in both permissive (SGR) and non-permissive (SD) media, when at the stationary phase of growth. It is likely that this accounts for the minor differences in the labelled profile. The identities of the base resistant sphingolipid species (indicated left) were established by comparison with the S. cerevisiae IPT1 mutant strain RCD113 which is deficient in M(IP)2C synthesis and accumulates MIPC (45).IPC - inositol phosphorylceramide; MIPC - mannose inositol phosphorylceramide; M(IP)2C - mannose (inositol phosphorus)2 ceramide; PI - phosphatidylinositol; X - unknown; O - origin; SD - synthetic minimal media with glucose; SGR - synthetic minimal media with galactose.
Biochemical analysis of the YPH499-HIS-GAL-AUR1 strain showed that suppression of AUR1 expression by incubation in non-permissive SD media leads to significantly reduced incorporation of tritiated inositol into base-resistant lipid species. This strongly suggests that IPC synthase activity is down-regulated under these conditions (figure 2B). The auxotrophic mutant strain transformed with empty pRS426MET25 vector was grown for 20 hours in either SD or permissive SGR media. At this point, both sets of cells were in the stationary phase of growth and 100% viable (as shown by a trypan blue exclusion assay and plating on permissive media; data not shown). Furthermore when grown in non-permissive media YPH499-HIS-GAL-AUR1 demonstrated cell clumping (data not shown) perhaps mirroring the pleomorphic defects in cell morphology and division previously observed in AUR1 mutants of S. cerevisiae and S. pombe (31,32). Analysis of the complemented YPH499-HIS-GAL-AUR1 line confirmed LmIPCS as a functional orthologue of AUR1p. YPH499-HIS-GAL-AUR1 rescued using either pRS426 ScAUR1 or pRS426 LmIPCS and labelled with [3H]-inositol demonstrated a base-resistant sphingolipid profile identical to that observed in the wild type (figure 2B). However, in the LmIPCS complemented line the relative quantity of labelled mannose inositol phosphorylceramide (MIPC) was significantly diminished. In addition, an accumulation of base-sensitive lyso-PI and lipid X was evident (figure 2B). A yeast mutant deficient in ceramide synthesis, with a consequently low level of IPC synthase activity, exhibits a similar phenotype (33), suggesting that LmIPCS does not function optimally in this system. This conclusion is supported by the relatively slow growth of the LmIPCS complemented yeast (data not shown). Despite this phenotype, these complementation data establish LmIPCS as a functional orthologue of S. cerevisiae AUR1p, the putative IPC synthase.
The Leishmania AUR1 orthologue defines a new class of sphingolipid synthase
Based on primary sequence identity, orthologues of LmIPCS were identified in the related kinetoplastids, T. cruzi and T. brucei (see Experimental Procedures), suggesting that IPC synthase is a ubiquitous feature of this important group of flagellated parasites. Like Leishmania spp., T. cruzi synthesizes IPC as its predominant complex sphingolipid and this parasite is known to possess IPC synthase activity (17). Therefore the 2 closely related T. cruzi predicted proteins, sharing 52-53% sequence identity and 69-70% similarity with the Leishmania orthologue, were provisionally designated TcIPCS1 and TcIPCS2. There are no published data indicating whether T. brucei produces IPC or SM. Given this uncertainty, the 4 predicted orthologues were provisionally designated as T. brucei sphingolipid synthase (TbSLS) 1-4. The 4 TbSLS predicted proteins share 43-44% identity and 61-63% similarity with LmIPCS. The high level of conservation within these kinetoplastid species is illustrated in figure 3A, together with the 7 TMDs identified during topology prediction of LmIPCS and those regions largely conserved with respect to the animal SM synthases (29). SM synthase motifs D3 (C-G-D-X3-S-G-H-T) and D4 (H-Y-T-X-D-V-X3-Y-X6-F-X2-Y-H) are similar to the C2 and C3 motifs in LPP (29), as are domains 3 and 4 of the fungal AUR1p (34). These regions contain the histidine and aspartate residues (underlined) that comprise the catalytic triad that mediates nucleophilic attack on lipid phosphate ester bonds (29,35). Mutagenesis of this triad has been shown to inactivate fungal IPC synthase activity (36). Huitema et al (2004) predict that the D1 (P-L-P-D) and D2 (R-R-X8-Y-X2-R-X6-T) motifs are entirely unique to the SM synthases.
Figure 3.
- The predicted kinetoplastid AUR1p orthologues are highly conserved. CLUSTALW alignment. TM - predicted LmIPCS transmembranes domains; D - conserved SM synthase domains. Shaded black - 100% identical; shaded grey - 100% similarity.
- The predicted kinetoplastid proteins share a high level of similarity with the SM synthases with respect to 3 domains. Edited CLUSTALW alignments of the conserved SM synthase domains D1, D3 and D4. Equivalent to LmIPCS amino acids 67-70, 212-221 and 264-284 respectively. Shaded black - 50% identical; shaded grey - 50% similarity. * - conserved histidine and aspartate residues that form the catalytic triad.
- The kinetoplastid proteins define a new class of sphingolipid synthases. An evolutionary tree constructed using maximum parasimony of sequence equivalent to LmIPCS amino acids 77-313, a region conserved between the Metazoa, Fungi, Apicomplexa and Kinetoplastidae and including the domains within which the amino acids forming the catalytic triad are present. The sequence of the human lipid phosphate phosphatase 1 (LPP1) was the designated outgroup. Bootstrap scores >60 indicated.
T. brucei SLS1-4 accession numbers: Tb09.211.1020, Tb09.211.1000, Tb09.211.1030, Tb09.211.1010; T. cruzi IPCS1&2: Tc00.1047053506885.124, Tc00.1047053510729.290; L. major IPCS: LmjF35.4990; Aspergillus fumigatus AUR1p: AAD22750; Candida albicans AUR1p: AAB67233; Pneumocystis carinii AUR1p: CAH17867; Saccharomyces cerevisiae AUR1p: NP_012922; Schizosaccharomyces pombe AUR1p: Q10142; Plasmodium falciparum SMS 1&2: MAL6P1.178, MAL6P1.177; Caenorhabditis elegans SMS1-3: Q9U3D4, AAA82341, AAK84597; Homo sapiens SMS1&2: AB154421, Q8NHU3; Mus musculus SMS1&2: Q8VCQ6, Q9D4B1; Homo sapiens LPP1 (outgroup): O14494.
Figure A above .

Figure B above.

Figure C above.

Analysing the conserved domains D1, D3 and D4 in isolation (figure 3B), it is clear that the predicted kinetoplastid proteins possess the histidine and aspartate residues forming the catalytic triad. However, it is also evident that they demonstrate a higher degree of similarity in these regions with the animal SM synthases than with the fungal AUR1p proteins. This is clearly illustrated in the 100% identity of D1, positioned between the first two TMDs of both the animal and kinetoplastid proteins, a domain with no equivalent in the fungal enzyme. Notably, both mammal (29) and Leishmania (37) sphingolipid synthases utilise ceramide (rather than phytoceramide as in fungi) so perhaps the conserved D1 domain functions in the preferential binding of this substrate. Unlike the SM synthases (29) the protozoan D1 is predicted to face the cytosol (figure 4D). However, there is no experimental evidence to support this prediction and, furthermore, ceramide is delivered to both the lumenal and cytosolic faces of the Golgi apparatus (38). Conversely, the protozoan proteins do not possess a motif with similarity to SM synthase D2.
Figure 4.

- LmIPCS activity in mammalian HEK293 cells. HEK293 transiently expressing LmIPCS from pcDNA3.1 synthesized a base resistant, inositol-labelled lipid species (σ) that co-migrated with L. major inositol phosphorylceramide. This lipid was absent in cells treated identically but expressing lacZ only. Following base treatment (BASE +) both lines demonstrated inositol-labelled lipid species without equivalents in untreated cells (*). These were assumed to be breakdown products of base sensitive (BASE -) inositol lipid species. O - origin; PI - L. major phosphatidylinositol; IPC - L. major inositol phosphorylceramide; PIP - L. major phosphatidylinositol phosphate.
- LmIPCS activity in mammalian HEK293 cells. HEK293 cells expressing LmIPCS incorporated NBD ceramide into a novel lipid species (sample 4) absent in control cells expressing lacZ (sample 5). This sphingolipid co-migrated with NBD labelled IPC from both S. cerevisiae (sample 2) and L. major (sample 3) as demonstrated by fractionation of the mammalian together with either the yeast or protozoan material (samples 2+4 and 3+4 respectively). 1 - NBD ceramide; 2 - S. cerevisiae YPH499; 3 - L. major; 4- HEK293 pCDNA3.1 LmIPCS; 5 - HEK293 pCDNA3 lacZ; O - origin; SM - NBD labelled sphingomyelin inferred from relative mobility with respect to IPC (29); IPC - NBD labelled inositol phosphorylceramide; CER - NBD ceramide.
- LmIPCS localises to the Golgi apparatus in mammalian HEK293 cells. i) DAPI stained; ii) Human ARF1- GFP; iii) LmIPCS; iv) merge of i-iii.
- The catalytic triad of amino acids is orientated towards the Golgi lumen. Predicted membrane topology of LmIPCS.
These data indicate that the protozoan enzymes are not closely related to fungal AUR1p proteins, despite exhibiting an equivalent function at least in the case of Leishmania. Further, phylogenetic analysis of both predicted SM and IPC synthase sequences using maximum parsimony demonstrated that the kinetoplastid proteins form a distinct clade and as such are proposed to define a new class of sphingolipid synthases (figure 3C). This analysis failed to place the putative SM synthases of the malaria parasite, Plasmodium falciparum (PfSMS) (29), raising a question over their classification and evolutionary origin.
Functional analyses of LmIPCS activity in a mammalian cell line
As described above, mammalian cells do not possess either IPC synthase or IPC (figure 1). In order to assay the activity of LmIPCS in a mammalian system, an epitope-tagged gene copy was ectopically expressed in HEK293 cells and the cells subsequently labelled with [3H]-inositol to screen for the synthesis of inositol-containing lipids or NBD-ceramide for the analysis of sphingolipid synthesis. Fractionation of inositol labelled lipids by thin-layer chromatography demonstrate the presence of a base-resistant inositol-containing lipid co-migrating with Leishmania IPC (figure 4A). The negative control, HEK293 cells transfected with lacZ, do not synthesize this lipid species. Leishmania spp. IPC predominantly contains C16 and C18 sphinganine and fatty acid moieties (37,39,40), more similar to the sphingomyelin in mammalian cells (41) than the very long chain fatty acid group predominant in S. cerevisiae IPC species (42). Additional analysis of NBD ceramide labelled cells demonstrated that expression of LmIPCS in the mammalian cell line leads to production of a ceramide-containing species that co-migrates with both Leishmania and yeast IPC (figure 4B). These results strongly indicate that the Leishmania LmIPCS gene encodes a protein that can constitute IPC synthase activity in a null background.
AUR1p has been localised to the Golgi apparatus in S. cerevisiae (36), whilst in mammalian cells the 2 distinct isoforms of SM synthase have been localised to the Golgi and the plasma membrane (29). Immunolocalisation of epitope-tagged LmIPCS in HEK293 cells demonstrated that the protozoan enzyme is concentrated within the Golgi apparatus as identified by the co-localisation of a GFP-tagged marker, ARF 1 (figure 4C). In addition, minor quantities of the protein also localise to the plasma membrane when the expression levels are high (figure 4C). Topology prediction using the PHD package indicates that the residues predicted to form the LmIPCS catalytic triad are orientated towards the Golgi lumen (or the cell surface) in a conformation identical to that predicted for both fungal AUR1p and animal SM synthases (29,36) (figure 4D). These observations suggest that, as in animals and fungi, kinetoplastid sphingolipid synthesis takes place in the lumen of the Golgi.
Specific inhibition of Leishmania IPC synthase
The expression of functional protozoan orthologues in the mutant yeast system described (figure 2) provided an ex vivo system in which to screen the existing fungal IPC synthase inhibitors against the functional Leishmania orthologue. In this study, the diffusion assay system previously described, with the sphingolipid bypass mutant (AGD) as a negative control, was utilised (11,30). The conditional AUR1 mutant strain rescued with ScAUR1 was, like the wild type control, sensitive to cycloheximide (an inhibitor of protein translation), myriocin (an inhibitor of serine palmitoyltransferase, SPT, which mediates the first step in sphingolipid biosynthesis) and Aureobasidin A (an inhibitor of fungal IPC synthase (11), to which mutations in AUR1 confer resistance (10)). The mutant rescued with LmIPCS, while sensitive to cycloheximide and myriocin, was resistant to Aureobasidin A at 25μM (figure 5A). However, by increasing the concentration of the IPC synthase inhibitor 4 fold (to 100μM) sensitivity of LmIPCS to the drug was detected (figure 5B). The sphingolipid bypass strain AGD is able to grow without synthesizing sphingolipids and so is able to tolerate loss-of-function mutations in both SPT and IPC synthase (11). Therefore AGD is resistant to anti-fungals targeting either of these enzymes, provided these agents have a specific mode of action (30). The lack of an inhibitory effect of Aureobasidin A at 100μM on the growth of AGD, together with its action against the rescued mutant, demonstrates that the drug specifically inhibits LmIPCS as well as ScAUR1p. The LmIPCS rescued mutant showed increased sensitivity to myriocin (an inhibitor of SPT) when compared with either the wild-type or the mutant rescued with ScAUR1 (figure 5A). The reason for this is unclear, but this observation correlates with the generally fragile nature of the cells complemented with the protozoan orthologue that results in a reduced rate of growth (data not shown).
Figure 5.

- Volumes of 1, 2 and 3 μl of 25μM Aureobasidin A (AbA), 1mM myriocin (MYR), 25μM cycloheximide (CYC) and DMSO as a control (CTL) spotted onto yeast plates prepared as described in Experimental Procedures.
- Volumes of 1, 2 and 3 μl of 100μM Aureobasidin A spotted onto yeast plates prepared as described in Experimental Procedures.
YPH499 - wild type yeast; ScAUR1 - YPH499-HIS-GAL-AUR1 pRS426 ScAUR1; LmIPCS - YPH499-HIS-GAL- AUR1 pRS426 LmIPCS; AGD - yeast sphingolipid bypass mutant, negative control.
Aureobasidin A (at 20μM) also exhibited an inhibitory effect against insect stage L. major in cell culture. However, in this system, the inhibitor was non-specific as demonstrated by an equivalent activity against parasites without an active sphingolipid biosynthetic pathway (14) in which LmIPCS would be predicted to be redundant (data not shown).
DISCUSSION
Sphingolipids are essential membrane components of eukaryotic cells. In mammalian systems, the major complex sphingolipid is sphingomyelin, whilst in the kinetoplastids, including Leishmania spp., it is inositol phosphorylceramide (IPC) (7). A similar situation exists in fungi where IPC is synthesized utilising the enzyme IPC synthase. The fungal IPC synthases are believed to be encoded by the AUR1 gene which has been demonstrated to be essential for viability (32,42). No close relatives of this gene can be identified in the kinetoplastid genome databases. However, in the study reported here, we have identified the single copy gene encoding the functional AUR1 orthologue from Leishmania major (LmIPCS), utilising a combination of bioinformatic and functional genetic methods. The protein encoded by this gene shares only limited amino acid sequence identity with fungal AUR1p but has closely related sequence orthologues in both the African and American Trypanosomes. Notably, the predicted kinetoplastid AUR1p orthologues demonstrate significant homology to 3 of the 4 conserved domains of the recently identified animal sphingomyelin (SM) synthases (29). This indicates that, despite possessing an equivalent function, the protozoan enzymes are not closely related to the fungal IPC synthases (AUR1p) but share more similarity, at least at a primary sequence level, with the animal SM synthases. Indeed, phylogenetic analyses indicated that the kinetoplastid proteins define a new class of sphingolipid synthases clustering with neither the fungal or animal sequences (figure 3C). The significance of this observation in evolutionary terms is unclear, although the identification of a plant IPC synthase will facilitate more informative analyses.
In addition to complementing an auxotrophic AUR1 yeast mutant, expression of LmIPCS in a mammalian cell line (null for IPC and IPC synthase activity) led to the synthesis of inositol and ceramide labelled lipids that co-migrated with Leishmania and yeast IPC. This strongly suggests that LmIPCS is able to constitute IPC synthase activity in an axenic system. LmIPCS is largely confined to the Golgi apparatus, as indicated by subcellular localisation, a situation similar to that for the proposed sphingolipid synthases of both fungi and animals (29,36). The association of LmIPCS with the Golgi membrane is predicted to be mediated by seven transmembrane helices, with the putative active site including the catalytic triad of histidine and aspartate residues orientated towards the lumen as in fungal AUR1p and mammalian sphingolipid synthase (29,36). Thus it is likely that the protozoan, mammalian and fungal enzymes possess a conserved mechanism of action similar to that described for LPPs (35) whereby, in the case of the yeast and kinetoplastids, IPC synthase catalyses the transfer of an inositol phosphate group from phosphatidylinositol to the 1-hydroxyl group of ceramide or phytoceramide releasing diacyl glycerol as a by-product. This is predicted to occur via a two-step process involving initial transfer of the inositol phosphate residue to an activated histidine in the active site, with the phosphate intermediate being subsequently subjected to nucleophilic attack by the oxygen of the (phyto)ceramide hydroxyl group resulting in transfer to the sphingoid base. Notably, mutation of His-294, part of catalytic triad in the yeast AUR1p, results in non-viable haploid cells (36). Taken together with the phylogenetic analysis in this study, the predicted conservation of the catalytic mechanism suggests that fungal, animal and protozoan sphingolipid synthases have evolved into 3 distinct classes from a common ancestor. Conversely, the soluble bacterial SM synthase lacks the motifs believed to be integral for activity of the eukaryotic enzymes (43) indicating that it arose independently.
The data presented here demonstrate that LmIPCS (like its fungal equivalent) is inhibited by Aureobasidin A. Given that IPC synthase activity is detectable in the pathogenic stages of all the parasitic kinetoplastids studied to date (15,17) the protozoan enzyme may represent a tractable drug target. The toxicity and expense of available treatments for leishmaniasis, African sleeping sickness and Chagas’ disease remain a major international health problem. These facts, coupled with the prevalence of drug resistant parasites, make the discovery of novel drug targets a priority. The identification and characterization of the kinetoplastid AUR1 orthologue, a probable IPC synthase with no functional equivalent in mammalian cells, will raise the possibility of developing specific inhibitors with little or no mammalian toxicity. In addition, the close evolutionary relationship observed between the kinetoplastid AUR1p orthologues indicates that the development of broad-spectrum inhibitors is a feasible objective, work which may lead to a new generation of anti-protozoals directed against the causative agents of a range of emerging diseases.
Footnotes
This work was funded by Royal Society (2005/R1) and Biotechnology and Biological Research Council (BB/D52396X/1) grants to PWD. HPP is funded by the Wellcome Trust (grant no. 077503 to DFS). This work was also supported in part by Deutsche Forschungsgemeinschaft, Bonn. We thank Melanie Sauer, Jörg Schmidt and Ulrike Bieker for expert technical assistance, Prof. Robert Dickson and Prof. Robert Lester (both University of Kentucky) for supplying AGD and RCD113 strains respectively, and Dr Patrick Steel and Dr David Hodgson (both Department of Chemistry, Durham University) for helpful discussions.
REFERENCES
- 1.Croft SL, Barrett MP, Urbina JA. Trends Parasitol. 2005;21:508–512. doi: 10.1016/j.pt.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 2.Nyame AK, Kawar ZS, Cummings RD. Arch Biochem Biophys. 2004;426:182–200. doi: 10.1016/j.abb.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Futerman AH, Hannun YA. EMBO reports. 2004;5:777–782. doi: 10.1038/sj.embor.7400208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown DA, London E. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 5.Magee T, Prinen N, Alder J, Pagakis SN, Parmryd I. Biol Res. 2002;35:127–131. doi: 10.4067/s0716-97602002000200003. [DOI] [PubMed] [Google Scholar]
- 6.Pierce SK. Nature Rev Immunol. 2002;2:96–105. doi: 10.1038/nri726. [DOI] [PubMed] [Google Scholar]
- 7.Lester RL, Dickson RC. Adv Lipid Res. 1993;26:253–274. [PubMed] [Google Scholar]
- 8.Sperling P, Heinz E. Biochim Biophys Acta. 2003;1632:1–15. doi: 10.1016/s1388-1981(03)00033-7. [DOI] [PubMed] [Google Scholar]
- 9.Becker GW, Lester RL. J Bacteriol. 1980;142:747–754. doi: 10.1128/jb.142.3.747-754.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heidler SA, Radding JA. Antimicrob Agents Chemother. 1995;39(12):2765–2769. doi: 10.1128/aac.39.12.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagiec MM, Nagiec EE, Baltisberger JA, Wells GB, Lester RL, Dickson RC. J Biol Chem. 1997;272(15):9809–9817. doi: 10.1074/jbc.272.15.9809. [DOI] [PubMed] [Google Scholar]
- 12.Georgopapadakou NH. Expert Opin Investig Drugs. 2000;9(8):1787–1796. doi: 10.1517/13543784.9.8.1787. [DOI] [PubMed] [Google Scholar]
- 13.Bromley PE, Li YO, Murphy SM, Sumner CM, Lynch DV. Arch Biochem Biophys. 2003;417(2):219–226. doi: 10.1016/s0003-9861(03)00339-4. [DOI] [PubMed] [Google Scholar]
- 14.Denny PW, Goulding D, Ferguson MA, Smith DF. Mol Microbiol. 2004;52(2):313–327. doi: 10.1111/j.1365-2958.2003.03975.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang K, Hsu FF, Scott DA, Docampo R, Turk J, Beverley SM. Mol Microbiol. 2005;55(5):1566–1578. doi: 10.1111/j.1365-2958.2005.04493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh S, Bhattacharyya S, Sirkar M, Sa GS, Das T, Majumdar D, Roy S, Majumdar S. Infect Immun. 2002;70(12):6828–6838. doi: 10.1128/IAI.70.12.6828-6838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Figueiredo JM, Dias WB, Mendonca-Previato L, Previato JO, Heise N. Biochem J. 2005;387(Pt 2):519–529. doi: 10.1042/BJ20041842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salto ML, Bertello LE, Vieira M, Docampo R, Moreno SN, de Lederkremer RM. Eukaryot Cell. 2003;2(4):756–768. doi: 10.1128/EC.2.4.756-768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luberto C, Toffaletti DL, Wills EA, Tucker SC, Casadevall A, Perfect JR, Hannun YA, Del Poeta MM. Genes Dev. 2001;15(2):201–212. doi: 10.1101/gad.856001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson M, Davis RW. Mol Cell Biol. 1984;4:1440–1448. doi: 10.1128/mcb.4.8.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shams-Eldin H, Azzouz N, Blaschke T, Hubel A, Kedees M, Schwarz RT. Yeast. 2001;18:33–39. doi: 10.1002/1097-0061(200101)18:1<33::AID-YEA648>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 22.Lorenz MC, Muir RS, Lim E, McElver J, Weber SC, Heitman J. Gene. 1995;158:113–117. doi: 10.1016/0378-1119(95)00144-u. [DOI] [PubMed] [Google Scholar]
- 23.Shams-Eldin H, Azzouz N, Kedees MH, Orlean P, Kinoshita T, Schwarz RT. Mol Biochem Parasitol. 2002;120(1):73–81. doi: 10.1016/s0166-6851(01)00434-0. [DOI] [PubMed] [Google Scholar]
- 24.Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ. Trends Biochem Sci. 1998;23(10):403–405. doi: 10.1016/s0968-0004(98)01285-7. [DOI] [PubMed] [Google Scholar]
- 25.Rost B. Methods Enzymol. 1996;266:525–539. doi: 10.1016/s0076-6879(96)66033-9. [DOI] [PubMed] [Google Scholar]
- 26.Segui B, Allen-Baume V, Cockcroft S. Biochem J. 2002;366:23–34. doi: 10.1042/BJ20020317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ralton JE, McConville MJ. J Biol Chem. 1998;273(7):4245–4257. doi: 10.1074/jbc.273.7.4245. [DOI] [PubMed] [Google Scholar]
- 28.Denny PW, Field MC, Smith DF. FEBS Lett. 2001;491(1-2):148–153. doi: 10.1016/s0014-5793(01)02172-x. [DOI] [PubMed] [Google Scholar]
- 29.Huitema K, van den Dikkenberg J, Brouwers JF, Holthuis JC. Embo J. 2004;23(1):33–44. doi: 10.1038/sj.emboj.7600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagiec MM, Young CL, Zaworski PG, Kobayashi SD. Biochem Biophys Res Commun. 2003;307(2):369–374. doi: 10.1016/s0006-291x(03)01164-1. [DOI] [PubMed] [Google Scholar]
- 31.Hashida-Okado T, Ogawa A, Endo M, Yasumoto R, Takesako K, Kato I. Mol Gen Genet. 1996;251(2):236–244. doi: 10.1007/BF02172923. [DOI] [PubMed] [Google Scholar]
- 32.Hashida-Okado T, Yasumoto R, Endo M, Takesako K, Kato I. Curr Genet. 1998;33(1):38–45. doi: 10.1007/s002940050306. [DOI] [PubMed] [Google Scholar]
- 33.Schorling S, Vallee B, Barz WP, Riezman H, Oesterhelt D. Mol Biol Cell. 2001;12:3417–3427. doi: 10.1091/mbc.12.11.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heidler SA, Radding JA. Biochim Biophys Acta. 2000;1500(1):147–152. doi: 10.1016/s0925-4439(99)00097-6. [DOI] [PubMed] [Google Scholar]
- 35.Neuwald AF. Protein Sci. 1997;6(8):1764–1767. doi: 10.1002/pro.5560060817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levine TP, Wiggins CA, Munro S. Mol Biol Cell. 2000;11(7):2267–2281. doi: 10.1091/mbc.11.7.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zufferey R, Allen S, Barron T, Sullivan DR, Denny PW, Almeida IC, Smith DF, Turco SJ, Ferguson MA, Beverley SM. J Biol Chem. 2003;278:44708–44718. doi: 10.1074/jbc.M308063200. [DOI] [PubMed] [Google Scholar]
- 38.Futerman AH, Riezman H. Trends Cell Biol. 2005;15:312–318. doi: 10.1016/j.tcb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 39.Kaneshiro ES, Jayasimhulu K, Lester RL. J Lipid Res. 1986;27:1294–1303. [PubMed] [Google Scholar]
- 40.Singh BN, Costello CE, Beach DH, Holz GG. Biochem Biophys Res Commun. 1988;157:1239–1246. doi: 10.1016/s0006-291x(88)81007-6. [DOI] [PubMed] [Google Scholar]
- 41.Liebisch G, Lieser B, Rathenberg J, Drobnik W, Schmitz G. Biochim Biophys Acta. 2004;1686:108–117. doi: 10.1016/j.bbalip.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Dickson RC, Lester RL. Biochim Biophys Acta. 1999;1426:347–357. doi: 10.1016/s0304-4165(98)00135-4. [DOI] [PubMed] [Google Scholar]
- 43.Luberto C, Stonehouse MJ, Collins EA, Marchesini N, El-Bawab S, Vasil AI, Vasil ML, Hannun YA. J Biol Chem. 2003;278(35):32733–32743. doi: 10.1074/jbc.M300932200. [DOI] [PubMed] [Google Scholar]
- 44.Denny PW, Smith DF. Mol Microbiol. 2004;53:725–733. doi: 10.1111/j.1365-2958.2004.04208.x. [DOI] [PubMed] [Google Scholar]
- 45.Dickson RC, Nagiec EE, Wells GB, Nagiec MM, Lester RL. J Biol Chem. 1997;272:29620–29625. doi: 10.1074/jbc.272.47.29620. [DOI] [PubMed] [Google Scholar]
- 46.Haak D, Gable K, Beeler T, Dunn T. J Biol Chem. 1997;272:29704–29710. doi: 10.1074/jbc.272.47.29704. [DOI] [PubMed] [Google Scholar]