

Abstract
Dietary Na+ intake influences β2-adrenergic receptor (β2AR) responsiveness. While receiving a normal Na+ diet (150 mmol day−1), subjects homozygous for glycine at amino acid 16 (Gly16) have greater forearm β2AR-mediated vasodilatation than subjects homozygous for arginine (Arg16), an effect that is mediated by endothelial NO. We tested the hypothesis that dietary Na+ restriction eliminates genotype differences in forearm and systemic β2AR-mediated dilatation in these groups. We measured heart rate, mean arterial pressure and cardiac output (CO, acetylene breathing) responses to administration of intravenous terbutaline (TRB) before and after 5 days of low dietary Na+ intake (10 mmol day−1) in healthy Gly16 (n = 17; age, 31 ± 7 year) and Arg16 homozygotes (n = 15; age, 29 ± 8 year). After the low-Na+ diet, a catheter was placed in the brachial artery to measure forearm blood flow (FBF, plethysmography) responses to administration of isoprenaline (isoproterenol) before and after NO inhibition with NG-mono-methyl-l-arginine (l-NMMA). In the Gly16 group, the low-Na+ diet decreased baseline CO from 6.4 ± 1.4 to 5.5 ± 1.2 l min−1 (P = 0.003, paired t test), tended to decrease stroke volume from 97.0 ± 20.6 to 86.9 ± 21.7 ml (P = 0.06) and increased peripheral resistance from 1106 ± 246 to 1246 ± 222 dynes s cm−5 (P = 0.02); significant effects of the low-Na+ diet were not observed in Arg16 subjects. In a repeated measures ANOVA, the responses of all cardiovascular measures to systemic administration of TRB were not influenced by genotype or diet. Additionally, the FBF response to incremenetal doses of isoprenaline did not differ between genotype groups before or after administration of l-NMMA. We conclude that dietary Na+ restriction blunted the increased forearm NO-mediated β2AR responsiveness in Gly16 homozygotes observed in a previous study after normal dietary Na+ intake, while baseline CO decreased and peripheral resistance increased in this group. This study provides evidence that dietary Na+ modulates effects of the Arg16Gly polymorphism on cardiovascular function.
Human and animal studies demonstrate interactions between genetic and environmental factors in the pathophysiology of hypertension. Of the environmental contributors, high dietary sodium intake is among the most prevalent in modern societies. The blood pressure response to changes in dietary sodium aggregates in families and is influenced by genetic factors (Miller et al. 1987). Evidence linking salt sensitivity (blood pressure response to intravenous sodium loading, followed by furosemide volume depletion) to the β2-adrenergic receptor (β2AR) locus has emerged from a linkage analysis of both hypertensive and normotensive siblings of hypertensive probands (Svetkey et al. 1997). Several studies have associated the Arg16Gly polymorphism in the β2AR with blood pressure, hypertension and family history of hypertension (Kotanko et al. 1997; Bray et al. 2000; Busjahn et al. 2000; Xie et al. 2000; Ranade et al. 2001; Herrmann et al. 2002; Jindra et al. 2002; Snieder et al. 2002; Ge et al. 2005). However, the relationship between specific polymorphisms in the β2AR gene and salt sensitivity is unknown.
In the ‘pregenomic’ era, intermediate physiological characteristics of β2AR function were described for both hypertension and dietary sodium intake. In individuals with essential hypertension, β2AR responsiveness is reduced (Feldman et al. 1984; Feldman, 1987); likewise, normotensive individuals at increased risk of developing hypertension have impaired vasodilator responses to β2AR agonists (Lang et al. 1995; Stein et al. 1995). Studies of the effects of sodium intake on sensitivity to β2AR-mediated vasodilatation have established that a normal response to increased sodium intake, consisting of increased sensitivity to β2AR-mediated vasodilatation, is decreased in hypertensive compared to normotensive individuals (Naslund et al. 1990). Furthermore, β2AR-mediated responsiveness is reduced selectively in peripheral veins of borderline hypertensive subjects, an effect that is potentially reversible by a low-Na+ diet (Feldman, 1990).
We previously reported that, in a group of healthy normotensive subjects, following 5 days of normal dietary Na+ intake (150 mmol day−1), Gly16 homozygotes demonstrated greater forearm arterial vasodilatation in response to isoprenaline (isoproterenol) than Arg16 homozygotes, and the effect appeared to be dependent on the generation of NO, while responses to acetylcholine (ACh) and sodium nitroprusside (SNP) were not different in the two genotype groups (Garovic et al. 2003). Additionally, increased β2AR-mediated venodilatation in response to isoprenaline is 3-fold greater in Gly16 than Arg16 subjects (Cockcroft et al. 2000). However, in contrast to regional vascular studies, systemic infusions of selective β2AR agonists have been shown to evoke greater systemic vasodilatation in Arg16 than Gly16 subjects receiving an unrestricted dietary sodium intake of 8–12 g salt (137–205 mmol sodium) per day (Gratze et al. 1999; Hoit et al. 2000).
With this information as background, the purpose of the present study was to investigate mechanistically the vasodilator pathways by which dietary sodium restriction might affect intermediate physiological characteristics and cardiovascular function in individuals homozygous for the Gly16 versus Arg16 polymorphism of the β2AR. We reasoned that effects of dietary sodium restriction would differ between genotypes of the β2AR. Since normotensive Gly16 subjects previously demonstrated augmented forearm β2AR-mediated vasodilatation while receiving a normal sodium dietary intake (Garovic et al. 2003), and dietary sodium intake has substantial effects on β2AR-mediated function, we tested the following hypotheses: (1) increased β2AR, NO-mediated forearm vasodilatation in Gly16 versus Arg16 homozygotes is dependent on normal dietary sodium intake, and therefore differences would not be apparent after a low-Na+ diet; (2) non-β2AR-mediated vasodilator responses to ACh and SNP in individuals homozygous for Gly16 and Arg16 would be similar after a low-Na+ diet; (3) systemic vasodilatation in response to intravenous β2AR agonist stimulation would be different in the Arg16 and Gly16 subjects before, but not after the low-Na+ diet; and (4) indices of myocardial function (cardiac output (CO) and stroke volume (SV)) would also be different in the Arg16 and Gly16 subjects before, but not after, receiving the low-Na+ diet. Our findings, and those in a companion paper in this issue by Snyder et al. (2006c) suggest the possibility that the Arg16Gly polymorphism influences salt sensitivity and cardiovascular function.
Methods
Study group
Thirty-two normotensive, unrelated, non-Hispanic white individuals in Rochester, MN, USA were recruited and grouped according to the Arg16Gly polymorphism, genotyped as previously described (Garovic et al. 2003). Men were under 40 years of age, women were under 50 years and premenopausal, and neither had a history of tobacco use or any acute or chronic disorder associated with alterations in cardiovascular structure or function. Subjects made up two genotype groups: homozygous for the Arg16 variant (n = 15) or homozygous for the Gly16 variant (n = 17). Nine of the Arg16 homozygotes and 12 of the Gly16 homozygotes had participated in a previous study following a normal sodium diet (Garovic et al. 2003).
One of the investigators reviewed each subject's medical history and performed a physical examination including blood pressure (by random zero sphygmomanometer). Three blood pressure readings taken 2 min apart were recorded after the subject had been seated quietly for at least 5 min. The second and third readings were averaged and used in the analyses. Women of childbearing potential were required to have a negative pregnancy test before participation. This study was performed in accordance with the Declaration of Helsinki, and the protocol was approved by the Institutional Review Board. Each participant gave written, informed consent.
Study protocol
Subjects fasted on the morning of study day 0, and checked into the General Clinical Research Center (GCRC) Physiology Core Laboratory (Fig. 1). Subjects were familiarized with the testing equipment while supine. After administration of subcutaneous local anaesthesia, an 18-gauge peripheral intravenous catheter was placed in either arm. CO was measured non-invasively by using an open-circuit acetylene gas method described in detail elsewhere (Johnson et al. 2000). Measurements of CO were acquired in duplicate for each experimental condition.
Figure 1. Timescale of protocol.
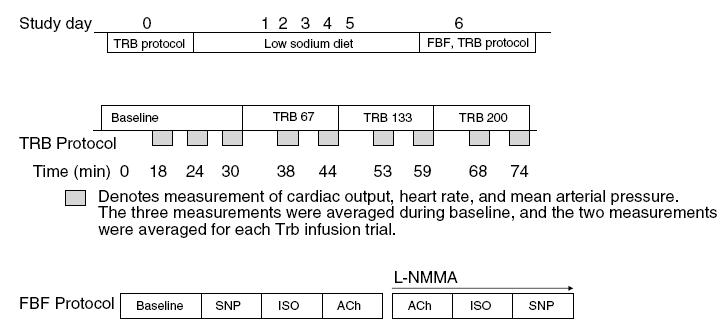
After an initial screening visit (top panel), subjects reported to the laboratory after their usual dietary sodium intake, designated as day 0, then underwent 5 days of dietary sodium restriction (10 mmol day−1). Middle panel denotes the systemic infusions of terbutaline (TRB), conducted on day 0 and day 6. The bottom panel shows the forearm blood flow (FBF) protocol that was conducted after the TRB trial on day 6. SNP, sodium nitroprusside; ISO, isoprenaline; ACh, acetylcholine.
Three CO measurements were obtained during the 30-min baseline. Terbutaline (TRB) infusions of 67, 133 and 200 ng kg −1 min−1 were administered for 15 min each, and CO was measured twice during each steady-state infusion. TRB infusion was discontinued and the subjects rested under observation until heart rate (HR) and blood pressure returned to resting values. The intravenous line was removed and subjects returned to the GCRC. Weight was recorded and a 24-h urine collection was begun to measure urine volume and Na+, K+ and creatinine (Cr) excretion. For the next 5 days, subjects were placed on a diet containing 10 mmol sodium per day. Food items remained the same throughout the week to provide constant daily amounts of protein (1.4 g (kg body weight)−1 day−1), potassium (100 mmol day−1) and calcium (1100 mg day−1). The caloric content of the diet was adjusted using the Harris Benedict equation to maintain constant body weight, and no more than 35% of calories were provided by fat. All meals were prepared in the diet kitchen of the GCRC where subjects ate two of their three meals each day. On day 5, a 24-h urine collection was repeated for measurement of sodium, potassium and creatinine excretion; that night, subjects slept in the GCRC. The next morning (study day 6) they remained fasting until the study measurements were completed.
On day 6, seated blood pressure measurements were repeated, followed by the forearm blood flow (FBF) protocol in the integrative physiology core of the GCRC. The volume of the non-dominant arm was measured by water displacement, followed by placement of a 20-gauge, 4.45-cm catheter in the brachial artery under aseptic conditions after administration of local anaesthetic (2% lidocaine). A three-port connector was placed in series with a catheter–transducer system so that drugs could be infused and arterial pressure measured simultaneously. FBF was measured using venous occlusion plethysmography with a mercury-in-silastic strain gauge placed around the non-dominant forearm at its greatest circumference (Greenfield et al. 1963). During recording, a wrist cuff was continuously inflated to suprasystolic pressure (250 mmHg) to occlude arterial blood flow to the hand while a venous occlusion cuff around the upper arm was inflated to 50 mmHg for 7.5 s of every 15 s, providing one blood flow measurement every 15 s. FBF values were expressed as ml (100 ml limb volume)−1 min−1.
Drug infusion protocol and drug doses
All drug infusions were administered at rates of 2–3 ml min−1. Infusions of SNP, isoprenaline and ACh were administered before and after NG-mono-methyl-l-arginine (l-NMMA). Each dose of each drug was administered for 2–3 min and the dose–response determinations were separated by a period of quiet rest for 20 min. To test NO-mediated endothelium-independent vasodilatation, the NO donor SNP was infused for 2 min at 1.0 μg (100 ml limb volume)−1 min−1. To test β2AR-mediated vasodilatation, isoprenaline was infused for 2 min at 1.0, 3.0, 6.0 and 12.0 ng (100 ml limb volume)−1 min−1. To test endothelium-dependent vasodilatation, ACh was infused for 2 min at 4.0 μg (100 ml limb volume)−1 min−1.
After the initial drug infusions and return of FBF to baseline levels, the wrist cuff was re-inflated and the NO synthase (NOS) inhibitor l-NMMA (50 mg) was infused over 10 min, followed by a ‘maintenance’ dose of l-NMMA (1 mg min−1) throughout the remainder of the protocol. Administration of the three vasodilator drugs was then repeated in reverse order (ACh, isoprenaline and SNP). We reasoned that if any differences between the groups were due to NO production, then the responses after administration of l-NMMA would be similar. The order of drug administration was not randomized so that the repeat doses of ACh could be given immediately after l-NMMA, to assess the magnitude of the NOS inhibition. Following FBF measurements, the arterial catheter was removed. Subjects walked to the GCRC Physiology Core Laboratory for placement of an intravenous catheter for systemic TRB infusion and the CO protocol was repeated exactly as described above for day 0 of the study.
Data analysis
Data were digitized at 200 Hz and stored on computer. Data were analysed off-line with signal processing software (Windaq, Dataq Instruments, Akron, OH, USA). FBF was determined from the derivative of the forearm plethysmogram during the last minute of each drug dose. FBF was reported rather than forearm vascular conductance because it was the primary measured variable in the pharmacological dose–response curves and because there were negligible changes in arterial pressure during the isolated forearm drug infusions. HR was derived from the electrocardiogram waveform, and mean arterial pressure (MAP) was derived from the arterial pressure waveform during plethysmography, and from an automated oscillometric brachial cuff during the TRB trials.
Statistical analysis
Baseline patient characteristics were summarized by calculating means and standard deviations for continuous variables and proportions for categorical variables. These characteristics were compared between Arg16 and Gly16 groups using the two-sample t test for continuous variables, and Fisher's exact test for categorical traits (sex). For MAP, HR, SV, CO and systemic vascular resistance (SVR), the paired t test was used to compare baseline values between day 0 (normal sodium) and day 6 (low sodium). In addition, repeated measures ANOVA models were used to assess differences between genotype groups for the within-subject responses to increasing doses of TRB. Separate models were used to analyse data collected on day 0 (normal Na+) and day 6 (low Na+) diets and an overall model was used to assess whether genotype effects were dependent on diet. For FBF, analogous models were used to assess differences between genotype groups for the within-subject responses to increasing doses of ACh, SNP or isoprenaline, both before and after administration of l-NMMA. In addition, an overall model was fitted for each of the individual drugs in which l-NMMA was included as a subject effect to assess whether the effect of l-NMMA was dependent on genotype. The sample size for this study was determined to provide statistical power of 80% to detect differences in the FBF response to isoprenaline based on the Arg16Gly polymorphism, consistent with previous observations (Garovic et al. 2003). Data are presented as means ± s.d. In all cases, two-tailed P values of 0.05 were considered statistically significant.
Results
There were no differences between genotype groups in the subject characteristics (Table 1). However, average seated systolic blood pressure obtained by random zero sphygmomanometry decreased from day 0 (normal sodium diet) to day 6 (low-Na+ diet) only in the Gly16 homozygotes (P = 0.003, paired t test). There was a main effect of diet on weight loss, urine volume and 24-h urinary excretion of sodium; however, there was no evidence that genotype or a genotype-by-diet interaction influenced these measurements (Table 2). Figure 2 shows MAP and HR at rest and during TRB infusion, before and after dietary sodium restriction. Before dietary sodium restriction, resting MAP prior to infusion of TRB was 81.1 ± 8.9 and 83.8 ± 6.4 mmHg in the Arg16 and Gly16 groups, respectively (P = 0.33). Dietary sodium restriction did not affect resting MAP in either group (Arg16, 81.8 ± 8.8 mmHg; Gly16, 83.2 ± 9.6 mmHg). TRB infusion decreased MAP before and after dietary sodium restriction (P < 0.001), and there was no evidence that sodium restriction or genotype influenced the MAP response to different doses of TRB (P = 0.80, genotype-by-dose interaction; P = 0.33, diet-by-dose interaction; P = 0.51 genotype-by-diet-by-dose interaction).
Table 1.
Subject characteristics
| Characteristic | Arg16 (n = 15) | Gly16 (n = 17) | P |
|---|---|---|---|
| Sex (female/male) | 8/7 | 7/10 | 0.72 |
| Age (years) | 29 ± 8 | 31 ± 7 | 0.36 |
| Height (cm) | 174 ± 8 | 173 ± 11 | 0.84 |
| Weight (kg) | 71 ± 11 | 74 ± 10 | 0.48 |
| HR (beats min−1) | 66 ± 6 | 67 ± 9 | 0.90 |
| BMI (kg m−2) | 22.9 | 23.9 | 0.30 |
| BSA (m2) | 1.8 ± 0.2 | 1.9 ± 0.2 | 0.58 |
| Cholesterol (mmol l−1) | 4.14 ± 0.81 | 4.61 ± 0.80 | 0.11 |
| Triglycerides (mmol l−1) | 2.00 ± 0.70 | 2.53 ± 0.29 | 0.13 |
| HDL (mmol l−1) | 1.33 ± 0.38 | 1.37 ± 0.32 | 0.94 |
| LDL (mmol l−1) | 2.41 ± 0.78 | 2.74 ± 0.70 | 0.17 |
| SBP (mmHg) | 102.7 ± 10.1 | 108.0 ± 9.0 | 0.13 |
| Low-Na+ SBP (mmHg) | 102.0 ± 9.4 | 100.6 ± 7.4* | 0.64 |
| DBP (mmHg) | 69.0 ± 7.7 | 70.9 ± 8.1 | 0.50 |
| Low-Na+ DBP (mmHg) | 67.5 ± 12.1 | 71.6 ± 5.6 | 0.23 |
| MAP (mmHg) | 80.1 ± 7.9 | 83.3 ± 8.1 | 0.28 |
| Low-Na+ MAP (mmHg) | 79.0 ± 11.0 | 81.2 ± 5.5 | 0.47 |
Values are means ± s.d. All values were recorded during the screening visit, except for the low-Na+ blood pressure values, which were measured by random zero sphygmomanometry on the morning of study day 6 after 5 days of low-Na+ diet (10 mmol day−1). The groups were compared by using the Wilcoxin rank-sum for all variables except gender, which was compared by Fischer's exact test. BMI, body mass index; BSA, body surface area; DBP, diastolic blood pressure; HDL, high density lipoprotein; LDL, low density lipoprotein; SBP, systolic blood pressure.
Gly16 group before versus after diet demonstrated a significant decrease in SBP after the low-Na+ diet (P = 0.003, paired t test).
Table 2.
Weight loss and urinary indices before and after dietary sodium restriction
| Na+ intake | Arg16 | Gly16 | Pdiet | Pgenotype | Pinteraction |
|---|---|---|---|---|---|
| Weight (kg) | |||||
| NL | 70.8 ± 11.1 | 74.0 ± 10.3 | < 0.001 | 0.40 | 0.35 |
| Low | 69.1 ± 10.7 | 72.1 ± 10.2 | |||
| Urine volume (ml) | |||||
| NL | 1562 ± 791 | 1636 ± 632 | 0.02 | 0.80 | 0.89 |
| Low | 1806 ± 699 | 1854 ± 709 | |||
| 24 h Na+ exc (mmol) | |||||
| NL | 176.6 ± 95.2 | 187.0 ± 56.5 | < 0.001 | 0.70 | 0.73 |
| Low | 30.1 ± 20.3 | 31.1 ± 12.4 | |||
| 24 h K+ exc (mmol) | |||||
| NL | 57.3 ± 21.7 | 71.7 ± 28.3 | < 0.001 | 0.31 | 0.18 |
| Low | 90.4 ± 21.0 | 90.4 ± 26.5 | |||
| 24 h Cr exc (mg) | |||||
| NL | 1644.3 ± 346.0 | 1678.0 ± 510.5 | 0.28 | 0.88 | 0.27 |
| Low | 1758.5 ± 386.2 | 1677.0 ± 549.6 | |||
Values are means ± s.d. Cr, creatinine; exc, excretion; Pdiet, statistical significance of dietary sodium intake on indices; Pgenotype, statistical significance of genotype effect on indices; Pinteraction, statistical significance of interaction effect on indices (i.e. genotype-by-diet interaction effect on weight loss).
Figure 2. Mean arterial pressure and heart rate at baseline and in response to intravenous terbutaline before (left panel) and after (right panel) dietary sodium restriction in Gly16 and Arg16 subjects.
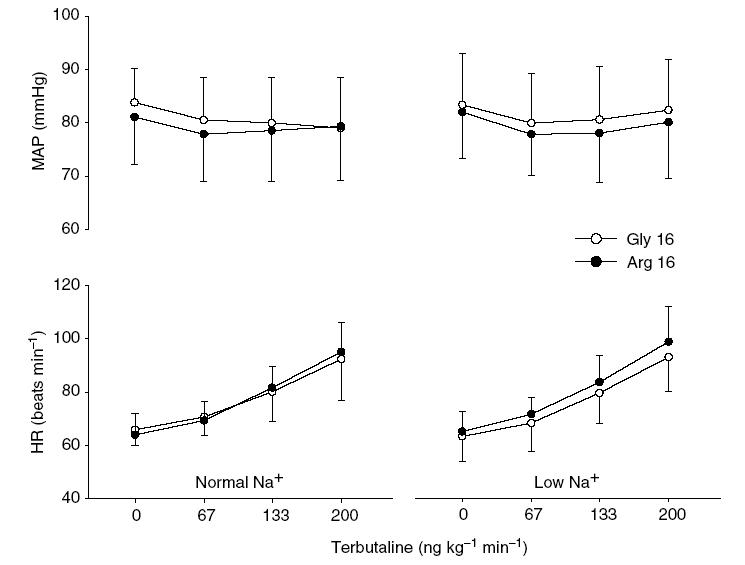
There were no baseline differences in the mean arterial pressure (MAP) and heart rate (HR) before and after the low-Na+ diet. Terbutaline decreased MAP (P < 0.001), and there was no evidence that sodium restriction or genotype influenced the MAP response. Terbutaline increased HR (P < 0.001), and there was no evidence that sodium restriction or genotype influenced the HR response. Error bars denote s.d.
Before dietary sodium restriction, resting HR prior to infusion of TRB was 63.9 ± 8.1 and 65.9 ± 6.1 beats min−1 in the Arg16 and Gly16 groups, respectively (P = 0.45). Dietary sodium restriction did not affect resting HR in either group (Arg16, 65.2 ± 7.5 beats min−1; Gly16, 63.4 ± 9.4 beats min−1). TRB infusion increased HR before and after dietary sodium restriction (P < 0.001), and there was no evidence that sodium restriction or genotype influenced the HR response to TRB (P = 0.43, genotype-by-dose interaction; P = 0.77, diet-by-dose interaction; P = 0.89, genotype-by-diet-by-dose interaction).
As shown in Fig. 3, resting CO was 5.8 ± 1.3 and 6.4 ± 1.4 l min−1 in the Arg16 and Gly16 groups, respectively, before dietary sodium restriction (P = 0.24). Following dietary sodium restriction, CO was 5.6 ± 1.0 and 5.5 ± 1.2 l min−1 in the Arg16 and Gly16 groups, respectively, representing a significant decrease in the Gly16 group (P = 0.003) but not in the Arg16 group (P = 0.38). TRB increased CO before and after dietary sodium restriction (P < 0.001). However, there was no evidence that diet or genotype affected the response to TRB (P = 0.33, genotype-by-dose interaction; P = 0.87, diet-by-dose interaction; P = 0.65, genotype-by-diet-by-dose interaction).
Figure 3. Cardiac output, stroke volume and systemic vascular resistance at baseline and in response to intravenous terbutaline before (left panel) and after (right panel) dietary sodium restriction in Gly16 and Arg16 subjects.
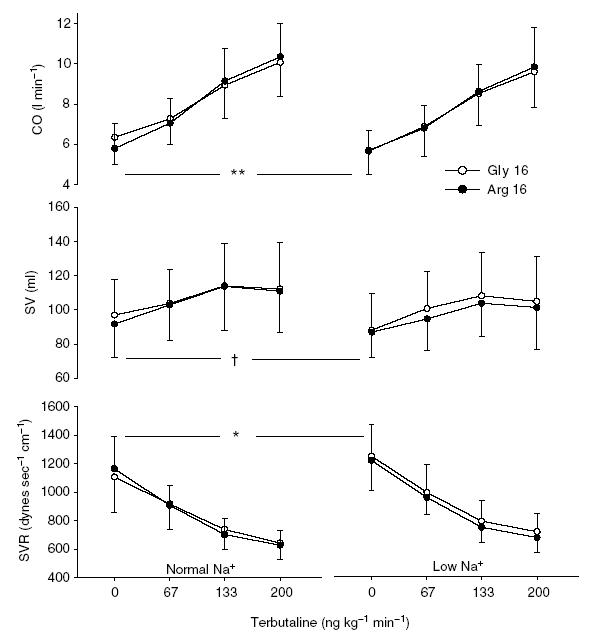
In the Gly16 group, dietary sodium restriction significantly decreased baseline cardiac output (CO, **P = 0.003), tended to decrease stroke volume (SV, †P = 0.06) and increased systemic vascular resistance (SVR, *P = 0.02), whereas these baseline indices were unaffected in the Arg16 group. Terbutaline increased CO and SV, and decreased SVR (P < 0.001), but there were no differences in the responses to terbutaline based on diet or genotype. Error bars denote s.d.
Resting SV was 91.7 ± 19.4 and 97.0 ± 20.6 ml in the Arg16 and Gly16 groups, respectively (P = 0.46), before the low-Na+ diet. Afterwards, baseline SV was reduced in all subjects (new baseline SV: Arg16, 85.7 ± 14.8 ml; Gly16, 86.9 ± 21.7 ml; P = 0.016) representing a tendency towards a decrease in the Gly16 group (P = 0.06) but not in the Arg16 group (P = 0.11). TRB increased SV before and after dietary sodium restriction (P < 0.001), and there was no evidence that sodium restriction or genotype influenced the SV response to TRB (P = 0.99, genotype-by-dose interaction; P = 0.83, diet-by-dose interaction; P = 0.24, genotype-by-diet-by-dose interaction).
Before dietary sodium restriction, resting SVR was 1165 ± 226 and 1106 ± 246 dynes s cm−5 in the Arg16 and Gly16 groups, respectively (P = 0.49). Following dietary sodium restriction, SVR was 1216 ± 209 and 1246 ± 222 dynes s cm−5 in the Arg16 and Gly16 groups, respectively, representing a significant increase in the Gly16 group (P = 0.02) but not in the Arg16 group (P = 0.47). TRB decreased SVR in all subjects before and after dietary sodium restriction (P < 0.001). However, there was no evidence that diet or genotype affected the dose–responses to TRB (P = 0.623, genotype-by-dose interaction; P = 0.66, diet-by-dose interaction; P = 0.72, genotype-by-diet-by-dose interaction).
Baseline FBF did not differ between groups (Arg16, 2.28 ± 0.92 ml (100 ml limb volume)−1 min−1; Gly16, 1.93 ± 0.43 ml (100 ml limb volume)−1 min−1; P = 0.17). SNP evoked an increase in FBF (P < 0.001), but there was no evidence of a difference between genotype groups (P = 0.76, main effect of genotype; P = 0.45, genotype-by-SNP interaction). l-NMMA significantly decreased baseline FBF (Arg16, 1.73 ± 0.48 ml (100 ml limb volume)−1 min−1, P = 0.04; Gly16, 1.49 ± 0.38 ml (100 ml limb volume)−1 min−1, P = 0.002), while the difference was not dependent on genotype (P = 0.67). l-NMMA did not affect the vasodilator responses to SNP (P = 0.07, SNP-by-l-NMMA interaction; P = 0.29, genotype-by-SNP-by-l-NMMA interaction).
Isoprenaline caused an increase in FBF (P < 0.001, main effect of isoprenaline dose) that did not differ between groups (P = 0.51, main effect of genotype; P = 0.72, genotype-by-dose interaction, Fig. 4). For example, the response to isoprenaline at 12 ng (dl limb volume)−1 min−1 was 10.3 ± 5.5 ml (100 ml limb volume)−1 min−1 in the Arg16 group and 12.0 ± 4.4 ml (100 ml limb volume)−1 min−1 in the Gly16 group (P = 0.33, unpaired t test). l-NMMA significantly blunted the vasodilator response to isoprenaline (P < 0.001), but there was no evidence that this effect was influenced by genotype (P = 0.89, genotype-by-dose-by-l-NMMA interaction).
Figure 4. Forearm blood flow responses to administration of isoprenaline via the brachial artery in Gly16 and Arg16 subjects following dietary sodium restriction.
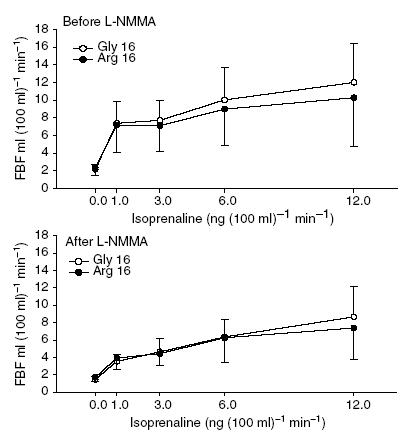
Baseline FBF did not differ between groups. Isoprenaline caused an increase in FBF that was not different in the two genotype groups (P = 0.51, main effect of genotype; P = 0.72, genotype-by-isoprenaline interaction). NOS inhibition with l-NMMA (lower panel) decreased baseline FBF, and significantly blunted the response to isoprenaline, but there was no evidence to suggest the responses were influenced by genotype (P = 0.89, genotype-by-isoprenaline-by-l-NMMA interaction).
ACh caused an increase in FBF (P < 0.001) that was not affected by genotype (P = 0.82, main effect of genotype; P = 0.83, genotype-by-ACh interaction). The response to ACh was 16.9 ± 11.3 ml (100 ml limb volume)−1 min−1 in the Arg16 group and 17.7 ± 8.5 ml (100 ml limb volume)−1 min−1 in the Gly16 group (P = 0.82). l-NMMA blunted the vasodilator response to ACh (P < 0.001). It is interesting to note that there was some evidence that l-NMMA affected the response to ACh differentially according to genotype (P = 0.057, genotype-by-ACh-by-l-NMMA interaction). In the presence of l-NMMA, the response to ACh was 13.0 ± 8.4 ml (100 ml tissue)−1 min−1 in the Arg16 group and 9.9 ± 6.9 ml (100 ml tissue−1 min−1) in the Gly16 group (P = 0.263).
As nine Arg16 and 12 Gly16 homozygotes were participants in a preceding study (Garovic et al. 2003), we performed an analysis of the effect of dietary sodium restriction on these individuals (Fig. 5). In the previous study, with a normal dietary sodium intake, the FBF responses of the Gly16 subjects to isoprenaline at 3, 6 and 12 ng (100 ml limb volume)−1 min−1 were 9.6 ± 3.6, 11.7 ± 4.6 and 15.9 ± 6.4 ml (100 ml limb volume)−1 min−1, respectively. In the present study, the FBF responses to the same doses in these individuals were 7.3 ± 2.6, 9.4 ± 4.1 and 11.2 ± 4.9 ml (100 ml limb volume)−1 min−1, respectively (P = 0.04, 0.08, 0.007, respectively, paired t test). Furthermore, the Arg16 subjects did not display a significant change in the FBF response to isoprenaline at any of the dosages after the low-Na+ diet (P ≥ 0.2 for all).
Figure 5. FBF dose responses to isoproterenol in the individuals who also participated in the previous study following normal sodium intake (150 mmol day−1).
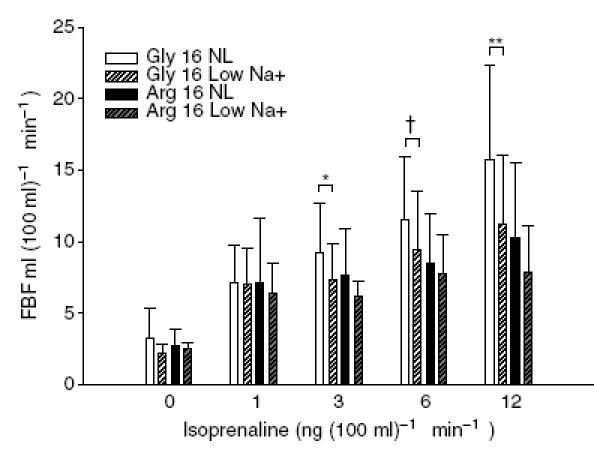
FBF response to isoprenaline was found to be dependent on genotype (P = 0.02, genotype-by-dose interaction) and diet (P = 0.02, diet-by-dose interaction). Supplemental genotype-specific analysis demonstrates that the 12 Gly16 homozygotes had a reduction in the FBF response to isoprenaline following 5 days of dietary sodium restriction at 3, 6 and 12 ng (100 ml tissue−1 min−1) (*P = 0.04, †P = 0.08 and **P = 0.007, respectively, paired t test). The FBF responses in the Arg16 group were also decreased with dietary sodium restriction, but this was not significant (P = 0.18 for all). From an analysis restricted to the maximum dose of isoprenaline, significant main effects were detected for diet (P = 0.006) and genotype (P = 0.044) with non-significant genotype-by-diet interaction (P = 0.364). Error bars denote s.d.
Discussion
This is the first study to examine the influence of a common polymorphism in the β2AR on forearm vasodilator function after dietary sodium restriction, and on systemic cardiovascular responses to a β2AR agonist before and after dietary sodium restriction in normotensive humans. There are several new findings in this study. First, there was no significant difference in NO-mediated, β2AR-mediated forearm vasodilatation between Gly16 and Arg16 homozygotes following 5 days of dietary sodium restriction. Second, non-β2AR-mediated vasodilator responses to ACh and SNP were not different in the two genotype groups after the low-Na+ diet. Third, contrary to what we would have expected based on our hypothesis, the cardiovascular responses to systemic administration of TRB did was not affected by genotype before or after the dietary sodium restriction. Fourth, also contrary to what we would have expected based on our hypothesis, CO and SV did not differ between the two genotype groups before the low-Na+ diet. However, dietary sodium restriction did affect baseline myocardial function in Gly16 homozygotes, as resting CO decreased, SV tended to decrease, systolic blood pressure decreased and SVR increased in the Gly16 homozygotes after the low-Na+ diet, whereas these indices were essentially unaffected by the low-Na+ diet in the Arg16 group.
Clinical significance of the Arg16Gly polymorphism has been demonstrated in genomic analyses of hypertension, but as yet there is no conclusive evidence that the Gly16 rather than the Arg16 variant is most important (Kotanko et al. 1997; Bray et al. 2000; Busjahn et al. 2000; Xie et al. 2000; Ranade et al. 2001; Herrmann et al. 2002; Jindra et al. 2002; Snieder et al. 2002; Ge et al. 2005). Despite this, recent studies have suggested that the Gly16 allele may actually be favourable in cardiovascular health. A study of 5888 people 65 years of age or older, suggested a decreased risk of coronary events in Gly16 compared to Arg16 carriers (Heckbert et al. 2003). A prospective cohort study of patients with acute coronary syndrome who received β-blockers, predicted lower 3-year cumulative mortality in Gly16 than in Arg16 subjects (Lanfear et al. 2005). Bao et al. (2005) reported that the haplotype containing the Gly16 allele was found to be protective against hypertension in European American adults below age 50. Healthy Gly16 homozygotes have enhanced left ventricular function compared to Arg16 homozgygotes (Tang et al. 2003; Eisenach et al. 2004, 2005; Snyder et al. 2006a), while decreases in ejection fraction increase the odds ratio of death in patients with acute coronary syndrome with non-ST-segment elevation (Bosch & Theroux, 2005). Taken together, the Arg16Gly polymorphism appears to have powerful pharmacogenetic and disease-modifying implications.
Because essential hypertension is a heterogeneous disorder caused by both genetic and environmental factors, we sought to determine whether intermediate physiological characteristics of cardiovascular function would clarify the understanding of the functional relevance of β2AR gene variation. Along these lines, we previously reported that Gly16 subjects demonstrated a greater vasodilator response to isoprenaline than Arg16 subjects, independent of amino acid position 27, and the difference was dependent on endothelial production of NO (Garovic et al. 2003); that study was performed after a controlled normal sodium diet (150 mmol day−1) for 5 days. As β2AR function in the vasculature (Feldman, 1990) and lymphocytes (Feldman, 1987) is affected by dietary sodium intake, we hypothesized that dietary sodium restriction would blunt the greater β2AR-mediated vasodilator responsiveness seen in Gly16 than in Arg16 subjects.
Naslund et al. (1990) placed normotensive subjects on a low-Na+ diet and found decreased forearm vasodilator sensitivity in response to isoprenaline, a decreased lymphocyte receptor affinity for isoprenaline, and a decrease in the proportion of lymphocyte β2AR binding agonists with high affinity, implying a decrease in adenylate cyclase coupling but not a difference in absolute receptor number. However, specific to the Arg16Gly polymorphism, lymphocyte assays have demonstrated a greater β2AR density in individuals homozygous for Gly16 (Snyder et al. 2006b) or in carriers of the most common haplotype associated with Gly16 in European Americans (Bao et al. 2005). Therefore, the present finding that Gly16 subjects no longer possessed the greater β2AR-mediated vasodilator function after low dietary sodium intake may suggest that low sodium intake affected either β2AR density, basal NO levels, or adenylate cyclase function. The blunted β2AR function in the forearm may also relate to the finding that baseline β2AR function in the heart was reduced after the low-Na+ diet, as CO decreased, systolic blood pressure decreased, SV tended to decrease and SVR increased in the Gly16 group. That ACh-evoked vasodilatation was similar in the groups before administration of l-NMMA, but tended to be blunted to a greater degree in the Gly16 group, also suggests that the polymorphism affects endothelial regulation of NO, but this was unaffected by dietary sodium, as similar findings were present in the previous study after normal dietary sodium intake (Garovic et al. 2003). Moreover, arterial vasodilator responses to ACh and SNP were unaffected by low dietary sodium, which is consistent with experiments performed in Sprague Dawley rats (Sofola et al. 2002).
Another potential explanation for the salt-sensitive β2AR responsiveness in Gly16 homozygotes may be dependence of the physiological stress response on low sodium intake. Formative work by Green and colleagues (1994) demonstrated in Chinese hamster fibroblasts that the Gly16 allele was associated with agonist-promoted down-regulation or sequestration of the β2AR. In this context, the sympatho-excitatory effect of dietary sodium restriction increases circulating catecholamines (Naslund et al. 1990; Grassi et al. 2002). Conceivably, a low-Na+ diet may result in down-regulation of the β2AR in Gly16 homozygotes, thereby decreasing the availability of β2ARs on the forearm vascular endothelium and abrogating the greater β2AR-mediated dilatation previously seen while receiving a normal sodium diet. We speculate that the desensitization of β2ARs in this group may differ in the setting of low dietary sodium for 5 days compared to chronic β2-agonist exposure. Bruck et al. (2003a, b) reported that following oral administration of the β2-agonist TRB for 2 weeks, desensitization of cardiac β2AR responses and down-regulation of lymphocyte β2ARs were not significantly different in Arg16 compared with Gly16 subjects. Furthermore, these same authors have shown that TRB-induced venodilatation in the dorsal hand vein following 2 weeks of oral TRB evoked the greatest extent of desensitization in the Arg16 homozygotes (Bruck et al. 2005). It remains unclear whether the polymorphisms affect myocardial responses, forearm arterial vasodilatation, and venodilatation differentially, although our study suggests that low dietary sodium affects myocardial function and arterial vasodilatation to a significant extent in Gly16 homozygotes.
Aside from the changes in baseline CO and SVR in the Gly16 group, the similar cardiovascular responses to TRB in the groups were not expected from our hypothesis and are not consistent with prior studies examining the Arg16Gly polymorphism. Gratze et al. (1999) showed that during systemic infusion of salbutamol, the total peripheral resistance index in Arg16 homozygotes decreased to a greater extent than in Gly16 homozygotes, implying a greater systemic vasodilator response in the Arg16 group; this group also had a greater increase in HR, cardiac index and stroke index. Similarly, Hoit et al. (2000) reported that during administration of a high systemic dose of TRB, calf blood flow was lower and SVR was greater in the Gly16 group, whereas HR and blood pressure responses were similar. Finally, Lee et al. (2004) reported that administration of inhaled salbutamol in asthmatics evoked a larger decrease in diastolic blood pressure in Arg16 + Gln27 (glutamine) homozygotes when compared to Gly16 + Glu27 (glutamate) homozygotes. The most likely explanation for the disparities between the findings of their studies and ours is the inability to isolate vascular responsiveness when counter-regulatory baroreflexes are engaged. Further analyses of systemic vasodilatation may require concomitant baroreflex inhibition with Nn-cholinergic blockade to measure adrenergic receptor sensitivity without the confounding influence of baroreflexes (Shannon et al. 1998; Jordan et al. 2002; Jones et al. 2003).
The functional relevance of β2AR polymorphism and its interaction with sodium intake is also highlighted in the companion paper in this issue by Snyder et al. (2006c). Compared to Arg16 homozygotes, the Gly16 group had a greater baseline CO and lower SVR before an acute intravenous saline infusion that remained following the saline load; furthermore, MAP increased to a greater extent and sodium excretion was less in the Gly16 group after the saline challenge (Snyder et al. 2006c). Whereas the Gly16 subjects who participated in our study did not have a significantly greater CO or lower SVR than the Arg16 subjects at rest, the dietary sodium restriction affected these indices in a direction opposite to that in our sodium loading study (Snyder et al. 2006c). Together, these findings suggest that increases in sodium balance may augment effects of the Gly16 allele on cardiovascular and renal phenotypes, and decreases in sodium balance may reduce β2AR-mediated cardiovascular function for the Gly16 allele. From an evolutionary viewpoint, the low-Na+ diet in the present study is probably closer to ‘normal’ when compared to the current sodium intake in modern society (Lev-Ran & Porta, 2005). In this context, we postulate that the intermediate physiological characteristics seen in Gly16 individuals are protective while receiving a normal to high-Na+ diet, but return to values similar to those of the Arg16 individuals after the low-Na+ diet. Thus, these studies provide evidence that the Arg16Gly polymorphism may be a genetic marker of salt sensitivity.
Limitations
The main experimental limitation involves the lack of forearm vascular response measurements prior to the low-Na+ diet, which is an important consequence of the inability to safely catheterize the non-dominant brachial artery in human volunteers both before and after a 5-day dietary intervention. From a study design perspective, it would have been ideal to perform these measurements before and after the low-Na+ diet. It was therefore necessary to perform a subanalysis of the 9 Arg16 and 12 Gly16 individuals who participated in the prior study approximately 1–2 years earlier following a controlled sodium diet (Garovic et al. 2003) (see Fig. 5). Moreover, characterization of position 16 in combination with position 27 will require larger sample sizes to associate salt sensitivity with composite haplotypes. Finally, similar studies will be necessary in older adults to extend the functional relevance of our findings to ageing and cardiovascular disease.
In summary, this is the first study to demonstrate that the Arg16Gly polymorphism modulates the effect of dietary sodium restriction on cardiovascular function in healthy, normotensive individuals. We conclude that dietary sodium restriction blunts the increased forearm NO-mediated, β2AR responsiveness in Gly16 subjects, as previously demonstrated for subjects receiving a normal sodium diet. The low-Na+ diet evoked a baseline decrease in systolic function and increase in peripheral resistance in the Gly16 group, providing evidence that sodium intake affects the influence of the Arg16Gly β2AR polymorphism on cardiovascular indices.
Acknowledgments
This work was supported by US National Institutes of Health grants U01 HL54464, HL 63328, NCRR K23-17520 and M01-RR00585. We thank all of our subjects for their enthusiastic participation, and the study coordinators for their tireless effort.
References
- Bao X, Mills PJ, Rana BK, Dimsdale JE, Schork NJ, Smith DW, Rao F, Milic M, O'Connor DT, Ziegler MG. Interactive effects of common beta2-adrenoceptor haplotypes and age on susceptibility to hypertension and receptor function. Hypertension. 2005;46:301–307. doi: 10.1161/01.HYP.0000175842.19266.95. [DOI] [PubMed] [Google Scholar]
- Bosch X, Theroux P. Left ventricular ejection fraction to predict early mortality in patients with non-ST-segment elevation acute coronary syndromes. Am Heart J. 2005;150:215–220. doi: 10.1016/j.ahj.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Bray MS, Krushkal J, Li L, Ferrell R, Kardia S, Sing CF, Turner ST, Boerwinkle E. Positional genomic analysis identifies the beta2-adrenergic receptor gene as a susceptibility locus for human hypertension. Circulation. 2000;101:2877–2882. doi: 10.1161/01.cir.101.25.2877. [DOI] [PubMed] [Google Scholar]
- Bruck H, Leineweber K, Beilfuss A, Weber M, Heusch G, Philipp T, Brodde OE. Genotype-dependent time course of lymphocyte beta 2-adrenergic receptor down-regulation. Clin Pharmacol Ther. 2003a;74:255–263. doi: 10.1016/S0009-9236(03)00188-7. [DOI] [PubMed] [Google Scholar]
- Bruck H, Leineweber K, Buscher R, Ulrich A, Radke J, Insel PA, Brodde OE. The Gln27Glu beta2-adrenoceptor polymorphism slows the onset of desensitization of cardiac functional responses in vivo. Pharmacogenetics. 2003b;13:59–66. doi: 10.1097/00008571-200302000-00001. [DOI] [PubMed] [Google Scholar]
- Bruck H, Leineweber K, Park J, Weber M, Heusch G, Philipp T, Brodde OE. Human beta2-adrenergic receptor gene haplotypes and venodilation in vivo. Clin Pharmacol Ther. 2005;78:232–238. doi: 10.1016/j.clpt.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Busjahn A, Li GH, Faulhaber HD, Rosenthal M, Becker A, Jeschke E, Schuster H, Timmermann B, Hoehe MR, Luft FC. Beta-2 adrenergic receptor gene variations, blood pressure, and heart size in normal twins. Hypertension. 2000;35:555–560. doi: 10.1161/01.hyp.35.2.555. [DOI] [PubMed] [Google Scholar]
- Cockcroft JR, Gazis AG, Cross DJ, Wheatley A, Dewar J, Hall IP, Noon JP. Beta2-adrenoceptor polymorphism determines vascular reactivity in humans. Hypertension. 2000;36:371–375. doi: 10.1161/01.hyp.36.3.371. [DOI] [PubMed] [Google Scholar]
- Eisenach JH, Barnes SA, Pike TL, Sokolnicki LA, Masuki S, Dietz NM, Rehfeldt KH, Turner ST, Joyner MJ. The Arg16/Gly beta2-adrenergic receptor polymorphism alters the cardiac output response to isometric exercise. J Appl Physiol. 2005;99:1776–1781. doi: 10.1152/japplphysiol.00469.2005. [DOI] [PubMed] [Google Scholar]
- Eisenach JH, McGuire AM, Schwingler RM, Turner ST, Joyner MJ. The Arg16/Gly beta2-adrenergic receptor polymorphism is associated with altered cardiovascular responses to isometric exercise. Physiol Genomics. 2004;16:323–328. doi: 10.1152/physiolgenomics.00152.2003. [DOI] [PubMed] [Google Scholar]
- Feldman RD. Beta-adrenergic receptor alterations in hypertension – physiological and molecular correlates. Can J Physiol Pharmacol. 1987;65:1666–1672. doi: 10.1139/y87-261. [DOI] [PubMed] [Google Scholar]
- Feldman RD. Defective venous beta-adrenergic response in borderline hypertensive subjects is corrected by a low sodium diet. J Clin Invest. 1990;85:647–652. doi: 10.1172/JCI114487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman RD, Limbird LE, Nadeau J, Robertson D, Wood AJ. Leukocyte beta-receptor alterations in hypertensive subjects. J Clin Invest. 1984;73:648–653. doi: 10.1172/JCI111255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garovic VD, Joyner MJ, Dietz NM, Boerwinkle E, Turner ST. Beta2-adrenergic receptor polymorphism and nitric oxide-dependent forearm blood flow responses to isoproterenol in humans. J Physiol. 2003;546:583–589. doi: 10.1113/jphysiol.2002.031138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge D, Huang J, He J, Li B, Duan X, Chen R, Gu D. Beta2-adrenergic receptor gene variations associated with stage-2 hypertension in northern Han Chinese. Ann Hum Genet. 2005;69:36–44. doi: 10.1046/j.1529-8817.2003.00093.x. [DOI] [PubMed] [Google Scholar]
- Grassi G, Dell'oro R, Seravalle G, Foglia G, Trevano FQ, Mancia G. Short- and long-term neuroadrenergic effects of moderate dietary sodium restriction in essential hypertension. Circulation. 2002;106:1957–1961. doi: 10.1161/01.cir.0000033519.45615.c7. [DOI] [PubMed] [Google Scholar]
- Gratze G, Fortin J, Labugger R, Binder A, Kotanko P, Timmermann B, Luft FC, Hoehe MR, Skrabal F. Beta-2 adrenergic receptor variants affect resting blood pressure and agonist-induced vasodilation in young adult Caucasians. Hypertension. 1999;33:1425–1430. doi: 10.1161/01.hyp.33.6.1425. [DOI] [PubMed] [Google Scholar]
- Green SA, Turki J, Innis M, Liggett SB. Amino-terminal polymorphisms of the human beta 2-adrenergic impart distinct agonist-promoted regulatory properties. Bio Chem. 1994;33:9414–9419. doi: 10.1021/bi00198a006. [DOI] [PubMed] [Google Scholar]
- Greenfield ADM, Whitney RJ, Mowbray JF. Methods for the investigation of peripheral blood flow. Br Med Bull. 1963;19:101–109. doi: 10.1093/oxfordjournals.bmb.a070026. [DOI] [PubMed] [Google Scholar]
- Heckbert SR, Hindorff LA, Edwards KL, Psaty BM, Lumley T, Siscovick DS, Tang Z, Durda JP, Kronmal RA, Tracy RP. Beta2-adrenergic receptor polymorphisms and risk of incident cardiovascular events in the elderly. Circulation. 2003;107:2021–2024. doi: 10.1161/01.CIR.0000065231.07729.92. [DOI] [PubMed] [Google Scholar]
- Herrmann SM, Nicaud V, Tiret L, Evans A, Kee F, Ruidavets JB, Arveiler D, Luc G, Morrison C, Hoehe MR, Paul M, Cambien F. Polymorphisms of the beta2-adrenoceptor (ADRB2) gene and essential hypertension: the ECTIM and PEGASE studies. J Hypertens. 2002;20:229–235. doi: 10.1097/00004872-200202000-00012. [DOI] [PubMed] [Google Scholar]
- Hoit BD, Suresh DP, Craft L, Walsh RA, Liggett SB. Beta2-adrenergic receptor polymorphisms at amino acid 16 differentially influence agonist-stimulated blood pressure and peripheral blood flow in normal individuals. Am Heart J. 2000;139:537–542. doi: 10.1016/s0002-8703(00)90099-1. [DOI] [PubMed] [Google Scholar]
- Jindra A, Horky K, Peleska J, Jachymova M, Bultas J, Umnerova V, Heller S, Hlubocka Z. Association analysis of Arg16Gly polymorphism of the beta2-adrenergic receptor gene in offspring from hypertensive and normotensive families. Blood Press. 2002;11:213–217. doi: 10.1080/08037050213761. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Beck KC, Proctor DN, Miller J, Dietz NM, Joyner MJ. Cardiac output during exercise by the open circuit acetylene washin method: comparison with direct Fick. J Appl Physiol. 2000;88:1650–1658. doi: 10.1152/jappl.2000.88.5.1650. [DOI] [PubMed] [Google Scholar]
- Jones PP, Christou DD, Jordan J, Seals DR. Baroreflex buffering is reduced with age in healthy men. Circulation. 2003;107:1770–1774. doi: 10.1161/01.CIR.0000057811.86187.88. [DOI] [PubMed] [Google Scholar]
- Jordan J, Tank J, Shannon JR, Diedrich A, Lipp A, Schroder C, Arnold G, Sharma AM, Biaggioni I, Robertson D, Luft FC. Baroreflex buffering and susceptibility to vasoactive drugs. Circulation. 2002;105:1459–1464. doi: 10.1161/01.cir.0000012126.56352.fd. [DOI] [PubMed] [Google Scholar]
- Kotanko P, Binder A, Tasker J, Defreitas P, Kamdar S, Clark AJ, Skrabal F, Caulfield M. Essential hypertension in African Caribbeans associates with a variant of the beta2-adrenoceptor. Hypertension. 1997;30:773–776. doi: 10.1161/01.hyp.30.4.773. [DOI] [PubMed] [Google Scholar]
- Lanfear DE, Jones PG, Marsh S, Cresci S, McLeod HL, Spertus JA. Beta2-adrenergic receptor genotype and survival among patients receiving beta-blocker therapy after an acute coronary syndrome. JAMA. 2005;294:1526–1533. doi: 10.1001/jama.294.12.1526. [DOI] [PubMed] [Google Scholar]
- Lang CC, Stein CM, Brown RM, Deegan R, Nelson R, He HB, Wood M, Wood AJ. Attenuation of isoproterenol-mediated vasodilatation in blacks. N Engl J Med. 1995;333:155–160. doi: 10.1056/NEJM199507203330304. [DOI] [PubMed] [Google Scholar]
- Lee DK, Bates CE, Lipworth BJ. Acute systemic effects of inhaled salbutamol in asthmatic subjects expressing common homozygous beta2-adrenoceptor haplotypes at positions 16 and 27. Br J Clin Pharmacol. 2004;57:100–104. doi: 10.1046/j.1365-2125.2003.01978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Ran A, Porta M. Salt and hypertension: a phylogenetic perspective. Diabetes Metab Res Rev. 2005;21:118–131. doi: 10.1002/dmrr.539. [DOI] [PubMed] [Google Scholar]
- Miller JZ, Weinberger MH, Christian JC, Daugherty SA. Familial resemblance in the blood pressure response to sodium restriction. Am J Epidemiol. 1987;126:822–830. doi: 10.1093/oxfordjournals.aje.a114719. [DOI] [PubMed] [Google Scholar]
- Naslund T, Silberstein DJ, Merrell WJ, Nadeau JH, Wood AJ. Low sodium intake corrects abnormality in beta-receptor-mediated arterial vasodilation in patients with hypertension: correlation with beta-receptor function in vitro. Clin Pharmacol Ther. 1990;48:87–95. doi: 10.1038/clpt.1990.121. [DOI] [PubMed] [Google Scholar]
- Ranade K, Shue WH, Hung YJ, Hsuing CA, Chiang FT, Pesich R, et al. The glycine allele of a glycine/arginine polymorphism in the beta2-adrenergic receptor gene is associated with essential hypertension in a population of Chinese origin. Am J Hypertens. 2001;14:1196–1200. doi: 10.1016/s0895-7061(01)02213-0. [DOI] [PubMed] [Google Scholar]
- Shannon JR, Jordan J, Black BK, Costa F, Robertson D. Uncoupling of the baroreflex by NN-cholinergic blockade in dissecting the components of cardiovascular regulation. Hypertension. 1998;32:101–107. doi: 10.1161/01.hyp.32.1.101. [DOI] [PubMed] [Google Scholar]
- Snieder H, Dong Y, Barbeau P, Harshfield GA, Dalageogou C, Zhu H, Carter ND, Treiber FA. Beta2-adrenergic receptor gene and resting hemodynamics in European and African American youth. Am J Hypertens. 2002;15:973–979. doi: 10.1016/s0895-7061(02)02991-6. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Beck KC, Dietz NM, Eisenach JH, Joyner MJ, Turner ST, Johnson BD. Arg16Gly polymorphism of the beta2-adrenergic receptor is associated with differences in cardiovascular function at rest and during exercise in humans. J Physiol. 2006a;571:121–130. doi: 10.1113/jphysiol.2005.098558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Hulsebus ML, Turner ST, Joyner MJ, Johnson BD. Genotype related differences in beta-2 adrenergic receptor density influence cardiac function in healthy humans. Med Sci Sports Exerc. 2006b;38(5):882–886. doi: 10.1249/01.mss.0000218144.02831.f6. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Turner ST, Joyner MJ, Eisenach JH, Johnson BD. The Arg16Gly polymorphism of the β2-adrenergic receptor and the natriuretic response to rapid saline infusion in humans. J Physiol. 2006c;574:947–954. doi: 10.1113/jphysiol.2006.107672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofola OA, Knill A, Hainsworth R, Drinkhill M. Change in endothelial function in mesenteric arteries of Sprague-Dawley rats fed a high salt diet. J Physiol. 2002;543:255–260. doi: 10.1113/jphysiol.2002.022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CM, Nelson R, Deegan R, He H, Wood M, Wood AJ. Forearm beta adrenergic receptor-mediated vasodilation is impaired, without alteration of forearm norepinephrine spillover, in borderline hypertension. J Clin Invest. 1995;96:579–585. doi: 10.1172/JCI118070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svetkey LP, Chen YT, McKeown SP, Preis L, Wilson AF. Preliminary evidence of linkage of salt sensitivity in black Americans at the beta 2-adrenergic receptor locus. Hypertension. 1997;29:918–922. doi: 10.1161/01.hyp.29.4.918. [DOI] [PubMed] [Google Scholar]
- Tang W, Devereux RB, Kitzman DW, Province MA, Leppert M, Oberman A, Hopkins PN, Arnett DK. The Arg16Gly polymorphism of the beta2-adrenergic receptor and left ventricular systolic function. Am J Hypertens. 2003;16:945–951. doi: 10.1016/s0895-7061(03)01001-x. [DOI] [PubMed] [Google Scholar]
- Xie HG, Stein CM, Kim RB, Gainer JV, Sofowora G, Dishy V, Brown NJ, Goree RE, Haines JL, Wood AJ. Human beta2-adrenergic receptor polymorphisms: no association with essential hypertension in black or white Americans. Clin Pharmacol Ther. 2000;67:670–675. doi: 10.1067/mcp.2000.106293. [DOI] [PubMed] [Google Scholar]