

Abstract
Meiotic cell cycle progression during vertebrate oocyte maturation requires the correct temporal translation of maternal mRNAs encoding key regulatory proteins. The mechanism by which specific mRNAs are temporally activated is unknown, although both cytoplasmic polyadenylation elements (CPE) within the 3′-untranslated region (3′-UTR) of mRNAs and the CPE-binding protein (CPEB) have been implicated. We report that in progesterone-stimulated Xenopus oocytes, the early cytoplasmic polyadenylation and translational activation of multiple maternal mRNAs occur in a CPE-and CPEB-independent manner. We demonstrate that polyadenylation response elements, originally identified in the 3′-UTR of the mRNA encoding the Mos proto-oncogene, direct CPE- and CPEB-independent poly-adenylation of an early class of Xenopus maternal mRNAs. Our findings refute the hypothesis that CPE sequences alone account for the range of temporal inductions of maternal mRNAs observed during Xenopus oocyte maturation. Rather, our data indicate that the sequential action of distinct 3′-UTR-directed translational control mechanisms coordinates the complex temporal patterns and extent of protein synthesis during vertebrate meiotic cell cycle progression.
Early developmental cell fate decisions are regulated through the translational activation of maternally derived mRNAs. Meiotic cell cycle progression during vertebrate oocyte maturation requires the correct temporal translation of mRNAs encoding key regulatory proteins (1–9). The translational induction of many maternal mRNAs has been correlated with an increase in mRNA poly(A) tail length. This evolution-arily conserved process, termed cytoplasmic polyadenylation, requires a polyadenylation hexanucleotide sequence (typically AAUAAA) as well as additional 3′-UTR1 regulatory sequences, including cytoplasmic polyadenylation elements (CPE) (10). CPE sequences have been shown to repress mRNA translation in immature oocytes and to direct cytoplasmic polyadenylation and translational activation in maturing oocytes. Both aspects of CPE function require the CPE-binding protein (CPEB) (9, 11–23).
Using progesterone-stimulated oocytes from the frog Xenopus laevis, it has been demonstrated that individual mRNAs are polyadenylated and translationally activated at different times following meiotic cell cycle resumption (24–26). Because all regulated mRNAs hitherto examined contain CPE sequences, the basis of this temporal order of translational activation was unclear. Various mechanisms been proposed to account for both early and late mRNA cytoplasmic polyadenylation and translational activation profiles during progesterone-stimulated meiotic cell cycle progression. These include the precise nucleotide sequence of the CPE, the number of CPEs, and/or the position of the CPEs within the 3′-UTR (24, 25, 27, 28). In addition, the cytoplasmic polyadenylation and translational activation of maternal mRNAs has been previously classified as being independent of (class I), or dependent on (class II), translation of the mRNA encoding the Mos proto-oncogene (24, 25).
A generalized understanding of the molecular mechanisms governing the temporal control of maternal mRNA translational activation has been confounded by a number of inconsistencies in assignment of CPE-containing mRNAs to particular classes, as well as differences in temporal regulation within class groups. Furthermore, the conversion of CPEB from a translational repressor to a translational activator appears to occur too late to regulate early mRNA cytoplasmic polyadenylation and translational activation. CPEB conversion involves both Aurora A/Eg2- and cdc2-dependent phosphorylation (28, 29). Cdc2 activation occurs subsequent to the translational activation of the temporally early Mos mRNA (30), and although initial studies suggested Aurora A/Eg2 activation occurred early during maturation (29, 31), subsequent studies (32–34) have concluded that Eg2 activation is temporally late and perhaps dependent on prior cdc2 activation. The recent identification of a CPE- and CPEB-independent regulatory mechanism that controls the early translational activation of the Mos mRNA (30) suggests that CPE- and CPEB-independent regulatory mechanisms may be more generally utilized to generate the range of temporal profiles of maternal mRNA translational activation observed during meiotic cell cycle progression.
The limited number of endogenous mRNAs or reporter RNAs that have been studied simultaneously has hampered elucidation of the mechanisms controlling the timing of maternal mRNA translational activation. Moreover, many of the 3′-UTR reporter constructs used previously were not full length thus compromising the generality of the conclusions that could be drawn. To circumvent these prior problems, we have utilized RNA ligation-coupled PCR (35) from the same oocyte samples to investigate the temporal kinetics of cytoplasmic polyadenylation of multiple endogenous mRNAs during oocyte meiotic maturation. We report that contrary to current dogma, CPE-and CPEB-dependent mechanisms do not control temporally early maternal mRNA polyadenylation and translational activation during Xenopus oocyte maturation. We define a new class of Xenopus maternal mRNAs, which undergo temporally early CPE- and CPEB-independent cytoplasmic polyadenylation and translational activation. We demonstrate that poly-adenylation response elements (PREs) present in the 3′-UTRs of early class mRNAs function to direct temporally early cytoplasmic polyadenylation and translational activation. By contrast, the cytoplasmic polyadenylation of CPE- and CPEB-dependent mRNAs is a temporally late event. Our findings suggest that the sequential action of distinct 3′-UTR-directed translational control mechanisms coordinates the complex temporal patterns and extent of protein synthesis during vertebrate meiotic cell cycle progression.
EXPERIMENTAL PROCEDURES
DNA Constructs
The 211-nt D7 3′-UTR, the 146-nt G10 3′-UTR, and the 71-nt histone-like B4 3′-UTR were isolated from immature Xenopus oocytes by RT-PCR using primers containing 5′ XbaI sites and 3′ SalI sites and cloned into pGEM GST (9) for polyadenylation and translation studies, or into pGEM4Z (Promega) for RNA EMSAs. It should be noted that the D7 3′-UTR was larger than predicted and was found to encode an additional 21 nucleotides of 3′ sequence not present in the GenBank™ entry, including a canonical AAUAAA polyadenylation hexanucleotide. The histone-like B4 CPE was changed from uuuuuaau to uuuuGGau; the D7 CPEs were changed from uuuuuuaa, uuuuaaca, uuuuuaca, and uuuuuau to uuuuuGGa, uuuGGaca, uuuuG-Gca, and uuuuGGu; and the G10 CPEs were changed from uuuuaau and uuuuuuau to uuuGGau and uuuuuGGu using QuikChange (Stratagene). The D7 UTR was truncated using standard PCR mutagenesis to remove the two 5′-most CPEs, while leaving the PRE intact (see Fig. 7A for PRE boundary). The PRE in the D7 UTR was deleted by using QuikChange. The B4, D7, and G10 3′-UTR constructs were linearized with SalI prior to in vitro transcription. QuikChange was used to replace β-globin sequence with the histone-like B4 and D7 PREs in pGEM GST β-globin UTR (9), at the respective distance from the polyadenylation hexanucleotide as they are found in their native mRNAs. Because the D7 PRE spans a CPE, the CPE was disrupted as described above to eliminate any possible contribution to cytoplasmic polyadenylation. pGEM GST CPEB-AA construction and RNA transcription have been described before (30). pGEM GST XeCPEB has been described (9).
Fig. 7. PRE sequences in 3′-UTRs direct temporally early, progesterone-stimulated cytoplasmic poly-adenylation.
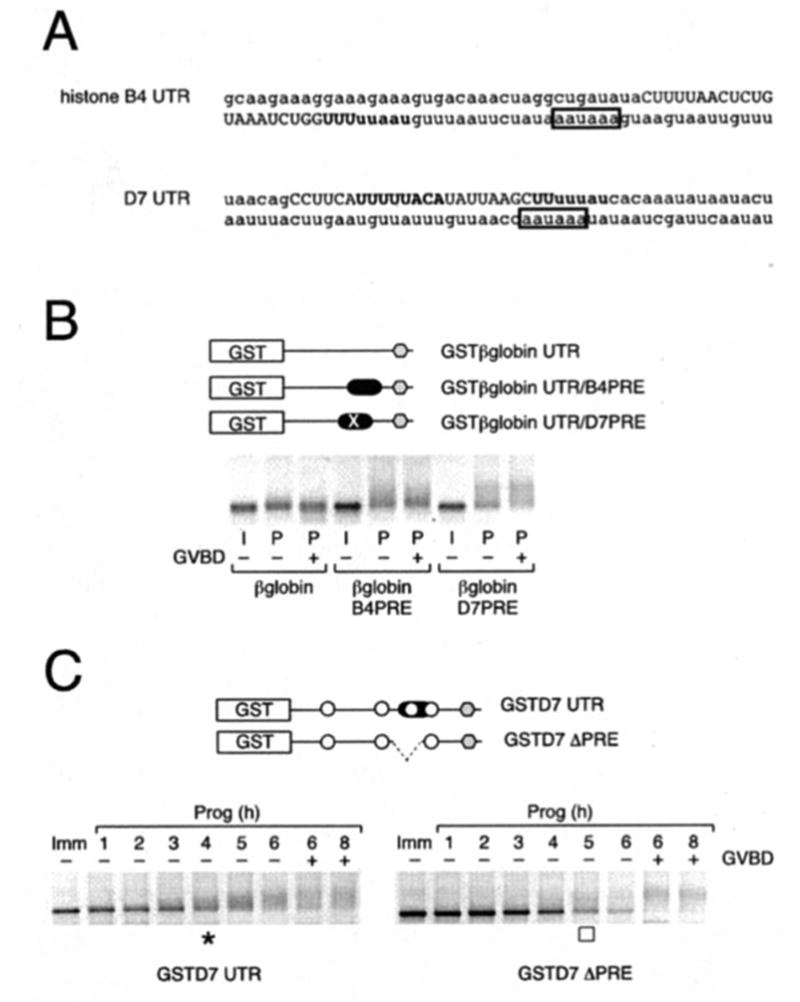
The indicated GST reporter RNA constructs were injected into immature oocytes. A, the last 100 nt of the histone-like B4 and D7 mRNAs are shown, with the PRE in uppercase, the CPEs in boldface, and the polyadenylation hexanucleotide in a box. B, histone-like B4 and D7 PRE sequences were inserted into the heterologous β-globin 3′-UTR. Polyadenylation of reporter constructs was assessed by RT-PCR using a GST forward primer. Progesterone-stimulated (P) samples were prepared at GVBD50 (see “Experimental Procedures”), segregated based on GVBD status (− or +), and shown relative to a time-matched sample prepared from immature (I) oocytes. Reporter constructs are schematically represented with the position of the histone-like B4 and D7 PREs (filled oblong) indicated. The PRE in D7 spans a CPE. To eliminate any possible contribution from the CPE, it was disrupted in the reporter RNA (denoted by ×). C, GST reporter RNA constructs fused to either the wild-type or PRE-deleted D7 3′-UTR were injected into immature oocytes. Poly-adenylation of reporter constructs was assessed by RT-PCR using a GST forward primer at the indicated times. GVBD50 (see “Experimental Procedures”) occurred at 6 h. Initiation of wild-type (asterisk, left panel) and PRE-deleted (open square, right panel) D7 3′-UTR-directed poly-adenylation is indicated. Reporter constructs are schematically represented with the position of CPEs (open circle), PRE (filled oblong), and deleted PRE (dashed line) indicated. − or + indicates GVBD status of harvested oocyte samples at the indicated times.
Oocyte Isolation, Culture, Microinjection, and Lysate Preparation
Xenopus oocyte isolation and culture has been described previously (36). Oocytes were induced to mature with 2 μg/ml progesterone, and pools of 5 or 6 oocytes were taken at the indicated times. Where synthetic RNA constructs were injected, collection of immature controls was performed in concert with time-matched progesterone-stimulated samples. Germinal vesicle breakdown (GVBD) was used as an indicator of maturation and assessed by the presence of a white spot on the animal pole. Where indicated, oocytes were segregated at the time when 50% of the oocyte population had reached GVBD (GVBD50) based on whether they had (+) or had not (−) completed GVBD. RNA was transcribed in vitro with SP6 and injected at concentrations described previously (9). RNA and protein were extracted from the same pool of oocytes (30). Total RNA was isolated using RNA STAT-60 (Tel-Test) (3).
Polyadenylation Assays
RNA ligation-coupled RT-PCR (see Fig. 1A) was a modified version of the technique described previously (35). 4 μg of total oocyte RNA, from pools of 5 or 6 oocytes, was ligated to 0.4 μg of P1 anchor primer (35), in a 10-μl reaction using T4 RNA ligase (New England Biolabs) according to the manufacturer’s directions. The whole 10-μl RNA ligation reaction was used in a 50-μl reverse transcription reaction using Superscript III (Invitrogen), according to manufacturer’s directions using 0.4 μg of P1′ as the reverse primer (35). 1 μl of this cDNA preparation was used in each 50-μl PCR using Platinum Taq (Invitrogen), according to the manufacturer’s directions. PCR was performed for 40 cycles, using a 56 °C annealing temperature and 1.5 mM final concentration of Mg2+, except the FGFR1 reaction which required 2.5 mM Mg2+. The specific primers used were designed to be 70–110 nucleotides from the poly(A) addition site according to sequence information available in GenBank™, and the accession numbers are as follows: X13855 (B4 mRNA for histone H1-like protein); XLD7 (maternal mRNA D7); Z17206 (Aurora A/Eg2); U24491 (FGF receptor 1); XLG10 (maternal G10 mRNA); X13311 (p39mos); J03166 (cyclin B1); AJ304991 (cyclin B4); U13962 (Xe-Wee1A); X53745 (cyclin A1); and M24769 (cytoskeletal actin type 5). The primers used are as follows: histone-like B4, AGT GAC AAA CTA GGC TGA TAT ACT; cyclin A1, CAT TGA ACT GCT TCA TTT TCC CAG; cyclin B1, GTG GCA TTC CAA TTG TGT ATT GTT; cyclin B4, CAT AGG ACA CTT GTT ATA TTG TAG; D7, TGT TGT GAA GTT GCC ATT TAG TAT; Aurora A/Eg2, GTT TCA ATC TTG TAT GTC CTT TTA; FGFR1, TTT GCT ATG TTT TCA GTT TGT ATT; G10, TAA GGC CGG CGA CTG AAA TTG TGT; Mos, GTT GCA TTG CTG TTT AAG TGG TAA; and Wee1, GGC CTG GAC AAA AAC TTT ATA ATT. To verify that an increase in PCR product size in stimulated oocytes is indicative of extension of the poly(A) tail, treatment of the initial RNA samples with oligo(dT) and RNase H (to degrade all poly(A) tails (3) prior to RNA ligation with the P1 DNA oligonucleotide) did reduce the size of the PCR product in progesterone-treated samples to a size comparable with that from immature oocyte samples (data not shown). Direct sequencing of representative PCR products also confirmed the increase in PCR product size observed in progesterone-stimulated oocytes was due to poly(A) tail extension.
Fig. 1. Temporal classification of endogenous mRNA cytoplasmic polyadenylation during oocyte maturation.
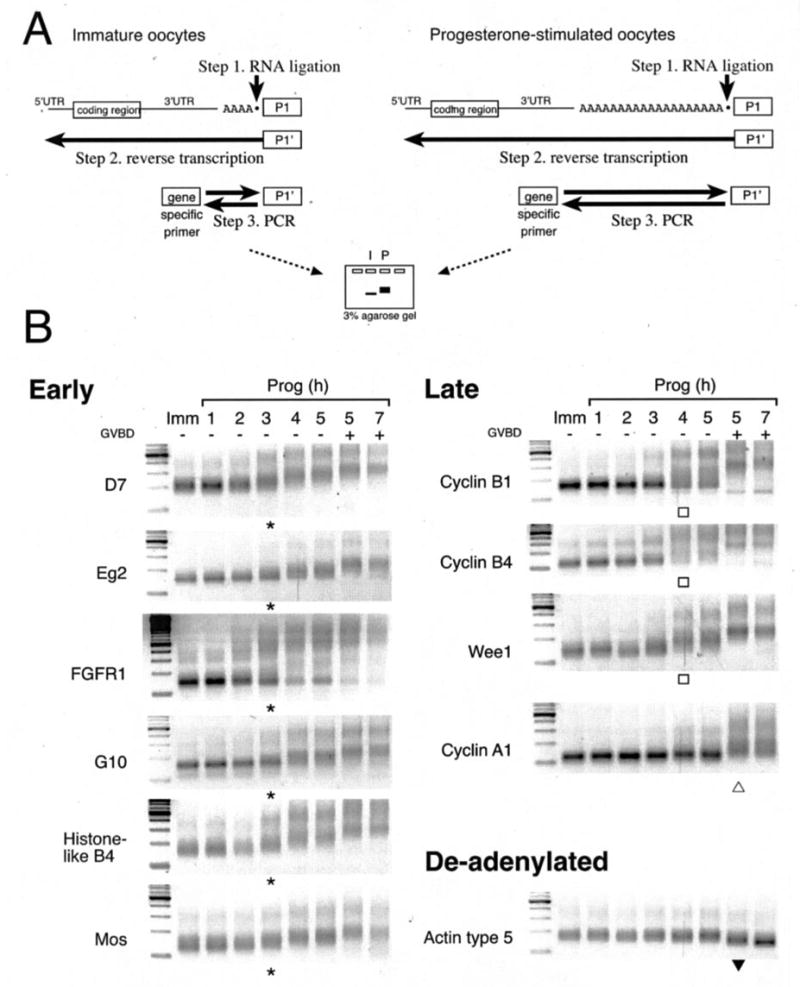
A, schematic of the RNA ligation-coupled RT-PCR technique (35) utilized in this study to assess poly(A) tail length. Retarded migration indicates polyadenylation. B, time course of polyadenylation of endogenous mRNAs using RT-PCR with different gene-specific primers from the same cDNA preparation. On the left of each gel is the 100-bp ladder (New England Biolabs). GVBD50 (see “Experimental Procedures”) occurred at 5 h in this experiment. Asterisks and open squares indicate initiation of early and late polyadenylation, respectively. Within the late class, initiation of cyclin A1 polyadenylation occurred predominantly after GVDB (open triangle). Deadenylation of the actin mRNA is indicated by a closed triangle. Similar results were obtained in eight separate experiments using oocytes derived from different animals. In all cases the polyadenylation of the early mRNAs was consistently initiated 2–3 h prior to GVBD, no matter how long it took the oocytes to reach GVBD. Polyadenylation of the late class was initiated up to 1 h prior GVBD, and typically 1 h after initiation of the early class mRNAs.
RNA Electrophoretic Mobility Shift Assays
The amount of GST and GST-XeCPEB protein expressed in rabbit reticulocyte lysates was normalized prior to use, and the RNA EMSAs were performed as described previously (9). The histone-like B4 and truncated D7 probes were run on 3.75% acrylamide gels, the G10 on a 3.5% gel, and the full-length D7 probe on a 3.25% gel.
GST Reporter mRNA Translation Assays
To measure the effect of wild-type and mutant 3′-UTRs on GST reporter mRNA translation, GST accumulation was visualized and quantitated by ECL Western blotting as described previously (9, 30) using ChemiGlow West, a ChemiImager 5500 and AlphaEaseFC software (AlphaInnotech Corp.).
RESULTS
To address the role of CPEs and CPEB in the control of maternal mRNA cytoplasmic polyadenylation and translational activation, we performed a comparative study of the polyadenylation of multiple endogenous mRNAs from the same sample sets utilizing an RNA ligation-coupled PCR technique (35). Briefly, a DNA oligonucleotide (P1) is ligated directly onto the 3′ end of all the RNAs in the sample preparation (Fig. 1A, step 1). An antisense DNA oligonucleotide (P1′), complementary to the RNA-ligated P1 oligonucleotide, is then used as a primer for reverse transcription (Fig. 1A, step 2). From this pool of cDNAs, the polyadenylation status of individual mRNAs was assessed by PCR using a gene-specific forward primer and the P1′ reverse primer (Fig. 1A, step 3) enabling a large number of endogenous mRNAs to be analyzed from the same sample preparation.
Because we have shown previously that the Mos mRNA undergoes temporally early CPE- and CPEB-independent translational activation (30), we wished to determine which Xenopus maternal mRNAs undergo similar temporally early cytoplasmic polyadenylation in progesterone-stimulated oocytes. 11 maternal mRNAs were analyzed utilizing the RNA ligation-coupled PCR technique from the same total RNA sample preparations (Fig. 1B). We identify two classes of maternal mRNAs based on the timing of their induced polyadenylation profiles, designated herein as early (where polyadenylation is initiated 2–3 h prior to oocyte germinal vesicle breakdown (GVBD), a marker of meiotic cell cycle progression) and late (where polyadenylation is initiated either 1 h before, or coincident with, GVBD). The early class of mRNAs include histone-like B4, D7, Eg2, FGFR1, G10, and Mos and consistently undergo coordinated initiation of polyadenylation 2–3 h prior to GVBD in oocytes from all animals tested. The polyadenylation of these mRNA populations occurs in a stepwise fashion up until GVBD, at which time the mRNA populations achieve maximal poly(A) tail length. The late class of mRNAs include cyclin B1, cyclin B4, Wee1, and cyclin A1. The mRNA populations of the late class mRNAs behave heterogeneously in terms of when polyadenylation is initiated for oocytes from any given animal. Generally, late class polyadenylation occurred coincident with or after GVBD. However, in some animals (as represented in Fig. 1B) some late class mRNA polyadenylation can be observed up to 1 h prior to GVBD, although this initiation occurred later than that seen with the early class mRNAs, and the entire population of each late class mRNA does not become fully polyadenylated until GVBD. Indeed, whereas cyclin A1 polyadenylation occurs predominantly after GVBD (Fig. 1B, open triangle), a proportion of the cyclin B1, B4, and Wee1 mRNA populations were polyadenylated an hour prior to GVBD (Fig. 1B, open boxes). In all animals examined so far, initiation of cyclin A1 polyadenylation was always the last to occur. The cytoskeletal actin mRNA, which lacks CPE sequences in the 3′-UTR, is utilized as a control (Fig. 1B, actin type 5) because it is not polyadenylated in response to progesterone stimulation and is deadenylated at GVBD (26). The timing of maternal mRNA polyadenylation characterized here generally agrees with previous studies that have reported that histone-like B4, G10, D7, FGF receptor 1, and Mos are early (11, 13, 24–26, 30, 37, 38), whereas cyclin A1, cyclin B1, and Wee1 are temporally late (9, 24–26). However, this is the first time it has been shown that the early endogenous mRNAs undergo coordinate initiation. The timing of cyclin B4 and Aurora A/Eg2 mRNA cytoplasmic polyadenylation has not been established previously.
Because the temporally early polyadenylation of the Mos mRNA occurs in a CPEB-independent manner (30), we next wished to determine whether Aurora A/Eg2-mediated CPEB phosphorylation was necessary for the progesterone-stimulated cytoplasmic polyadenylation of the other members of the temporally early class of maternal mRNAs. To this end we employed the expression of a dominant negative form of CPEB (CPEB-AA) (29) that has alanine residue substitutions in the Aurora A/Eg2 consensus phosphorylation sites. We demonstrate that in addition to the Mos mRNA, the mRNAs encoding histone-like B4, D7, Aurora A/Eg2, FGFR1, and G10 undergo progesterone-stimulated polyadenylation in the presence of CPEB-AA. It should be noted, however, that the expression of CPEB-AA reduced the overall length of progesterone-stimulated poly(A) tail extension (Fig. 2). Because the reduced poly(A) tail still allows for PRE-directed translational activation of the Mos mRNA in CPEB-AA expressing oocytes (30), it is likely that the reduced poly(A) tail added to the histone-like B4, D7, Aurora A/Eg2, FGF receptor 1, and G10 mRNAs is sufficient for translational activation. By contrast, polyadenylation of the cyclin B1 (30), cyclin B4, cyclin A1, and Wee1 mRNAs was completely abolished in CPEB-AA expressing oocytes (Fig. 2). These results demonstrate that there are distinct CPEB-independent and CPEB-dependent classes of Xenopus maternal mRNAs. The CPEB-independent class corresponds to the temporally early mRNAs, whereas the CPEB-dependent class correlates with the temporally late mRNAs shown in Fig. 1.
Fig. 2. Progesterone-stimulated cytoplasmic polyadenylation of multiple maternal mRNAs is CPEB-independent.
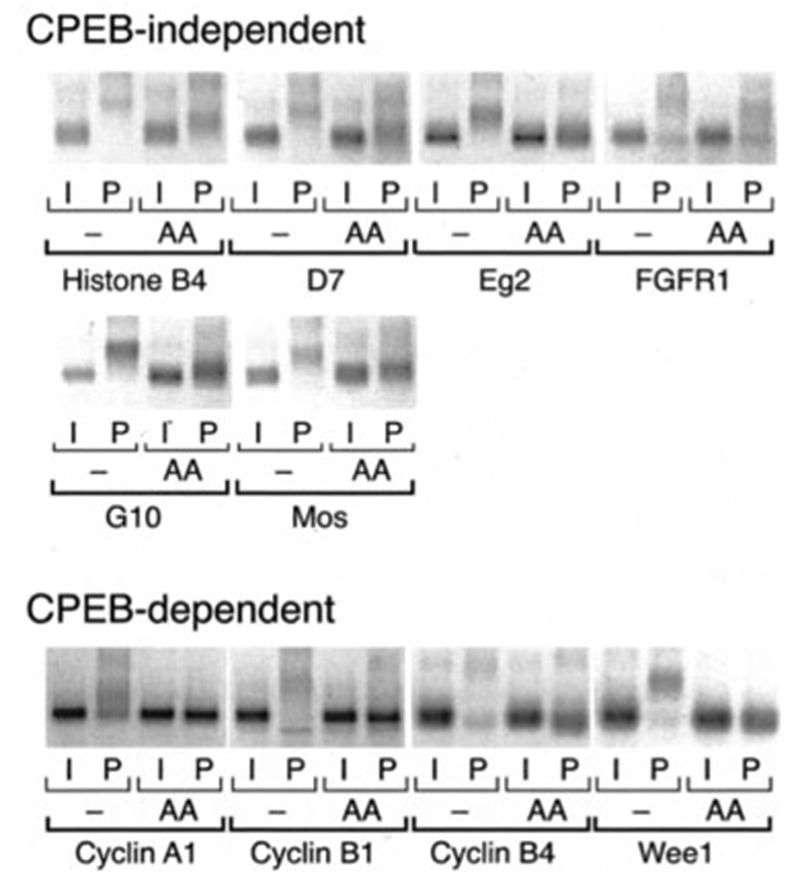
Where indicated, immature oocytes were injected with RNA encoding CPEB-AA (AA) and left 20 h at 21 °C to express the protein prior to stimulation with progesterone. CPEB-AA significantly delayed oocyte maturation as expected (29, 30). When all the control oocytes had matured (6 h in this experiment), samples were prepared from control and CPEB-AA expressing oocytes (with (P) or without (I) progesterone stimulation), and polyadenylation was assessed by RT-PCR. The mRNAs are categorized based on whether expression of CPEB-AA reduced but did not eliminate polyadenylation (CPEB-independent) or ablated polyadenyl-ation (CPEB-dependent). Of the latter class, CPEB-AA expression prevented cyclin A1 and cyclin B1 polyadenylation in progesterone-stimulated oocytes and led to a slight deadenylation of the cyclin B4 and Wee1 mRNAs relative to the poly(A) tail present in immature oocytes.
As noted in the Introduction, the sequence, number, and/or position of CPEs have been proposed previously to encode the temporal polyadenylation characteristics of maternal mRNAs. Although there is some variability in precise sequence, Xenopus CPE sequences generally conform to the consensus U4–5A1–3U and function up to 100 nucleotides 5′ of the polyadenylation hexanucleotide (10, 39). A terminal U is not always required for CPE function because UUUUAACA has been implicated in cyclin B1 repression (22). We find no apparent correlation between temporally early or late class mRNAs and a specific CPE sequence. Indeed, the different CPE sequence permutations (e.g. UUUUAU, UUUUUAAU, and UUUUAACA) can be found in both early and late class mRNAs analyzed in this study (Fig. 3). We also found no general correlation between temporally early or late class mRNAs and CPE number because both classes contain mRNAs with one or more CPEs (Fig. 3). However, CPE position did appear to correlate with the classification of temporally late mRNAs analyzed because they all contain a CPE in the 3′-UTR that overlaps the polyadenylation hexanucleotide.
Fig. 3. Temporal control of cytoplasmic polyadenylation does not correlate with CPE number or sequence.
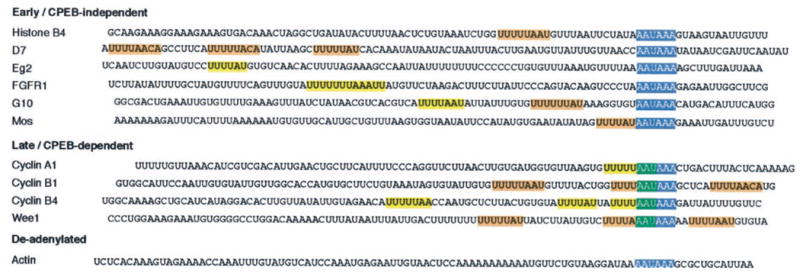
The last 100 nucleotides of the mRNAs used in this study are shown. Polyadenylation hexanucleotides are shown in blue boxes; putative CPEs are shown in yellow boxes, and CPEs that have been experimentally determined by mutational analysis are shown in orange boxes: histone-like B4 (14, 43); D7 (this study); G10 ((13) this study); Mos (30, 44); cyclin B1 (18, 20, 22); and Wee1 (9). The overlap of a CPE with the polyadenylation hexanucleotide is noted in green. It should be noted that the D7 RT-PCR product contained 21 bp of extra 3′ sequence not reported previously (45), including a canonical polyadenylation hexanucleotide.
Because the temporally early mRNAs examined herein do contain one or more CPE sequences but none of the CPEs overlap the polyadenylation hexanucleotide, we next wanted to test whether CPE sequences 5′ of the polyadenylation hex-anucleotide were responsible for the cytoplasmic polyadenylation of the early class of mRNAs. The full-length 3′-UTRs of histone-like B4, D7, and G10 mRNAs were isolated from Xeno-pus oocytes, and the consensus CPE sequences in each 3′-UTR were mutationally disrupted with dinucleotide substitutions (Fig. 4A). To verify that the mutations had indeed disrupted the CPEs, the binding to CPEB in vitro was assessed by RNA electrophoretic mobility shift assays (EMSAs). For these assays, GST and chimeric GST Xenopus CPEB (GST CPEB) fusion proteins were prepared by coupled in vitro transcription and translation in rabbit reticulocyte lysates as described previously (9). The levels of GST and GST-CPEB were normalized in the reticulocyte lysate prior to incubation in an EMSA (data not shown).
Fig. 4. Xenopus CPEB does not bind to CPE-disrupted histone-like B4, D7, or G10 3′-UTRs.
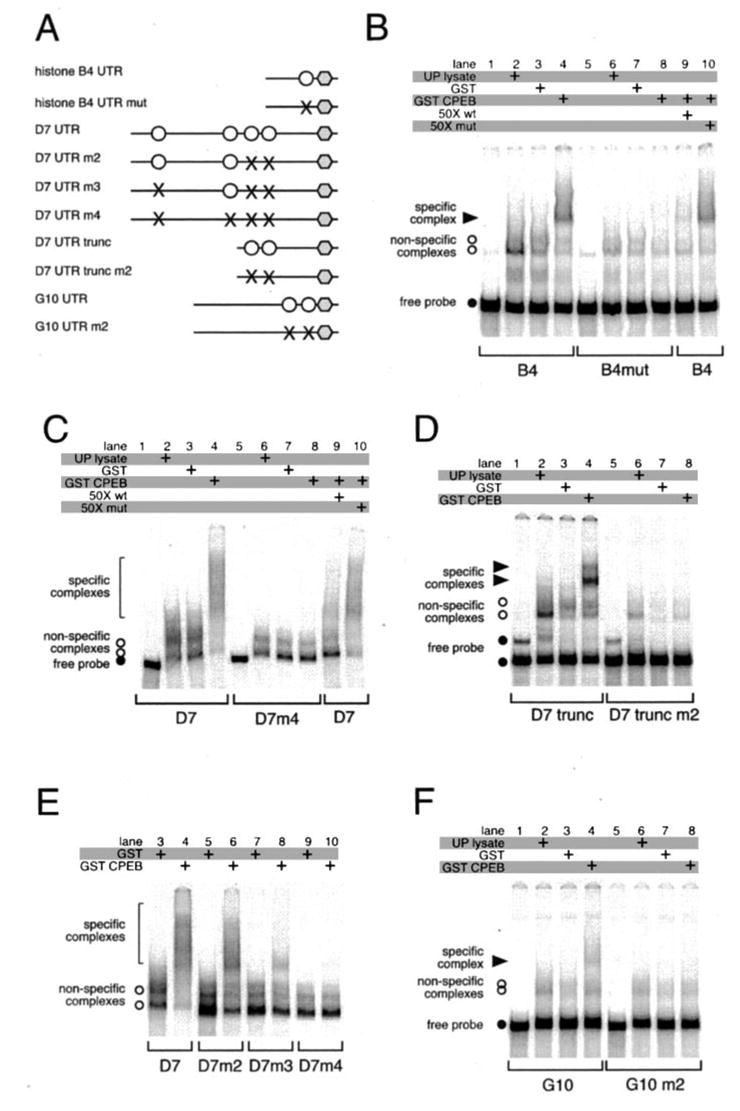
A, schematic showing constructs used in the study. Polyadenyl-ation hexanucleotides are represented by gray hexagons, CPEs by open circles, and disrupted CPEs by ×. The D7 3′-UTR has another CPE-like sequence 121 nt back from the polyadenylation hexanucleotide, and so was not noted in Fig. 3. B–F, RNA EMSAs. The indicated probes were incubated in the presence of unprogrammed lysate, or GST, or GST-XeCPEB expressing reticulocyte lysates, and specific complex formation was assessed. B, either wild-type (lanes 1–4, 9, and 10) or CPE-disrupted (lanes 5–8) B4 3′-UTR probes were used. Where indicated, 50-fold molar excess of unlabeled wild-type (wt) (specific) or CPE-disrupted (mut) (non-specific) competitor B4 probe was included in the binding reactions. C, wild-type D7 3′-UTR probe (lanes 1–4, 9, and 10) or a mutant D7 probe that had all four CPEs disrupted (lanes 5–8) were used. Where indicated, 50-fold molar excess of unlabeled wild-type or CPE-disrupted competitor D7 probe was included. D, two complexes are formed on the truncated D7 UTR demonstrating that there are at least two CPEs present. E, identification of all four CPEs in the D7 3′-UTR. F, either wild-type (lanes 1–4) or CPE-disrupted (lanes 5–8) G10 3′-UTR probes were used.
Incubation of the wild-type histone-like B4 probe with reticulocyte lysate expressing the GST CPEB fusion protein resulted in formation of a specific complex (solid arrowhead) (Fig. 4B, lane 4). No specific complex was formed when the CPE-disrupted UTR probe was used (Fig. 4B, lane 8). To confirm the specificity of CPEB complex formation with the wild-type UTR probe, the complex formation was challenged with 50-fold molar excess of unlabeled RNA probe competitor. Upon addition of unlabeled, wild-type histone-like B4 UTR probe (Fig. 4B, lane 9), the formation of the specific complex was abolished. Specific complex formation was not abolished when challenged with a 50-fold molar excess of CPE-disrupted UTR competitor probe (Fig. 4B, lane 10). Nonspecific complex formation was observed with the histone-like B4 probe either upon addition of un-programmed reticulocyte lysate or lysate expressing the GST moiety alone (Fig. 4B, lanes 2, 3, 6, and 7). The formation of nonspecific complexes has been observed previously with rabbit reticulocyte lysates in EMSAs when wild-type or CPE-disrupted Xenopus Mos and Wee1 3′-UTRs were employed (9, 40).
We found that the D7 3′-UTR contained four CPE sequences that interacted with CPEB (Fig. 4, C and E). Incubation of the wild-type D7 probe with reticulocyte lysate expressing the GST CPEB fusion protein resulted in formation of specific complexes (bracket) (Fig. 4C, lane 4). No specific complex was formed when the mutant probe was used that had disruptions in all 4 CPEs (Fig. 4C, lane 8). To confirm the specificity of CPEB complex formation with the wild-type UTR probe, the complex formation was challenged with 50-fold molar excess of unlabeled RNA probe competitor (Fig. 4C, lane 9). Specific complex formation was not abolished when challenged with a 50-fold molar excess of CPE-disrupted UTR competitor probe (Fig. 4C, lane 10). Fig. 4D shows that a truncated D7 UTR containing only the two 3′ CPEs forms two specific complexes with CPEB, whereas the CPE-disrupted truncated D7 probe does not bind CPEB. Fig. 4E illustrates the process of identification of all 4 CPEs in the D7 UTR using a series of CPE-disrupted mutants that have progressively fewer intact CPEs.
Incubation of the wild-type G10 probe with reticulocyte ly-sate expressing the GST CPEB fusion protein resulted in formation of a specific complex (solid arrowhead) (Fig. 4F, lane 4). No specific complex was formed when the CPE disrupted UTR probe was used (Fig. 4F, lane 8). Taken together, these results show that the introduced mutations disrupt the CPEs and ablate CPEB binding to the histone-like B4, D7, and G10 3′-UTRs.
To determine whether the CPEs were dispensable for CPEB-independent cytoplasmic polyadenylation, the wild-type and CPE-disrupted histone-like B4, D7, and G10 3′-UTRs were fused to a GST reporter RNA (Fig. 5A). Progesterone-stimulated polyadenylation of the injected RNAs was assessed by RNA ligation-coupled PCR using a GST forward primer common to all constructs. Polyadenylation of the wild-type B4 UTR was initiated 3 h after progesterone stimulation (Fig. 5B). In the absence of the CPE, polyadenylation of the mutant B4 UTR was initiated at the same time (or perhaps slightly earlier than) as the wild-type B4 UTR. This finding indicates that similar to the Mos mRNA (30), temporally early cytoplasmic polyadenylation of the B4 mRNA occurs in a CPE-independent manner. These findings are consistent with the observed CPEB independence of the B4 and Mos mRNAs (Fig. 2). The overall poly(A) tail length was shorter in the absence of the CPE, suggesting that the CPE may contribute to B4 mRNA poly(A) tail length in maturing oocytes. Similarly, although a consistent reduction of poly(A) tail extension was observed, disruption of the CPEs in the D7 or G10 3′-UTRs did not prevent early cytoplasmic polyadenylation of the GST reporter RNAs (Fig. 5B). To assess the effects of disrupting the CPEs on translational activation, the accumulation of GST protein from each of the reporter RNAs was assessed by Western blot. The temporally early polyadenylation directed by wild-type and CPE-disrupted B4, D7, and G10 UTRs correlated with reporter RNA translational activation and GST protein accumulation prior to GVBD (−2-fold, n = 2; Fig. 5C), indicating that temporally early mRNA cytoplasmic polyadenylation and translational induction occur via a CPE-independent mechanism. At later times the overall levels of GST accumulation were similar in the wild-type and the CPE-disrupted reporter RNAs. These findings suggest that whereas the poly(A) tail added to the CPE-disrupted B4, D7, and G10, 3′-UTRs was reduced compared with the wild-type 3′-UTRs, the reduced poly(A) tail was nonetheless sufficient to mediate translational activation of the GST reporter RNAs. The data further suggest that the shorter poly(A) tails of the temporally early class mRNAs observed in CPEB-AA-expressing oocytes (where CPE-dependent poly-adenylation is also ablated) would likely be sufficient to mediate translational activation (see Fig. 2).
Fig. 5. The initiation of CPEB-independent class mRNA polyadenylation and translational activation occurs in a CPE-independent manner.
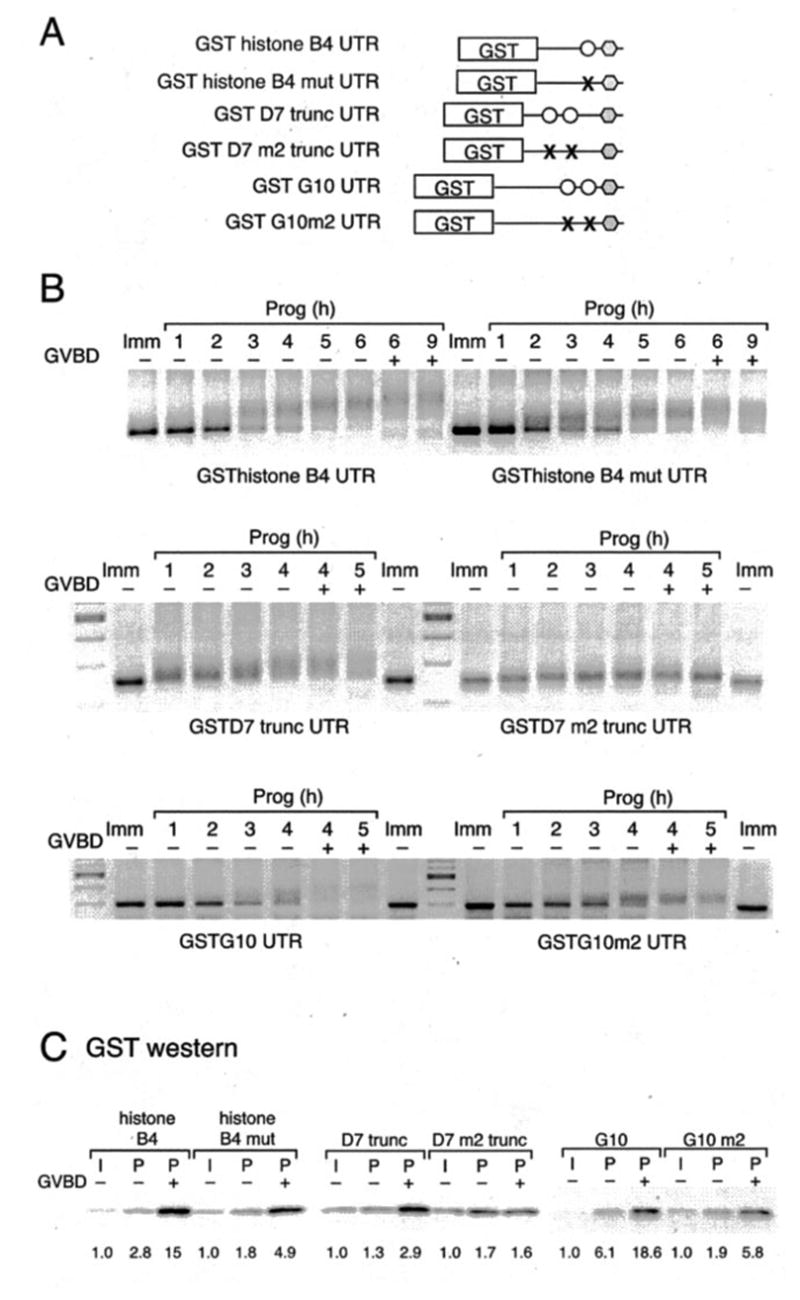
A, schematic of the reporters used in this study. The histone-like B4, D7, and G10 3′-UTRs from Fig. 4 were fused to the GST coding region. B and C, wild-type and mutant histone-like B4, D7, or G10 reporter constructs were injected into immature oocytes. B, polyadenylation of reporter constructs after progesterone stimulation was assessed by RT-PCR using a GST forward primer. In this experiment, the truncated D7 UTR was used because the PCR product from the GST forward primer is considerably smaller than that obtained with the full-length D7 UTR, and hence the change in poly(A) tail length is more readily observed. C, accumulation of GST protein after progester-one stimulation was assessed by Western blot with anti-GST antibodies. Samples were prepared at GVBD50 (see “Experimental Procedures”), segregated based on GVBD status (− or +) and shown relative to a time-matched sample prepared from immature oocytes. The numbers below the panels indicate the fold increase in GST accumulation over that seen in the immature oocyte control sample for each RNA reporter construct.
If a CPE- and CPEB-dependent mechanism does not control early class mRNA translational activation, what regulatory process could be involved? We have reported previously (30) that a novel PRE is both necessary and sufficient to regulate the early, CPE-, and CPEB-independent, translational activation of the Mos mRNA prior to GVBD. An analysis of the 3′-UTRs of the temporally early, CPEB-independent class of mRNAs examined in this study revealed that they all contain PRE-like sequences (Fig. 6). No matches to the PRE consensus sequence (Fig. 6) were identified in the temporally late, CPEB-dependent class of mRNAs. Interestingly, with the exception of the FGFR1 3′-UTR, the PRE-like sequences partially overlap a 3′ CPE sequence, similar to the arrangement reported for the Xenopus Mos PRE (30).
Fig. 6. Sequence alignment comparison of PRE-like sequences in the 3′-UTRs of early class mRNAs.

Below the alignment, the derivative consensus PRE sequence is shown using the single letter code: V, A or C or G; N, A or G or C or T; Y, C or T; H, A or C or T; W, A or T; D, A or G or T; R, A or G; B, C or G or T; K, G or T.
To determine directly whether these were bona fide regulatory elements, the histone-like B4 and D7 PRE-like sequences (Fig. 7A, uppercase) were inserted into the heterologous β-globin 3′-UTR at the same positions relative to the polyadenylation hexanucleotide, which they are found in their native 3′-UTRs (Fig. 7B). The β-globin 3′-UTR is not subject to regulated cytoplasmic polyadenylation during oocyte maturation (9, 30, 41). When the histone-like B4 and D7 PREs were inserted into the β-globin 3′-UTR, they directed cytoplasmic polyadenylation in response to progesterone (Fig. 7B). To establish whether the PRE was necessary for temporally early poly-adenylation, the PRE in the D7 UTR was deleted, while leaving the 5′ and 3′ CPEs intact (Fig. 7C). In contrast to disruption of the D7 CPE sequences that did not significantly alter the kinetics of polyadenylation (Fig. 5), deletion of the D7 PRE resulted in at least a 1–2-h delay to reporter RNA polyadenylation in progesterone-stimulated oocytes. A similar delay to Mos 3′-UTR-directed polyadenylation was observed when the Mos PRE was deleted (30). These findings suggest that PRE sequences present in the temporally early class of mRNAs direct CPEB-independent, temporally early cytoplasmic poly-adenylation in response to progesterone stimulation.
DISCUSSION
Our primary finding is that temporally early translational activation of multiple maternal mRNAs is regulated by a CPE-and CPEB-independent mechanism during progesterone-stimulated Xenopus oocyte maturation. By contrast, temporally late mRNA translational activation is regulated by a CPE- and CPEB-dependent mechanism. Temporally early mRNA translational activation is defined as initiating 2 or more hours prior to GVBD, while temporally late is defined as initiating up to an hour prior to, or coincident with, GVBD. Based on our data, we propose that the default timing for translational activation of CPE containing mRNAs is temporally late unless the mRNA 3′-UTR contains an early acting element that can direct CPE-independent control. Our studies suggest that earlier classifications of maternal mRNA translational activation need to be revised to include CPE-independent regulatory components. In particular, we define a class of PRE-containing mRNAs that are polyadenylated and translationally activated temporally early during maturation in a CPE- and CPEB-independent manner. In addition to the Mos mRNA characterized previously (30), we now report the presence of PREs in the early class mRNAs encoding D7, Aurora A/Eg2, FGF receptor 1, G10, and histone-like B4. Our data strongly suggest that these Xenopus PRE sequences direct temporally early cytoplasmic polyadenylation and translational activation during oocyte maturation. An analysis of the 3′-UTR data base (42) indicates that the PRE consensus sequence (Fig. 6) is found in 2–3% of 3′-UTR sequences of mRNAs from Xenopus, Caenorhabditis elegans, Drosophila, rodents, and humans. It is thus likely that PRE-directed mRNA translational regulation may be evolutionarily conserved. For comparison, the UUUUU(U/A)AU CPE sequence is more common and found in ~7–11% of Xenopus, C. elegans, Drosophila, rodent, and human 3′-UTR data base entries.
We present multiple lines of evidence to support the contention that the translational regulation of the temporally early, PRE-containing mRNAs occurs in a CPE- and CPEB-independent manner. First, we show that insertion of the histone-like B4 or D7 PREs into the heterologous β-globin 3′-UTR is sufficient to confer temporally early polyadenylation (Fig. 7). Second, deletion of the PRE sequence in the D7 3′-UTR shifts the timing of polyadenylation from temporally early to temporally late (Fig. 7). Third, mutational disruption of CPE sequences within the D7, G10, or histone-like B4 3′-UTRs does not affect the kinetics of progesterone-stimulated cytoplasmic poly-adenylation and translational activation (Fig. 5). Mutational disruption of these CPE sequences in the 3′-UTR ablates interaction with the CPEB protein, indicating that the observed early translational activation is both CPE- and CPEB-independent (Fig. 4). Fourth, overexpression of a dominant inhibitory form of CPEB (CPEB-AA), which prevents CPE-dependent cytoplasmic polyadenylation of the late class of mRNAs, does not prevent early polyadenylation of the PRE-containing class of mRNAs (Fig. 2). Taken together, our findings indicate that temporally early mRNA translational activation can be mediated by PRE sequences, independently of CPE- and CPEB-mediated control.
All temporally early, PRE-containing mRNAs appear to initiate polyadenylation at about the same time in response to progesterone stimulation (Fig. 1, asterisks), suggesting that a common signaling pathway may target the PRE-containing class of early mRNAs. The PRE has been shown previously (30) to be MAP kinase-responsive, whereas CPEs are MPF-responsive. An important issue that now needs to be addressed relates to whether MAP kinase coordinates the temporally early progesterone-dependent “trigger” activation of the PRE-containing mRNAs or if MAP kinase only mediates Mos-dependent feedback amplification (3) once early PRE-dependent mRNA translational activation has been independently initiated. Within the PRE-containing mRNA class, there are differences in the length of poly(A) tail added to individual mRNAs. It is possible that sequence differences between the PREs of the various mRNA 3′-UTRs may account for different efficiency of polyadenylation. In contrast to the PRE-mediated translational activation of histone-like B4 described in this study, an apparently contradictory CPE-dependent regulation of histone-like B4 translation has been reported previously (14, 43). These former studies, however, utilized a very truncated form of the 3′-UTR that lacked the 5′ PRE identified in our study. Whereas G10 polyadenylation has also been reported to be CPE-dependent (13), a truncated 3′-UTR reporter was utilized that lacked the 5′ PRE identified in this study. Similarly, although the Mos 3′-UTR was shown recently to contain a PRE (30), the use of truncated 3′-UTR reporter constructs that lacked the PRE sequence may have contributed to prior misconceptions that the initial polyadenylation of the Mos mRNA was CPE-dependent (29). Collectively, our findings caution against the use of truncated 3′-UTR reporter constructs, unless complementary analyses of the full-length 3′-UTR or corresponding endogenous mRNA are also performed in the same study.
This study challenges the premise that CPE number alone can control the timing of mRNA translational activation. For example, the Mos 3′-UTR contains a single CPE and is polya-denylated early, whereas the 3′-UTRs of cyclin B1 and Wee1 contain multiple CPEs and are polyadenylated later in maturation (9, 22, 24–26). However, the observation that cyclin A1 has a single CPE and is polyadenylated late in maturation (Fig. 1) (26) and the D7 and G10 3′-UTRs have multiple CPEs and are polyadenylated early (Fig. 1) (11, 13) is not consistent with the idea that a single CPE directs early polyadenylation and multiple CPEs direct late polyadenylation. Rather, our findings indicate that distinct regulatory elements, such as the PRE, direct temporally early mRNA polyadenylation and translational activation in response to progesterone stimulation. Indeed, we show here that temporally early translational activation occurs in a CPE- and CPEB-independent manner. Temporally late polyadenylation correlates with CPE and CPEB dependence. There does appear to be variation in the timing of initiation of late class mRNA polyadenylation. If a 3′-UTR contains a single CPE that overlaps the polyadenylation hexanucleotide, then the majority of the mRNA population is polyadenylated at or after GVBD (e.g. cyclin A1, Fig. 1). If the 3′-UTR contains additional CPE(s) then the timing of initiation may be slightly advanced (e.g. cyclin B1, Fig. 1) but never precedes initiation early class mRNA polyadenylation. Consistent with this observation, disruption of the 5′ CPE in the cyclin B1 3′-UTR (while retaining the polyadenylation hex-anucleotide overlapping CPE) delays initiation of polyadenylation until after GVBD, as observed with the endogenous cyclin A1 mRNA (data not shown). Although not directly addressing temporal control, a positional affect of CPE sequences within the 3′-UTR has been proposed previously (25) to determine Mos dependence.
Previous studies (24, 25) have classified Xenopus maternal mRNA cytoplasmic polyadenylation and translational activation based upon distinction between a requirement for prior Mos translation (class II) or no dependence for prior Mos translation (class I). However, several problems exist with this classification system. First, there are various inconsistencies in timing and signaling dependences within the same class groups. For example, the originally defined class II mRNAs (cyclin B1, D7, and histone-like B4 (24)) contain temporally late (e.g. cyclin B1) and temporally early mRNAs (e.g. D7 and his-tone-like B4) as shown in this study. Similarly, controversy existed as to whether cyclin A1 was defined as a class I or class II mRNA (24, 25). Second, the Mos-independent and Mos-dependent classifications are too ambiguous to serve as operational definitions. Because Mos protein can lead to MAP kinase and subsequent MPF activation, Mos dependence does not adequately distinguish between a MAP kinase dependence, an MPF dependence, or a dual requirement for both MAP kinase and MPF activation. Finally, microinjection of morpholino antisense Mos oligonucleotides has recently challenged the dogma that Mos mRNA translation was a prerequisite for activation of MPF and progression to GVBD. Unlike standard antisense oligonucleotide ablation of Mos mRNA translation, which had significant nonspecific inhibitory effects on MPF activation and oocyte maturation, morpholino antisense Mos oligonucleotides ablated Mos mRNA translation but only delayed MPF activation and progression to GVBD (8). It is thus unclear if the reported Mos dependence for class II polyadenylation really reflects an a priori requirement for Mos mRNA translation at all, or if it is attributable to nonspecific inhibition of MPF.
For these reasons we favor a revision of the prior classification of Xenopus maternal mRNA translational activation (24). In such a revised classification, class I mRNAs would be defined as being polyadenylated and translationally activated in a CPE- and CPEB-independent manner as a temporally early response to progesterone stimulation. In this study we identified PRE elements in the early mRNAs that direct CPE- and CPEB-independent polyadenylation. We do not, however, exclude the possibility that other maternal mRNAs may contain novel CPE-independent elements that can direct temporally early cytoplasmic polyadenylation and mRNA translational activation. In the revised classification, polyadenylation of the class II mRNAs would be defined as being CPE- and CPEB-dependent with initiation occurring after early class I polyadenylation. The new assignment of maternal mRNAs based on CPE and CPEB dependence proposed here agrees to a certain extent with the earlier signaling-dependent categorizations but also serves to resolve many of the prior inconsistencies in temporal assignment.
If CPE-independent processes control activation of the early class of mRNAs, then why do these mRNAs also contain CPE sequences in their 3′-UTR? Based on the findings presented in this study, we speculate that the early mRNAs may be subject to sequential translational regulation. In this model, early PRE-directed polyadenylation results in a short poly(A) tail and initiation of protein translation. Subsequent CPE-directed polyadenylation leads to an increased length of poly(A) tail and/or maintenance of the extended poly(A) tail and continued protein synthesis. Consistent with this model, whereas CPEB-AA did not block progesterone-stimulated polyadenylation of the early class mRNAs, CPEB-AA expression did attenuate the length of tail that was added (Fig. 2). We propose that the sequential action of CPE-independent and CPE-dependent regulatory processes allow for coordination of the complex temporal patterns and extent of protein synthesis during vertebrate meiotic cell cycle progression.
Acknowledgments
We thank Dr. Melanie C. MacNicol for insightful discussions and critical reading of the manuscript.
The abbreviations used are
- 3′-UTR
3′-untranslated region
- CPE
cytoplasmic polyadenylation element
- CPEB
cytoplasmic polyadenylation element-binding protein
- PRE
polyadenylation response element
- GST
glutathione S-transferase
- EMSA
electrophoretic mobility shift assay
- MAP kinase
mitogen-activated protein kinase
- MPF
maturation/M-phase- promoting factor
- GVBD
germinal vesicle breakdown
- nt
nucleotide
- RT
reverse transcriptase
- FGF
fibroblast growth factor
- FGFR
FGF receptor
Footnotes
This work was supported by grants from the National Institutes of Health (to A. M. M.), American Cancer Society (to A. C.), and the Arkansas Biosciences Institute (to A. M. M.). The costs of publication of this article were defrayed in part by the payment of page charges.
References
- 1.Hochegger H, Klotzbucher A, Kirk J, Howell M, le Guellec K, Fletcher K, Duncan T, Sohail M, Hunt T. Development. 2001;128:3795–3807. doi: 10.1242/dev.128.19.3795. [DOI] [PubMed] [Google Scholar]
- 2.Nakajo N, Yoshitome S, Iwashita J, Iida M, Uto K, Ueno S, Okamoto K, Sagata N. Genes Dev. 2000;14:328–338. [PMC free article] [PubMed] [Google Scholar]
- 3.Howard EL, Charlesworth A, Welk J, MacNicol AM. Mol Cell Biol. 1999;19:1990–1999. doi: 10.1128/mcb.19.3.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheets MD, Wu M, Wickens M. Nature. 1995;374:511–516. doi: 10.1038/374511a0. [DOI] [PubMed] [Google Scholar]
- 5.Gross SD, Schwab MS, Taieb FE, Lewellyn AL, Qian YW, Maller JL. Curr Biol. 2000;10:430–438. doi: 10.1016/s0960-9822(00)00425-5. [DOI] [PubMed] [Google Scholar]
- 6.Ferby I, Blazquez M, Palmer A, Eritja R, Nebreda AR. Genes Dev. 1999;13:2177–2189. doi: 10.1101/gad.13.16.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sagata N, Oskarsson M, Copeland T, Brumbaugh J, Vande Woude GF. Nature. 1988;335:519–525. doi: 10.1038/335519a0. [DOI] [PubMed] [Google Scholar]
- 8.Dupre A, Jessus C, Ozon R, Haccard O. EMBO J. 2002;21:4026–4036. doi: 10.1093/emboj/cdf400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charlesworth A, Welk J, MacNicol A. Dev Biol. 2000;227:706–719. doi: 10.1006/dbio.2000.9922. [DOI] [PubMed] [Google Scholar]
- 10.Richter JD. In: Translational Control of Gene Expression. Sonenberg N, Hershey J, Mathews MB, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 785–805. [Google Scholar]
- 11.Fox CA, Sheets MD, Wickens MP. Genes Dev. 1989;3:2151–2162. doi: 10.1101/gad.3.12b.2151. [DOI] [PubMed] [Google Scholar]
- 12.McGrew LL, Dworkin-Rastl E, Dworkin MB, Richter JD. Genes Dev. 1989;3:803–815. doi: 10.1101/gad.3.6.803. [DOI] [PubMed] [Google Scholar]
- 13.McGrew LL, Richter JD. EMBO J. 1990;9:3743–3751. doi: 10.1002/j.1460-2075.1990.tb07587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paris J, Richter JD. Mol Cell Biol. 1990;10:5634–5645. doi: 10.1128/mcb.10.11.5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sallès FJ, Darrow AL, O’Connell ML, Strickland S. Genes Dev. 1992;6:1202–1212. doi: 10.1101/gad.6.7.1202. [DOI] [PubMed] [Google Scholar]
- 16.Standart N, Dale M. Dev Genet. 1993;14:492–499. doi: 10.1002/dvg.1020140610. [DOI] [PubMed] [Google Scholar]
- 17.Gebauer F, Xu W, Cooper GM, Richter JD. EMBO J. 1994;13:5712. doi: 10.1002/j.1460-2075.1994.tb06909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stebbins-Boaz B, Hake LE, Richter JD. EMBO J. 1996;15:2582–2592. [PMC free article] [PubMed] [Google Scholar]
- 19.Stutz A, Conne B, Huarte J, Gubler P, Volkel V, Flandin P, Vassalli JD. Genes Dev. 1998;12:2535–2548. doi: 10.1101/gad.12.16.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Moor CH, Richter JD. EMBO J. 1999;18:2294–2303. doi: 10.1093/emboj/18.8.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minshall N, Walker J, Dale M, Standart N. RNA (N Y) 1999;5:27–38. doi: 10.1017/s1355838299981220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barkoff AF, Dickson KS, Gray NK, Wickens M. Dev Biol. 2000;220:97–109. doi: 10.1006/dbio.2000.9613. [DOI] [PubMed] [Google Scholar]
- 23.Tay J, Hodgman R, Richter JD. Dev Biol. 2000;221:1–9. doi: 10.1006/dbio.2000.9669. [DOI] [PubMed] [Google Scholar]
- 24.Ballantyne S, Daniel DLJ, Wickens M. Mol Biol Cell. 1997;8:1633–1648. doi: 10.1091/mbc.8.8.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Moor CH, Richter JD. Mol Cell Biol. 1997;17:6419–6426. doi: 10.1128/mcb.17.11.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheets MD, Fox CA, Hunt T, Vande Woude G, Wickens M. Genes Dev. 1994;8:926–938. doi: 10.1101/gad.8.8.926. [DOI] [PubMed] [Google Scholar]
- 27.Mendez R, Richter JD. Nat Rev Mol Cell Biol. 2001;2:521–529. doi: 10.1038/35080081. [DOI] [PubMed] [Google Scholar]
- 28.Mendez R, Barnard D, Richter JD. EMBO J. 2002;21:1833–1844. doi: 10.1093/emboj/21.7.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mendez R, Hake LE, Andresson T, Littlepage LE, Ruderman JV, Richter JD. Nature. 2000;404:302–307. doi: 10.1038/35005126. [DOI] [PubMed] [Google Scholar]
- 30.Charlesworth A, Ridge JA, King LA, MacNicol MC, MacNicol AM. EMBO J. 2002;21:2798–2806. doi: 10.1093/emboj/21.11.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrèsson T, Ruderman JV. EMBO J. 1998;17:5627–5637. doi: 10.1093/emboj/17.19.5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maton G, Thibier C, Castro A, Lorca T, Prigent C, Jessus C. J Biol Chem. 2003;278:21439–21449. doi: 10.1074/jbc.M300811200. [DOI] [PubMed] [Google Scholar]
- 33.Frank-Vaillant M, Haccard O, Thibier C, Ozon R, Arlot-Bonnemains Y, Prigent C, Jessus C. J Cell Sci. 2000;113:1127–1138. doi: 10.1242/jcs.113.7.1127. [DOI] [PubMed] [Google Scholar]
- 34.Castro A, Mandart E, Lorca T, Galas S. J Biol Chem. 2003;278:2236–2241. doi: 10.1074/jbc.M207894200. [DOI] [PubMed] [Google Scholar]
- 35.Rassa JC, Wilson GM, Brewer GA, Parks GD. Virology. 2000;274:438–449. doi: 10.1006/viro.2000.0494. [DOI] [PubMed] [Google Scholar]
- 36.Machaca K, Haun S. J Cell Biol. 2002;156:75–85. doi: 10.1083/jcb.200110059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paris J, Philippe M. Dev Biol. 1990;140:221–224. doi: 10.1016/0012-1606(90)90070-y. [DOI] [PubMed] [Google Scholar]
- 38.Culp PA, Musci TJ. Dev Biol. 1998;193:63–76. doi: 10.1006/dbio.1997.8785. [DOI] [PubMed] [Google Scholar]
- 39.Richter JD. Microbiol Mol Biol Rev. 1999;63:446–456. doi: 10.1128/mmbr.63.2.446-456.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welk J, Charlesworth A, Smith GD, MacNicol AM. Gene (Amst) 2001;263:113–121. doi: 10.1016/s0378-1119(00)00588-6. [DOI] [PubMed] [Google Scholar]
- 41.Hyman LE, Wormington WM. Genes Dev. 1988;2:598–605. doi: 10.1101/gad.2.5.598. [DOI] [PubMed] [Google Scholar]
- 42.Pesole G, Liuni S, Grillo G, Licciulli F, Mignone F, Gissi C, Saccone C. Nucleic Acids Res. 2002;30:335–340. doi: 10.1093/nar/30.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paris J, Swenson K, Piwnica-Worms H, Richter JD. Genes Dev. 1991;5:1697–1708. doi: 10.1101/gad.5.9.1697. [DOI] [PubMed] [Google Scholar]
- 44.Fox CA, Sheets MD, Wahle E, Wickens M. EMBO J. 1992;11:5021–5032. doi: 10.1002/j.1460-2075.1992.tb05609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith RC, Dworkin MB, Dworkin-Rastl E. Genes Dev. 1988;2:1296–1306. doi: 10.1101/gad.2.10.1296. [DOI] [PubMed] [Google Scholar]