

Abstract
Spinal motoneurons, like many neurons, respond with repetitive spiking to sustained inputs. The afterhyperpolarization (AHP) that follows each spike, however, decays relatively slowly in motoneurons. The slow depolarization during this decay should allow sodium (Na+) channel inactivation to keep up with its activation and thus should prevent initiation of the next spike. We hypothesized that the persistent component of the total Na+ current provides the mechanism that generates a rate of rise sufficiently rapid to generate a spike. In large cultured spinal neurons, presumed to be primarily motoneurons, inhibition of persistent sodium current (NaP) by the drug riluzole at low concentrations resulted in a loss of repetitive firing. However, cells remained fully capable of producing spikes to transient inputs. These effects of riluzole were not due to insufficient depolarization, enhancement of the AHP, or sustained Na+ channel inactivation. To further test this hypothesis, computer simulations were performed with a kinetic Na+ channel model that provided greater independent control of NaP relative to transient Na+ current (NaT) than that provided by riluzole administration. The model was tuned to generate substantial NaP and exhibited good repetitive firing to slowly rising inputs. When NaP was sharply reduced without significantly altering NaT, the model reproduced the effects of riluzole administration, inducing failure of repetitive firing but allowing single spikes in response to sharp transients. These results strongly support the essential role of NaP in spike initiation to slow inputs in spinal neurons. NaP may play a fundamental role in determining how a neuron responds to sustained inputs.
The basic features of action potential initiation are well understood (Hille, 2001). A rapidly depolarizing input allows activation of the Na+ channel to escape its inactivation and provide the regenerative depolarization that initiates the spike. The rate of rise of the input, however, plays a key role: activation can only escape inactivation if this rate is rapid. Thus, when a step of current of sufficient magnitude is injected into a cell, the sharp onset of the step initiates a spike. If the step is prolonged, action potentials will continue to be initiated at a more or less steady rate in many types of neurons, even though the input stays constant and contains no subsequent transients (Calvin, 1975; Powers & Binder, 2001; Steriade, 2001). During this repetitive firing, each spike is followed by an afterhyperpolarization (AHP). As the AHP decays, however, the resulting depolarization is relatively slow compared to the time constant for inactivation of the Na+ channel. Consequently, in the fraction of channels that open in this membrane potential range, Na+ channel inactivation should keep up with activation. In other words, the rate of depolarization falls well short of the level needed to allow activation to escape inactivation, and spike initiation should fail to occur. What mechanism then provides the fast rate of rise for initiation of these spikes?
In the work presented here, we tested the hypothesis that the persistent component of the total Na+ current is essential for spike initiation during steady or slowly rising inputs. Persistent Na+ current (NaP) is a key element in depolarizing the cell to its threshold for action potential generation, due to its role in providing subthreshold depolarization and in amplifying synaptic inputs (Schwindt & Crill, 1995; Stuart & Sakmann, 1995; Uteshev et al. 1995; Fleidervish & Gutnick, 1996; Lipowsky et al. 1996; Baker & Bostock, 1997; Hsiao et al. 1998; Butera et al. 1999; Lee & Heckman, 1999; Bennett et al. 2000; Urbani & Belluzzi, 2000; Agrawal et al. 2001; Taddese & Bean, 2002; Li & Bennett, 2003; Wu et al. 2004). Although NaP constitutes only a small fraction of the total Na+ current, it is nonetheless a major component of the currents active in the voltage range for spike initiation during repetitive firing (Taddese & Bean, 2002; Do & Bean, 2003). The sustained depolarization provided by NaP is thus critically important in controlling cell excitability. The hypothesis tested here focuses on an additional role for NaP: accelerating depolarization sufficiently to allow inactivation to escape activation, for even very slowly rising inputs.
Previously, we evaluated this hypothesis with indirect methods in vivo (Lee & Heckman, 2001). Here, this hypothesis is tested more directly via five specific predictions (see Results) for spinal neurons in cell culture, where drugs to alter Na+ channel behaviour can be readily administered. In addition, we also employed computer simulations that incorporate a kinetic model of the Na+ channel based on the work of Taddese & Bean (2002).
Methods
Cell culture
The dissociation and culturing techniques have been previously described (Kuo et al. 2004, 2005). All procedures were approved by the Northwestern University Animal Care and Use Committee. Briefly, each spinal cord was removed from embryonic mice (day 12–14) and sliced into 400 μm slices. Cells were isolated by mechanical trituration and plated at approximately 133 000 cells cm−2 on poly d-lysine-coated glass coverslips. The medium was changed after the second day in vitro and then twice per week. The cultures were used between 10 and 30 days in vitro. The mice used in this study were either C57BL6 or transgenic mice expressing the human SOD1 gene (Kuo et al. 2004, 2005). Animals with the mutant version of the SOD1 gene are a standard model of amyotrophic lateral sclerosis (Rosen et al. 1993) and we have previously shown that cells from these animals have significantly larger amplitudes for NaP than cells from animals with wild-type SOD1 genes or from control animals (Kuo et al. 2004, 2005). Despite this difference, the role of NaP in spike initiation in mutant SOD1 cells, as determined from the effect of administration of the drug riluzole, was the same as in wild-type and control cells. Thus data from all cells were combined for the present study.
Electrophysiology
Electrodes were typically 3–4 MΩ in resistance when filled with (mm) 145 potassium gluconate, 0.1 CaCl2, 1.1 EGTA, 5 Hepes, 2 MgCl2, and 5 ATP-Mg2+ with a pH of 7.3. Artificial CSF (aCSF) contained (mm) 126 NaCl, 26.2 NaHCO3, 3 KCl, 1.5 MgSO4, 2.5 CaCl2, 10 Glucose, 0.1 picrotoxin (Sigma), 0.05 d-AP5, 0.01 NBQX (Tocris), and 0.01 strychnine (Sigma) with a pH of 7.4 when bubbled with 95% O2–5% CO2. In experiments examining the small conductance calcium-mediated potassium current (IAHP) the synaptic blockers (picrotoxin, d-AP5, NBQX, and strychnine) were removed and the following were added to the aCSF (mm): 1 TEA-Cl, 0.05 4-AP, and 0.001 TTX. TTX was added to the aCSF as a 1 μm TTX–aCSF solution and riluzole was dissolved in the aCSF using DMSO at 0.05% or less. No differences were observed with the vehicle controls. All aCSF and drug solutions were bath applicated using a gravity-fed Automate drug perfusion system. Data were acquired at 10 kHz using a 1401 Plus (Cambridge Electronic Design) and digitally filtered offline.
Whole-cell recordings
Multipolar cells with soma diameters >20 μm were chosen for study. Most were likely to be motoneurons (Carriedo et al. 1996), but large interneurons may have also been included in the sample. Whole-cell patch-clamp measurements were performed at room temperature in current- and voltage-clamp mode using the Axoclamp 2A or Multiclamp 700A amplifier (Axon Instruments, Union City, CA, USA). Rhythmic firing was generated using current-step or triangular-ramp protocols. Current responses in voltage-clamp mode were elicited using a depolarizing voltage-ramp protocol. The relation between the action potential firing frequency and injected current (F–I relation) was measured using a triangular current-ramp injection protocol. Synaptic activity was approximated by injecting random current noise. This signal was generated from a white noise input, filtered at 10 kHz by the output of the data acquisition system D/A converter, and then much more strongly filtered by the time constants of the electrodes (≪ 1 ms) and motoneurons (typically 10–20 ms). The amplitude of the noise input could be adjusted across a wide range. Typically, the amplitudes we used produced a peak-to-peak depolarization between 10 and 15 mV. For the IAHP experiments (see above), cells were held at −40 mV and then stepped to 0 mV for 50 ms. IAHP was calculated as the integral of the current, after the offset of the step command, from the peak of the outward current to 50 ms after the peak. No differences were observed in the time to the peak (time to peak riluzole/time to peak control = 0.996 ± 0.156; n = 4).
Computer simulations
The primary goal of the simulations was to evaluate the effect of NaP on spike initiation. The Na+ channel was thus modelled in a relatively detailed fashion using a multiple-state kinetic scheme (Taddese & Bean, 2002). Otherwise the model was simple, including only the minimal structure that produced a reasonable replication of repetitive firing of the cultured motoneurons providing the experimental data. All simulations were performed using NEURON 5.8 (Hines & Carnevale, 1997). The model consisted of a single compartment (a cylinder 6.4 μm in diameter and 100 μm in length). In addition to the Na+ channel, this model cell included a leak channel, a voltage-sensitive K+ (the delayed rectifier), a high-voltage-activated Ca2+ channel, a Ca2+-activated K+ channel and a pump and buffer mechanism for controlling intracellular Ca2+. The voltage-sensitive K+ and Ca2+ channels were modelled via the standard Hodgkin–Huxley formulation.
Na+ current
The sodium current was modelled using a kinetic scheme, which is based on the model of Taddese & Bean (2002). The state model is reproduced in Fig. 1.
Figure 1. Kinetic model of the Na+ channel used in the computer simulations.

Values for all the constants shown are given in Table 1. Based on the model developed by (Taddese & Bean, 2002) but adapted to match the transient behaviour for young rat motoneurons (Safronov & Vogel, 1995).
C, I and O represent closed, inactivated and open states, respectively. α, β, γ, δ, Con, Coff, Ooff and Oon are the rate constants. The factors a and b are defined as a = (Oon/Con)¼ and b = (Ooff/Coff)¼, respectively. The rate constants of this model were adjusted to generate two basic types of Na+ current: transient only and transient plus persistent. We first adjusted all rate constants to approximate the sodium current behaviour as a function of voltage from the experimental data reported by Safronov & Vogel, (1995) (see Table 1 for the obtained values). These results did not contain significant persistent Na+ current. Thus to simulate the motoneurons in the present study, we increased the rate constant Ooff from 0.015 ms−1 to 0.03 ms−1 in order to produce a persistent component that is around 4.3% of the total Na+ current (see Table 1; as measured from a step from −120 mV to −10 mV; see Fig. 11). In all simulations, Na+ current was calculated using Ohm's law, multiplying the occupancy of O by the driving force and Gmax(0.01487 S cm−2). The reversal potential ENa was set at +50 mV.
Table 1.
| Motoneuron | Taddese & Bean (2002) | |
|---|---|---|
| α(ms−1) | 14αexp((V+ 27)/27.5) | 140αexp(V/27) |
| β (ms−1) | 7.45αexp(−(V+ 27)/27.5) | 10⋆exp(−V/27) |
| γ (ms−1) | 1880 | 150 |
| δ (ms−1) | 32 | 40 |
| Con(ms−1) | 0.026 | 0.004 |
| Coff (ms−1) | 1.235 | 9 |
| Oon (ms−1) | 0.84173 | 0.8 |
| Ooff (ms−1) | 0.03 (0.015α) | 0.004 |
| a | 1.88 | 1.88 |
| b | 13.8 | 13.8 |
Value for simulations with low persistent Na+ current.
Figure 11. The behaviour of the Na+ channel model in response to a series of voltage steps.

A, Na+ currents generated by steps from −120 mV to −60 mV, −50 mV, −40 mV, and −10 mV. The dashed line shows the behaviour of the reduced NaP version of the model to the step to −10 mV. At this and all other voltage steps, the Na+ current of the reduced NaP model was virtually superimposed on the standard model for the first few milliseconds. The lower level of NaP is evident after the decay of the most of NaT. B, same as A, but on an expanded vertical scale to more clearly reveal differences in NaP. C, steady-state activation and inactivation curves for the standard (▾) and reduced (×) NaP models.
Voltage-gated potassium current
The voltage-gated K+ current was matched to data on the delayed rectifier current (IKdr; Safronov & Vogel, 1995) and modelled using the formalism of Hodgkin & Huxley (1952). The potassium channel activity is dependent on the activation gate m,
 |
The steady-state activation m∞ satisfies the Boltzmann equation:
The potassium current was calculated from Ohm's law:
where the delayed rectifier conductance GKdr was set to 0.002 S cm−2 and Ek was set to −70 mV.
High-voltage-activated Ca2+ current
The model of the high-voltage-activated Ca2+ current was based on the data from Booth (1997), where the activation of this calcium current was modelled with a single activation gate. The steady state of the activation is:
And the time constant τm was set to 20 ms.
Calcium-activated K+ current
Calcium-activated K+ current was integrated into the model to generate the afterhyperpolarization (AHP). For the calcium-dependent gate m, the steady-state activation and time constant were set to according to data from Destexhe (1997):
where the [Ca2+]i is a millimolar concentration. The internal calcium concentration changed according to:
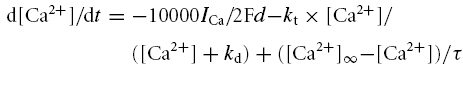 |
where F has its usual value, d is depth in micrometres, kt is the time constant of the pump, kd is the equilibrium calcium value, [Ca2+]∞ is the equilibrium intracellular calcium value (usually in the range 200–300 nm), and τ is the time constant of calcium removal.
Leak current
The leak current was modelled as a linear conductance independent of the voltage. It was calculated by following Ohm's law, with the reversal potential was set as −90 mV. The conductance density gleak was set to 0.0005 S cm−2, giving a total input resistance of 100 MΩ.
Data analysis
Data were analysed offline using IGOR Pro software (Wavemetrics, Lake Oswego, OR, USA). The input conductance of the cell was determined by fitting a linear regression to the I–V relation in its linear region, subthreshold to the voltage-sensitive conductances (Powers & Binder, 2001). The leak conductance was subtracted from each waveform to produce the leak-subtracted I–V relation. The total persistent inward current was defined as the downward deviation in the leak-subtracted I–V relation. NaP was isolated by subtracting the leak-subtracted TTX I–V relation from the non-TTX leak-subtracted I–V relation, with the standard assumption that all Na+ current responding to the slow ramp was persistent (Crill, 1996). Some cells exhibited ‘breakthrough’ spikes as NaP neared its peak amplitude, a phenomenon also seen in other studies where external Na+ concentration was kept at normal levels (e.g. Powers & Binder, 2003). These spikes appeared to initiate well away from the soma, because the voltage command showed little or no deviation due to a loss of clamp control. The breakthrough region was avoided in our quantification of the amplitude of NaP, which was assessed as the average current amplitude in a voltage range spanning 2–10 mV depolarized to the NaP onset threshold. This range also corresponded to the voltage range for spike initiation during repetitive firing. The slope of the linear regression of the F–I relation fit to the ascending phase of the ramp defined the F–I gain, which provided an overall measure of neuronal steady-state excitability. The current onset was the absolute current amplitude on the initial spike of the ascending phase of the current ramp protocol. The spike threshold was defined as the voltage 0.5 ms before the peak of the second derivative.
A two-tailed Student's t test was used to test for a significant difference of the means. A probability of <0.05 was accepted as significant.
Results
To test the hypothesis that NaP acts as the spike-generating mechanism to slow or steady inputs, five predictions were tested: (1) a substantial reduction of NaP should prevent spike initiation to slowly rising inputs; (2) reduced NaP should not prevent spikes merely by reducing the net depolarizing drive; therefore depolarizing the cell well above the voltage threshold for spike initiation should not restore repetitive firing; (3) a reduction in NaP sufficient to eliminate repetitive firing should not prevent cells from generating a normal spike to a rapidly rising input, such as the onset of a current step; (4) graded reductions in NaP should produce graded reductions in firing rate, so that the amplitude of NaP should correlate with the gain of the relationship between repetitive firing frequency and magnitude of injected current; (5) NaP should be the dominant current near spike voltage threshold, overcoming leak current combined with any voltage-sensitive outward currents. These predictions were tested in the experiments using the drug riluzole to block NaP. Since riluzole also has an effect on the transient component of the total Na current (NaT) (Huang et al. 1997; Ptak et al. 2005), the above hypotheses were also evaluated with computer simulations based on a kinetic model of the Na+ channel (Taddese & Bean, 2002). In this model, NaP and NaT could be manipulated with a high degree of independence, even though both were generated by the same channel.
Cultured motoneurons exhibit strong repetitive firing and significant Nap
All cells (n = 57) exhibited strong repetitive firing in response to slow ramp inputs or prolonged steps (1–2 s) (Fig. 2A). This repetitive firing stayed consistent during repeated trials for the entire recording period (as long as 90 min), indicating that the currents responsible for repetitive firing were not subject to significant run down during the recordings. The relationship between firing frequency and injected current (i.e. the F–I function) was usually approximately linear (Fig. 2B). The F–I gain (i.e. slope) of the F–I functions averaged 0.205 ± 0.113 spikes s−1 pA−1 (n = 57) and the current threshold for onset of firing averaged 26.8 ± 51.4 pA (n = 57). With a slowly rising voltage command, the current generated by most cells revealed a downwards inflection that resulted in a region of negative slope in the I–V function (Fig. 2C, D). This negative slope was eliminated by TTX (Fig. 2C, D grey traces; Fig. 3A bold, dotted trace). Subtraction of the trial with TTX from the control trial revealed NaP (bold black trace, Fig. 3B). The average amplitude of NaP calculated in this manner was −182.8 ± 51.4 pA (n = 32).
Figure 2. Frequency–current (F–I) and current–voltage (I–V) relations.

A, the cell responded to a slow-depolarizing current ramp (bottom trace) with sustained repetitive firing (top trace). B, the F–I relation (ascending ramp only) is plotted for the cell in A. The slope of the linear regression (black line) defined the F–I gain for the cell. C, a slow-depolarizing voltage-ramp command (bottom trace) was used for all voltage-clamp protocols. The current response in control conditions (black line) and in the presence of 1 μm TTX (grey line) is shown. D, the I–V relations (ascending ramp only) for the control (black trace) and TTX (grey trace) trial are shown. NaP was measured as the difference between these two I–V functions.
Figure 3. Reduction of NaP with riluzole.
The I–V relations for NaP are shown for control conditions and for riluzole at three different doses (0.1, 1.0 and 5.0 μm) (thick grey line, thin grey line, dotted line), and after TTX administration (thick dotted line). A, Raw I–V functions. B, The control and riluzole currents after subtraction of the TTX I–V function. Increasing riluzole concentrations dose-dependently inhibited NaP.
Riluzole is relatively specific to Na+ current
As in previous studies (Urbani & Belluzzi, 2000; Del Negro et al. 2002, 2005; Spadoni et al. 2002), riluzole suppressed NaP in a dose-dependent manner in our cultured motoneurons (Fig. 3). On average, riluzole (5 μm) reduced NaP by 73 ± 7% and a concentration of 10 μm reduced it by 82 ± 8% (full dose–response relation provided in Fig. 6).
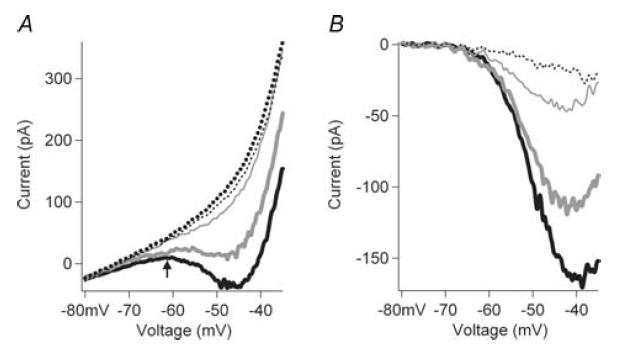
Figure 6. Dose-dependent riluzole inhibition of excitability and NaP.
A, the F–I relations and linear regressions are shown for control (•), 0.1 mm riluzole (□), and 1.0 mm riluzole (▵). The F–I gain is reduced and the current threshold for the onset of firing is increased with increasing riluzole concentrations. B, the dose–response curve for all cells is shown for the effect of riluzole on the F–I gain (grey circle) for 0.1 mm riluzole (n = 11), 0.5 mm riluzole (n = 10), 1 mm riluzole (n = 8), 2 mm riluzole (n = 5), 5 mm riluzole (n = 5), and 10 mm riluzole (n = 5). The effect of riluzole on for NaP (▪) is also shown 0.1 mm riluzole (n = 6), 0.5 mm riluzole (n = 5), 1 mm riluzole (n = 7), 2 mm riluzole (n = 5), 5 mm riluzole (n = 5) and 10 mm riluzole (n = 5). The EC50 for riluzole inhibition of the F–I gain (grey arrow) was 1.1 mm and NaP (black arrow) was 1.8 mm. Riluzole also dose-dependently increased the current threshold for firing (right side, bars; mean ±s.e.m. shown for B). Threshold amplitudes could not be measured above 2 mm riluzole because spiking behaviour became very irregular.
The specificity of riluzole to Na+ is an important concern for this study. Based on dose–response studies of the effects of riluzole on NaP in previous studies (Urbani & Belluzzi, 2000) and in this study (presented below; see Fig. 6), we administered riluzole at a concentration of 5–10 μm. Riluzole has been reported to influence both Ca2+ and K+ currents as well as the currents underlying the AHP (IAHP). These actions, however, primarily occur at concentrations greater than 5–10 μm (Huang et al. 1997; Song et al. 1997; Zona et al. 1998; Urbani & Belluzzi, 2000; Cao et al. 2002). To confirm the specificity of riluzole to NaP, we examined its effect on IAHP in our cultured motoneurons. IAHP was isolated using a depolarized voltage-clamp step protocol and pharmacological agents (see Methods). Riluzole (10 μm) had a minimal effect on the magnitude of IAHP (riluzole IAHP/control IAHP= 0.989 ± 0.218; not significant, t test, P > 0.2; n = 4). We also assessed the amplitude of the initial AHP following the first spike to steps of injected current. All trials used identical step-pulse amplitudes initiated from the same membrane potential. With 10 mm riluzole, a small tendency for an increased AHP amplitude was observed (control: 13 ± 4 mV, riluzole: 15 ± 4 mV, n = 20), but was not significant (t test P > 0.1).
Another point to consider is the relative effects of riluzole on NaPversus NaT. Spike overshoot was assessed for 10 μm riluzole, and a small but significant reduction was detected (control: 31 ± 9 mV, riluzole: 27 ± 10, n = 20, t test P < 0.001), consistent with a previous study (Del Negro et al. 2002). Although this result suggests minimal impact on NaT, the relationship between spike height and NaT is highly non-linear (Swensen & Bean, 2003), such that an approximately 50% reduction in NaT may produce only a 10–15 mV reduction in spike height. Thus the approximately 4 mV reduction for 10 μm riluzole in our results may reflect a significant reduction in NaT. Thus, we assume that riluzole preferentially affects NaP compared to NaT, not that it has no effect on NaT. Instead, our fundamental argument is that riluzole blocks spiking to slow inputs, but that spikes to fast inputs can still occur despite any reduction in NaT. To further support this argument, the same basic protocols were performed with computer simulations that provided fully independent control of NaP and NaT.
Reduction of NaP eliminates repetitive firing but allows spikes to transient inputs
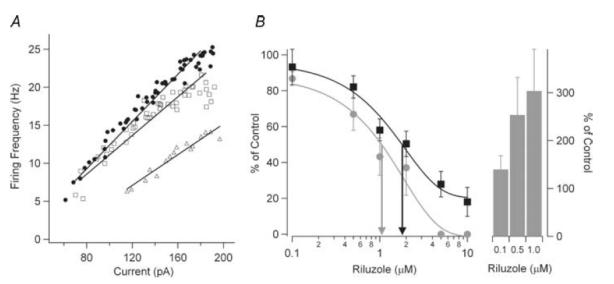
Figures 4 and 5 illustrate results supporting our first three predictions: reducing NaP eliminates repetitive firing; further depolarization does not restore this firing; and spikes can still be generated to transient inputs. For the slowly rising ramps of injected current, administration of 5–10 μm riluzole completely blocked repetitive firing in all cells tested (n = 20), as illustrated by the example in Fig. 4A (compare thin and thick black traces, upper panel). This failure persisted even though the ramp depolarized the cell well above the spike voltage threshold (see the later parts of Fig. 4A, upper panel). Superimposition of a series of brief current steps (5 Hz) on the ramp (lower panel, grey trace), however, did evoke spikes at the onset of each superimposed step (middle panel, grey trace). Similar results were obtained in five cells. Pulses were also applied at a faster rate in three cells, and spikes followed the pulses well up to 10 Hz. Finally, if spikes can still be initiated from transients, then synaptic noise should also restore a significant degree of spiking once NaP is blocked by riluzole. Synaptic noise was mimicked by noisy current injection (see Methods) in four cells. We found that when the peak-to-peak amplitude of the noise exceeded 10 mV, a significant number of spikes occurred to ramps (Fig. 4B, middle trace) and steps once firing was completely blocked by riluzole. The substantial reduction in spike height for the pulses (Fig 4A) and noise (Fig 4B) presumably reflect the combined effects of reduced NaT and greater overall inactivation due to the depolarized membrane potential baseline compared to that produced by the successive AHPs in control conditions. However, the restoration of spiking by transient inputs also shows that the mechanism underlying spike failure was not due to the Na+ channel entering a prolonged state of inactivation.
Figure 4. Restoration of firing from transient inputs.
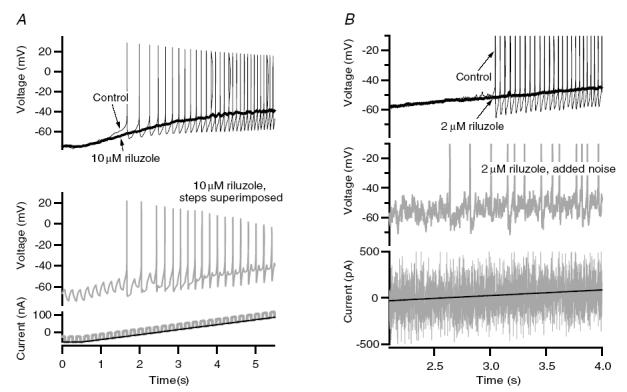
A, under control conditions (top panel, thin black trace), a motoneuron responded to a depolarizing current ramp injection (bottom panel, black trace) with repetitive firing. The addition of 10 μm riluzole (top panel, thick black trace) inhibited all firing to the same amplitude current injection, even at voltages depolarized compared to spike threshold. However, superimposing a 5 Hz current step on top of the current ramp (bottom panel, grey trace) resulted in a spike at the onset of virtually all superimposed pulses (middle panel, grey trace). B, in a different cell than in A, sustained repetitive firing was again observed to a current ramp injection (top panel, thin black trace). In the presence of 2 μm riluzole, all firing was inhibited (top panel, thick black trace). However, superimposition of current noise on the current ramp injection (bottom panel, grey trace restored significant firing (middle panel, grey trace).
Figure 5. Effect of increased current amplitude on firing.

A, in control conditions, repetitive firing (top panel, black trace) was observed to a current step (bottom panel, black trace). Riluzole (10 μm) blocked all firing to the current step except at its onset (top panel, thick black trace). Increasing the current amplitude (bottom panel, grey traces) did not restore firing (middle panel). B, the first 50 ms of the step from A is shown. Riluzole (10 μm) did not increase the AHP (upper panel, compare thick and thin black traces), and increased step sizes depolarized the cell well beyond spike threshold without restoring repetitive firing (middle panel, grey traces).
Figure 5 shows analogous results for step inputs instead of ramps. Riluzole (10 μm) eliminated repetitive firing during steps (compare thin and thick black traces, upper panels in both Fig. 5A and B). A single spike invariably occurred at step onset (upper panel, thick trace, Fig 5B), consistent with our third prediction. This behaviour, failure of repetitive firing but preservation of a single spike at onset, was observed in every cell tested with steps (n = 14). Similar to the ramps, increasing step amplitudes to depolarize the membrane potential well beyond spike threshold did not restore sustained firing (middle panel, grey traces in Fig. 5A and B). Single spikes at step onset remained, and a decrease in the time to initial spike onset was observed with increasing step amplitudes (Fig. 5B shows the first 50 ms after step onset). Figure 5B also illustrates the lack of effect of riluzole on the AHP noted above (compare thin and thick black traces, upper panel) as well as the slight reduction in initial spike amplitude. Overall, these results strongly support the first three predictions.
The gain of repetitive firing is influenced by NaP
The fourth prediction we formulated was that, if elimination of NaP prevents repetitive firing, then a graded reduction of NaP should produce a graded decline in repetitive firing capacity. The effect of a graded reduction of NaP was studied by examining the corresponding effect on the gain of the F–I function. The cell shown in Fig. 6A illustrates the typical effect of increasing concentrations of riluzole on both the F–I gain and current threshold. The control trial had an F–I gain of 0.157 Hz pA−1 and generated 56 action potentials with an F–I current onset of 61.3 pA during the ascending ramp. Riluzole decreased the F–I gain and number of spikes to the ascending ramp, and increased the F–I current onset in a dose-dependent manner (0.1 μm: 0.134 Hz pA−1, 47 spikes, 70.22 pA; 0.5 μm: 0.117 Hz pA−1, 27 spikes, 96.7 pA; 1.0 μm: 0.101 Hz pA−1, 16 spikes, 116.48 pA). In this cell, repetitive firing failed completely at 2 μm riluzole, and was partially restored with riluzole washout (not shown). The effect of riluzole on F–I gain was not due to changes in input conductance, which was not altered by riluzole concentrations of up to 10 μm (P > 0.4, t test).
The dose-dependent effect of riluzole on NaP, F–I gain, and F–I current threshold on a sample of cells is shown in Fig. 6B. The EC50 of NaP inhibition was approximately 1.8 μm, while EC50 for F–I gain inhibition was about 1.1 μm (Fig. 6B, arrows), values very similar to the results obtained in cortical cells (Urbani & Belluzzi, 2000). It should be noted that even with approximately 30% NaP remaining (5 μm), all firing was lost. A significant correlation between the relative magnitude of riluzole-inhibited NaP and F–I gain (riluzole trial/control trial) was observed (n = 10 cells, 30 data points, P < 0.001). In addition, the current amplitude for the threshold of the F–I function increased from 140% (0.1 μm) to 253% (0.5 μm) to 304% (1.0 μm) of control levels (Fig. 6B, bars). These results on current threshold for firing further support previous studies (Taddese & Bean, 2002; Do & Bean, 2003) showing that NaP provides an important depolarizing drive for neurons. The parallel effects of riluzole on NaP and F–I gain strongly support our fourth prediction. In addition, these results show that NaP plays an essential role not only in setting the threshold but also in the gain of the F–I function of the cell, as hypothesized by Lee & Heckman (2001).
Nap generates a negative I–V slope to initiate spikes during repetitive firing
Our fifth prediction was that, if NaP plays an essential role in spike initiation, it must be the dominant current in the voltage range where initiation occurs. In other words, NaP must be large enough to overcome leak currents and any voltage-sensitive outward currents active near spike voltage threshold. If this prediction is correct, the I–V relation should exhibit a negative slope (indicating dominance by an inward current) in the spike voltage region, and this negative slope should be eliminated by TTX. This is indeed what occurred in most of our cells (Fig. 2D, compare black to grey I–V functions; Fig. 3A, compare black to dotted functions). The voltage range for the negative slope region corresponded to the voltage range for spike initiation during repetitive firing. Spike initiation typically began with an ‘upswing’ phase, as shown in Fig. 7 (arrow on thin trace). The upswing was invariably blocked by 5–10 μm riluzole or 1 μm TTX (Fig. 7, bold trace), and thus was probably due to NaP. The rate of rise of this NaP-dependent upswing typically ranged from about 20–30 mV s−1 near its onset to over 200 mV s−1 at spike onset. The average voltage for the onset of the upswing during firing in current clamp (−50.6 ± 4.8 mV) was similar to that for NaP onset during voltage clamp (−53.0 ± 5.0 mV; these values were not significantly different, t test, P > 0.05). The voltage for the onset of the negative slope in the I–V function, i.e. the voltage at which NaP was large enough to overcome the leak conductance was a bit more depolarized (−46.3 ± 6.7 mV; significantly more depolarized than the upswing onset and NaP onset, P < 0.01 in both cases, paired t test). Even within these rather restricted ranges of values, a significant correlation between the voltages for onset of the upswing during firing and onset of the negative slope region in the I–V function (r = 0.88, n = 9, P < 0.01) as well as for onset of NaP (r = 0.76, n = 9, P < 0.05) were observed. Thus, NaP was activated in the voltage range for spike initiation (Taddese & Bean, 2002; Do & Bean, 2003) and dominated all other currents in this region.
Figure 7. Inhibition of voltage upswing.

In control conditions (thin trace), repetitive firing was generated from a depolarizing current ramp injection (spikes are shown truncated). Prior to spike initiation, an upswing the membrane potential was observed (arrow). The addition of 1 mm TTX (bold trace) blocked all firing and the voltage upswing to the current stimulus, strongly suggesting that the upswing was due to NaP.
Nap does not always impart a negative slope during slow voltage command
Although results thus far support our predictions, in some cells (15 of a total of 55), NaP was insufficient to overcome leak and other outward currents and induced only an inflection in the I–V function that did not result in a negative slope. The cell shown in Fig. 8B provides an example of this. Yet cells such as this were usually capable of repetitive firing (Fig. 8A). A similar lack of negative I–V slope coupled with significant repetitive firing was often seen at moderate doses of riluzole (0.5–1.0 μm).
Figure 8. No negative slope region.
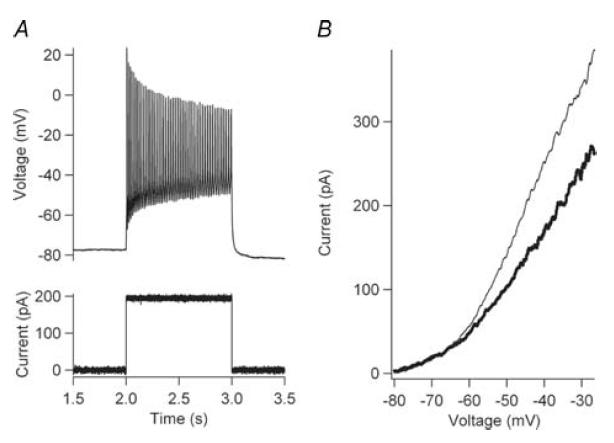
A, in control aCSF, repetitive firing (top panel) was observed to a current step injection (bottom panel). B, in control aCSF (thick trace), no negative slope region was observed to a voltage ramp command, even though sustained repetitive firing was observed. The addition of 1 mm TTX (thin trace) inhibited all Na+, and a larger outward current was observed.
The explanation for this discrepancy with our predictions lies in the rate of rise of voltage that we used to generate the I–V function, which was only 8–10 mV s−1. In contrast, the rate of change of voltage as the AHP decays following a spike is at least 10-fold higher, at ∼50–100 mV s−1 in our cultured motoneurons. Thus NaP may have undergone significant slow inactivation (Crill, 1996) or be masked by slowly activating outward currents. Consequently, a negative I–V slope may become apparent with faster rates of rise of voltage. The effect of increasing ramp speed from 10 to 200 mV s−1 was investigated in five cells. In all of these cells, increasing the speed of the voltage ramp command, which increased the voltage rate of rise, resulted in a larger inward current (Fig. 9A). The amplitude of NaP calculated from TTX subtraction also sharply increased with the voltage rate of rise (Fig. 9B), suggesting the presence of significant slow inactivation at the slower rates of voltage rise.
Figure 9. Effect of the rate of rise of the voltage command.
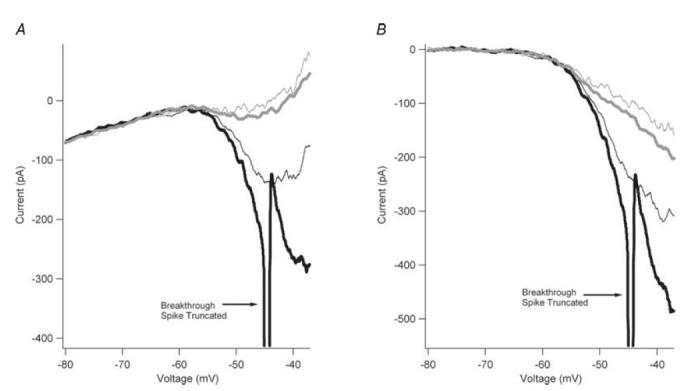
A, the I–V relations are shown for trials with rates of rise of 9 mV s−1 (thin grey trace), 18 mV s−1 (thick grey trace), 90 mV s−1 (thin black trace), and 180 mV s−1 (thick black trace). A single breakthrough spike (shown truncated) was observed with the trial using a voltage command of 180 mV s−1. B, the traces from A are shown leak- and TTX-subtracted to reveal the effect of rate of rise on NaP. By increasing the rate of rise of the voltage command, NaP was markedly increased, suggesting that NaP does slowly inactivate.
NaP accelerates membrane potential depolarization during current clamp
From the foregoing, it is clear that the rate of depolarization during the decay of the AHP is sufficiently fast to strongly activate NaP in all cells, regardless of whether the I–V function to very slow voltage ramps exhibits a net negative slope. One further point remains: initiation of the first spike during a current ramp, which produces a very slow rate of rise of voltage. Motoneurons are in fact known to initiate spikes no matter how slow the rate of rise of input, i.e. they exhibit no accommodation during slowly rising inputs (Schlue et al. 1972; Calvin, 1975; Powers & Binder, 2001). The key is the ‘upswing’ noted above in Fig. 7, i.e. the acceleration of voltage preceding spike initiation. Figure 10 shows that the I–V function corresponding to the rate of change of voltage during the portion of the current ramp before the upswing did not have a negative slope (Fig. 10, red trace). A rate of rise of voltage approximating the average change during the upswing phase (Fig. 10 inset), however, produced an I–V function with a very clear negative slope region (Fig. 10, blue trace). Thus, during slow current injection, when the voltage level reaches NaP threshold, there is a progressive increase in NaP activation, accelerating the rate of change of voltage until it becomes rapid enough to generate a negative slope and hence the initial spike in the train.
Figure 10. Effect of voltage upswing on NaP.
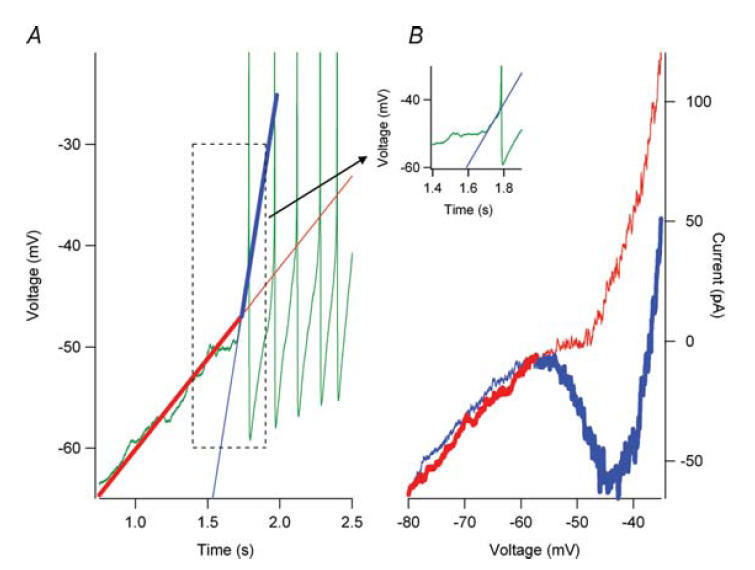
In the presence of 0.5 mm riluzole, repetitive firing (A, green trace) was observed in response to a depolarizing current ramp injection. In response to a voltage-clamp ramp command (A, red trace) that approximated the subthreshold rate of rise to the current ramp command, no negative slope region was observed in the I–V relation (B, red trace). However, prior to spike initiation, an upswing in the membrane potential (inset and Figure 7) was observed. (The inset shows the boxed region in A.) A voltage-clamp command that approximated this upswing (A, blue trace) generated a robust persistent inward current (B, blue trace). Therefore, during current-clamp conditions, the voltage transitions from the slow voltage command (A, bold red trace) to the fast voltage command (A, bold blue trace) when the voltage upswing begins. The current response to the voltages transitions from the bold red trace (B) to the bold blue trace (B) and the negative slope region to generate a spike is now present. The thin red lines (A and B) represent the slow voltage command and current response after the voltage upswing. The thin blue lines (A and B) represent the fast voltage command and current response before the voltage upswing.
A kinetic model of the Na+ channel supports the role of NaP in spike initiation
Computer simulations were performed to allow a higher degree of independent control of NaP and NaT than provided by riluzole. The Na+ channel in our simulations was based on a kinetic model developed by Taddese & Bean, 2002) for tuberomammillary cells but with rate constants adjusted to fit the voltage-clamp data of Safronov & Vogel (1995) for Na+ currents in young rat motoneurons (see Methods). Figure 11A shows this model's Na+ channel behaviour in response to voltage steps from −120 mV to various levels (the model also included a standard set of K+ and Ca2+ channels to produce repetitive firing – see Methods). After each step, NaT rapidly inactivated to a small but steady level of NaP (seen most clearly in the expansion in Fig. 11B). NaP was about 4.3% of peak NaT with the voltage step to −10 mV. In the minimal NaP version of the model (dashed line in Fig 11A and B), constructed by halving the rate constant between the inactivated and open states (see Table 1), behaviour of NaT was virtually identical to that in the standard model, but NaP was reduced to about 1.6% of peak NaT (−10 mV step). The difference in NaP between the two models was not accompanied by any significant change in the activation of NaT. Figure 11C shows the expected change in inactivation due to increased NaP (i.e. an upward shift in the hyperpolarized portion of the steady-state inactivation function), but activation curves are virtually superimposed.
The normal motoneuron model exhibited good tonic firing to both current ramps and steps (Fig. 12A and B, black traces). Just as for cultured motoneurons in the presence of riluzole (Figs 4 and 5), the reduced NaP motoneuron model was incapable of rhythmic firing (Fig. 12A and B, thick black traces) and further increasing input amplitude did not restore firing (Fig. 12B, grey trace). Similarly, spikes could readily be generated when pulses or noise were added (Fig. 12A, grey trace). These simulation results show that a reasonably realistic Na+ channel model reproduces the basic experimental results obtained with riluzole administration, and thus support the essential role of NaP in spike initiation during repetitive firing.
Figure 12. Repetitive firing behaviour of the model motoneuron.
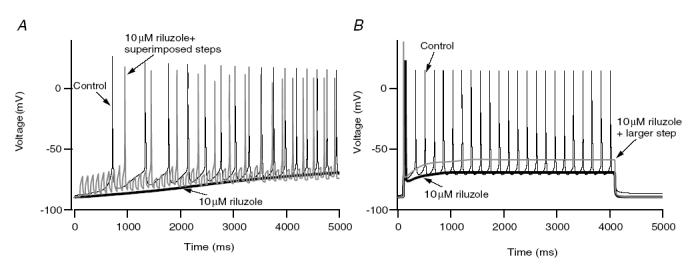
A, with a linearly rising ramp, the model with the standard level of NaP exhibited good repetitive firing (thin black trace) to a ramp of injected current. The reduced NaP model failed to generate any spike to this same input (thick black trace). However, adding a series of transient current pulses on top of the ramp, generated spikes in the reduced NaP model (grey trace), showing it was still capable of initiating spikes to transient inputs. B, behaviour to step inputs: good repetitive firing in the standard model (thin black trace), loss of all but the initial spike in the reduced NaP model (thick black trace), and failure to restore repetitive firing by increasing the amplitude of the step (grey trace). These simulation results parallel the experimental results for riluzole administration.
We also examined the behaviour of motoneuron models with amplitudes of NaP intermediate between the two shown in Fig. 12. In each case, current ramps of varying rates of rise were applied, and the minimal input rate, defined as the lowest rate of rise of the ramp that evoked at least five spikes, was measured. Figure 13 shows that the smaller NaP was, the greater the minimal rate. This result is consistent with the essential role of NaP in spike initiation to slow inputs, and further suggests that the amplitude of NaP can control the degree to which a neuron responds to slow versus fast inputs.
Figure 13. Effect of varying amplitude of NaP on the rate of rise of input required for repetitive firing.

In the model motoneuron, varying the rate constant Ooff (see Fig. 1) from 0.015 (reduced NaP model) to 0.03 (standard model) decreased the rate of rise of a linear current ramp needed to generate repetitive firing. The minimal rate of rise that produced at least five spikes is plotted versus NaP expressed as a percentage of NaT for a step from −120 mV to −10 mV.
Discussion
Our experimental results in large cultured spinal neurons and our simulation results based on motoneurons strongly support the hypothesis that NaP provides an essential step in spike generation to slowly rising inputs. This spike initiation function probably works in conjunction with the important other established functions of NaP, including subthreshold depolarization, membrane potential oscillation, and synaptic amplification (Schwindt & Crill, 1995; Stuart & Sakmann, 1995; Uteshev et al. 1995; Fleidervish & Gutnick, 1996; Lipowsky et al. 1996; Baker & Bostock, 1997; Hsiao et al. 1998; Butera et al. 1999; Lee & Heckman, 1999; Bennett et al. 2000; Urbani & Belluzzi, 2000; Agrawal et al. 2001; Taddese & Bean, 2002; Li & Bennett, 2003; Li et al. 2004; Wu et al. 2004).
The role for NaP in spike initiation probably does not apply solely to spinal motoneurons. A previous study in cultured cortical cells (Urbani & Belluzzi, 2000), while not focusing on spike-initiating mechanisms, obtained results that supported three of our five predictions: riluzole blocked repetitive firing (prediction 1), yet allowed a single spike at step onset (prediction 3) and graded administration of riluzole had graded effects on both NaP and number of spikes fired per step (prediction 4). Additionally, elimination of sustained repetitive firing but preservation of a single spike to a step onset has been observed in cultured dorsal column neurons following riluzole administration (Reboreda et al. 2003). Although these previous studies did not perform the key experiment of verifying that increasing step amplitude did not restore firing, their results are otherwise analogous to those presented here. Finally, reduction or suppression of tonic firing by riluzole has also been seen in several other types of neurons (Del Negro et al. 2002; Rybak et al. 2003; Darbon et al. 2004; Kononenko et al. 2004; Pena et al. 2004).
Our results ‘however’, do not exclude other mechanisms for spike initiation to slowly rising inputs. The relationship between amplitude of NaP and rate of rise shown in Fig. 13 suggests that the faster the rate of decay of the AHP following each spike, the less the requirement for NaP to initiate spikes. In addition, the time constant for inactivation of NaT tends to increase at hyperpolarizing levels (e.g. Safronov & Vogel, 1995; Taddese & Bean, 2002). A long time constant in the region for spike initiation might provide a sufficient duration of Na current to initiate spikes. Probably all that is required is that a small fraction of the total Na+ channels remain non-inactivated for long enough to impart a significant acceleration to the membrane potential. This acceleration then converts the slow rate of rise of the slow input to one sufficiently fast to allow activation to escape from inactivation in the rest of the Na+ channels. In our model, the inactivation time constant increased from about 1 ms for steps to −10 mV, to about 15 ms at −60 mV. Since this model failed to generate repetitive firing with low NaP, it is likely that the inactivation time constant has to be substantially longer than 15 ms to provide sufficient open time to accelerate the membrane potential and generate spikes. The resurgent Na+ current that is seen in some types of neurons (Raman & Bean, 1997; Do & Bean, 2003; Khaliq et al. 2003) may be sufficiently persistent in this sense to assist in spike initiation, but the resurgent state can only be entered once a channel is open, and thus probably could not produce an acceleration in membrane potential needed to produce the initial spike to a ramp current input.
NaP as a primary determinant of neuronal processing of transient versus sustained synaptic inputs
Our results imply that NaP may play a major role in producing differences in spiking patterns evoked by synaptic input seen in different types of neurons. At one end of a continuum, cells like spinal motoneurons with substantial NaP should respond to all components of the synaptic input: sharp transients, slow changes and steady baseline levels. High-NaP cells may also exhibit spontaneous firing due to potent subthreshold depolarization provided by NaP. For example, Purkinje cells, subthalamic neurons and some suprachiasmatic neurons exhibit both spontaneous firing and high levels of NaP (Kay et al. 1998; Do & Bean, 2003; Kononenko et al. 2004). A further important question is whether NaP enhances the response to transient inputs, so that neurons with high NaP not only track the baseline of synaptic input, but also are more sensitive to transients than low-NaP neurons. Previous studies established that NaP acts to amplify synaptic potentials (Schwindt & Crill, 1995; Stuart & Sakmann, 1995), clearly implying enhanced transient response. Moreover, dendritic NaP may play a critical role in determining whether dendritic spikes occur, due to backpropagation from the soma or directly from dendritic synaptic inputs (Turner et al. 1991; Stuart et al. 1997; Gasparini et al. 2004; Larkum et al. 2004). The effectiveness of NaP in providing a good response to transients will, however, depend on its interaction with other low-threshold currents (cf. Prescott & De Koninck, 2005).
Neurons with low intrinsic NaP levels should respond only to transient inputs. In fact, in the spinal cord, some interneurons produce only a single spike to current pulses (Garraway & Hochman, 2001; Prescott & De Koninck, 2002; Ruscheweyh & Sandkuhler, 2002; Szucs et al. 2003; Theiss and Heckman, 2005). This is exactly how cells behave when NaP is reduced with riluzole, and therefore we hypothesize that the primary mechanism for the single spiking in these cells is an intrinsically low level of NaP compared to tonic firing cells. This hypothesis is consistent with the recent results of Prescott & De Koninck (2005) who showed that only tonic-firing interneurons exhibited greater NaP than single-spiking interneurons. Single-spiking neurons largely ignore the baseline upon which the transients are superimposed, and have been proposed to be coincidence detectors for synaptic inputs (Prescott & De Koninck, 2002). Other types of spinal interneurons exhibit phasic bursts of spikes to steps of injected current (Garraway & Hochman, 2001; Grudt & Perl, 2002; Prescott & De Koninck, 2002; Ruscheweyh & Sandkuhler, 2002; Szucs et al. 2003). These cells may have moderate levels of NaP, in addition to other currents, but this remains to be investigated.
Effects of riluzole on pre-Bötzinger complex neurons
Differences in NaP amplitude may also underlie the diversity in responses to riluzole administration seen in recent studies of repetitive firing and rhythmic behaviour in pre-Bötzinger complex neurons (Del Negro et al. 2002, 2005; Pena et al. 2004). In some cases, riluzole effectively suppressed repetitive firing, as in motoneurons in our study. Yet, some pre-Botzinger neurons continued to exhibit good repetitive firing post-riluzole administration. In part, this failure may reflect a high degree of synaptic noise (e.g. Fig. 3C in Pena et al. (2004), Fig. 3A in Del Negro et al. (2002), and Fig. 1C in Del Negro et al. (2005)). In contrast, other recordings show strong repetitive firing in response to injected current steps without noise, persisting at riluzole concentrations that suppress 90% of NaP (Fig. 5 in Del Negro et al. (2002)), suggesting perhaps that the rate of decay of the AHP in these cells may be much faster than in the motoneurons studied here.
NaP, excitability and neurodegeneration
Urbani & Belluzzi, 2000) pointed out that the low micromolar riluzole inhibition of firing and NaP is important because riluzole is used for the treatment of amyotrophic lateral sclerosis (ALS). The calculated concentration in patients administered riluzole treatment (50 mg, 2 times a day) was 0.9–1.6 μm (Lacomblez et al. 1996), correlating to a greater than 50% inhibition of sustained repetitive firing to slow or steady inputs (Fig. 5B). By decreasing neuronal excitability, a greater depolarization or faster rising input would be necessary to achieve similar firing frequencies. Motoneurons cultured from SOD1 transgenic mice, a standard model of ALS, exhibit significantly higher F–I gains (Pieri et al. 2003; Kuo et al. 2004). We have recently shown that the primary source of this enhanced excitability in SOD1 motoneurons is elevated levels of NaP (Kuo et al. 2005). The concentration of riluzole achieved by oral administration in ALS patients is roughly 1 μm (Urbani & Belluzzi, 2000), which would reduce NaP by about 40% and F–I gain by about 60% (see Fig. 6A). Thus therapeutic administration of riluzole should significantly reduce motoneuron excitability. Also, recent studies by Bennett and colleagues (Li & Bennett, 2003; Li et al. 2004) show that the changes in motoneuron excitability during chronic spinal cord injury were due to a marked upregulation of NaP, along with an equally strong increase in an L-type Ca2+ current. Thus NaP not only plays a fundamental role in spike initiation during the normal state but also is subject to upregulation in both disease and injury states.
Acknowledgments
This work was supported by the NIH/NINDS NS034382 (C.J.H.) and NS045199 (R.H.L.). J.J.K. was supported by the Les Turner ALS Foundation.
References
- Agrawal N, Hamam BN, Magistretti J, Alonso A, Ragsdale DS. Persistent sodium channel activity mediates subthreshold membrane potential oscillations and low-threshold spikes in rat entorhinal cortex layer V neurons. Neuroscience. 2001;102:53–64. doi: 10.1016/s0306-4522(00)00455-3. [DOI] [PubMed] [Google Scholar]
- Baker MD, Bostock H. Low-threshold, persistent sodium current in rat large dorsal root ganglion neurons in culture. J Neurophysiol. 1997;77:1503–1513. doi: 10.1152/jn.1997.77.3.1503. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Callaway JC, Wilson CJ. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J Neurosci. 2000;20:8493–8503. doi: 10.1523/JNEUROSCI.20-22-08493.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth V, Rinzel J, Kiew O. Compartmental Model of Vertebrate Motoneurons for Ca2+-dependent Spiking and plateau potentials under pharmacological treatment. J Neurophysiol. 1997;78:3371–3385. doi: 10.1152/jn.1997.78.6.3371. [DOI] [PubMed] [Google Scholar]
- Butera RJ, Jr, Rinzel J, Smith JC. Models of respiratory rhythm generation in the pre-Botzinger complex. I. Bursting pacemaker neurons. J Neurophysiol. 1999;82:382–397. doi: 10.1152/jn.1999.82.1.382. [DOI] [PubMed] [Google Scholar]
- Calvin WH. Generation of spike trains in CNS neurons. Brain Res. 1975;84:1–22. doi: 10.1016/0006-8993(75)90796-9. [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Yin HZ, Weiss JH. Motor neurons are Selectively Vulnerable to AMPA a/Kainate receptor-Mediated injury in vitro. J Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao YJ, Dreixler JC, Couey JJ, Houamed KM. Modulation of recombinant and native neuronal SK channels by the neuroprotective drug riluzole. Eur J Pharmacol. 2002;449:47–54. doi: 10.1016/s0014-2999(02)01987-8. [DOI] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- Darbon P, Yvon C, Legrand JC, Streit J. INaP underlies intrinsic spiking and rhythm generation in networks of cultured rat spinal cord neurons. Eur J Neurosci. 2004;20:976–988. doi: 10.1111/j.1460-9568.2004.03565.x. [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Morgado-Valle C, Feldman JL. Respiratory rhythm: an emergent network property? Neuron. 2002;34:821–830. doi: 10.1016/s0896-6273(02)00712-2. [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Morgado-Valle C, Hayes JA, Mackay DD, Pace RW, Crowder EA, Feldman JL. Sodium and calcium current-mediated pacemaker neurons and respiratory rhythm generation. J Neurosci. 2005;25:446–453. doi: 10.1523/JNEUROSCI.2237-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A. Conductance-based integrate-and-fire models. Neural Comput. 1997;9:503–514. doi: 10.1162/neco.1997.9.3.503. [DOI] [PubMed] [Google Scholar]
- Do MT, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron. 2003;39:109–120. doi: 10.1016/s0896-6273(03)00360-x. [DOI] [PubMed] [Google Scholar]
- Fleidervish IA, Gutnick MJ. Kinetics of slow inactivation of persistent sodium current in layer V neurons of mouse neocortical slices. J Neurophysiol. 1996;76:2125–2130. doi: 10.1152/jn.1996.76.3.2125. [DOI] [PubMed] [Google Scholar]
- Garraway SM, Hochman S. Modulatory actions of serotonin, norepinephrine, dopamine, and acetylcholine in spinal cord deep dorsal horn neurons. J Neurophysiol. 2001;86:2183–2194. doi: 10.1152/jn.2001.86.5.2183. [DOI] [PubMed] [Google Scholar]
- Gasparini S, Migliore M, Magee JC. On the initiation and propagation of dendritic spikes in CA1 pyramidal neurons. J Neurosci. 2004;24:11046–11056. doi: 10.1523/JNEUROSCI.2520-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudt TJ, Perl ER. Correlations between neuronal morphology and electrophysiological features in the rodent superficial dorsal horn. J Physiol. 2002;540:189–207. doi: 10.1113/jphysiol.2001.012890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;116:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic channels of Excitable Membranes. Sunderland, MA: Sinauer Assoc. Inc; 2001. [Google Scholar]
- Hines ML, Carnevale NT. The NEURON stimulation environment. Neural Comput. 1997;9:1179–1209. doi: 10.1162/neco.1997.9.6.1179. [DOI] [PubMed] [Google Scholar]
- Hsiao CF, Del Negro CA, Trueblood PR, Chandler SH. Ionic basis for serotonin-induced bistable membrane properties in guinea pig trigeminal motoneurons. J Neurophysiol. 1998;79:2847–2856. doi: 10.1152/jn.1998.79.6.2847. [DOI] [PubMed] [Google Scholar]
- Huang CS, Song JH, Nagata K, Yeh JZ, Narahashi T. Effects of the neuroprotective agent riluzole on the high voltage-activated calcium channels of rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 1997;282:1280–1290. [PubMed] [Google Scholar]
- Kay AR, Sugimori M, Llinas R. Kinetic and stochastic properties of a persistent sodium current in mature guinea pig cerebellar Purkinje cells. J Neurophysiol. 1998;80:1167–1179. doi: 10.1152/jn.1998.80.3.1167. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci. 2003;23:4899–4912. doi: 10.1523/JNEUROSCI.23-12-04899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononenko NI, Shao LR, Dudek FE. Riluzole-sensitive slowly inactivating sodium current in rat suprachiasmatic nucleus neurons. J Neurophysiol. 2004;91:710–718. doi: 10.1152/jn.00770.2003. [DOI] [PubMed] [Google Scholar]
- Kuo JJ, Schonewille M, Siddique T, Schults AN, Fu R, Bar PR, Anelli R, Heckman CJ, Kroese AB. Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol. 2004;91:571–575. doi: 10.1152/jn.00665.2003. [DOI] [PubMed] [Google Scholar]
- Kuo JJ, Siddique T, Fu R, Heckman CJ. Increased persistent Na+ current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol. 2005;563:843–854. doi: 10.1113/jphysiol.2004.074138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacomblez L, Bensimon G, Leigh PN, Guillet P, Powe L, Durrleman S, Delumeau JC, Meininger V. A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology. 1996;47:S242–S250. doi: 10.1212/wnl.47.6_suppl_4.242s. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Senn W, Luscher HR. Top-down dendritic input increases the gain of layer 5 pyramidal neurons. Cereb Cortex. 2004;14:1059–1070. doi: 10.1093/cercor/bhh065. [DOI] [PubMed] [Google Scholar]
- Lee RH, Heckman CJ. Paradoxical effect of QX-314 on persistent inward currents and bistable behavior in spinal motoneurons in vivo. J Neurophysiol. 1999;82:2518–2527. doi: 10.1152/jn.1999.82.5.2518. [DOI] [PubMed] [Google Scholar]
- Lee RH, Heckman CJ. Essential role of a fast persistent inward current in action potential initiation and control of rhythmic firing. J Neurophysiol. 2001;85:472–475. doi: 10.1152/jn.2001.85.1.472. [DOI] [PubMed] [Google Scholar]
- Li Y, Bennett DJ. Persistent sodium and calcium currents cause plateau potentials in motoneurons of chronic spinal rats. J Neurophysiol. 2003;90:857–869. doi: 10.1152/jn.00236.2003. [DOI] [PubMed] [Google Scholar]
- Li Y, Gorassini MA, Bennett DJ. Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J Neurophysiol. 2004;91:767–783. doi: 10.1152/jn.00788.2003. [DOI] [PubMed] [Google Scholar]
- Lipowsky R, Gillessen T, Alzheimer C. Dendritic Na+ channels amplify EPSPs in hippocampal CA1 pyramidal cells. J Neurophysiol. 1996;76:2181–2191. doi: 10.1152/jn.1996.76.4.2181. [DOI] [PubMed] [Google Scholar]
- Pena F, Parkis MA, Tryba AK, Ramirez JM. Differential contribution of pacemaker properties to the generation of respiratory rhythms during normoxia and hypoxia. Neuron. 2004;43:105–117. doi: 10.1016/j.neuron.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Pieri M, Albo F, Gaetti C, Spalloni A, Bengtson CP, Longone P, Cavalcanti S, Zona C. Altered excitability of motor neurons in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett. 2003;351:153–156. doi: 10.1016/j.neulet.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Powers RK, Binder MD. Input–output functions of mammalian motoneurons. Rev Physiol Biochem Pharmacol. 2001;143:137–263. doi: 10.1007/BFb0115594. [DOI] [PubMed] [Google Scholar]
- Powers RK, Binder MD. Persistent sodium and calcium currents in rat hypoglossal motoneurons. J Neurophysiol. 2003;89:615–624. doi: 10.1152/jn.00241.2002. [DOI] [PubMed] [Google Scholar]
- Prescott SA, De Koninck Y. Four cell types with distinctive membrane properties and morphologies in lamina I of the spinal dorsal horn of the adult rat. J Physiol. 2002;539:817–836. doi: 10.1113/jphysiol.2001.013437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SA, De Koninck Y. Integration time in a subset of spinal lamina I neurons is lengthened by sodium and calcium currents acting synergistically to prolong subthreshold depolarization. J Neurosci. 2005;25:4743–4754. doi: 10.1523/JNEUROSCI.0356-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak K, Zummo GG, Alheid GF, Tkatch T, Surmeier DJ, McCrimmon DR. Sodium currents in medullary neurons isolated from the pre-Botzinger complex region. J Neurosci. 2005;25:5159–5170. doi: 10.1523/JNEUROSCI.4238-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboreda A, Sanchez E, Romero M, Lamas JA. Intrinsic spontaneous activity and subthreshold oscillations in neurones of the rat dorsal column nuclei in culture. J Physiol. 2003;551:191–205. doi: 10.1113/jphysiol.2003.039917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Ruscheweyh R, Sandkuhler J. Lamina-specific membrane and discharge properties of rat spinal dorsal horn neurones in vitro. J Physiol. 2002;541:231–244. doi: 10.1113/jphysiol.2002.017756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak IA, Shevtsova NA, St-John WM, Paton JF, Pierrefiche O. Endogenous rhythm generation in the pre-Botzinger complex and ionic currents: modelling and in vitro studies. Eur J Neurosci. 2003;18:239–257. doi: 10.1046/j.1460-9568.2003.02739.x. [DOI] [PubMed] [Google Scholar]
- Safronov BV, Vogel W. Single voltage-activated Na+ and K+ channels in the somata of rat motoneurones. J Physiol. 1995;487:91–106. doi: 10.1113/jphysiol.1995.sp020863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlue WR, Richter DW, Mauritz KH, Nacimiento AC. Accomodation and Na-inactivation in cat spinal motor neurons. Pflugers Arch. 1972;332(Suppl.):R383. [PubMed] [Google Scholar]
- Schwindt PC, Crill WE. Amplification of synaptic current by persistent sodium conductance in apical dendrite of neocortical neurons. J Neurophysiol. 1995;74:2220–2224. doi: 10.1152/jn.1995.74.5.2220. [DOI] [PubMed] [Google Scholar]
- Song JH, Huang CS, Nagata K, Yeh JZ, Narahashi T. Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J Pharmacol Exp Ther. 1997;282:707–714. [PubMed] [Google Scholar]
- Spadoni F, Hainsworth AH, Mercuri NB, Caputi L, Martella G, Lavaroni F, Bernardi G, Stefani A. Lamotrigine derivatives and riluzole inhibit INa,P in cortical neurons. Neuroreport. 2002;13:1167–1170. doi: 10.1097/00001756-200207020-00019. [DOI] [PubMed] [Google Scholar]
- Steriade M. Impact of network activities on neuronal properties in corticothalamic systems. J Neurophysiol. 2001;86:1–39. doi: 10.1152/jn.2001.86.1.1. [DOI] [PubMed] [Google Scholar]
- Stuart G, Sakmann B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron. 1995;15:1065–1076. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- Stuart G, Spruston N, Sakmann B, Hausser M. Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends Neurosci. 1997;20:125–131. doi: 10.1016/s0166-2236(96)10075-8. [DOI] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szucs P, Odeh F, Szokol K, Antal M. Neurons with distinctive firing patterns, morphology and distribution in laminae V–VII of the neonatal rat lumbar spinal cord. Eur J Neurosci. 2003;17:537–544. doi: 10.1046/j.1460-9568.2003.02484.x. [DOI] [PubMed] [Google Scholar]
- Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- Theiss RD, Heckman C J. Systematic variation in effects of Serotonin and norepinephrine on repetitive firing properties of ventral horn neurons. Neurosci. 2005;134:803–815. doi: 10.1016/j.neuroscience.2005.04.041. [DOI] [PubMed] [Google Scholar]
- Turner RW, Meyers DE, Richardson TL, Barker JL. The site for initiation of action potential discharge over the somatodendritic axis of rat hippocampal CA1 pyramidal neurons. J Neurosci. 1991;11:2270–2280. doi: 10.1523/JNEUROSCI.11-07-02270.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbani A, Belluzzi O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci. 2000;12:3567–3574. doi: 10.1046/j.1460-9568.2000.00242.x. [DOI] [PubMed] [Google Scholar]
- Uteshev V, Stevens DR, Haas HL. A persistent sodium current in acutely isolated histaminergic neurons from rat hypothalamus. Neuroscience. 1995;66:143–149. doi: 10.1016/0306-4522(94)00593-t. [DOI] [PubMed] [Google Scholar]
- Wu N, Enomoto A, Tanaka S, Hsiao CF, Nykamp DQ, Izhikevich E, Chandler SH. Persistent sodium currents in mesencephalic V neurons participate in burst generation and control of membrane excitability. J Neurophysiol. 2005;93:2710–2722. doi: 10.1152/jn.00636.2004. [DOI] [PubMed] [Google Scholar]
- Zona C, Siniscalchi A, Mercuri NB, Bernardi G. Riluzole interacts with voltage-activated sodium and potassium currents in cultured rat cortical neurons. Neuroscience. 1998;85:931–938. doi: 10.1016/s0306-4522(97)00604-0. [DOI] [PubMed] [Google Scholar]