

Abstract
Exerting its actions pre-, post- and peri-synaptically, brain-derived neurotrophic factor (BDNF) is one of the most potent modulators of hippocampal synaptic function. Here, we examined the effects of BDNF on a rapidly recycling pool (RRP) of vesicles within excitatory synapses. First, we estimated vesicular release in hippocampal cultures by performing FM4-64 imaging in terminals impinging on enhanced green fluorescent protein (eGFP)-labelled dendritic spines – a hallmark of excitatory synapses. Consistent with a modulation of the RRP, BDNF increased the evoked destaining rate of FM4-64 only during the initial phase of field stimulation. Multiphoton microscopy in acute hippocampal slices confirmed these observations by selectively imaging the RRP, which was loaded with FM1-43 by hyperosmotic shock. Slices exposed to BDNF showed an increase in the evoked and spontaneous rates of FM1-43 destaining from terminals in CA1 stratum radiatum, mostly representing excitatory terminals of Schaffer collaterals. Variance-mean analysis of evoked EPSCs in CA1 pyramidal neurons further confirmed that release probability is increased in BDNF-treated slices, without changes in the number of independent release sites or average postsynaptic quantal amplitude. Because BDNF was absent during dye loading, imaging, destaining and whole-cell recordings, these results demonstrate that BDNF induces a long-lasting enhancement in the probability of transmitter release at hippocampal excitatory synapses by modulating the RRP. Since the endogenous BDNF scavenger TrkB-IgG prevented the enhancement of FM1-43 destaining rate caused by induction of long-term potentiation in acute hippocampal slices, the modulation of a rapidly recycling vesicle pool may underlie the role of BDNF in hippocampal long-term synaptic plasticity.
Brain-derived neurotrophic factor (BDNF) has been consistently shown to modify excitatory synaptic transmission and long-term synaptic plasticity in a variety of preparations (Lessmann et al. 1994; Kang & Schuman, 1995; Levine et al. 1995; Figurov et al. 1996; Akaneya et al. 1997; Carmignoto et al. 1997; Gottschalk et al. 1998; Huber et al. 1998; Messaoudi et al. 1998). The proposed mechanisms underlying the actions of BDNF include the immediate and long-term modulation of both pre- and postsynaptic function (Poo, 2001). Regarding presynaptic function at CNS synapses, BDNF rapidly increases the frequency of spontaneous miniature excitatory postsynaptic currents (mEPSCs), without affecting their amplitude or kinetics (Lessmann et al. 1994; Carmignoto et al. 1997; Lessmann & Heumann, 1998; Li et al. 1998a; Schinder et al. 2000). In addition, BDNF increases the variance of evoked EPSC amplitudes (Lessmann & Heumann, 1998), modulates paired-pulse facilitation, and attenuates synaptic fatigue during high-frequency stimulation (Figurov et al. 1996; Gottschalk et al. 1998). Mice with a constitutive deletion of the Bdnf gene exhibit several presynaptic impairments, including pronounced synaptic fatigue, fewer docked vesicles, and reduced expression levels of synaptobrevin and synaptophysin (Pozzo-Miller et al. 1999), two vesicle proteins involved in their mobilization and docking (Sudhof, 2004). In addition, direct measurements of glutamate concentration have also confirmed that BDNF enhances K+-evoked transmitter release from cultured neurons and isolated synaptosomes (Numakawa et al. 1999; Jovanovic et al. 2000). Lastly, using restricted genetic deletions it was demonstrated that BDNF is selectively required for a form of long-term potentiation (LTP) of synaptic strength that recruits a presynaptic module of expression (Zakharenko et al. 2003). Taken together, these observations provide consistent evidence that BDNF modulates the efficiency of vesicular release during exocytosis of excitatory neurotransmitter. However, much less is known about the specific steps in the synaptic vesicle cycle where BDNF exerts its effects.
Quantitative optical imaging of the uptake and release of amphipathic fluorescent styryl dyes such as FM1-43 and FM4-64, have been used to directly detect the endocytosis and exocytosis of synaptic vesicles at small presynaptic nerve terminals (Betz et al. 1992), greatly enhancing our understanding of the vesicular trafficking events underlying transmitter release (Cochilla et al. 1999; Rizzoli & Betz, 2005). Using FM-imaging, it has been shown that BDNF enhances vesicle exocytosis evoked by high-K+ depolarization in cultured embryonic cortical neurons (Bradley & Sporns, 1999). However, this report did not determine if the FM1-43-labelled presynaptic terminals were of inhibitory or excitatory nature. To examine the effects of BDNF on transmitter release at excitatory synapses, we performed confocal microscopy of FM4-64 within individually identified presynaptic nerve terminals apposing dendritic spines in cultured hippocampal neurons. In addition, multiphoton excitation microscopy was employed to image individual FM1-43-labelled presynaptic terminals in acute hippocampal slices, where afferent excitatory terminals can be easily identified by their characteristic laminar distribution (Stanton et al. 2001, 2003, 2005; Zakharenko et al. 2001; Axmacher et al. 2004). Our data demonstrate that BDNF causes a long-lasting facilitation of evoked and spontaneous transmitter release at excitatory presynaptic terminals by selectively modulating a rapidly recycling pool (RRP) of synaptic vesicles, and that this modulation is necessary for the expression of presynaptic changes after induction of LTP.
Methods
Primary cultures of dissociated neurons, eGFP transfection, and BDNF treatment
Postnatal hippocampal neurons were dissociated from newborn (postnatal day 0 or 1, P0–1) Sprague-Dawley rats following standard procedures (Brewer, 1997). After 12 days in vitro (div), neurons were transfected with cDNA coding for enhanced green fluorescent protein (eGFP; Clontech, Mountain View, CA, USA) using a modified calcium phosphate transfection procedure (Kohrmann et al. 1999). Cultures were then either maintained in serum-free control culture medium (Neurobasal plus B-27 supplement; Invitrogen, Carlsbad, CA, USA), or exposed for either 3 or 72 h to human recombinant BDNF (250 ng ml−1; provided by Amgen, Thousand Oaks, CA, USA). All experimental procedures followed national and international ethics guidelines, being performed in completely anaesthetized animals using isoflurane by inhalation to effect in a saturation chamber, in accordance with animal use protocols approved by the UAB Institutional Animal Care and Use Committee (UAB-IACUC).
Imaging of FM-labelled presynaptic terminals on eGFP-labelled spines of cultured hippocampal neurons
Coverslips with 15 div-cultured neurons were transferred to a custom-made perfusion chamber on the stage of an upright microscope (Olympus BX50WI; Melville, NY, USA). Cultures were continuously perfused (∼1 ml min−1; 24°C) with a modified Tyrode's solution containing (mm): 119 NaCl, 5 KCl, 2.5 CaCl2, 2.5 MgCl2, 25 Hepes, and 30 glucose, pH 7.3; 310 mosmol l−1. Action potential-dependent loading of presynaptic terminals with the styryl dye FM4-64 or FM1-43 (15 μm; Molecular Probes, Eugene, OR, USA) was achieved by field stimulation through parallel platinum using 1 ms square pulses (80 V cm−1; S48 stimulator; Grass Medical Instruments, Quincy, MA, USA), as described (Waters & Smith, 2000). Repeated spaced electrical stimulation was used to load the recycling pool of synaptic vesicles. To load the total recycling pool of synaptic vesicles we used a strong stimulation protocol (80 V cm−1, 1 ms pulses, 10 Hz for 90 s = 900 action potentials, APs), whereas weaker stimulation was used to load a fraction of the recycling pool (80 V cm−1, 1 ms pulses, 10 Hz for 30 s = 300 APs). Alternatively, incubation with Tyrode's solution containing 60 mm K+ for 60 s was used as a maximum loading stimulus in order to determine the fraction of vesicles loaded using electrical stimulation protocols. FM1-43 or FM4-64 was present from 20 s before to 60 s after field stimulation to allow dye equilibration on membranes and its uptake by the ensuing vesicle endocytosis, respectively. Following the poststimulation incubation period, cultures were washed for 10 min in dye-free Tyrode's solution to reduce non-specific staining before image acquisition. Destaining of FM-labelled terminals was performed with field stimulation (80 V cm−1, 1 ms pulses at 10 Hz for 120 s) during the entire image acquisition period. To prevent network activity from accelerating FM dye release, synaptic activity was blocked throughout the experiments with the AMPA, NMDA and GABAA receptor antagonists CNQX (10 μm), d,l-2-aminophosphonovaleric acid (d,l-APV) (80 μm), and picrotoxin (50 μm), respectively.
Laser scanning confocal imaging of FM4-64 and eGFP fluorescence was performed using a modified FV300 scan-head (Olympus) and a ×60 water immersion lens (0.9 NA; Olympus). Simultaneous dual-channel fluorescence imaging was achieved by using the 488 nm line of an Ar laser, and the 568 nm line of a He–Ne laser to excite eGFP and FM4-64, respectively. Full-frame images (512 × 512) were acquired every 3 s during baseline acquisition (five frames) and throughout the subsequent field stimulation. Laser intensity and PMT sensitivity settings (voltage, gain and offset) remained constant throughout all experiments. Destaining profiles from a population of excitatory terminals were quantified by drawing regions of interest (ROI) around fluorescent puncta (∼1 μm2) superimposed on eGFP-labelled dendritic spines (Fig. 1D). The raw fluorescence intensity within each ROI was measured for every image acquired across time. The average fluorescent intensity of each ROI was then normalized to the background-subtracted mean baseline fluorescent intensity. The change in normalized pixel intensity from baseline for every time point where data were acquired was calculated for each ROI. Alternatively, FM1-43 imaging in cultured hippocampal neurons was performed by standard wide-field fluorescence microscopy techniques where data acquisition and stimulation were controlled using the imaging software SLIDEBOOK (Intelligent Imaging Innovations, Santa Monica, CA, USA). FM1-43 fluorescence was excited by light from a Xe lamp filtered at wavelength 470–490 nm. Emission was collected at wavelength 505–545 nm by a cooled charge-coupled device camera (CCD; PCO Sensicam, Cooke Corporation, Romulus, MI, USA) on an Olympus inverted microscope (IX 70) with an oil-immersion lens (×60, 1.25 NA, Olympus).
Figure 1. BDNF persistently accelerates the initial phase of evoked release from presynaptic terminals on dendritic spines of eGFP-transfected cultured hippocampal neurons.
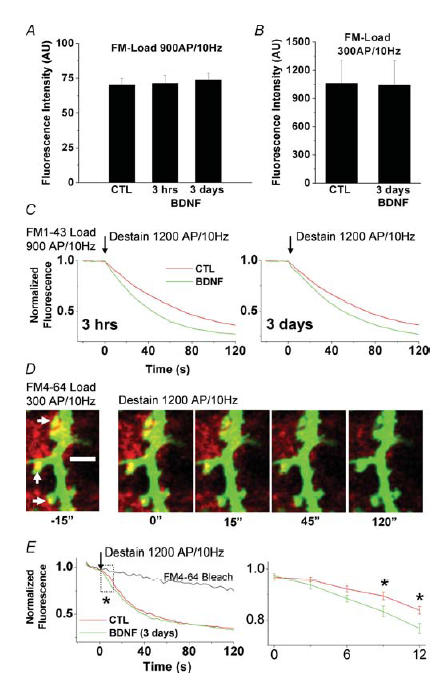
A, initial raw fluorescent intensities of hippocampal presynaptic terminals from control and brain-derived neurotrophic factor (BDNF)-treated cultures (3 h and 3 days) following FM1-43 staining with a strong stimulus (900 APs at 10 Hz) loading ∼80% of the total recycling pool, as estimated by high K+ stimulation (Student's t test, P > 0.05). B, initial raw fluorescent intensities of hippocampal excitatory presynaptic terminals on dendritic spines of eGFP-transfected control and BDNF-treated neurons (3 days) following FM4-64 staining with a weaker stimulus (300 APs at 10 Hz) loading ∼40% of the total recycling pool, as estimated by high K+ stimulation (Student's t test, P > 0.05). C, either 3 h or 3 days exposure to BDNF accelerated FM1-43 destaining evoked by field electrical stimulation (1200 APs at 10 Hz). D, time-lapse confocal images of individual hippocampal excitatory presynaptic nerve terminals (FM4-64, red puncta) overlapping onto dendritic spines of a control postsynaptic neuron (eGFP, green). Time-lapse images were acquired for baseline, as well as during the delivery of the field stimulation protocol used to evoke synaptic vesicle exocytosis. Note the decrease in brightness of red puncta across time (left to right) corresponding to a decrease in FM4-64 fluorescence as a result of exocytosis. E, background-subtracted line plots of mean normalized pixel intensities during baseline acquisition, and during field stimulation, for terminals from control (red) and BDNF-treated cultures (green). The area in the box is enlarged in the plot to the right to highlight the effect of BDNF on FM destaining during the initial phase of the stimulation protocol. Asterisks indicate significant differences (ANOVA followed by Scheffé's procedure for multiple comparisons, P < 0.05).
Acute slice preparation and exposure to BDNF or TrkB-IgG for FM1-43 multiphoton imaging
After deep ether anaesthesia, P17 Wistar rats were rapidly decapitated and transverse hippocampal slices (300 μm) were prepared using a vibratome, as described (Stanton et al. 2003). Slices were allowed to recover for at least 1 h at room temperature in an interface holding chamber filled with artificial cerebrospinal fluid (aCSF, see below for composition) gassed with 95% O2/5% CO2. After recovery, half of the slices from each dissection were exposed to BDNF (250 ng ml−1), or the endogenous BDNF scavenger TrkB-IgG (2 μg ml−1; provided by Regeneron, Tarrytown, NY, USA) (Shelton et al. 1995) for an additional ∼2 h. To facilitate penetration into the slice, a 10 μl droplet of BDNF or TrkB-IgG final dilution was also gently applied to the top of each slice while in the recovery chamber. The inhibitor k-252a was used at a concentration (200 nm) known to be selective for plasma membrane receptor tyrosine kinase, while not affecting soluble tyrosine kinases (Knusel & Hefti, 1992; Tapley et al. 1992).
FM1-43 loading and multiphoton imaging of the RRP pool in acute slices
After recovery and exposure to BDNF or TrkB-IgG (>3 h), individual slices were transferred to a submersion chamber on the stage of an upright microscope (DM-LFS E; Leica, Nussloch, Germany), and continuously perfused with aCSF (1.5 ml min−1) containing (mm): 129 NaCl, 3 KCl, 1.8 MgSO4, 1.6 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, pH 7.4 when gassed with 95% O2/5% CO2 at 24°C. Field excitatory postsynaptic potentials (fEPSPs) were evoked in CA1 stratum radiatum by Schaffer collateral stimulation using a stainless-steel bipolar electrode, and recorded with an aCSF-filled glass electrode. After recording a stable baseline of half-maximal amplitude fEPSPs (∼1 mV), the AMPA receptor antagonist CNQX (10 μm), or the nonselective glutamate ionotropic receptor antagonist kynurenic acid (KYN, 2 mm), were included in the aCSF for the remaining of the imaging session to prevent network activity from accelerating FM1-43 release. In some experiments, LTP was induced by high-frequency afferent stimulation (four 500 ms 100 Hz tetani at 30 s intervals) before blocking excitatory synaptic transmission. After 5 min incubation with FM1-43 (5 μm) to allow for dye equilibration, a RRP of vesicles was selectively labelled by rapid bath application of hypertonic (800 mosmol l−1) aCSF (supplemented with sucrose and FM dye) for 25 s, followed by a rapid return to normal aCSF (with FM1-43). This hyperosmotic shock preferentially releases a pool of quanta classically defined as the readily releasable pool (Rosenmund & Stevens, 1996). Alternatively, the total recycling pool of vesicles was loaded by incubation with aCSF containing 40 mm K+ (equimolar substitution with NaCl) for 15 min, followed by a rapid return to normal aCSF (with FM1-43). After 2 min in FM-containing normal aCSF to allow dye uptake by the ensuing endocytosis, slices were perfused with dye-free aCSF for at least 30 min to reduce extracellular fluorescence, prior to multiphoton excitation imaging.
FM1-43 fluorescence was imaged with a ×40 water-immersion objective (0.8 NA; Leica) in the upright microscope (Leica DM-LFS E) coupled to a Leica multispectral confocal laser scan unit (TCS SP2). The excitation light source was a Ti:sapphire laser (Tsunami) pumped by a diode laser (5 W Millenia; both from Spectra-Physics, Mountain View, CA, USA), which provided ∼130 fs pulses at 82 MHz, and was tuned to 840 nm centre wavelength. Bandpass-filtered FM1-43 fluorescence emission was detected in non-descanned mode by two external PMTs, one behind the objective and the other behind an oil-immersion condenser (1.3 NA). Fluorescence signals were optimized for signal over background (540–600 nm) based on spectral analysis with the confocal laser scanhead with the pinhole maximally open. Laser intensity was controlled with a variable-beam splitter exploiting laser light polarization, and neutral density filters. Four images (512 × 512 pixels; 0.15 μm pixel−1 in x–y axes) were acquired in rapid succession and averaged (peak-to-maximum filter) to obtain a single time point image. For spontaneous FM1-43 destaining time courses, images were acquired every 30 s for 10 min. In addition, FM1-43 destaining was evoked by 2 s, 10 Hz bursts of afferent stimulation, synchronized with the end of image acquisition every 30 s for 20 min. We have refrained from using continuous afferent stimulation during image acquisition in acute slices due to significant movement artifacts that preclude subsequent ROI analyses of the small FM puncta throughout the time-series image sequences. Delivering the afferent stimulation at the end of image acquisition yielded stable fluorescence image sequences without stimulus-evoked movement artifact. In offline analyses, rectangular ROIs (∼2–4 μm2) were selected around the centre of bright punctate fluorescence spots, as well as over neighbouring non-fluorescence regions of the slice for background subtraction. Fluorescence puncta that displayed lateral displacement beyond the enclosing ROI were discarded. The time course and amplitude of evoked or spontaneous destaining of FM1-43 was generated by normalizing each ROI time course to the baseline intensity, averaging normalized background ROIs to produce a photobleaching time course, and then dividing each ROI by the bleaching at the corresponding time points. Only puncta that exhibited stimulus-dependent destaining following a single exponential decay after photobleaching correction were analysed in the evoked and spontaneous data sets, since both processes were acquired in every slice.
Acute slice preparation and BDNF treatment for whole-cell recordings
Transverse hippocampal slices (400 μm thick) were prepared from P12–19 Sprague-Dawley rats. Animals were deeply anaesthetized with isoflurane and then decapitated. The brains were quickly removed, immersed into ice-cold (2–4°C) oxygenated (95% O2/5% CO2) standard aCSF, and sliced with a vibratome; aCSF contained (mm): 126 NaCl, 3 KCl, 2.5 CaCl2, 1.3 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 11 glucose. Slices were incubated at 32 ± 1°C in a holding chamber for 1 h, and then kept at room temperature for the remainder of the recovery period (6–8 h). Slices were treated with BDNF (2–3 h; 250 ng ml−1), as described above for FM1-43 multiphoton imaging. Individual slices were transferred into an interface chamber, maintained at 33 ± 1°C and continuously perfused with bubbled (95% O2/5% CO2) aCSF at a rate ∼4 ml min−1. Since aCSF contained 25 μm bicuculline, a cut was made between the CA1 and CA3 regions to prevent recurrent excitation. A bipolar stimulating electrode (FCH, Bowdoinham, ME, USA) was used to activate Schaffer collateral/commissural fibre synapses onto CA1 pyramidal cells.
Whole-cell recording of evoked EPSCs and variance-mean analysis
Whole-cell recordings were performed from CA1 pyramidal neurons according to standard techniques using a MultiClamp 700B amplifier (Axon Instruments, Union City, CA, USA). Membrane currents were filtered at 3 kHz and digitized at 10 kHz using Clampex (v.9, Axon Instruments). Pipette resistance was 3–4 MΩ when filled with intracellular solution containing (mm): 135 CsMeSO3, 8 NaCl, 10 Hepes, 2 MgATP, 0.3 NaGTP, 0.5 EGTA, and 1 QX-314; 275 mosmol l−1, pH 7.25 adjusted with CsOH. Access resistance was carefully monitored, and only cells with stable access resistance were included in analyses. CA1 pyramidal cell membrane potential was held at −60 mV throughout the experiment. Extracellular stimuli used to evoke EPSCs were triggered by the MultiClamp and delivered to the bipolar stimulating electrode via a stimulus-isolator (ISO-Flex; API, Jerusalem, Israel).
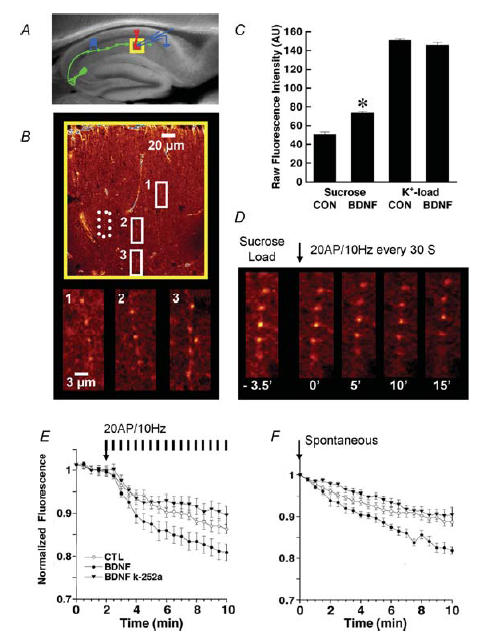
The variance-mean analysis was applied to steady-state sequences of evoked EPSCs recorded at different extracellular [Ca2+]o, and analysing the variance of EPSC amplitudes at different EPSC mean amplitudes. We assume that the mean amplitude of the EPSC (Imean) is:
where N represents the total number of release sites, P the release probability, and Q the quantal amplitude of the postsynaptic response. Following the binomial model, the variance of EPSC amplitude is given by:
and the parabolic relationship between variance and Imean is defined as:
Q and N can be obtained from the initial slope and the width of a parabolic fit to a variance-mean plot. Alternatively, this parabola can be written in linear form as:
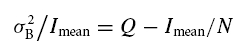 |
and Q and N determined from the y-axis intercept and slope, respectively, of a linear fit to a variance/mean–mean plot. Since this analysis requires that postsynaptic AMPA receptors be responding to a submaximal non-saturating concentration of glutamate, so that EPSC amplitude accurately reflects concentration of glutamate released, we conducted all experiments in a low concentration of the selective AMPA receptor antagonist DNQX (100 nm). To modulate the probability of transmitter release, we used three different ratios of [Ca2+]o/[Mg2+]o in the recording aCSF (4/1 mm, 2/2 mm, and 1/4 mm). A typical experiment began with perfusing the slice with aCSF at the 4/1 [Ca2+]o/[Mg2+]o ratio after the whole-cell configuration was established. CA1 neurons were voltage clamped at −65 mV, and EPSCs were evoked by 100 μs current pulses delivered to Schaffer collateral/commissural fibres every 10 s. Stable recordings of 8–10 min were made before switching to aCSF with a 1/4 [Ca2+]o/[Mg2+]o ratio. After the amplitude of EPSCs decreased and stabilized at a lower level, another 10–12 min of recordings were made. Lastly, slices were switched to the aCSF at a 2/2 [Ca2+]o/[Mg2+]o ratio for the last 8–10 min recording epoch. This procedure yielded 60–70 EPSCs at each [Ca2+]o, with two runs performed on each neuron.
Statistical analyses
The unit of observations used for statistical analyses (n) is the number of coverslips for FM dye experiments in primary cultures, the number of slices for FM dye experiments and field potential recordings in acute slices, and the number of neurons for whole-cell recordings in acute slices. The number of FM dye fluorescence puncta (i.e. ROIs around them) is given for information purposes, and was used to calculate a statistical mean within each coverslip with cultured neurons or acute slice. All data were analysed by analysis of variance (ANOVA), or independent Student's t test using SPSS software (SPSS, Inc., Chicago, IL, USA). Significance level was preset to P < 0.05. Data are presented as means ± s.e.m. across different experiments.
Results
BDNF persistently accelerates the initial phase of evoked release from presynaptic terminals on dendritic spines of eGFP-transfected cultured hippocampal neurons
Although it is well established that BDNF enhances spontaneous and evoked transmitter release in populations of excitatory synapses (reviewed by Poo, 2001; Tyler et al. 2002b), it has yet to be determined if this increase is actually due to the facilitation of transmitter release from individual excitatory presynaptic nerve terminals. To test this hypothesis directly, we used the styryl dyes FM1-43 and FM4-64 to estimate transmitter release from cultured hippocampal neurons (>14 div). We first aimed to determine the effect of BDNF-treatment on the size of the recycling pool of synaptic vesicles. The fluorescence intensity measured on individual FM-positive puncta following electrical field stimulation can be used as a quantitative estimate of the number of vesicles endocytosed during synaptic vesicle recycling, and thus of the size of the pool of recycling vesicles (reviewed by Murthy, 1999; Ryan, 2001).
Using two different BDNF (250 ng ml−1)-treatment time courses (3 h or 3 days), we found that BDNF does not affect the size of the recycling pool of synaptic vesicles as indicated by FM1-43 staining. Following a standard electrical stimulation FM-loading protocol (10 Hz, 90 s field stimulation), which loaded ∼80% of the pool compared with maximal loading obtained with a high-K+-loading protocol, there were no differences in the raw fluorescent intensities of individually FM1-43-positive hippocampal presynaptic terminals among the treatment conditions (control, 70.03 ± 4.89 arbitrary units (AU) of fluorescence, n = 4 cultures, 118 terminals; BDNF, 3 h, 71.32 ± 5.66 AU, n = 3 cultures, 104 terminals; BDNF, 3 days 73.82 ± 4.76 AU, n = 3 cultures, 123 terminals; Student's t test P > 0.05; Fig. 1A). In an additional set of experiments, we transfected hippocampal cultures with eGFP to identify dendritic spines, one of the hallmarks of excitatory Type-I synapses (Gray, 1959). We identified functional excitatory presynaptic terminals by their FM4-64 fluorescence and apposition to eGFP-labelled dendritic spines by dual-channel confocal fluorescence microscopy after activity-dependent loading of FM4-64 (Fig. 1D). Here, we implemented a submaximal electrical stimulation FM-loading protocol (field stimulation 10 Hz, 30 s), which loaded a fraction (∼40%) of the recycling pool compared to maximal loading obtained with a high-K+-loading protocol. In these experiments we examined the effects of 3 day BDNF treatment. Again, we found no differences in the initial raw fluorescence intensities between excitatory presynaptic terminals on eGFP-labelled spines from control cultures and those treated with BDNF (control, 1060.42 ± 245.77 AU, n = 4 cultures, 53 terminals, versus BDNF, 1039.07 ± 262.55 AU, n = 5 cultures, 42 terminals; Student's t test P > 0.05; Fig. 1B). Note that the different arbitrary values of fluorescence (AU) between these two experiments arise from the different detectors used, i.e. PMTs in the laser-scanning confocal experiments versus a cooled CCD in the wide-field fluorescence experiments. Taken together, these results demonstrate that BDNF does not persistently alter the total size of recycling pools of vesicles at excitatory synapses on hippocampal neurons, as estimated by electrically evoking transmitter release with sustained electrical stimulation followed by a 60 s period of vesicle endocytosis.
Next, in order to evaluate the modulation of neurotransmitter release kinetics at hippocampal synapses by BDNF, we analysed the fluorescence destaining profiles during vesicle exocytosis, evoked by field stimulation (10 Hz, 120 s), from hippocampal terminals loaded with FM1-43. Indeed, BDNF treatment for both 3 h (n = 3, 104 terminals) and 3 days (n = 3, 123 terminals) induced a significant increase in the rate of FM dye destaining compared with control cultures (n = 4, 118 terminals) during field stimulation (ANOVA for multiple comparisons, P < 0.001; Fig. 1C).
In order to evaluate the modulation of transmitter release at identified excitatory synapses by BDNF, we analysed the fluorescence destaining profiles of FM4-64 positive puncta apposed to eGFP positive dendritic spines. The fluorescent intensity of individual FM4-64 puncta adjacent to eGFP spines decayed during field stimulation in a manner indicative of synaptic vesicle exocytosis, whereas spine eGFP fluorescence was unaffected (Fig. 1D). These observations illustrate that it is possible to estimate the kinetics of evoked transmitter release from individually identified excitatory presynaptic terminals apposing dendritic spines by FM4-64 and eGFP imaging in cultured hippocampal neurons. We loaded a small fraction of the rapidly recycling pool (∼40%) of terminals with FM4-64 using field stimulation (10 Hz, 30 s; Fig. 1D), then, following the washout period, we monitored the destaining rates of FM4-64-loaded terminals during vesicle exocytosis evoked by field stimulation (10 Hz, 120 s; Fig. 1E). Indeed, there was a significantly greater stimulus-evoked reduction in FM4-64 intensity from baseline in BDNF-treated cultures compared with controls at the 9 s (BDNF, −0.17 ± 0.02 normalized pixel intensity, versus control, −0.12 ± 0.01) and the 12 s poststimulus time points (BDNF, −0.24 ± 0.02 normalized pixel intensity, versus control, −0.17 ± 0.01; 42 terminals from n = 5 BDNF-treated cultures, 53 terminals from n = 4 control cultures; ANOVA followed by Scheffé's procedure for multiple comparisons, P < 0.05; Fig. 1E). Since no further differences were observed at individual time points later in the destaining trial, these observations are consistent with BDNF producing a selective increase in the rate of release from an immediately available pool of quanta, which is likely to be the classically defined readily releasable pool (Rosenmund & Stevens, 1996). The statistical differences only within the initial 9–12 s of field stimulation are consistent with the observation that the time course for recovery of transmitter release after depletion of the readily releasable pool in this type of hippocampal cultures is between 7 and 12 s (Stevens & Tsujimoto, 1995; Rosenmund & Stevens, 1996; Stevens & Wesseling, 1998; Pyle et al. 2000). These results demonstrate that BDNF enhances evoked vesicle release in excitatory synapses in a manner consistent with the modulation of an immediately available pool of vesicles. Taken together, these data demonstrate that BDNF persistently enhances evoked vesicle release from individual excitatory presynaptic terminals on dendritic spines, consistent with its modulation of presynaptic structure and function (reviewed by Poo, 2001; Tyler et al. 2002b).
BDNF persistently accelerates both evoked and spontaneous transmitter release from the RRP within presynaptic terminals in CA1 stratum radiatum of hippocampal slices
The results described above demonstrate that BDNF enhances evoked vesicle release from an immediately available vesicle pool in excitatory spine synapses between cultured hippocampal neurons. However, short-term (minutes to hours) exposure to BDNF is also known to enhance excitatory synaptic transmission and facilitate the induction of LTP of excitatory synapses in hippocampal slices (reviewed by Poo, 2001; Tyler et al. 2002b; Lu, 2003). To test whether BDNF can also modulate vesicle release in acute hippocampal slices, we took advantage of multiphoton excitation microscopy (Stanton et al. 2001; Zakharenko et al. 2001; Winterer et al. 2006). Hippocampal slices preserve the distribution of CA3 excitatory afferent fibres within CA1 stratum radiatum, thus allowing us to identify individual excitatory presynaptic terminals labelled with FM1-43 (Fig. 2A and B). To selectively label an immediately available vesicle pool with FM1-43, we used hyperosmotic solutions, a manipulation that evokes the selective release of the readily releasable pool of quanta (Rosenmund & Stevens, 1996; Rizzoli & Betz, 2005). By exposing slices for 25 s to hyperosmotic aCSF (800 mosmol l−1 with sucrose) containing FM1-43 (5 μm, with CNQX or KYN), brightly fluorescent puncta could be imaged by multiphoton microscopy in the apical dendritic field (stratum radiatum) of area CA1. Figure 2B illustrates a representative field of fluorescent puncta in CA1 stratum radiatum after loading FM1-43 with hyperosmotic solution. It is noteworthy that because exchange rates between vesicle pools are much slower in acute slices (1.7 h; Stanton et al. 2005) compared with cultured neurons (1–2 min; Pyle et al. 2000), most vesicles loaded during the hyperosmotic shock are retained within the same vesicle pool during the dye wash period (∼30 min), allowing the estimation of its size and release kinetics in the following experiments. For consistency with our previous work (Stanton et al. 2005), we shall refer to this sucrose-labelled pool as the rapidly recycling pool (RRP) because this terminology describes more specifically the origin of this vesicle pool, i.e. it was labelled with FM dye by recycling endocytosis during a brief exocytosis stimulus.
Figure 2. BDNF persistently enhances the evoked and spontaneous destaining rate of FM1-43 from a rapidly recycling pool (RRP) in CA1 stratum radiatum of acute hippocampal slices visualized by multiphoton microscopy.
A, schematic representation of the stimulation of CA3 afferent fibres and recording of extracellular fEPSPs in CA1 stratum radiatum superimposed on a bright-field image of an acute hippocampal slice. The yellow box represents the area imaged by multiphoton microscopy. B, multiphoton excitation (840 nm) fluorescence images of FM1-43 loaded into RRP vesicles by hyperosmotic shock (25 s, 800 mosmol l−1 aCSF with sucrose) within stratum radiatum of area CA1 (yellow box in A) of a control hippocampal slice. Note the row of CA1 pyramidal cell bodies at the top of the image. The areas within the solid white boxes are shown at higher magnification in the panels below. Note the tip of the stimulating electrode in the middle left of the top panel (as a triangular non-fluorescent object). The activity-dependent FM1-43 destaining of terminals within the area denoted by the dotted white line is shown in D. C, initial raw fluorescent intensities of RRP-labelled terminals from control and BDNF-treated slices following FM1-43 staining using sucrose to load the RRP or high K+ to load the total recycling pool of vesicles (Student's t test, *P < 0.05). D, activity-dependent destaining of FM1-43 from RRP-labelled terminals within the dotted box shown in B (numbers represent time in minutes; 0′ is the beginning of the unloading stimulation, i.e. 10 Hz 2 s bursts every 30 s). E, time course of stimulus-evoked FM1-43 destaining from RRP-labelled terminals in control (open circles) versus BDNF-treated (filled circles) slices showing enhanced vesicular release from the RRP after BDNF (background-corrected and normalized averaged pixel intensity within multiple ROIs centred over stable fluorescent puncta). The vertical bars indicate the stimulation to the afferent fibres consisting of 10 Hz 2 s bursts every 30 s throughout and synchronized with image acquisition. The enhanced rate of activity-dependent release from the RRP after BDNF exposure is completely prevented by coapplication of the Trk inhibitor k-252a (filled triangles). F, time course of spontaneous FM1-43 destaining from RRP-labelled terminals in control (open circles) versus BDNF-treated (filled circles) slices showing enhanced vesicular release from the RRP after BDNF. The enhancement of spontaneous FM1-43 destaining is also blocked by coapplication of k-252a (filled triangles).
The fluorescence intensity of hyperosmotic shock-loaded FM1-43 puncta prior to destaining was 45% higher in slices exposed to BDNF for 2–3 h (250 ng ml−1), compared with control slices (BDNF, 74 ± 1.6 AU, 121 puncta from n = 6 slices, versus control, 51 ± 2.3 AU, 89 puncta from n = 6 slices; Student's t test P < 0.05; Fig. 2C). On the other hand, BDNF had no effect on the size of the total vesicle recycling pool, as estimated by the fluorescence intensity of FM1-43 puncta loaded with 40 mm K+ aCSF (BDNF, 146 ± 3 AU, 36 puncta from n = 6 slices, versus control, 151.34 ± 1.1 AU, 36 puncta from n = 6 slices, Student's t test P > 0.05; Fig. 2C). The conclusion that hyperosmotic shock preferentially loads the RRP is further supported by the fact that fluorescence intensity after sucrose loading is ∼34% of that after high K+ loading, a fraction of the total recycling pool consistent with earlier FM1-43 measurements (Pyle et al. 2000; Stevens & Williams, 2000), as well as electron microscopic localization of photoconverted FM1-43 in vesicles either docked or within 200 nm of the active release zone (Stanton et al. 2003). It is important to note that BDNF increased the fraction of the RRP with respect to the total recycling pool (34% in control, versus 51% after BDNF), consistent with the increased density of docked vesicles in BDNF-treated slice cultures (Tyler & Pozzo-Miller, 2001), and their reduced density in Bdnf knockout mice (Pozzo-Miller et al. 1999). Taken together, these results support the view that BDNF selectively modulates the size of the RRP, without affecting the total recycling pool within CA1 excitatory synapses.
To measure the evoked destaining rate from the RRP, afferent burst stimulation was delivered to Schaffer collaterals once every 30 s (2 s, 10 Hz), synchronized with image acquisition. Figure 2D shows the progressive destaining of FM1-43 puncta within the box delimited by the dotted line in Fig. 2B. Pre-treatment with BDNF produced a significant increase in the rate of FM1-43 destaining compared to control slices (1/t½ of the single-exponential fit to the first 3 min of destaining: BDNF, 145 ± 10 × 10−4 s−1, 121 puncta from n = 6 slices, versus control, 63 ± 7 × 10−4 s−1, 89 puncta from n = 5 slices; Student's t test P < 0.05; Fig. 2E). This effect of BDNF was prevented by coincubation with the receptor tyrosine kinase inhibitor k-252a (1/t½ = 65 ± 9 × 10−4 s−1, 79 puncta from n = 6 slices, versus BDNF, 145 ± 10 × 10−4 s−1; Student's t test P < 0.05; Fig. 2E). At the concentration used (200 nm), k-252 is selective for membrane-associated receptor tyrosine kinases without affecting soluble tyrosine kinases (Knusel & Hefti, 1992; Tapley et al. 1992), thus implicating neurotrophin Trk receptors in these effects of BDNF.
In addition to the effect on evoked vesicular release, the rate of spontaneous FM1-43 destaining following RRP loading (in aCSF with either CNQX or KYN to reduce network activity) was significantly faster in BDNF-treated slices compared with controls (BDNF 1/t½ = 6.4 ± 0.2 × 10−4 s−1, 121 puncta from n = 6 slices, versus control, 3.8 ± 0.12 × 10−4 s−1, 89 puncta from n = 5 slices, Student's t test p < 0.05; Fig. 2F). This effect was also blocked by k-252a (1/t½ = 2.65 ± 0.21 × 10−4 s−1, 79 puncta from n = 6 slices, versus BDNF, 1/t½ = 6.4 ± 0.2 × 10−4 s−1; Student's t test P < 0.05; Fig. 2F). Together with its effect on the initial phase of evoked FM dye destaining from excitatory terminals between cultured hippocampal neurons (Fig. 1E), the observations in acute slices demonstrate that BDNF enhances transmitter release from an immediately available, rapidly recycling pool of synaptic vesicles, the RRP.
BDNF increases release probability at CA3–CA1 excitatory synapses in hippocampal slices, without affecting the number of independent release sites or quantal amplitude
To further confirm the mechanisms of BDNF action on transmitter release at CA3–CA1 synapses, we used the variance-mean technique assuming a binomial model of synaptic transmission (Silver, 2003). Excitatory postsynaptic currents (EPSCs) were evoked at different extracellular Ca2+ concentrations (1, 2 and 4 mm) in the presence of a submaximal concentration of the AMPA receptor antagonist DNQX (100 nm) to prevent receptor saturation.
Figure 3A illustrates the variance of typical collections of successful EPSCs at each [Ca2+]o, and the corresponding single Gaussian distributions of histograms of EPSC peak amplitudes at the same three [Ca2+]o (synaptic transmission failures were not included in the analysis). Parabolic relations for all data are plotted for untreated control and BDNF-treated slices (Fig. 3B). The good superposition of the two parabolic relations, with data from BDNF-treated slices moving the variance-mean points to the right along a single parabola, indicates that the action of BDNF is associated with a purely presynaptic increase in release probability (Pr). Figure 3C illustrates the mean Pr derived from the parabolic fits as a function of [Ca2+]o, which was enhanced by BDNF pre-exposure at all three concentrations (Table 1; ANOVA for multiple measures, *P < 0.05; n = 9 BDNF cells, eight control cells). As indicated by the overlapping parabolic fits from BDNF-treated and control cells, there were no significant differences in either quantal amplitude (3.5 ± 0.36 versus 3.12 ± 0.25 pA; Student's t test P > 0.05) or number of independent release sites (100 ± 24.2 versus 114 ± 18.8; Student's t test P > 0.05). These results are in agreement with the BDNF-induced increase in Pr observed in cultured neurons (Lessmann & Heumann, 1998; Berninger et al. 1999; Schinder et al. 2000). It is important to note that the observed actions of BDNF on release probability and destaining of FM dyes from excitatory terminals must be long lasting, because the neurotrophin was not present during the whole-cell recordings or imaging sessions.
Figure 3. BDNF increases the probability of transmitter release at CA3–CA1 synapses.
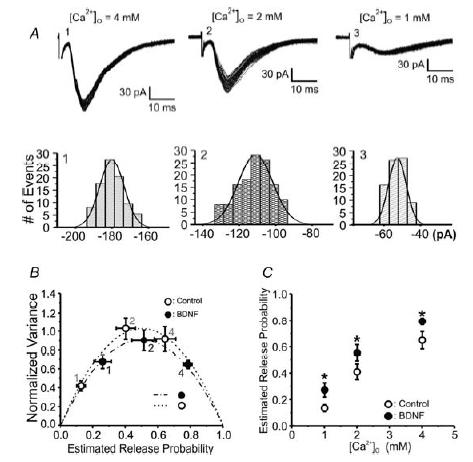
A, superimposed evoked EPSCs (no failures) recorded in CA1 pyramidal neurons from a control slice at three different concentrations of extracellular Ca2+ (4, 2 and 1 mm). Panels below the traces show the frequency distribution of EPSC amplitudes at each extracellular Ca2+ concentration fitted with Gaussian distributions. B, variance-mean analysis assuming a binomial model of synaptic transmission (Silver, 2003). Plot of the normalized variance of EPSC amplitudes versus the estimated release probability fitted by single parabolas in control (open circles) and BDNF-treated (filled circles) slices. EPSCs were evoked at different extracellular Ca2+ concentrations in the presence of a submaximal concentration of the AMPA receptor antagonist DNQX (100 nm) to prevent receptor saturation. As indicated by the overlapping parabolic fits from BDNF-treated and control cells, there were no significant differences in either quantal amplitude or number of independent release sites (Student's t test P > 0.05). C, mean probability of release (Pr) values derived from the parabolic fits plotted as a function of [Ca2+]o. Pr was enhanced by BDNF pre-exposure (filled circles) at all three extracellular Ca2+ concentrations (ANOVA for multiple measures, *P < 0.05; see also Table 1).
Table 1.
Probability of transmitter release at different extracellular Ca2+ concentrations, as estimated by EPSC mean-variance analysis (see Methods)
| [Ca2+]o (mm) | Control (n = 7) | BDNF (n = 9) |
|---|---|---|
| 1 | 0.13 ± 0.03 | 0.26 ± 0.05* |
| 2 | 0.40 ± 0.06 | 0.55 ± 0.06* |
| 4 | 0.64 ± 0.07 | 0.79 ± 0.03* |
BDNF, brain-derived neurotrophic factor. Data are expressed as means ± s.e.m. ANOVA for multiple measures
P < 0.05.
The BDNF scavenger TrkB-IgG prevents the enhancement of FM1-43 destaining from the RRP following LTP induction in area CA1 of hippocampal slices
Our data suggest that short-term (2–3 h) BDNF exposure increases both the size and the rate of release from the RRP, as well as the probability of transmitter release in area CA1 of acute hippocampal slices. In order to test the role of endogenous BDNF signalling in these observations, we next examined the consequence of scavenging BDNF released during high-frequency afferent activity using a ‘receptor-body’– TrkB-IgG – containing the extracellular binding domain of TrkB receptors fused with the heavy chain of an IgG to make it soluble (Shelton et al. 1995). This BDNF scavenger, as well as function-blocking anti-BDNF antibodies, prevented the induction of LTP of EPSPs in hippocampal slices (Figurov et al. 1996; Kang et al. 1997; Chen et al. 1999; Kossel et al. 2001). Because the induction of LTP is associated with an accelerated rate of FM1-43 destaining from the total vesicular pool (Zakharenko et al. 2001), we asked whether endogenous BDNF released during the LTP-inducing stimuli also modulates release from the RRP.
LTP of excitatory synaptic transmission in the CA1 region of hippocampal slices was induced with a strong stimulus (4 × 100 Hz 500 ms bursts, every 30 s), 30 min before loading Schaffer collaterals with FM1-43 using a hyperosmotic shock (see above). This stimulus protocol elicited a 187 ± 6.9% increase in fEPSP amplitude, measured 15 min after the conditioning stimuli (n = 5 slices). Indeed, induction of LTP increased the rate of FM1-43 destaining from the RRP (LTP 1/t½ = 115 ± 13 × 10−4 s−1, 122 puncta from n = 6 slices versus control, 63 ± 7 × 10−4 s−1, 89 puncta from n = 5 slices; Student's t test P < 0.05; Fig. 4A), consistent with our previous observations (Stanton et al. 2005). As we and others have previously shown (Figurov et al. 1996; Kang et al. 1997), the scavenger TrkB-IgG (2 μg ml−1, 2–3 h pre-exposure) reduced the amplitude of burst-induced LTP (128.5 ± 3.2%; n = 4 slices). Consistent with the modulation of the RRP by BDNF, TrkB-IgG also completely prevented the enhancement of the rate of FM1-43 destaining from the RRP observed after induction of LTP (TrkB-IgG 1/t½ = 31 ± 6 × 10−4 s−1, 95 puncta from n = 5 slices, versus LTP 1/t½ = 115 ± 13 × 10−4 s−1; Student's t test P < 0.05; Fig. 4B). In fact, the destaining rate in the presence of TrkB-IgG was even slower than that observed in control slices, suggesting that extracellular levels of endogenous BDNF are facilitating vesicular release during burst-type high frequency afferent stimulation (20 APs at 10 Hz).
Figure 4. The BDNF scavenger TrkB-IgG prevents the enhancement of FM1-43 release from the RRP after LTP induction.
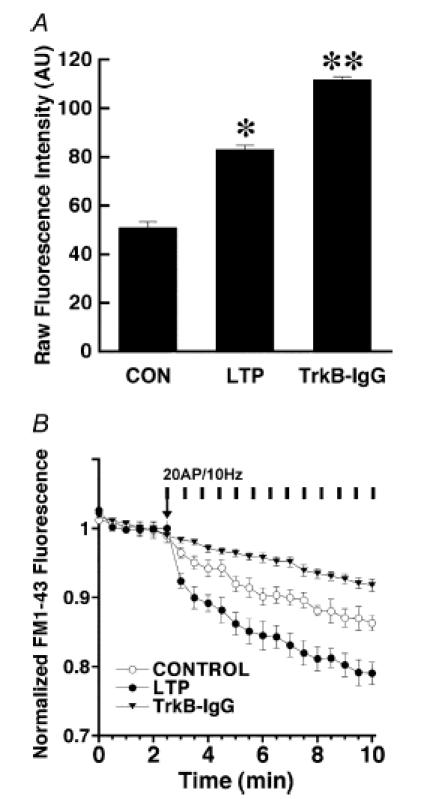
A, average initial raw FM1-43 fluorescence intensities of RRP-labelled terminals from control slices, and in slices after LTP induction in the absence and presence of the BDNF scavenger TrkB-IgG. FM1-43 staining was performed using sucrose to selectively load the RRP (Student's t test, *P < 0.05, **P < 0.001). B, time course of evoked FM1-43 destaining from the RRP in control slices (open circles) versus slices where LTP was induced (filled circles), showing the enhanced vesicular release from the RRP after LTP induction in area CA1. The vertical bars indicate the stimulation to the afferent fibres consisting of 10 Hz 2 s bursts every 30 s throughout and synchronized with image acquisition. The enhanced rate of activity-dependent release from the RRP after LTP induction is completely prevented by application of the BDNF scavenger TrkB-IgG before the LTP-inducing stimuli (filled triangles).
In addition to enhance the RRP destaining rate, LTP induction caused an increase in the size of the RRP, as estimated by FM1-43 fluorescence intensity after labelling the RRP by hyperosmotic shock (LTP 83 ± 2 AU, 122 puncta from n = 6 slices, versus control 51 ± 2.3 AU, n = 89 puncta from n = 5 slices; Student's t test P < 0.05; Fig. 4B). The effect of the LTP-inducing protocol on RRP size is similar to that of BDNF exposure, strengthening the view that BDNF is necessary for long-term changes in synaptic transmission (reviewed by Poo, 2001; Tyler et al. 2002a; Bramham & Messaoudi, 2005). Intriguingly, conditioning afferent stimulation in the presence of TrkB-IgG increased the initial FM1-43 intensity after hyperosmotic shock (TrkB-IgG 112 ± 1.6, 95 puncta from n = 5 slices, versus LTP 83 ± 2 AU; Student's t test P < 0.05; Fig. 4B). Together with the lack of enhancement of FM1-43 destaining after LTP induction in presynaptic-targeted conditional Bdnf knockout mice (Zakharenko et al. 2003), our results further demonstrate the requirement of endogenous BDNF released during high-frequency afferent activity for the induction and/or expression of long-term synaptic plasticity in the hippocampus.
Discussion
Our study provides three novel insights into the modulation of presynaptic transmitter release by the neurotrophin BDNF, in addition to a direct consequence of such modulation for hippocampal long-term synaptic plasticity. First, the results presented here directly demonstrate that BDNF enhances vesicular release from individual presynaptic terminals impinging on dendritic spines, a morphological hallmark of excitatory synapses. Second, our findings provide direct support for BDNF-induced modulation of a rapidly recycling pool of vesicles at excitatory CA3–CA1 synapses, as indirectly suggested in our prior studies of its effects on docked vesicles, mEPSC frequency, and presynaptic short-term plasticity (Figurov et al. 1996; Gottschalk et al. 1998; Pozzo-Miller et al. 1999; Tartaglia et al. 2001; Tyler & Pozzo-Miller, 2001; Tyler et al. 2002b). Third, we show that BDNF increases the probability of transmitter release at excitatory hippocampal CA3–CA1 synapses, without affecting the number of independent release sites or the quantal amplitude, consistent with earlier observations in cultured embryonic neurons (Lessmann & Heumann, 1998; Schinder et al. 2000). Since BDNF was absent during the loading and destaining of FM dyes in the imaging experiments on cultured cells and acute slices, as well as during whole-cell recordings in acute slices, we conclude that the effects observed in pretreated neurons are long lasting, at least for the duration of the imaging and recording sessions (1–2 h). This enhancement of the release capability at excitatory terminals is likely to represent one of the mechanisms by which BDNF mediates long-lasting synaptic plasticity. Indeed, enhancement of vesicular release normally observed after the induction of LTP (Zakharenko et al. 2001; Stanton et al. 2005) is prevented by endogenous BDNF quenching during the conditioning high-frequency afferent stimulus. Thus, the cellular mechanisms described here represent excellent prospects for mediating the fundamental role(s) of BDNF in hippocampal-dependent learning and memory.
It had been previously reported that acute BDNF application to cultured cortical neurons increased FM1-43 destaining rate, but from unidentified terminals modestly stimulated with high K+ solutions for loading and destaining (Bradley & Sporns, 1999). Here, using hippocampal primary cultures we examined the effects of both short-term (3 h) and chronic (3 days) BDNF-treatment on synaptic vesicle exocytosis using FM-dye staining/destaining evoked by electrical field stimulation. Further, in order to determine the effects of BDNF on individually identified excitatory synapses, we used the red-shifted dye FM4-64 in combination with eGFP imaging to identify presynaptic nerve terminals on dendritic spines. First, we observed no differences in the fluorescent intensity between control and BDNF-treated cultures following activity-dependent FM dye loading using sustained stimulus trains (10 Hz for 30 or 90 s) or high K+ stimulation, suggesting that BDNF does not modulate the size of the total recycling pool of vesicles, either after short-term or chronic treatment. Interestingly, synaptic vesicles clustered away from the active zone, presumed ultrastructural correlates of the recycling and resting pools of synaptic vesicles, are not affected in either Bdnf knockout mice (Pozzo-Miller et al. 1999) or in BDNF-treated slice cultures (Tartaglia et al. 2001; Tyler & Pozzo-Miller, 2001). While we did not observe an effect of BDNF-treatment on the size of the total recycling pool of synaptic vesicles, we did observe that both short-term and chronic BDNF application accelerated activity-dependent FM-destaining kinetics.
Following two different electrical stimulation protocols (900 and 300 APs; 10 Hz each) to load synaptic vesicles with FM dyes, we observed that BDNF treatment significantly accelerated electrically evoked destaining (1200 APs; 10 Hz) of FM-positive puncta. Moreover, our results demonstrate that BDNF treatment facilitates synaptic vesicle release during the initial phase of exocytosis from identified hippocampal excitatory synapses. These data are consistent with the modulation of an immediately available pool of vesicles by BDNF signalling. It is interesting to note that we observed a persistent increase in the rate of destaining following BDNF treatment throughout the entire destaining trial (1200 APs; 10 Hz) when synaptic vesicles were loaded with FM dyes using 900 APs. However, BDNF treatment only produced a significant increase in the rate of FM4-64 destaining during the initial period (9–12 s) of field stimulation used to evoke synaptic vesicle exocytosis (1200 APs; 10 Hz) when implementing a 300 APs FM-dye-loading protocol. In cultured hippocampal neurons, the rate of mixing between vesicles in the readily releasable pool and the total recycling pool is very fast (τ∼100–140 s; Murthy & Stevens, 1999; Pyle et al. 2000) compared with the 10 min FM-dye washout period we implemented here. Given a high rate of mixing among these vesicle pools in cultured neurons, and the long FM-dye washout period we implemented, it is expected to observe differential destaining effects when using different loading protocols. Furthermore, recent evidence demonstrates that synaptic vesicles in hippocampal terminals are differentially partitioned and recycled to different pools in a use-dependent fashion, which makes them more or less likely to be available for subsequent reuse, depending on the history of activity experienced by the terminal (Vanden Berghe & Klingauf, 2006). Whether or not BDNF treatment exerts an effect on the mixing of distinct synaptic vesicle pools in cultured neurons remains to be established. Despite these considerations and regardless of the dye-loading protocol, BDNF consistently increased the rate of FM-dye destaining, which was observable during the initial phase of synaptic vesicle exocytosis triggered by electrical stimulation.
In order to investigate the effects of BDNF on vesicular release at excitatory synapses in intact tissue, we loaded a RRP of vesicles within hippocampal CA3–CA1 terminals with FM dyes in acute brain slices using hyperosmotic shock followed by multiphoton imaging. BDNF accelerated activity-dependent FM1-43 destaining when this RRP was selectively labelled by hyperosmotic shock, the manipulation originally used to define the size and release kinetics of the readily releasable pool of quanta within hippocampal synapses (Stevens & Tsujimoto, 1995; Rosenmund & Stevens, 1996). We have shown previously in acute hippocampal slices that this protocol selectively loads a fraction (∼28%) of the total recycling pool of vesicles, as estimated by depolarization with 45 mm K+ aCSF (Stanton et al. 2003). Additionally, the vesicles in this sucrose-stainable recycling pool are either in contact (docked) or within 200 nm of the active zone, as determined by electron microscopy after FM1-43 photoconversion (Stanton et al. 2003). Further, the rate of evoked FM1-43 destaining from these terminals, i.e. vesicular release, is much faster than release from depolarization-loaded terminals (Stanton et al. 2003; Winterer et al. 2006), consistent with the properties of the classically defined readily releasable pool (Rosenmund & Stevens, 1996). Notably, the vesicle pool labelled with sucrose in preterminals of acute slices mixed with other pools with exchange rates much slower (Stanton et al. 2005) than those observed in cultured neurons, as discussed above (Pyle et al. 2000). Furthermore, maximally loading the recycling pool in acute slices with high K+ stimulation failed to reveal any differences in the size of the total recycling pool between control and BDNF-treated slices, as observed in cultured hippocampal neurons. Finally, in two different model preparations (hippocampal primary cultures and acute hippocampal slices) using several different FM-dye-loading protocols, the effects of BDNF were evident within the first 90–120 stimuli, again consistent with the engagement of a immediately available set of synaptic vesicles. Taken together, these observations provide strong evidence that BDNF enhances transmitter release by modulating an immediately available set of vesicles, which undergo exocytosis in response to stimulation, a pool we referred to as the rapidly recycling pool. It is important to note that this rapidly recycling pool may include multiple classically defined readily releasable pools, as suggested by our EM observation of sucrose-loaded vesicles up to 200 nm away from the active zone (Stanton et al. 2003).
Modulation of a rapidly recycling vesicle pool by BDNF has been suggested by several earlier observations. First, short-term application of BDNF (3–4 h) to acute hippocampal slices prevented synaptic fatigue during high-frequency stimulation, thus facilitating long-term potentiation (LTP) induction (Figurov et al. 1996; Gottschalk et al. 1998, 1999). Second, slices from Bdnf knockout mice exhibited more pronounced synaptic fatigue during high-frequency stimulation, as well as reduced LTP success rate and LTP magnitude (when successful) compared with slices from their wild-type littermates (Pozzo-Miller et al. 1999). Lastly, scavenging endogenous BDNF with TrkB-IgG not only prevented LTP induction, but also enhanced synaptic fatigue during the conditioning stimulus (Figurov et al. 1996). Considering that synaptic fatigue during sustained high-frequency stimulation is thought to result from vesicle depletion from a readily available vesicle pool followed by fast vesicle recycling (Model et al. 1975; Dickinson-Nelson & Reese, 1983; Dobrunz & Stevens, 1997; Sara et al. 2002; Zucker & Regehr, 2002), those earlier physiological observations and the present optical approach provide functional evidence supporting the hypothesis that BDNF modulates the size and/or the rate of release from a rapidly recycling pool of vesicles, including the classically defined readily releasable pool.
Quantitative electron microscopy of synaptic vesicle distributions within excitatory presynaptic terminals on CA1 pyramidal neuron spines further supports this view. The structural correlate of the readily releasable pool is considered to be represented by those vesicles docked at the active zone (Harris & Sultan, 1995; Schikorski & Stevens, 1997, 2001), although this view has been recently revised for large terminals such as the neuromuscular junction (Rizzoli & Betz, 2004, 2005). Consistent with their pronounced synaptic fatigue during high-frequency stimulation, Bdnf knockout mice had fewer docked vesicles at the active zones of CA1 spine synapses than their wild-type littermates (Pozzo-Miller et al. 1999). Similarly, Trkb knockout mice were also found to have fewer docked vesicles at various hippocampal synapses, including those in CA1 stratum radiatum (Martinez et al. 1998). Furthermore, we have shown that long-term BDNF exposure increases the number of docked vesicles at the active zones of CA1 spine synapses in hippocampal slice cultures (Tartaglia et al. 2001; Tyler & Pozzo-Miller, 2001). Similar effects on synaptic vesicle density have been reported in dissociated cultures of hippocampal and cortical neurons (Takei et al. 1997; Collin et al. 2001). Collectively, these results provide structural evidence that BDNF enhances transmitter release by either increasing the number of docked vesicles (and thus the size) or the rate of release from a readily releasable or rapidly recycling pool of vesicles.
In parallel with well-characterized postsynaptic changes (Malinow & Malenka, 2002), induction of LTP is accompanied by an enhancement of FM1-43 destaining from the total recycling vesicle pool within CA1 terminals (Zakharenko et al. 2001). Intriguingly, this enhancement is absent in acute slices from conditional Bdnf knockout mice (Zakharenko et al. 2003). We show here that the BDNF scavenger TrkB-IgG prevented a similar potentiation of FM1-43 release from the RRP after induction of LTP in hippocampal slices. It is worth noting that this consequence of LTP induction is completely confined to the RRP, as the release from the reserve pool of vesicles is neither affected by LTP nor sensitive to the receptor tyrosine kinase inhibitor k-252a (Stanton et al. 2005). Interestingly, TrkB-IgG reduced the rate of FM destaining below control values, suggesting that endogenous BDNF levels are necessary to maintain a certain level of activity-dependent vesicular release. In addition, the RRP size, as estimated from the initial FM intensity, was increased in slices exposed to TrkB-IgG, suggesting that RRP vesicles accumulated due to the reduction in spontaneous vesicular release in the absence of endogenous BDNF signalling. Additional experiments are underway to further characterize these effects of endogenous BDNF levels. Taken altogether, the increase in docked vesicles induced by BDNF seems to be translated functionally into an increased RRP size, and ultimately, the Pr of evoked transmitter release from hippocampal excitatory synapses. In fact, our observations here using mean-variance analysis of evoked EPSCs at CA3–CA1 synapses in acute hippocampal slices directly demonstrate that BDNF-treated synapses have a higher probability of transmitter release, but similar mean quantal amplitude. It is important to note that BDNF enhances transmitter release from GABAergic inhibitory terminals following a seemingly different strategy than that observed here for excitatory terminals. In the case of inhibitory terminals between cultured hippocampal neurons, chronic BDNF exposure (3–4 weeks) increased both the Pr per terminal as well as per vesicle (vesicle replenishment rate), without major effects on quantal size, number of release sites, readily releasable pool size, or Ca2+ dependence of transmitter release (Baldelli et al. 2005). For excitatory hippocampal synapses, we have previously demonstrated that chronic BDNF treatment increases the number of release sites (i.e. N) without affecting the quantal size (Tyler & Pozzo-Miller, 2001). In addition, these effects of BDNF on inhibitory terminals are accompanied by a redistribution leading to an enhanced overlapping of N- and P/Q-types of Ca2+ channels to vesicle release sites. It remains to be tested whether similar redistribution of presynaptic Ca2+ channels contribute to the BDNF effects on glutamatergic excitatory terminals.
Which are the potential downstream effectors of BDNF actions on presynaptic terminals? Consistent with the modulation of high-frequency synaptic depression and vesicle docking by BDNF, it is notable that cultured neurons from Rab3A knockout mice do not exhibit the characteristic increase in presynaptic transmitter release induced by BDNF (Thakker-Varia et al. 2001; Alder et al. 2005). Intriguingly, Rab3A and its effector RIM1α are necessary for a delayed component of LTP (Huang et al. 2005), which may represent the presynaptic plasticity module that requires BDNF for its induction (Zakharenko et al. 2003). On the other hand, BDNF is completely ineffective at enhancing glutamate release from synaptosomes prepared from synapsin-I/II-deficient mice (Jovanovic et al. 2000). Since both rab3A and the synapsins have been implicated in vesicle trafficking from the vesicle cluster to the active zone (Sudhof, 2004), these observations suggest that BDNF facilitates the mobilization from a reserve pool to a readily releasable and fast recycling pool of synaptic vesicles.
Current models of neurotrophin actions at synapses have begun to differentiate between acute (seconds to minutes), short-term (minutes to hours) and chronic (hours to days) effects (Poo, 2001; Tyler et al. 2002b; Lu, 2003; Bramham & Messaoudi, 2005). Well-established acute effects, such as increased frequency of synaptic currents, have been attributed to a direct modulation of presynaptic Ca2+ levels (Stoop & Poo, 1996; Li et al. 1998b). On the other hand, long-term effects are thought to represent molecular and/or structural modifications of transmitter release sites, such as increased Ca2+ channel expression and/or coupling to release machinery (Baldelli et al. 2000, 2005), and docked vesicle density at the active zone (Collin et al. 2001; Tyler & Pozzo-Miller, 2001). At this point, it is worth mentioning that the effects of BDNF in acute slices, such as the facilitation of LTP induction and the prevention of synaptic fatigue during high-frequency stimulation, required a ∼3 h exposure (Figurov et al. 1996; Gottschalk et al. 1998, 1999), similar to that used in the present experiments in cultured neurons and acute slices. Intriguingly, the effect of BDNF on FM1-43 destaining in cultured cortical neurons was evident only after 3 h because it required protein translation (Bradley & Sporns, 1999), an effect consistent with the increased expression of presynaptic vesicle proteins induced by BDNF (Tartaglia et al. 2001). Since long-term (days) BDNF exposure increases the number of excitatory synapses in vitro (Vicario-Abejon et al. 1998; Collin et al. 2001; Tyler & Pozzo-Miller, 2001), it is difficult to conclude from electrophysiological recordings of populations of synapses that BDNF modulates transmitter release from individual excitatory nerve terminals. However, quantitative EM has directly demonstrated that BDNF increases the number of docked synaptic vesicles at the active zone (Collin et al. 2001; Tartaglia et al. 2001; Tyler & Pozzo-Miller, 2001). The visualization of vesicular release from individual excitatory presynaptic terminals on dendritic spines of cultured neurons and within CA1 stratum radiatum of hippocampal slices using FM dyes provides direct support to the hypothesis that BDNF enhances transmitter release from individual excitatory terminals.
In summary, we have presented direct evidence that BDNF enhances vesicular release from identified excitatory synapses by modulating the size of a readily releasable pool of synaptic vesicles and the probability of transmitter release. This modulation has direct consequences for the expression of a presynaptic consequence of LTP induction, providing further support to the notion that neurotrophins exert their role in hippocampal-dependent learning and memory by modulating fundamental mechanisms of synaptic transmission and plasticity at hippocampal synapses.
Acknowledgments
We would like to thank Drs Robin Lester (UAB) and John Hablitz (UAB) for insightful discussions and comments on the manuscript. Supported by NIH-NS40593 (L.P.-M., UAB), P30-HD38985 (UAB Mental Retardation Research Center), PO1-HD38760 (Neurobiology, UAB), Civitan International Foundation (L.P.-M.), NIH-NS44421 (P.K.S., NYMC), DFG-Mu-809/7–2 (W.M., Humboldt Berlin), DFG-GRK 238 (J.W., Humboldt Berlin). We also thank Amgen and Regeneron for the generous supply of BDNF and TrkB-IgG, respectively. L.P.-M. is a McNulty Civitan Scientist.
References
- Akaneya Y, Tsumoto T, Kinoshita S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J Neurosci. 1997;17:6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder J, Thakker-Varia S, Crozier RA, Shaheen A, Plummer MR, Black IB. Early presynaptic and late postsynaptic components contribute independently to brain-derived neurotrophic factor-induced synaptic plasticity. J Neurosci. 2005;25:3080–3085. doi: 10.1523/JNEUROSCI.2970-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axmacher N, Winterer J, Stanton PK, Draguhn A, Muller W. Two-photon imaging of spontaneous vesicular release in acute brain slices and its modulation by presynaptic GABAA receptors. Neuroimage. 2004;22:1014–1021. doi: 10.1016/j.neuroimage.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Baldelli P, Forni PE, Carbone E. BDNF, NT-3 and NGF induce distinct new Ca2+ channel synthesis in developing hippocampal neurons. Eur J Neurosci. 2000;12:4017–4032. doi: 10.1046/j.1460-9568.2000.00305.x. [DOI] [PubMed] [Google Scholar]
- Baldelli P, Hernandez-Guijo JM, Carabelli V, Carbone E. Brain-derived neurotrophic factor enhances GABA release probability and nonuniform distribution of N- and P/Q-type channels on release sites of hippocampal inhibitory synapses. J Neurosci. 2005;25:3358–3368. doi: 10.1523/JNEUROSCI.4227-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berninger B, Schinder AF, Poo MM. Synaptic reliability correlates with reduced susceptibility to synaptic potentiation by brain-derived neurotrophic factor. Learn Mem. 1999;6:232–242. [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Bewick GS. Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci. 1992;12:363–375. doi: 10.1523/JNEUROSCI.12-02-00363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J, Sporns O. BDNF-dependent enhancement of exocytosis in cultured cortical neurons requires translation but not transcription. Brain Res. 1999;815:140–149. doi: 10.1016/s0006-8993(98)01112-3. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods. 1997;71:143–155. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol. 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19:7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochilla AJ, Angleson JK, Betz WJ. Monitoring secretory membrane with FM1-43 fluorescence. Annu Rev Neurosci. 1999;22:1–10. doi: 10.1146/annurev.neuro.22.1.1. [DOI] [PubMed] [Google Scholar]
- Collin C, Vicario-Abejon C, Rubio ME, Wenthold RJ, McKay RD, Segal M. Neurotrophins act at presynaptic terminals to activate synapses among cultured hippocampal neurons. Eur J Neurosci. 2001;13:1273–1282. doi: 10.1046/j.0953-816x.2001.01500.x. [DOI] [PubMed] [Google Scholar]
- Dickinson-Nelson A, Reese TS. Structural changes during transmitter release at synapses in the frog sympathetic ganglion. J Neurosci. 1983;3:42–52. doi: 10.1523/JNEUROSCI.03-01-00042.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Gottschalk WA, Jiang H, Tartaglia N, Feng L, Figurov A, Lu B. Signaling mechanisms mediating BDNF modulation of synaptic plasticity in the hippocampus. Learn Mem. 1999;6:243–256. [PMC free article] [PubMed] [Google Scholar]
- Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray E. Electron microscopy of synaptic contacts on dendritic spines of the cerebral cortex. Nature. 1959;183:1592–1594. doi: 10.1038/1831592a0. [DOI] [PubMed] [Google Scholar]
- Harris KM, Sultan P. Variation in the number, location and size of synaptic vesicles provides an anatomical basis for the nonuniform probability of release at hippocampal CA1 synapses. Neuropharmacology. 1995;34:1387–1395. doi: 10.1016/0028-3908(95)00142-s. [DOI] [PubMed] [Google Scholar]
- Huang YY, Zakharenko SS, Schoch S, Kaeser PS, Janz R, Sudhof TC, Siegelbaum SA, Kandel ER. Genetic evidence for a protein-kinase-A-mediated presynaptic component in NMDA-receptor-dependent forms of long-term synaptic potentiation. Proc Natl Acad Sci U S A. 2005;102:9365–9370. doi: 10.1073/pnas.0503777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Sawtell NB, Bear MF. Brain-derived neurotrophic factor alters the synaptic modification threshold in visual cortex. Neuropharmacology. 1998;37:571–579. doi: 10.1016/s0028-3908(98)00050-1. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Knusel B, Hefti F. K-252 compounds: modulators of neurotrophin signal transduction. J Neurochem. 1992;59:1987–1996. doi: 10.1111/j.1471-4159.1992.tb10085.x. [DOI] [PubMed] [Google Scholar]
- Kohrmann M, Haubensak W, Hemraj I, Kaether C, Lessmann VJ, Kiebler MA. Fast, convenient, and effective method to transiently transfect primary hippocampal neurons. J Neurosci Res. 1999;58:831–835. [PubMed] [Google Scholar]
- Kossel AH, Cambridge SB, Wagner U, Bonhoeffer T. A caged Ab reveals an immediate/instructive effect of BDNF during hippocampal synaptic potentiation. Proc Natl Acad Sci U S A. 2001;98:14702–14707. doi: 10.1073/pnas.251326998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. Neuroreport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- Lessmann V, Heumann R. Modulation of unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived neurotrophic factor in hippocampal microcultures: presynaptic enhancement depends on pre-established paired-pulse facilitation. Neuroscience. 1998;86:399–413. doi: 10.1016/s0306-4522(98)00035-9. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci U S A. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 1998a;95:10884–10889. doi: 10.1073/pnas.95.18.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998b;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Martinez A, Alcantara S, Borrell V, Del Rio JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, Soriano E. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci. 1998;18:7336–7350. doi: 10.1523/JNEUROSCI.18-18-07336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi E, Bardsen K, Srebro B, Bramham CR. Acute intrahippocampal infusion of BDNF induces lasting potentiation of synaptic transmission in the rat dentate gyrus. J Neurophysiol. 1998;79:496–499. doi: 10.1152/jn.1998.79.1.496. [DOI] [PubMed] [Google Scholar]
- Model PG, Highstein SM, Bennett MV. Depletion of vesicles and fatigue of transmission at a vertebrate central synapse. Brain Res. 1975;98:209–228. doi: 10.1016/0006-8993(75)90002-5. [DOI] [PubMed] [Google Scholar]
- Murthy VN. Optical detection of synaptic vesicle exocytosis and endocytosis. Curr Opin Neurobiol. 1999;9:314–320. doi: 10.1016/s0959-4388(99)80046-4. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Stevens CF. Reversal of synaptic vesicle docking at central synapses. Nat Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- Numakawa T, Takei N, Yamagishi S, Sakai N, Hatanaka H. Neurotrophin-elicited short-term glutamate release from cultured cerebellar granule neurons. Brain Res. 1999;842:431–438. doi: 10.1016/s0006-8993(99)01867-3. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, Du McDermott KJ, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Piedras-Rentería ES, Tsien RW. Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron. 2000;28:221–231. doi: 10.1016/s0896-6273(00)00098-2. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. The structural organization of the readily releasable pool of synaptic vesicles. Science. 2004;303:2037–2039. doi: 10.1126/science.1094682. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Ryan TA. Presynaptic imaging techniques. Curr Opin Neurobiol. 2001;11:544–549. doi: 10.1016/s0959-4388(00)00247-6. [DOI] [PubMed] [Google Scholar]
- Sara Y, Mozhayeva MG, Liu X, Kavalali ET. Fast vesicle recycling supports neurotransmission during sustained stimulation at hippocampal synapses. J Neurosci. 2002;22:1608–1617. doi: 10.1523/JNEUROSCI.22-05-01608.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci. 2001;4:391–395. doi: 10.1038/86042. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Berninger B, Poo M. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron. 2000;25:151–163. doi: 10.1016/s0896-6273(00)80879-x. [DOI] [PubMed] [Google Scholar]
- Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll K, Spencer SD, Levinson AD. Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA. Estimation of nonuniform quantal parameters with multiple-probability fluctuation analysis: theory, application and limitations. J Neurosci Methods. 2003;130:127–141. doi: 10.1016/j.jneumeth.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Heinemann U, Muller W. FM1-43 imaging reveals cGMP-dependent long-term depression of presynaptic transmitter release. J Neurosci. 2001;21:RC167. doi: 10.1523/JNEUROSCI.21-19-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Bailey CP, Kyrozis A, Raginov I, Laube G, Veh RW, Nguyen CQ, Muller W. Long-term depression of presynaptic release from the readily releasable vesicle pool induced by NMDA receptor-dependent retrograde nitric oxide. J Neurosci. 2003;23:5936–5944. doi: 10.1523/JNEUROSCI.23-13-05936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Zhang XL, Muller W. Imaging LTP of presynaptic release of FM1-43 from the rapidly recycling vesicle pool of Schaffer collateral-CA1 synapses in rat hippocampal slices. Eur J Neurosci. 2005;22:2451–2461. doi: 10.1111/j.1460-9568.2005.04437.x. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci U S A. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Williams JH. ‘Kiss and run’ exocytosis at hippocampal synapses. Proc Natl Acad Sci U S A. 2000;97:12828–12833. doi: 10.1073/pnas.230438697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoop R, Poo MM. Synaptic modulation by neurotrophic factors: differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci. 1996;16:3256–3264. doi: 10.1523/JNEUROSCI.16-10-03256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Takei N, Sasaoka K, Inoue K, Takahashi M, Endo Y, Hatanaka H. Brain-derived neurotrophic factor increases the stimulation-evoked release of glutamate and the levels of exocytosis-associated proteins in cultured cortical neurons from embryonic rats. J Neurochem. 1997;68:370–375. doi: 10.1046/j.1471-4159.1997.68010370.x. [DOI] [PubMed] [Google Scholar]
- Tartaqlia N, Du J, Tyler WJ, Neale E, Pozzo-Miller L, Lu B. Protein synthesis-dependent and independent regulation of hippocampal synapses by brain-derived neurotrophic factor. J Biol Chem. 2001;276:37585–37593. doi: 10.1074/jbc.M101683200. [DOI] [PubMed] [Google Scholar]
- Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- Thakker-Varia S, Alder J, Crozier RA, Plummer MR, Black IB. Rab3A is required for brain-derived neurotrophic factor-induced synaptic plasticity: transcriptional analysis at the population and single-cell levels. J Neurosci. 2001;21:6782–6790. doi: 10.1523/JNEUROSCI.21-17-06782.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Alonso M, Bramham CR, Pozzo-Miller LD. From acquisition to consolidation: on the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn Mem. 2002a;9:224–237. doi: 10.1101/lm.51202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Perrett SP, Pozzo-Miller LD. The role of neurotrophins in neurotransmitter release. Neuroscientist. 2002b;8:524–531. doi: 10.1177/1073858402238511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe P, Klingauf J. Synaptic vesicles in hippocampal boutons recycle to different pools in a use-dependent fashion. J Physiol. 2006;572:707–720. doi: 10.1113/jphysiol.2005.100842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario-Abejon C, Collin C, McKay RD, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters J, Smith SJ. Phorbol esters potentiate evoked and spontaneous release by different presynaptic mechanisms. J Neurosci. 2000;20:7863–7870. doi: 10.1523/JNEUROSCI.20-21-07863.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterer J, Stanton PK, Müller W. Direct monitoring of vesicular release and uptake in brain slices by multiphoton excitation of the styryl dye FM1-43. Biotechniques. 2006;40:343–351. doi: 10.2144/000112120. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1–CA3 synapses. Neuron. 2003;39:975–990. doi: 10.1016/s0896-6273(03)00543-9. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci. 2001;4:711–717. doi: 10.1038/89498. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]