

Abstract
Mitochondria sequester much of the Ca2+ that enters motor nerve terminals during repetitive stimulation at frequencies exceeding 10–20 Hz. We studied the post-stimulation extrusion of Ca2+ from mitochondria by measuring changes in matrix [Ca2+] with fluorescent indicators loaded into motor terminal mitochondria in the mouse levator auris longus muscle. Trains of action potentials at 50 Hz produced a rapid increase in mitochondrial [Ca2+] followed by a plateau, which was usually maintained after the end of the stimulus train and then slowly decayed back to baseline. Increasing the Ca2+ load delivered to the terminal by increasing the number of stimuli (from 500 to 2000) or the stimulation frequency (from 50 to 100 Hz), by increasing bath [Ca2+], or by prolonging the action potential with 3,4-diaminopyridine (100 μm) prolonged the post-stimulation decay of mitochondrial [Ca2+] without increasing the amplitude of the plateau during stimulation. Inhibiting the opening of the mitochondrial permeability transition pore with cyclosporin A (5 μm) had no significant effect on the decay of mitochondrial [Ca2+]. Inhibition of the mitochondrial Na+–Ca2+ exchanger with CGP-37157 (50 μm) dramatically prolonged the post-stimulation decay of mitochondrial [Ca2+], reduced post-stimulation residual cytosolic [Ca2+], and reduced the amplitude of endplate potentials evoked after the end of a stimulus train in the presence of both low and normal bath [Ca2+]. These findings suggest that Ca2+ extrusion from motor terminal mitochondria occurs primarily via the mitochondrial Na+–Ca2+ exchanger and helps to sustain post-tetanic transmitter release at mouse neuromuscular junctions.
Mitochondria temporarily sequester large, stimulation-induced calcium loads in neurons and other secretory cells (Friel & Tsien, 1994; Stuenkel, 1994; Herrington et al. 1996; Babcock et al. 1997; David et al. 1998; Pivovarova et al. 1999; David, 1999; Kaftan et al. 2000; Suzuki et al. 2002; David & Barrett, 2003). Mitochondrial Ca2+ uptake during repetitive stimulation of motor nerve terminals limits the increase in cytosolic [Ca2+] to a plateau level which is maintained until stimulation ceases. Inhibition of mitochondrial Ca2+ uptake results in a much greater elevation of cytosolic [Ca2+] during stimulation, accompanied by an increase in asynchronous neurotransmitter release and accelerated depression of phasic transmitter release (David & Barrett, 2000, 2003; Talbot et al. 2003). Thus mitochondrial Ca2+ uptake is important for maintaining neuromuscular transmission during repetitive stimulation. Following stimulation, the slow efflux of Ca2+ from mitochondria contributes to a residual post-stimulation ‘tail’ of elevated [Ca2+] in the cytosol (Werth & Thayer, 1994; Baron & Thayer, 1997; Colegrove et al. 2000a).
During repetitive stimulation of motor nerve terminals, the increase in matrix [Ca2+] is limited to 1–2 μm, even though mitochondrial Ca2+ uptake continues (David, 1999; David et al. 2003). Powerful buffering of mitochondrial Ca2+ has also been reported in other secretory cells (Kaftan et al. 2000; Warashina, 2006). One major form of mitochondrial Ca2+ buffering appears to involve reversible formation of an insoluble complex containing calcium and phosphate (Pivovarova et al. 1999; Chalmers & Nicholls, 2003; reviewed by Carafoli, 2003). After stimulation ends, matrix [Ca2+] decreases back to baseline over a time course of several minutes (David, 1999). In the work reported here, we determined how the post-stimulation decay of matrix [Ca2+] is affected by varying the amount of Ca2+ entering the nerve terminal during a stimulus train (the Ca2+ load).
Mitochondria return sequestered Ca2+ to the cytosol via several mechanisms. Mitochondrial Na+–Ca2+ and H+–Ca2+ exchangers exploit a favourable inward electrochemical gradient for Na+ and H+, respectively, to extrude Ca2+ from the matrix (reviewed by Gunter & Pfeiffer, 1990; Bernardi, 1999; Carafoli, 2003). In vertebrate neuronal mitochondria, Na+–Ca2+ exchange (followed by H+–Na+ exchange) appears to be more important than H+–Ca2+ exchange. Ca2+ can also exit mitochondria during opening of the mitochondrial permeability transition pore.
We studied mitochondrial Ca2+ extrusion mechanisms in mouse motor terminals, and how post-tetanic mitochondrial Ca2+ extrusion affects evoked neurotransmitter release. Many studies have linked short-term synaptic plasticity (facilitation, augmentation and post-tetanic potentiation (PTP)) with persisting elevations of cytosolic [Ca2+] in presynaptic terminals (reviewed by Zucker & Regehr, 2002; Millar et al. 2005). Tang & Zucker (1997) suggested that the persisting elevation of cytosolic [Ca2+] that mediates PTP is produced by mitochondrial Ca2+ extrusion, based on their finding that at crayfish motor terminals both these phenomena were abolished by agents that inhibit mitochondrial Ca2+ uptake and/or extrusion (ruthenium red, tetraphenylphosphonium (TPP+) and the mitochondrial depolarizing agent carbonyl cyanide m-chlorophenylhydrazone (CCCP)). Similar evidence for a mitochondrial contribution to various forms of synaptic potentiation has been presented for rat and toad motor nerve terminals and cultured cortical neurons (Hubbard & Gage, 1964; Yang et al. 2003; Storozhuk et al. 2005). However, Zhong et al. (2001) found that CGP-37157, which inhibits the mitochondrial Na+–Ca2+ exchanger, had no effect on residual [Ca2+] or PTP in crayfish motor terminals. This and other findings led them to conclude that the mitochondrial contribution to PTP in crayfish motor terminals must be via Na+-independent Ca2+ efflux mechanisms. We tested the effect of inhibiting the mitochondrial Na+–Ca2+ exchanger in mouse motor nerve terminals and, in contrast to results in crayfish motor terminals, found that CGP-37157 powerfully inhibits mitochondrial Ca2+ extrusion and decreases the amplitude of post-tetanic endplate potentials (EPPs). Thus, Ca2+ extruded via the mitochondrial Na+–Ca2+ exchanger helps to maintain transmitter release following repetitive stimulation in this mammalian neuromuscular preparation.
Methods
Preparation and solutions
We used neuromuscular junctions from the levator auris longus muscle (Angaut-Petit et al. 1987) of wild-type mice (C57BL/6, Jackson Laboratories, Bar Harbour, ME, USA) or transgenic mice that express yellow fluorescent protein (YFP) in neurons (B6.Cg-Tg(Thy1-YFP)16Jrs/J, Jackson Laboratories). YFP expression facilitated localization of motor terminals. Mice were killed with 100% CO2 using a procedure approved by the University of Miami Animal Care and Use Committee. The muscle with attached nerve was pinned down in a chamber with silicon walls constructed on top of a no. 1 glass coverslip. The preparation was bathed in physiological saline containing (mm): NaCl 137, NaHCO3 15, KCl 4, CaCl2 1.8, MgCl2 1.1, glucose 11.2 and NaH2PO4 0.33. Bath [Ca2+] was reduced in some experiments (Figs 3A and 7C and D). The pH of the solution was kept near 7.4 by gassing with 95% O2–5% CO2. Bath temperature (monitored with a thermistor) was maintained between 29 and 31°C by blowing hot air onto the underside of the chamber. Temperature was reduced to 22°C in the experiment shown in Fig. 7B. The motor nerve was stimulated via a suction electrode by applying suprathreshold depolarizing 0.3-ms pulses. Stimulus trains consisted of 200–2000 stimuli delivered at 50–100 Hz. The minimum interval between stimulus trains was ∼8 min, to allow sufficient time for mitochondrial [Ca2+] to return to baseline.
Figure 3. The post-stimulation decay of mitochondrial [Ca2+] is accelerated by reducing bath [Ca2+] (A) and slowed by 3,4-diaminopyridine (3,4-DAP) (B).
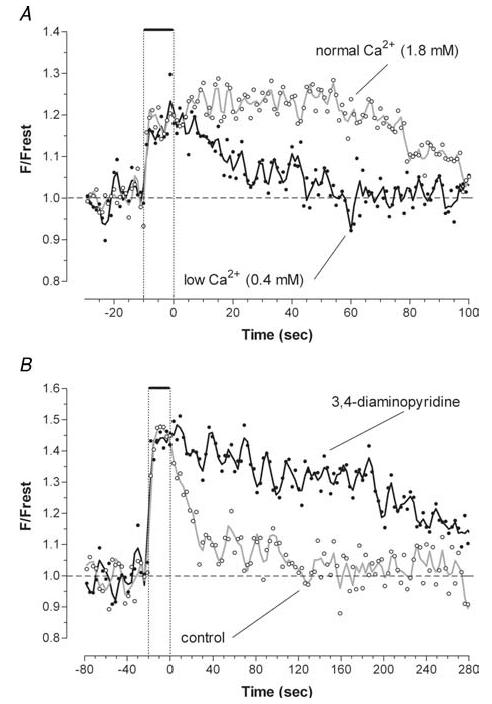
A, responses to trains of 500 stimuli at 50 Hz measured in a motor terminal in a bath [Ca2+] of 1.8 and 0.4 mm. Similar changes were seen in another terminal (not shown) in which bath [Ca2+] was changed from 2.4 to 0.4 mm. B, responses to trains of 1000 stimuli at 50 Hz measured in another terminal before and 24 min after adding 100 μm 3,4-DAP. The constant amplitude of the peak increase in mitochondrial [Ca2+] in different bath [Ca2+] and after addition of 3,4-DAP is consistent with the hypothesized formation of an insoluble calcium-containing complex that limits the increase in matrix [Ca2+] (see Introduction). X-rhod-1 was used in both experiments.
Figure 7. Inhibition of mitochondrial Ca2+ extrusion with CGP-37157 reduces post-tetanic evoked release in near-physiological conditions (A), with reduced temperature and fewer stimuli (B) and in low bath [Ca2+] (C).
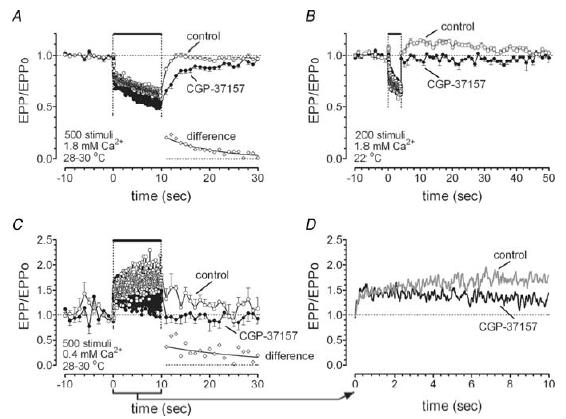
A–C, EPP amplitudes (normalized to EPP amplitude before the train) during and after 50-Hz stimulation. Error bars indicate s.d. Insets in the lower right of A and C show the difference between post-tetanic EPP amplitudes recorded in the absence and presence of CGP-37157. Mono-exponential decays (solid line) fitted to these data had time constants of 10.5 s in A (95% confidence interval, 8.8–12.1 s) and 22.6 s in C (95% confidence interval, 9.8–35 s). D, shows tetanic EPPs from C on an expanded time scale to show effects of CGP-37157 during the train. Curves are the weighted average of nine nearest neighbours. In all experiments muscle contractions were blocked with 2.5–4 μmμ-conotoxin GIIIB. Data in A were averaged from 17 trains recorded from 12 endplates in control (○), and from 12 trains in 10 terminals after addition of CGP-37157 (•). Data in B are the average of three control and six post-drug trains from the same terminal. Data in C and D are the average of 12 trains from 12 terminals in four animals (control) and 10 trains from 10 terminals (drug).
In most imaging experiments (Figs 1–6), muscle contractions were blocked using d-tubocurarine (10 mg l−1), which blocks muscle nicotinic acetylcholine (ACh) receptors thereby preventing entry of Ca2+ into the muscle end-plate via ACh receptors or depolarization-activated Ca2+ channels. Thus mitochondria in the muscle end-plate did not contribute to the recorded stimulation-induced changes in fluorescence of the indicator for mitochondrial [Ca2+] (see below). For Fig. 7, in which EPPs were recorded, muscle contractions were blocked with μ-conotoxin GIIIB (2.5–4 μm), which blocks muscle (but not axonal) Na+ channels (Hong & Chang, 1989). 3,4-diaminopyridine (3,4-DAP, 100 μm), which blocks certain depolarization-activated K+ channels in the motor terminal membrane (Tabti et al. 1989; Morita & Barrett, 1990), was used to prolong action potentials in Fig. 3B. The benzothiazepine CGP-37157 (50 μm) was used to inhibit the mitochondrial Na+–Ca2+ exchanger (Chiesi et al. 1988; Cox et al. 1993); this drug does not block the plasma membrane Na+–Ca2+ exchanger. Cyclosporin A (5 μm) was used to inhibit openings of the mitochondrial permeability transition pore (Fournier et al. 1987; Broekemeier et al. 1989).
Figure 1. Localization of stimulation-induced increases in X-rhod-5F fluorescence within a yellow fluorescent protein-expressing mouse motor nerve terminal.
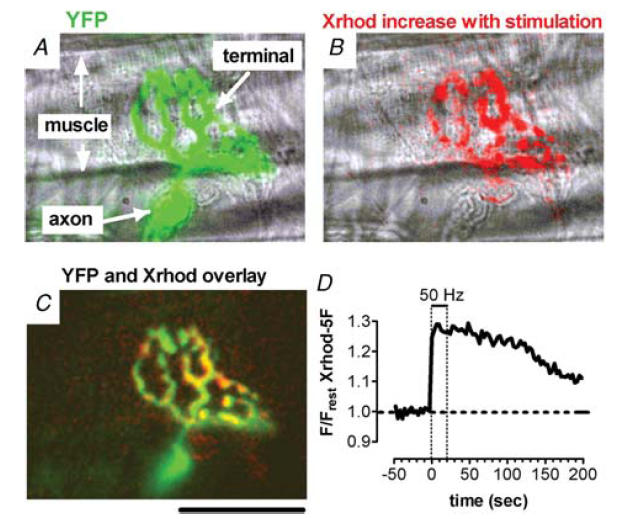
A, overlay of phase and yellow fluorescent protein (YFP; green) images. B, overlay of phase and X-rhod-5F difference (red) images, illustrating regions in which fluorescence increased in response to 50-Hz stimulation for 20 s. The difference image was created by subtracting the average prestimulation fluorescence (20 frames) from the average fluorescence during stimulation (six frames). C, overlay of YFP image and X-rhod-5F difference image. Calibration bar, 50 μm. D, time course of stimulation-induced X-rhod-5F fluorescence changes (plotted as F/Frest = Fnet/Frest) in this terminal. The dashed horizontal line indicates the baseline measured from prestimulation images. Dashed vertical lines indicate the duration of stimulation. For this record, as well as those in Figs 2–6, muscle contraction was blocked using d-tubocurarine (10 μg/ml). The plateau amplitude observed during and immediately following the stimulus train in this and subsequent figures is not an artifact due to dye saturation. As noted in the Introduction, the maximal stimulation-induced increase in matrix [Ca2+] in normal motor terminals is only 1–2 μm above a resting level estimated as 0.05–0.10 μm, and none of the rhod dyes used here would saturate over this range. Also, a similar stimulation-induced plateau of matrix [Ca2+] is evident with a very low affinity indicator (rhod-5N, Kd∼300 μm, David et al. 2003).
Figure 6. Residual cytosolic [Ca2+] after a tetanus is reduced after inhibition of the mitochondrial Na+–Ca2+ exchanger with CGP-37157.
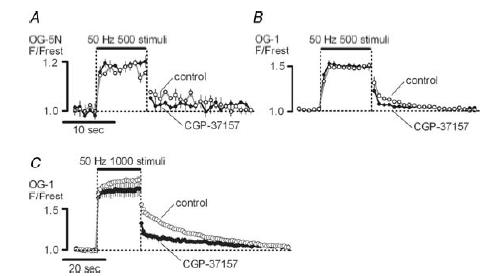
Cytosolic [Ca2+] was monitored as F/Frest for OG-5N (A) or OG-1 (B and C) loaded ion ophoretically into the axon. Each record shows the average of three to four stimulus trains recorded in the same terminal before and 15–80 min after addition of 50 μm CGP-37157; 500 stimuli in A and B; 1000 stimuli in C. A–C were from different terminals, all in 1.8 mm bath Ca2+. Error bars indicate s.e.m.
Imaging of Ca2+ indicator dyes
Stimulation-induced changes in mitochondrial [Ca2+] were measured using Ca2+-sensitive fluorescent indicators from the rhod family (rhod-2, Kd∼0.6 μm; X-rhod-1, Kd∼0.7 μm; X-rhod-5F, Kd∼1.6 μm). These Kd values are appropriate, because resting mitochondrial [Ca2+] is estimated to be 0.05–0.1 μm (reviewed by Gunter & Pfeiffer, 1990) and the maximal increase in mitochondrial [Ca2+] under physiological conditions is 1–2 μm (David, 1999; David et al. 2003). Preparations were exposed to the membrane-permeable, acetoxymethylester (AM) forms of these indicators (∼25 μg ml−1) for ∼30 min, followed by washout with indicator-free saline for ∼30 min. The AM moiety is cleaved by cytosolic and intramitochondrial esterases, converting the indicator into its charged, Ca2+-binding form and effectively trapping it in the compartment where the de-esterification occurs. During washout, the indicator in the cytosol is diluted by diffusion out of the terminal into the myelinated axon, leaving most of the remaining indicator in terminal mitochondria. Morphological and functional criteria used to verify intramitochondrial localization of the indicator are described by David (1999). The criterion most relevant to this study is that following the stimulus train, the initial decay of intramitochondrial [Ca2+] is much slower than that of cytosolic [Ca2+]. Rhod dyes were excited with a 568-nm argon–krypton laser line (Laser Physics, Salt Lake City, UT, USA) and the emitted light was filtered with a long-pass 590-nm filter (Chroma, Rockingham, VT, USA). YFP was excited with a 488-nm laser line, and emitted light was filtered with a 535-nm band-pass filter (bandwidth, 40 nm, Chroma).
Stimulation-induced changes in cytosolic [Ca2+] were monitored in separate experiments using mice that did not express YFP. The membrane-impermeable, hexapotassium salt forms of Oregon Green 488 BAPTA-1 (OG-1, Kd∼0.17 μm) or Oregon Green 488 BAPTA-5N (OG-5 N, Kd∼50 μm) were loaded ionophoretically via an electrode inserted into an axonal internode, using techniques described by David & Barrett (2000). These indicators were excited at 488 nm with emissions monitored at 535 nm.
The experimental chamber was placed on the stage of an inverted microscope in a confocal system that included a Yokogawa spinning disc (Solamere, Salt Lake City, UT, USA), a 60 × water immersion lens (NA 1.2, Olympus), and a Photometrics Cascade 512B CCD camera (Roper Scientific, Trenton, NJ, USA) that used on-chip multiplication gain to achieve greater sensitivity with low light intensities. A series of images were obtained before, during and after a train of action potentials; interimage intervals ranged from 1 to 3 s and exposure times ranged from 0.8 to 2 s. Data were recorded using IP Laboratory v3.61 software (Scanalytics, Inc., Fairfax, VA, USA) and analysed on a Pentium computer using V++ software (Digital Micro Optics, Auckland, New Zealand). Variability due to fluctuations in laser light intensity was minimized by correcting the recorded signal based on the simultaneously recorded signal from a fluorescent bead located close to the dichroic filter in the light path. Net fluorescence (Fnet) was calculated by averaging the total fluorescence (Ftotal) signals from regions of interest in the terminal, and then subtracting background fluorescence (Fbackground) from surrounding non-terminal regions (Fnet = Ftotal−Fbackground). Changes in fluorescence, representative of changes in mitochondrial or cytosolic [Ca2+], were plotted as Fnet/Frestversus time, where Frest is the average Fnet for 20 images obtained before stimulation began.
Analysis was restricted to terminals where the average fluorescence during stimulation exceeded twice the standard deviation of prestimulation values, and where the fluorescence responses remained relatively stable during repeated stimulus trains. The time integral of the post-stimulation decay of mitochondrial [Ca2+] (decay integral) was calculated from normalized data. Integration was stopped when fluorescence fell to prestimulation values, or (for very prolonged decays) when imaging stopped. Other measures of the decay of post-stimulation fluorescence, such as the time to decay to baseline or the half-decay time, were less useful because of signal noise. Most responses did not exhibit a simple exponential decay.
Electrophysiology
EPPs were recorded with a microelectrode (filled with 3 m KCl) inserted into the muscle fibre near the end-plate region, using standard intracellular recording techniques. EPP amplitudes were measured using Clampfit (Axon Instruments, Union City, CA, USA).
Reagent sources
μ-Conotoxin GIIIB was from Alomone Laboratories (Jerusalem, Israel), CGP-37157 was from Tocris (Ellisville, MO, USA) and the Ca2+-indicator dyes were from Invitrogen (Carlsbad, CA, USA). Other reagents were obtained from Sigma (St Louis, MO, USA).
Results
Increasing the Ca2+ load prolongs the post-stimulation decay of mitochondrial [Ca2+]
Figure 1A shows a fluorescence micrograph of a YFP-labelled motor nerve terminal (green) superimposed on a phase-contrast image. Figure 1B shows regions in which the fluorescence of the mitochondrially loaded Ca2+ indicator X-rhod-5F increased in this terminal during 50-Hz stimulation of the motor nerve (red). The overlaid fluorescence images in Fig. 1C show that the regions in which X-rhod-5F fluorescence increased were within the YFP-labelled motor terminal. Figure 1D shows the increase in X-rhod-5F fluorescence (Fnet/Frest) recorded in this terminal during and following a 50-Hz, 20-s train. During stimulation, mitochondrial [Ca2+] increased to a plateau value that persisted for ∼50 s after the end of stimulation and then decayed back to baseline over a time course of several minutes.
Figure 2A compares changes in F/Frest (= Fnet/Frest) recorded in a different terminal during and following trains of 1000 and 2000 stimuli delivered at 50 Hz. Records were aligned so that time 0 corresponds to the end of stimulation. Increasing the train duration prolonged the post-tetanic decay.
Figure 2. Increasing the duration of a 50-Hz stimulus train prolongs the post-stimulation decay of mitochondrial [Ca2+] measured by changes in the fluorescence of X-rhod-5F.
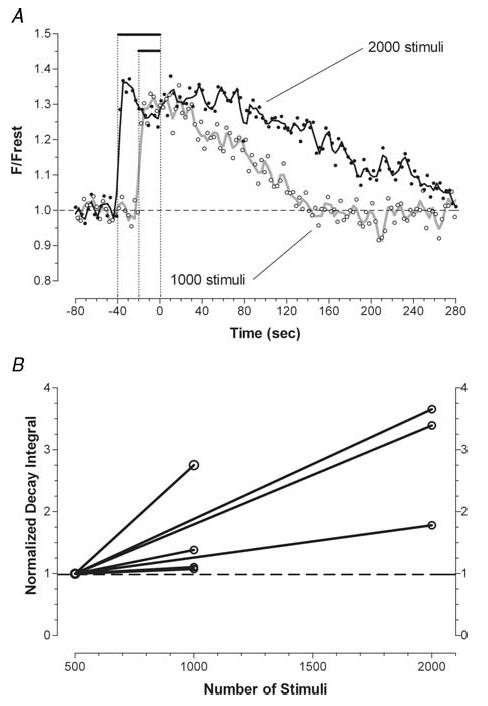
A, two trains (1000 and 2000 stimuli) were delivered sequentially to a single terminal. Curves drawn through the data points were calculated as the weighted average of the five nearest neighbours. The dotted vertical lines mark (from left to right) the beginning of the 2000 stimulus train, the beginning of the subsequent 1000 stimulus train, and the end of stimulation for both trains. B, pairwise comparison of the time integrals of the post-train decays of mitochondrial [Ca2+] measured after 1000 stimuli (n = 4 terminals) or 2000 stimuli (n = 3 terminals), normalized to those measured in the same terminal after 500 stimuli.
The time course of decay of mitochondrial [Ca2+] following a given stimulus train varied greatly from terminal to terminal. Thus to assess the effect of varying the Ca2+ load, we analysed responses recorded sequentially from single terminals (as in Fig. 2A). Figure 2B compares the time integral of the decay of mitochondrial [Ca2+] recorded after 500 stimuli to that recorded after 1000 or 2000 stimuli for five different terminals. These comparisons showed that increasing the number of stimuli always prolonged the post-stimulation decay of mitochondrial [Ca2+], but the percentage increase was variable. On average, increasing the stimulus train from 500 to 1000 stimuli or to 2000 stimuli increased the decay integral by 60% or 200%, respectively (Table 1). Similarly, Warashina (2006) reported that the decay of matrix [Ca2+] in chromaffin cells was prolonged by increasing the duration of a high K+-induced depolarization.
Table 1.
Increasing the Ca2+ load increases the time integral of the post-tetanic decay of mitochondrial [Ca2+]
| Manipulation | Ratio of decay integrals (post/pre) | % of expected |
|---|---|---|
| 500→1000 stimuli | 1.6 ± 0.4 (4) | 79* |
| 500→2000 stimuli | 3.0 ± 1.0 (3) | 74* |
| 0.4→1.8 mm Ca2+ | 3.5 | 78# |
| 0.4→2.4 mm Ca2+ | 3.9 | 65# |
| Add 3,4-DAP | 3.0 ± 0.2 (3) | — |
Number of tested terminals indicated in parenthesis. Values are means ± s.d.
Calculated assuming that the Ca2+ load is proportional to the number of stimuli in the train
calculated assuming that the Ca2+ load is proportional to bath [Ca2+].
The Ca2+ load delivered to the terminal was also varied by changing bath [Ca2+]. Figure 3A shows that decreasing bath [Ca2+] from the normal 1.8 mm to 0.4 mm shortened the decay of mitochondrial [Ca2+]. Table 1 shows quantitatively how changing bath [Ca2+] affected the decay integral.
An additional manipulation to vary the Ca2+ load was addition of 3,4-DAP (100 μm). This drug prolongs the duration of action potentials in motor terminals and thereby prolongs the amount of time that voltage-gated Ca2+ channels are open (Tabti et al. 1989; Morita & Barrett, 1990; Robitaille & Charlton, 1992). Figure 3B shows that 3,4-DAP prolonged the post-stimulation decay of mitochondrial [Ca2+] (also see Table 1). Note that neither reducing nor increasing the stimulation-induced Ca2+ load altered the plateau value of matrix [Ca2+] recorded during the stimulus train, a reflection of the powerful Ca2+ buffering system in the mitochondrial matrix.
Figure 4 shows that increasing the stimulation frequency also prolonged the post-stimulation decay of mitochondrial [Ca2+]. Increasing frequency while keeping the total number of stimuli constant would not be expected to change the total influx of Ca2+ into the terminal. However, delivering the same Ca2+ load in a shorter time increases the elevation of cytosolic [Ca2+] during stimulation (David & Barrett, 2000), which would increase the driving force for Ca2+ entry into mitochondria. Ca2+ influx via the mitochondrial uniporter is activated in a supralinear manner by increases in cytosolic [Ca2+] (reviewed by Gunter & Pfeiffer, 1990). Thus, one would expect a greater accumulation of mitochondrial Ca2+ at higher stimulation frequencies, consistent with the observed prolongation of the post-stimulation decay of mitochondrial [Ca2+].
Figure 4. Increasing the frequency of stimulation prolongs the post-stimulation decay of mitochondrial [Ca2+] measured using rhod-2.
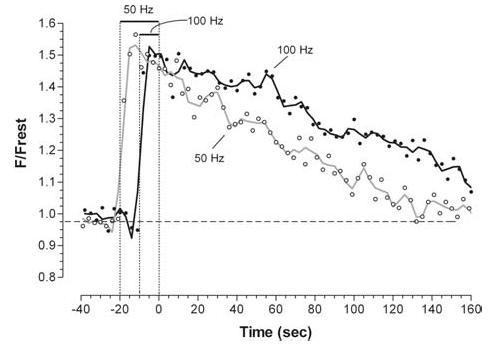
Six trains of 1000 stimuli were delivered sequentially to a single terminal, three at 50 Hz and three at 100 Hz, alternating the frequencies. The averages of each set of three trains are shown.
The mitochondrial Na+–Ca2+ exchanger is the principal pathway for mitochondrial Ca2+ extrusion
How is the Ca2+ that is taken up by mitochondria during repetitive stimulation returned to the cytosol after stimulation?Figure 5A shows that inhibiting the mitochondrial Na+–Ca2+ exchanger with 50 μm CGP-37157 markedly prolonged the decay of mitochondrial [Ca2+]. It has also been demonstrated that CGP-37157 inhibits Ca2+ extrusion by mitochondria in other vertebrate preparations including rat heart (Cox et al. 1993), rat chromaffin cells (Babcock et al. 1997; Warashina, 2006) and multiple types of neurons and other secretory cells (Babcock et al. 1997; Baron & Thayer, 1997; White & Reynolds, 1997; Colegrove et al. 2000a). In contrast, Fig. 5B shows that inhibiting openings of the permeability transition pore with 5 μm cyclosporin A had little effect on the post-stimulation decay of mitochondrial [Ca2+]. Thus the mitochondrial Na+–Ca2+ exchanger is a more important Ca2+ extrusion pathway for mouse motor terminal mitochondria than cyclosporin-inhibitable openings of the transition pore.
Figure 5. The mitochondrial Na+–Ca2+ exchanger contributes more to mitochondrial Ca2+ extrusion than openings of the mitochondrial permeability transition pore.
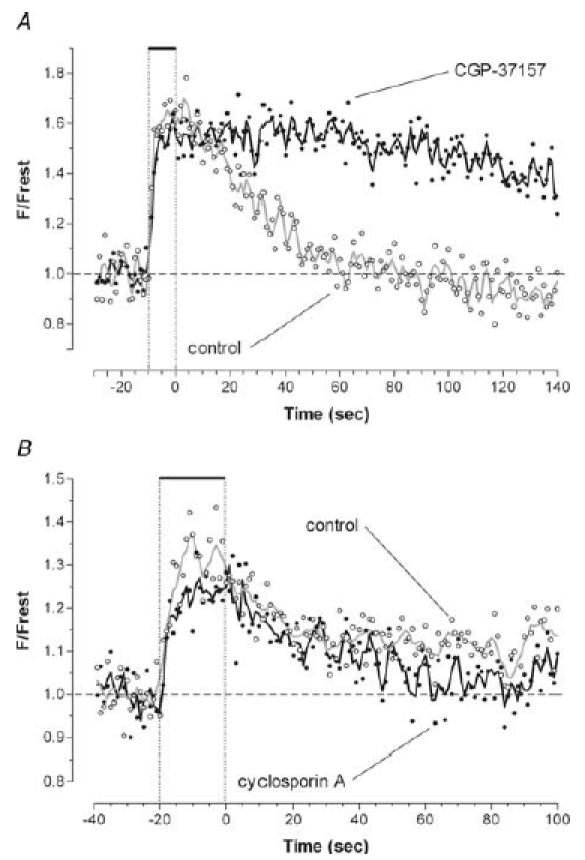
A, responses to 500 stimuli at 50 Hz recorded in a single terminal before and 22 min after addition of 50 μm CGP-37157, which blocks the mitochondrial Na+–Ca2+ exchanger. B, responses to 1000 stimuli at 50 Hz recorded in a different terminal before and 18 min after addition of 5 μm cyclosporin A, which inhibits some openings of the permeability transition pore. X-rhod-1 was used in both experiments.
Ca2+ extruded from mitochondria contributes to the slowly decaying tail of elevated cytosolic [Ca2+] that follows stimulation. Thus, one would predict that inhibition of the mitochondrial Na+–Ca2+ exchanger with CGP-37157 would reduce post-train residual [Ca2+]. This prediction is supported by the measurements in Fig. 6 showing changes in cytosolic [Ca2+] produced during and after stimulation at 50 Hz, monitored as F/Frest of ionophoretically injected OG-5N (Kd, ∼50 μm, Fig. 6A) or OG-1 (Kd, ∼0.17 μm, Fig. 6B and C). For the 500-stimulus trains in Fig. 6A and B, CGP-37157 reduced post-train [Ca2+] but had little effect on [Ca2+] during the train. This is similar to the results of David (1999) in lizard motor terminals (see also Colegrove et al. 2000a). The low-affinity indicator OG-5N in Fig. 6A allows a good estimate of the relative magnitude of the [Ca2+] elevations during and following stimulation. The high-affinity indicator OG-1 yields a better signal-to-noise ratio, but is more saturated with Ca2+ during the train than the low-affinity OG-5N. Thus the OG-1 records in Fig. 6B and C overestimate the ratio of [Ca2+] post train to [Ca2+] during the train. In Fig. 6C, the number of stimuli was increased to 1000 to improve resolution of post-train [Ca2+]. Single exponentials fit to these data (2–70 s post train) had magnitudes and time constants of 0.5 and 25 s in control, and 0.19 and 29 s in the presence of CGP-37157, respectively. Thus, this drug mainly affected the magnitude of post-train [Ca2+]. In Fig. 6C, CGP-37157 slightly reduced the elevation of cytosolic [Ca2+] during the latter half of the stimulus train. This effect is expected if mitochondrial Na+–Ca2+ exchange occurs during as well as after the stimulus train (see modelling study of Colegrove et al. 2000b). Activation of this exchanger, which increases cytosolic [Ca2+], would be expected to increase over the course of the train as mitochondrial [Ca2+] and intra-axonal [Na+] increase. Thus the reduction in cytosolic [Ca2+] produced by inhibiting this exchanger with CGP-37157 would be expected to become greater over the course of the train.
Inhibition of the mitochondrial Na+–Ca2+ exchanger reduces evoked transmitter release following high-frequency stimulation
During post-tetanic stimulation, the residual tail of cytosolic Ca2+ and action potential-induced Ca2+ influx summate, thus increasing post-tetanic evoked transmitter release (reviewed by Zucker & Regehr, 2002). To test whether mitochondrial Ca2+ extrusion increases post-tetanic release, we measured EPP amplitudes before, during and after 50-Hz stimulus trains in the presence and absence of CGP-37157 (Fig. 7). In these experiments, muscle contractions were blocked using μ-conotoxin GIIIB rather than d-tubocurarine, because during repetitive stimulation there is less depression of transmitter release in the presence of μ-conotoxin GIIIB than in d-tubocurarine (Hong & Chang, 1989). Changes in mitochondrial [Ca2+] during and after a 500-stimulus train delivered in the presence of μ-conotoxin GIIIB were similar to those recorded in the presence of d-tubocurarine; that is, attainment of a plateau during stimulation and a prolonged (> 1 min) post-stimulation decay.
Figure 7A shows that in physiological saline prior to addition of CGP-37157, post-tetanic EPP amplitudes returned to prestimulation values within less than 5 s; no augmentation or potentiation was evident. Hubbard et al. (1971) also reported minimal post-tetanic potentiation in rat motor terminals subjected to similar stimulus trains at 30°C (their Fig. 9). This lack of a post-tetanic increase in EPP amplitudes may have occurred because augmentation and potentiation were obscured by depression, as described by Kalkstein & Magleby (2004) in frog motor terminals. CGP-37157 markedly slowed the post-tetanic recovery of EPP amplitude, consistent with the hypothesis that mitochondrial extrusion of sequestered Ca2+ into the cytosol is important for accelerating the recovery of evoked transmitter release following repetitive stimulation. The time course of the difference between post-stimulation EPP amplitudes recorded in the presence and absence of CGP-37157 is plotted in the inset of Fig. 7A. This difference decayed with a time constant of ∼10 s (solid line).
As shown in Fig. 7A, CGP-37157 also increased the depression of EPP amplitudes recorded during the latter part of the tetanus. This result, considered together with the cytosolic [Ca2+] record in Fig. 6C, is consistent with the idea that mitochondrial Ca2+ extrusion via the Na+–Ca2+ exchanger can in some terminals increase cytosolic [Ca2+] and evoked release during as well as after the train. Another possible explanation of the increased depression in the presence of CGP-37157 is that this drug increased the initial (baseline) level of evoked release. This explanation seems unlikely, because CGP-37157 had no significant effect on either the muscle resting potential or the initial EPP amplitude.
We also tested the effects of CGP-37157 on tetanic and post-tetanic EPP amplitudes under two non-physiological experimental conditions designed to minimize tetanic depression and increase post-tetanic release. For the experiment shown in Fig. 7B, the duration of the stimulus train was reduced from 500 to 200 stimuli and the temperature was reduced from 30°C to 22°C. There was a pronounced and prolonged post-tetanic increase in EPP amplitudes in control solution, consistent with the demonstration by Hubbard et al. (1971) that lowering the temperature enhances post-tetanic release in rat motor terminals. This post-tetanic increase was abolished by CGP-37157. Under these conditions, CGP-37157 had no effect on EPP amplitudes during the stimulus train, indicating that inhibition of post-tetanic release by this drug can occur independent of any inhibition of release during the tetanus.
For the experiment shown in Fig. 7C and D, bath [Ca2+] was reduced from the normal 1.8 mm to 0.4 mm. Under this condition of reduced release, EPPs increased (rather than decreased) during the train, and remained above control levels for tens of seconds following the train (reviewed by Magleby, 2004). CGP-37157 abolished the post-tetanic increase in release, and the increase in EPP amplitudes during the stimulus train was less marked. The expanded time scale in Fig. 7D shows that this latter effect of CGP-37157 developed during the course of stimulation, similar to the delayed onset in Fig. 7A.
Thus under all the experimental conditions tested (Fig. 7), inhibition of mitochondrial Ca2+ extrusion with CGP-37157 reduced post-tetanic release.
Discussion
The post-tetanic decay of mitochondrial [Ca2+] depends on the mitochondrial Na+–Ca2+ exchanger and the Ca2+ load
Results presented here demonstrate that following 500–2000 action potentials delivered at 50–100 Hz, the [Ca2+] within mouse motor terminal mitochondria decays back to baseline over a time course of several minutes. This decay is greatly prolonged by inhibiting the mitochondrial Na+–Ca2+ exchanger with CGP-37157, consistent with evidence from isolated mitochondria and other intact neurons that this exchanger is the dominant mechanism for Ca2+ extrusion from vertebrate neuronal mitochondria (see Introduction). The increase in mitochondrial [Ca2+] during stimulation occurs rapidly, consistent with ion flux through a channel driven by a large electrochemical gradient. The slow post-stimulation decay of mitochondrial [Ca2+] is consistent with the slower turnover rate of transporters. As for matrix [Ca2+], [Na+] within the terminal remains elevated for several minutes following high-frequency stimulation, as assessed by a fluorescent Na+ indicator (Zhong et al. 2001) or by the duration of the Na+–K+-ATPase-mediated post-tetanic axonal hyperpolarization (Morita et al. 1993; Kiernan et al. 2004). Thus after a stimulus train, Ca2+ extrusion from the matrix is favoured by sustained elevations of both matrix [Ca2+] and cytosolic [Na+].
The post-stimulation decay of matrix [Ca2+] was minimally affected by cyclosporin A, suggesting that Ca2+ extrusion from the mitochondrial matrix does not occur via transient opening of the mitochondrial transition pore under these experimental conditions. This conclusion is consistent with the finding of minimal depolarization of the mitochondrial membrane potential during or after stimulation in lizard motor nerve terminals (David, 1999). Also, Chalmers & Nicholls (2003) found no evidence for opening of the transition pore in isolated brain mitochondria until matrix [Ca2+] exceeded 3–5 μm, whereas the elevation in matrix [Ca2+] in stimulated motor terminals is limited to 1–2 μm (David, 1999; David et al. 2003).
In all motor terminals, mitochondrial [Ca2+] reached a plateau level during the stimulus train, and the amplitude of the plateau was not changed by manipulations designed to vary the Ca2+ load. This finding is consistent with the powerful matrix buffering described in the Introduction. Assuming little change in matrix [inorganic phosphate], matrix [Ca2+] would be expected to remain ‘clamped’ at this plateau level as long as the (hypothesized) insoluble calcium–phosphate complexes remained. Thus one might expect that after stimulation stopped, matrix [Ca2+] would remain near the plateau level until all of the complex formed during stimulation had dissolved. Consistent with this idea, matrix [Ca2+] in many motor terminals exhibited an initial continuation of the plateau during the post-stimulation decay. The larger the Ca2+ load delivered to the terminal, the more likely the persistence of this post-stimulation plateau phase (e.g. compare 2000- and 1000-stimulus trains in Fig. 2A).
However in other terminals, matrix [Ca2+] began decaying immediately after stimulation stopped (e.g. see control record in Fig. 3B). We do not know why some terminals in which the matrix [Ca2+] exhibited a clear plateau during stimulation did not sustain that plateau during the initial phase of the post-stimulus decay. One possible explanation is that in these mitochondria the rate of Ca2+ efflux via the Na+–Ca2+ exchanger exceeded the rate at which the hypothesized calcium–phosphate complex could dissolve. Energy-dispersive X-ray microanalysis measurements of total calcium within the matrix of high K+-stimulated sympathetic ganglion neurons disclosed marked inter- and intramitochondrial heterogeneity (Pivovarova et al., 1999). Their results suggest that the hypothesized calcium–phosphate complexes are heterogeneously distributed among and within mitochondria.
Because strong buffering limits the increase in matrix [Ca2+] during stimulation, measurements of matrix [Ca2+] cannot be used to assess total mitochondrial Ca2+ uptake. However, because increasing the stimulation-induced Ca2+ influx into the terminal prolonged the post-stimulation decay of matrix [Ca2+], we tested whether the time integral of this decay might be proportional to the (presumed) mitochondrial Ca2+ load. The results shown in Table 1 indicate that the percentage increase in the time integral of post-stimulation matrix [Ca2+] decay was less than the percentage increase in the (presumed) Ca2+ load. The ratio of the average percentage increases in the decay integral and the estimated Ca2+ load was similar (65%–79%) whether the extra Ca2+ load was delivered without an extra Na+ load (as with increases in bath [Ca2+]), or with an extra Na+ load (as with increases in the number of stimuli). Thus changes in the post-train time integral of matrix [Ca2+] probably underestimated the percentage change in mitochondrial Ca2+ load.
Ca2+ extrusion from mitochondria helps sustain post-tetanic transmitter release
Kamiya & Zucker (1994) demonstrated that decreasing cytosolic [Ca2+] in crayfish motor terminals, by photo-activating a Ca2+ chelator at various times after tetanic stimulation, decreased facilitation, augmentation and potentiation. However, the decrease in potentiation was only transient, suggesting that the elevated cytosolic [Ca2+] that contributed to potentiation arose from a non-cytosolic source that could be replenished. One of these sources is mitochondria (Tang & Zucker, 1997). Another source is Ca2+ influx from the bath via reverse action of a plasmalemmal Na+–Ca2+ exchanger (Zhong et al. 2001).
In crayfish motor terminals, Ca2+ extrusion from mitochondria is Na+-independent; inhibiting the mitochondrial Na+–Ca2+ exchanger with CGP-37157 had no effect on residual [Ca2+] or post-tetanic potentiation (Zhong et al. 2001). In contrast, in mouse motor terminals CGP-37157 prolonged the post-tetanic decay of mitochondrial [Ca2+], reduced post-tetanic residual cytosolic [Ca2+] and reduced post-tetanic transmitter release at both normal and low quantal contents. These results demonstrate that in mouse motor terminals the mitochondrial Na+–Ca2+ exchanger contributes importantly to sustaining evoked release during the post-tetanic period. This drug offers a ‘cleaner’ method for testing the hypothesis of mitochondrial involvement in post-tetanic release than some previous strategies because (at least in the short term) it does not block mitochondrial Ca2+ uptake or interfere with mitochondrial energy production. Previously used agents that inhibit mitochondrial Ca2+ uptake (e.g. CCCP or inhibitors of the uniporter or electron transport chain) also reduce post-tetanic evoked release, but interpretation of this effect is complicated because inhibition of mitochondrial Ca2+ uptake results in greater elevations of cytosolic [Ca2+] during the tetanus, resulting in high levels of asynchronous release and faster depression of phasic release during the tetanus (Talbot et al. 2003; David & Barrett, 2003). The effects of CGP-37157 are easier to interpret because it did not increase cytosolic [Ca2+] during tetanic stimulation, and (in certain experimental conditions) its inhibition of post-tetanic evoked release could be observed without increased depression during the tetanus.
Under near-physiological conditions (normal bath [Ca2+] and 30°C), the time constant of the post-tetanic release component that was inhibited by CGP-37157 was ∼10 s (Fig. 7A), similar to time constants reported for augmentation. As mitochondrial Ca2+ extrusion under these conditions continued for at least 80 s, it probably also affected potentiation. Indeed, under altered experimental conditions designed to increase post-tetanic potentiation, CGP-37157 abolished all post-tetanic enhancement of release (Fig. 7B and C).
The contrasting physiology of crayfish and mouse motor nerve terminals may be related to the mechanisms by which they sustain the elevation of cytosolic [Ca2+] following tetanic stimulation. At crayfish exciter opener neuromuscular junctions there is marked potentiation of synaptic transmission during and following tetanic stimulation under physiological conditions, and this potentiation is physiologically important because contractions of the underlying muscle are graded with EPP amplitude. Thus it seems appropriate that the elevated residual [Ca2+] that sustains this potentiation comes both from mitochondria and from reverse operation of a plasmalemmal Na+–Ca2+ exchanger (Zhong et al. 2001). In contrast, at mouse and rat neuromuscular junctions neither tetanic nor post-tetanic potentiation is prominent under physiological conditions; rather the net result is usually depression (as in Fig. 7A; also Halstead et al. 2005; and for in vivo results see Argaw et al. 2004). Augmentation and potentiation dominate only under non-physiological conditions (e.g. low bath [Ca2+]). As contraction of the underlying muscle fibre is an all-or-nothing phenomenon, the physiologically important task is not to increase the amplitude of the post-tetanic EPP to above control levels, but rather simply to restore EPP amplitude to a value suprathreshold for muscle contraction as quickly as possible. Evidence presented in this paper demonstrates that mitochondrial extrusion of previously sequestered Ca2+ via the Na+–Ca2+ exchanger is a major mechanism for accomplishing this task.
Acknowledgments
This work was supported by National Institutes of Health grant RO1 NS 12404 and American Heart Association no. 455215B. Purchase of the confocal microscope was enabled by NIH Shared Equipment grant 1S10 RR16856. We thank Dr John Barrett for valuable comments concerning the manuscript, and Mr Conrado Freites for technical assistance.
References
- Angaut-Petit D, Molgo J, Connold AL, Faille L. The levator auris longus muscle of the mouse: a convenient preparation for studies of short- and long-term presynaptic effects of drugs or toxins. Neurosci Lett. 1987;82:83–88. doi: 10.1016/0304-3940(87)90175-3. [DOI] [PubMed] [Google Scholar]
- Argaw A, Desaulniers P, Gardiner PF. Enhanced neuromuscular transmission efficacy in overloaded rat plantaris muscle. Muscle Nerve. 2004;29:97–103. doi: 10.1002/mus.10535. [DOI] [PubMed] [Google Scholar]
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. J Cell Biol. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron KT, Thayer SA. CGP 37157 modulates mitochondrial Ca2+ homeostasis in cultured rat dorsal root ganglion neurons. Eur J Pharmacol. 1997;340:295–300. doi: 10.1016/s0014-2999(97)01433-7. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem. 1989;264:7826–7830. [PubMed] [Google Scholar]
- Carafoli E. Historical review: mitochondria and calcium: ups and downs of an unusual relationship. Trends Biochem Sci. 2003;28:175–181. doi: 10.1016/S0968-0004(03)00053-7. [DOI] [PubMed] [Google Scholar]
- Chalmers S, Nicholls DG. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem. 2003;278:19062–19070. doi: 10.1074/jbc.M212661200. [DOI] [PubMed] [Google Scholar]
- Chiesi M, Schwaller R, Eichenberger K. Structural dependency of the inhibitory action of benzodiazepines and related compounds on the mitochondrial Na+-Ca2+ exchanger. Biochem Pharmacol. 1988;37:4399–4403. doi: 10.1016/0006-2952(88)90623-5. [DOI] [PubMed] [Google Scholar]
- Colegrove SL, Albrecht MA, Friel DD. Dissection of mitochondrial Ca2+ uptake and release fluxes in situ after depolarization-evoked [Ca2+]i elevations in sympathetic neurons. J Gen Physiol. 2000a;115:351–369. doi: 10.1085/jgp.115.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrove SL, Albrecht MA, Friel DD. Quantitative analysis of mitochondrial Ca2+ uptake and release pathways in sympathetic neurons. Reconstruction of the recovery after depolarization-evoked [Ca2+]i elevations. J Gen Physiol. 2000b;115:371–388. doi: 10.1085/jgp.115.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DA, Conforti L, Sperelakis N, Matlib MA. Selectivity of inhibition of Na+-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J Cardiovasc Pharmacol. 1993;21:595–599. doi: 10.1097/00005344-199304000-00013. [DOI] [PubMed] [Google Scholar]
- David G. Mitochondrial clearance of cytosolic Ca2+ in stimulated lizard motor nerve terminals proceeds without progressive elevation of mitochondrial matrix [Ca2+] J Neurosci. 1999;19:7495–7506. doi: 10.1523/JNEUROSCI.19-17-07495.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett EF. Stimulation-evoked increases in cytosolic [Ca2+] in mouse motor terminals are limited by mitochondrial uptake and are temperature-dependent. J Neurosci. 2000;20:7290–7296. doi: 10.1523/JNEUROSCI.20-19-07290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett EF. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol. 2003;548:425–438. doi: 10.1113/jphysiol.2002.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett JN, Barrett EF. Evidence that mitochondria buffer physiological Ca2+ loads in lizard motor nerve terminals. J Physiol. 1998;509:59–65. doi: 10.1111/j.1469-7793.1998.059bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Talbot JD, Barrett EF. Quantitative estimate of mitochondrial [Ca2+] in stimulated motor nerve terminals. Cell Calcium. 2003;33:197–206. doi: 10.1016/s0143-4160(02)00229-4. [DOI] [PubMed] [Google Scholar]
- Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr. 1987;19:297–303. doi: 10.1007/BF00762419. [DOI] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Halstead SK, Morrison I, O'Hanlon GM, Humphreys PD, Goodfellow JA, Plomp JJ, Willison HJ. Anti-disialosyl antibodies mediate selective neuronal or Schwann cell injury at mouse neuromuscular junctions. Glia. 2005;52:177–189. doi: 10.1002/glia.20228. [DOI] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Chang CC. Use of geographutoxin II (μ-conotoxin) for the study of neuromuscular transmission in the mouse. Br J Pharmacol. 1989;97:934–940. doi: 10.1111/j.1476-5381.1989.tb12034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard JI, Gage PW. Abolition of post-tetanic potentiation. Nature. 1964;202:299–300. doi: 10.1038/202299a0. [DOI] [PubMed] [Google Scholar]
- Hubbard JI, Jones SF, Landau EM. The effect of temperature change upon transmitter release, facilitation and post-tetanic potentiation. J Physiol. 1971;216:591–609. doi: 10.1113/jphysiol.1971.sp009542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaftan EJ, Xu T, Abercrombie RF, Hille B. Mitochondria shape hormonally induced cytoplasmic calcium oscillations and modulate exocytosis. J Biol Chem. 2000;275:25465–25470. doi: 10.1074/jbc.M000903200. [DOI] [PubMed] [Google Scholar]
- Kalkstein JM, Magleby KL. Augmentation increases vesicular release probability in the presence of masking depression at the frog neuromuscular junction. J Neurosci. 2004;24:11391–11403. doi: 10.1523/JNEUROSCI.2756-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Zucker RS. Residual Ca2+ and short-term synaptic plasticity. Nature. 1994;371:603–606. doi: 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Lin CS, Burke D. Differences in activity-dependent hyperpolarization in human sensory and motor axons. J Physiol. 2004;558:342–349. doi: 10.1113/jphysiol.2004.063966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magleby KL. Neuromuscular transmission. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 2004. pp. 373–395. [Google Scholar]
- Millar AG, Zucker RS, Ellis-Davies GC, Charlton MP, Atwood HL. Calcium sensitivity of neurotransmitter release differs at phasic and tonic synapses. J Neurosci. 2005;25:3113–3125. doi: 10.1523/JNEUROSCI.4717-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, Barrett EF. Evidence for two calcium-dependent potassium conductances in lizard motor nerve terminals. J Neurosci. 1990;10:2614–2625. doi: 10.1523/JNEUROSCI.10-08-02614.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, David G, Barrett JN, Barrett EF. Posttetanic hyperpolarization produced by the electrogenic Na+-K+ pump in lizard axons impaled near their motor terminals. J Neurophysiol. 1993;70:1874–1884. doi: 10.1152/jn.1993.70.5.1874. [DOI] [PubMed] [Google Scholar]
- Pivovarova NB, Hongpaisan J, Andrews SB, Friel DD. Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: spatial and temporal characteristics. J Neurosci. 1999;19:6372–6384. doi: 10.1523/JNEUROSCI.19-15-06372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Charlton MP. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J Neurosci. 1992;12:296–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storozhuk MV, Ivanova SY, Balaban PM, Kostyuk PG. Possible role of mitochondria in posttetanic potentiation of GABAergic synaptic transmission in rat neocortical cell cultures. Synapse. 2005;58:45–52. doi: 10.1002/syn.20186. [DOI] [PubMed] [Google Scholar]
- Stuenkel EL. Regulation of intracellular calcium and calcium buffering properties of rat isolated neurohypophysial nerve endings. J Physiol. 1994;481:251–271. doi: 10.1113/jphysiol.1994.sp020436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Osanai M, Mitsumoto N, Akita T, Narita K, Kijima H, Kuba K. Ca2+-dependent Ca2+ clearance via mitochondrial uptake and plasmalemmal extrusion in frog motor nerve terminals. J Neurophysiol. 2002;87:1816–1823. doi: 10.1152/jn.00456.2001. [DOI] [PubMed] [Google Scholar]
- Tabti N, Bourret C, Mallart A. Three potassium currents in mouse motor nerve terminals. Pflugers Arch. 1989;413:395–400. doi: 10.1007/BF00584489. [DOI] [PubMed] [Google Scholar]
- Talbot JD, David G, Barrett EF. Inhibition of mitochondrial Ca2+ uptake has differential effects on phasic release from motor terminals in physiological and low bath [Ca2+] J Neurophysiol. 2003;90:491–502. doi: 10.1152/jn.00012.2003. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Warashina A. Mode of mitochondrial Ca2+ clearance and its influence on secretory responses in stimulated chromaffin cells. Cell Calcium. 2006;39:35–46. doi: 10.1016/j.ceca.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurones. J Physiol. 1997;498:31–47. doi: 10.1113/jphysiol.1997.sp021839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, He XP, Russell J, Lu B. Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na+-Ca2+ exchanger and protein kinase C. J Cell Biol. 2003;163:511–523. doi: 10.1083/jcb.200307027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N, Beaumont V, Zucker RS. Roles for mitochondrial and reverse mode Na+/Ca2+ exchange and the plasmalemma Ca2+ ATPase in post-tetanic potentiation at crayfish neuromuscular junctions. J Neurosci. 2001;21:9598–9607. doi: 10.1523/JNEUROSCI.21-24-09598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]