

Abstract
The human intestinal proton-coupled oligopeptide transporter hPEPT1 has been implicated in the absorption of pharmacologically active compounds. We have investigated the interactions between a comprehensive selection of drugs, and wild-type and variant hPEPT1s expressed in Xenopus oocytes, using radiotracer uptake and electrophysiological methods. The β-lactam antibiotics ampicillin, amoxicillin, cephalexin and cefadroxil, the antineoplastics δ-aminolevulinic acid (δ-ALA) and bestatin, and the neuropeptide N-acetyl-Asp-Glu (NAAG), were transported, as judged by their ability to evoke inward currents. When the drugs were added in the presence of the typical substrate glycylsarcosine (Gly-Sar), the inward currents were equal or less than that induced by Gly-Sar alone. This suggests that the drugs are transported at a lower turnover rate than Gly-Sar, but may also point towards complex interactions between dipeptides, drugs and the transporter. Gly-Sar and the drugs also modified the kinetics of hPEPT1 presteady-state charge movement, by causing a reduction in maximum charge (Qmax) and a shift of the midpoint voltage (V0.5) to more negative potentials. Our results indicate that the substrate selectivity of hPEPT1 is: Gly-Sar > NAAG, δ-ALA, bestatin > cefadroxil, cephalexin > ampicillin, amoxicillin. Based on steady-state and presteady-state analysis of Gly-Sar and cefadroxil transport, we proposed an extension of the 6-state kinetic model for hPEPT1 function that globally accounts for the observed presteady-state and steady-state kinetics of neutral dipeptide and drug transport. Our model suggests that, under saturating conditions, the rate-limiting step of the hPEPT1 transport cycle is the reorientation of the empty carrier within the membrane. Variations in rates of drug cotransport are predicted to be due to differences in affinity and turnover rate. Oral availability of drugs may be reduced in the presence of physiological concentrations of dietary dipeptides in the gut, suggesting that oral delivery drugs should be taken on an empty stomach. The common hPEPT1 single-nucleotide polymorphisms Ser117Asn and Gly419Ala retained the essential kinetic and drug recognition characteristics of the wild type, suggesting that neither variant is likely to have a major impact on oral absorption of drugs.
PEPT1 (gene SLC15A1) is the prototype member of the proton-coupled oligopeptide transporters superfamily (Daniel, 2004), and is expressed mainly in the brush-border membrane of enterocytes, renal proximal tubular cells and bile duct epithelial cells (Daniel & Kottra, 2004). The human isoform hPEPT1 (Fig. 1) is composed of 708 amino acid residues, and predicted to contain 12 membrane-spanning domains, with a large extracellular loop between the transmembrane regions 9 and 10, and with amino and carboxy termini facing the cytosol (Liang et al. 1995).
Figure 1. Membrane topology model of hPEPT1.

Most common non-synonymous single nucleotide polymorphisms Ser117Asn (S117N) and Gly419Ala (G419A), occurring with respective frequencies of 25% and 8% (http://pharmacogenetics.ucsf.edu), are highlighted.
Described as a low-affinity high-capacity system, PEPT1 mediates the transport of all possible di- and tripeptides, but not of free amino acids (Boll et al. 1994; Fei et al. 1994; Liang et al. 1995; Mackenzie et al. 1996a; Daniel, 2004). In addition, mammalian PEPT1 has been shown to recognize a wide array of chemically diverse compounds, including β-lactam antibiotics (Ganapathy et al. 1995, 1997; Wenzel et al. 1996; Terada et al. 1997; Bretschneider et al. 1999), inhibitors of the angiotensin converting enzyme (captopril, enalapril) (Boll et al. 1994; Temple & Boyd, 1998; Zhu et al. 2000), peptidomimetic compounds (bestatin, an antitumour agent) (Inui et al. 1992) and non-peptidic drugs (δ-aminolevulinic acid, or δ-ALA) (Doring et al. 1998) and prodrugs (valacyclovir, l-α-methyldopa) (Hu et al. 1989; Ganapathy et al. 1998). However, there is still a gap in our knowledge about drug absorption via human PEPT1, and most of the limited information available has been obtained by competition studies (Daniel, 2004).
Transport by hPEPT1 is electrogenic, proton coupled, and voltage dependent (Mackenzie et al. 1996a). In addition to the steady-state inward current induced by substrates, there is an hPEPT1-mediated transient presteady-state current following step jumps in membrane potential in the absence of substrates (Mackenzie et al. 1996a). This transient current has been postulated to be due to the movement of charged and polar residues in the membrane electric field, and is associated with the two voltage-dependent partial reactions of the transport cycle: the conformational change of the empty transporter between the external and internal membrane surfaces, and the H+ binding/dissociation (Mackenzie et al. 1996a).
In the present work, we used radiolabelled tracer uptake and electrophysiological measurements to investigate the molecular interactions between human PEPT1, the dipeptide glycylsarcosine and a comprehensive selection of drugs, including β-lactam antibiotics and antineoplastic agents. To gain insights into the mechanism of hPEPT1, we examined the kinetics of the steady-state and presteady-state currents in the presence of substrates. We revised and extended our 6-state kinetic model (Mackenzie et al. 1996a) to describe the global behaviour of the transporter in the presence of electroneutral substrates and drugs. Finally, we evaluated the functional implications of the most common genetic variants of hPEPT1, Ser117Asn and Gly419Ala (Fig. 1), which occur at a frequency of 25 and 8% (http://pharmacogenetics.ucsf.edu).
Methods
Chemicals
All unlabelled chemicals, reagent grade, were purchased from Sigma (St Louis, MO, USA), except glycylsarcosine (Gly-Sar), cephalexin and cefadroxil, which were from MP Biomedicals (Irvine, CA, USA). Amoxicillin, ampicillin, cefadroxil and cephalexin were used at concentrations up to their maximal solubility at pH 5.0 (5, 10, 10 and 20 mm, respectively). δ-ALA and bestatin were tested at 0.5 mm, whereas N-acetyl-Asp-Glu (NAAG) was used at 0.1 mm; higher concentrations of these three drugs induced non-specific current responses in control oocytes (not shown). [glycyl-2-3H]Gly-Sar (specific activity 60 Ci mmol−1) was obtained from American Radiolabelled Chemicals (St Louis, MO, USA). Restriction endonucleases were from New England Biolabs (Beverly, MA, USA).
Construction of variant hPEPT1s
Ser117Asn (S117N) and Gly419Ala (G419A) were obtained by cassette exchange mutagenesis, using the wild-type (WT) hPEPT1 (subcloned in pBluescript) (Mackenzie et al. 1996a) as receptor and cDNAs for each variant (subcloned in pGEM) as donors. The variants in pGEM were provided by the UCSF Pharmacogenetics Core Facility. Double digestion of each donor construct with Mfe I and Hpa I yielded a 1.5 kb cDNA fragment containing the desired mutation. These fragments were gel-purified using the QIAEX II kit (Qiagen, Valencia, CA, USA) and subcloned into the receptor plasmid by means of the Fast-Link kit (Epicentre, Madison, WI, USA), after removal of the equivalent 1.5 kb fragment. Competent XL1-Blue cells (Stratagene, La Jolla, CA, USA) were transformed by electroporation, and colonies were selected in a medium with ampicillin and tetracycline. Plasmid DNA was prepared using purification kits by Qiagen. The fidelity of the new clones was verified both by restriction analysis and automated sequencing.
cRNA synthesis
hPEPT1 plasmids were linearized with BamHI, and transcribed in vitro using the T7 MEGAScript kit and RNA cap analogue (Ambion, Austin, TX, USA). The cRNAs were prepared as described (Mackenzie et al. 1996a).
Expression of hPEPT1s in oocytes
Mature female Xenopus laevis were purchased from Nasco (Fort Atkinson, WI, USA). All animal protocols followed guidelines approved by the University of California Chancellor's Committee on Animal Research and the National Institutes of Health. Frogs were anaesthetized with 0.1% Tricaine (Sigma) buffered with 0.1% NaHCO3, a portion of the ovary was surgically removed, and the frogs were killed by an overdose of Nembutal (60 mg for 60 min). Stage V–VI oocytes were selected and maintained at 18°C in modified Barth's solution (Parent et al. 1992a) supplemented with 50 mg l−1 gentamicin (Sigma), 5.75 mg l−1 ciprofloxacin (Bayer, West Haven, CT, USA) and 100 mg l−1 streptomycin sulphate/100 000 units l−1 penicillin G sodium (Gibco, Invitrogen, Carlsbad, CA, USA).
Oocytes were injected 1 day after isolation with 50 ng of hPEPT1, S117N or G419A cRNA, and incubated at 18°C for 4–7 days. Experiments were performed at 20°C. Non-injected oocytes served as controls.
Gly-Sar uptake assays
Oocytes were incubated in the presence of 5 μm to 5 mm Gly-Sar (0.1 μm [3H]Gly-Sar) in a medium containing (mm): 100 NaCl or choline (Cho) chloride, 2 KCl, 1 MgCl2, 1 CaCl2, and 10 Hepes/Tris (pH 7.5) or 10 2-(N-morpholino)ethanesulphonic acid (Mes)/Tris (pH 5.0). After 30 min, oocytes were rinsed and assayed for radioactivity as described (Hediger et al. 1987). Competition studies were performed using the neuropeptide NAAG and selected cephalosporins (cefadroxil and cephalexin), penicillins (ampicillin and amoxicillin), peptidomimetic drugs (bestatin) and non-peptidic compounds (δ-ALA).
Electrophysiology
A two-microelectrode voltage-clamp system was used to measure substrate-induced steady-state currents in hPEPT1 expressing oocytes (Loo et al. 1993; Mackenzie et al. 1996a). Steady-state current–voltage relationships were measured in Na+ pH 5.0 buffer, in the absence and in the presence of Gly-Sar and/or drugs. A pulse protocol was applied in which membrane potential (Vm) of oocytes was held at −50 mV and stepped to a test value for 100 ms before returning to the holding potential. The test potential varied from +50 to −150 mV in 20 mV increments. Steady-state currents were recorded at the end of 100 ms. pClamp and Axoscope software (Axon Instruments, Union City, CA, USA) were used for pulse protocol application and data acquisition, and continuous current data were recorded with a chart recorder. Unless otherwise noted, experiments were repeated on at least three oocytes from different donor frogs.
Data analysis
The kinetic parameters of radiotracer uptake and substrate-related inward currents were calculated by nonlinear regression, using SigmaPlot 9.0 (Systat Software, Inc., Richmond, CA, USA). Data were fitted to eqn (1), for which J is the influx (or the evoked current I), Jmax is the derived maximum transport (or maximal current Imax), S is the substrate concentration, and is the substrate concentration at which transport is half-maximal.
| (1) |
Presteady-state currents in response to step jumps in membrane potential were isolated by fitting the total current across the oocyte membrane (It) to eqn (2) (Loo et al. 1993), where Im is the initial value of the membrane capacitance current, which has a time constant τm; t is time; Ipss is the initial hPEPT1 presteady-state currents with time constant τpss; and Iss is the steady-state current. Transporter-mediated transients (Ipssexp(−t/τpss) were determined by subtraction of the capacitive and steady-state components.
| (2) |
At each voltage, charge movements (Q) were calculated by integrating the presteady-state currents with time and fitted to the Boltzmann relation (eqn (3)), for which the apparent maximum charge Qmax = Qdep − Qhyp (Qdep and Qhyp represent Q at depolarizing and hyperpolarizing limits); z, the apparent valence of the movable charge (Loo et al. 1993), is 1; Vm is the test potential; V0.5 is the membrane potential at which half of the total charge has moved; F is the Faraday's constant; R is the universal gas constant; and T is the absolute temperature.
| (3) |
The results presented in Figs 6, 7 and 8 and Table 2 were obtained in individual WT-, S117N- and G419A-expressing oocytes from the same batch, and confirmed in at least another oocyte of each type from the same frog.
Figure 6. Presteady-state currents associated with hPEPT1.

A, carrier-mediated OFF transients in a representative oocyte expressing wild-type hPEPT1. Total currents were recorded at pH 5.0 as a series of voltage jumps (+50 to −150 mV) was applied to an oocyte clamped to −50 mV (see for example Fig. 4A); presteady-state currents were isolated from the total currents using the fitted method (eqn (2)). Continuous lines are the OFF hPEPT1-mediated transients for −10, −50 and −130 mV, and are displayed 5 ms after the voltage step; the trace at −50 mV (holding potential) represents zero current. Dashed lines are the predictions based on simulation of the model shown in Fig. 9. B, voltage dependence of the relaxation time constants (τ) in representative oocytes expressing WT (circle), S117N (triangle) or G419A (square) hPEPT1. The values of τ for the ON and OFF transients in a Na+ buffer at pH 5.0 were determined from the fit of the total currents to eqn (2). The bell-shaped τON−V relationships (filled symbols) were fitted to a Gaussian equation; values of the kinetic constants are given in Table 2. τOFF the time constant when the membrane was stepped from the test back to the holding potential (−50 mV), was independent of the test voltage; the values for −150 mV are shown (open symbols). C, charge–voltage (Q–V) relationships. Data are from the same representative oocytes as in B. Charge movements (Q) were determined by time integration of the carrier-mediated presteady-state currents for the ON and OFF transients. QON and QOFF were equal and opposite in sign; for clarity, only QOFF values are shown. Data were fitted a Boltzmann relation; values of the kinetic constants are given in Table 2. Lines in B and C are the predicted τON−V and Q–V relationships according to the model in Fig. 9; the values of the corresponding predicted kinetic constants are shown in Table 2.
Figure 7. Effect of Gly-Sar and cefadroxil on hPEPT1 charge movements.

A, Q–V relations in the absence (○) and in the presence of 1 mm Gly-Sar (•) and 2 mm cefadroxil (▪). B and C, dependence of reduction of Qmax on external concentration of Gly-Sar (•, B) and cefadroxil (▪, C). All experimental data (symbols) are from the same representative WT-expressing oocyte showed in Fig 6. Continuous lines are the predictions of the model described in Fig. 9 for all the Gly-Sar and cefadroxil concentrations tested; the dashed line in C is the extrapolation of the model for the cefadroxil concentration range 25–250 mm. B and C, the percentages of maximum charge decrease %ΔQmax were calculated as described in Results. When the %ΔQmax–[Gly-Sar] data set in B was fitted to a hyperbolic equation, the apparent affinity constant () for Gly-Sar was 0.6 ± 0.1 mm; the standard error of the fit is given. The model predicted a of 0.6 mm for Gly-Sar (B) and 2.5 mm for cefadroxil (C).
Figure 8. Effect of substrates on hPEPT1 charge distribution.

A, reduction in Qmax by Gly-Sar and selected drugs in representative oocytes expressing WT (black bar), S117N (grey bar) and G419A (white bar) hPEPT1. B, shift of V0.5 in the same oocytes. Data are from the same oocytes shown in Figs 6 and 7. The apparent maximum charge (Qmax) and the voltage for 50% charge movement (V0.5) in a Na+ buffer at pH 5.0 were determined from the Q–V relations and the Boltzmann equation (see Methods).
Table 2. Presteady-state parameters in individual oocytes expressing WT, S117N or G419A hPEPT1.
| Predicted | WT | S117N | G419A | |
|---|---|---|---|---|
| (ms) | 10.8 | 10.8 ± 0.2 | 10.4 ± 0.3 | 11.1 ± 0.3 |
| Vτmax (mV) | −36 | −36 ± 2 | −32 ± 3 | −33 ± 3 |
| (ms) | 10.7 | 10.1 ± 0.1 | 10.5 ± 0.3 | 11.1 ± 0.2 |
| Qmax (nC) | 10.6 | 10.4 ± 0.4 | 11.3 ± 0.8 | 10.9 ± 0.4 |
| Qmax, 10 mm GS (nC) | 1.2 | 1.4 ± 0.1 | 1.2 ± 0.1 | 1.6 ± 0.2 |
| Qmax, 10 mm CEF (nC) | 3.4 | 4.5 ± 0.3 | 5.1 ± 0.7 | 4.8 ± 0.4 |
| V0.5 (mV) | −27 | −27 ± 3 | −22 ± 3 | −24 ± 3 |
| V0.5, 10 mm GS (mV) | −67 | −74 ± 7 | −65 ± 8 | −73 ± 9 |
| V0.5, 10 mm CEF (mV) | −35 | −47 ± 5 | −46 ± 6 | −48 ± 4 |
Presteady-state currents at pH 5.0 were obtained from total currents by the fitted method (eqn (2)). Charge movements (Q) were obtained by integration of presteady-state currents with time and fitted to the Boltzmann relation (eqn (3)). Data are from the same oocytes shown in Figs 6, 7 and 8. The standard errors of the fits are given. , maximum value of the time constant for the ON presteady-state transient; Vτmax, voltage at which is achieved; , time constant for the OFF presteady-state transient at −150 mV (voltage independent); Qmax, apparent maximum charge; V0.5, voltage for 50% Qmax. Predicted values are those based on the model predictions in Fig. 9
Presteady-state data fitting was performed by means of Clampfit 8.2 (Axon Instruments). One-way analysis of variance and Tukey's test were applied to evaluate statistical differences between WT and variant hPEPT1s, by means of SigmaStat 3.1 (Systat Software, Richmond, CA, USA).
Mathematical modelling
The differential equation (eqn (4)) for the evolution of states for the 7-state kinetic model for hPEPT1 (Fig. 9) was solved using Berkeley Madonna 8.0.1: (http://www.berkeleymadonna.com).
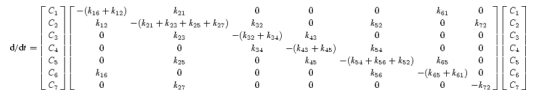 |
(4) |
where Ci is the occupancy probability in state C1, with . The current generated by the transporter is I = −FN[α(k12C1 − k21C2)−δ(k16C1 − k61C6)] (Parent et al. 1992b), where N is the number of transporters, and α and δ describe the fraction of the membrane electric field sensed by the binding of H+ and by the empty H+-binding site during membrane translocation.
Figure 9. Kinetic model for hPEPT1.

A 7-state model in which the empty carrier is negatively charged (apparent valence −1) and one H+ binds to the transporter (C) before the substrate (S). Carrier states at the outer side of the membrane are identified by prime and, at the inner side, by double prime. Details on the transport cycle are given in the Discussion. The partial reaction C2 ⇌ C5 represents the H+-leak pathway (5% of the transport at saturating substrate concentration). Presteady-state currents are due to the partial reactions C6 ⇌ C1 ⇌ C2, marked by the shaded region. Transitions between conformational states are assumed to be first order or pseudo first order, with rate constants kij representing the transition rates from Ci to Cj. Rate constants k12, k21, k23, k54, k65, k16, k61, k27 and k72 are described by voltage-independent values () modulated by voltage and/or ligand concentration, whereas k32, k34, k43, k52 and k56 have a fixed value, and k25 and k45 are obtained by microscopic reversibility (Parent et al. 1992b). The effect of membrane potential (Vm) in k12, k21 k16 and k61 is assumed to follow the Eyring rate theory with symmetric energy barriers (Parent et al. 1992b). α and δ are the fractional dielectric distance coefficients at α = 0.27 and δ = 0.73 (Mackenzie et al. 1996a). The voltage dependence of C2 ⇌ C7 is defined by the empirical coefficient 0.37. μ is the electrochemical potential FVm/RT. To simulate the presteady-state currents in the absence of substrate and the characteristics of Gly-Sar transport, the following set of parameters was used: = 1.5 × 108 m−1 s−1, = 550 s−1, = 105 m−1 s−1, = 200 s−1, = 600 s−1, = 2000 s−1, = 105 m−1 s−1, = 500 s−1, = 1.5 × 108 m−1 s−1, = 35 s−1, = 310 s−1, = 1 s−1, = 10−1 s−1, = 5 × 105 m−1 s−1 and = 800 s−1. The characteristics of cefadroxil transport were simulated by changing , , , and to 1.25 × 104m−1 s−1, 25 s−1, 250 s−1, 1.25 × 104m−1 s−1 and 3 × 106m−1 s−1, respectively. The total number of transporters, estimated from Qmax (Loo et al. 1993), was 7 × 1010, and the temperature was 20°C. The predictions of the model are shown by the curves in Figs 5–7, and given in Tables 1 and 2.
The SQUAREPULSE function of Berkeley Madonna was used to obtain the time course of occupancy probabilities (C1…C7) and total transporter current (I) for each external concentration of Gly-Sar and cefadroxil, with a set of rate constants (Fig. 9). In these simulations, the holding potential was −50 mV, and 100 ms test pulse Vm varied from +50 to −150 mV, in 2 mV increments. The pH of the cell interior was fixed to 7.5, and the pH of the external solution was 7.5 or 5.0. For analysis of the steady-state kinetics, the substrate-induced currents were taken as the difference between the steady-state current in the presence and in the absence of substrate. The steady-state current–voltage (I–V) relations were generated as the external concentration of substrate ([S]o) was varied. The dependence of the kinetic parameters Imax and K0.5 on Vm were obtained by fitting the I–[S]o curves to eqn (1). At each Vm, presteady-state currents for the ON- and OFF- pulses were simulated by fitting I to eqn (2), and integrated with time to obtain the predicted charge movement Q. The Q–V curves were fitted to the Boltzmann relation (eqn (3)) to obtain Qmax, the apparent valence z, and V0.5, the membrane voltage for 50% charge transfer. The time constants (τ) of transient current decay were obtained as described (Mackenzie et al. 1996b), as well as by fitting the simulated presteady-state currents.
Results
Uptakes
Figure 2A illustrates the influx of radiolabelled Gly-Sar into Xenopus laevis oocytes expressing WT, Ser117Asn (S117N) or Gly419Ala (G419A) hPEPT1 transporters. Both variants showed similar transport rates to the WT, under all the conditions tested, indicating similar levels of protein expression in the oocyte plasma membrane. In all cases, uptake of [3H]Gly-Sar occurred in a proton-dependent manner, as it increased up to 13-fold when the external pH was lowered from 7.5 to 5.0. Replacement of Na+ by Cho did not affect the uptake (data not shown). Levels of [3H]Gly-Sar influx in non-injected oocytes were 15% (at pH 7.5) or 1.5% (at pH 5.0) of those measured in cRNA-injected oocytes. In WT, the apparent affinity constant of Gly-Sar uptake () was 1.3 ± 0.3 mm, and maximal rate of influx () was 3970 ± 290 pmol h−1 oocyte−1.
Figure 2. Glycylsarcosine (Gly-Sar) uptake into Xenopus laevis oocytes expressing wild-type (WT), S117N or G419A hPEPT1.

A, influence of external pH. B, effect of selected drugs. Oocytes were injected with 50 ng of the corresponding transporter cRNA, and, after 5 days, uptake of 5 μm Gly-Sar (0.1 μm [3H]Gly-Sar) was measured in 100 mm Na+ buffer at pH 7.5 or 5.0, in the absence (A) and in the presence (B) of various compounds, for 30 min and at 20°C. Non-injected (NI) oocytes were used as controls. Data are shown as means ± s.e.m. for at least five oocytes, and are representative of three experiments. B, results were normalized to the uptake at pH 5.0 and in the absence of external inhibitors, as shown in A. *Significant difference between WT and variant hPEPT1s (P < 0.05).
We studied the effect of various representative drugs on radiolabelled Gly-Sar uptake (Fig. 2B). The compounds chosen were the β-lactam antibiotics ampicillin, amoxicillin (penicillins), cephalexin and cefadroxil (cephalosporins), the antineoplastic agents δ-ALA and bestatin, and the neuropeptide NAAG. In accordance with the of 1.3 mm, influx of 5 μm radiolabelled Gly-Sar dropped about 50% in the presence of 1 mm of non-labelled dipeptide. In all groups, Gly-Sar uptake decreased 50% upon addition of 10 mm cephalexin or 2 mm cefadroxil, whereas the inhibition caused by 0.5 mm δ-ALA was of 30% in the WT and of 15% in the variants. Bestatin (0.5 mm) reduced transport in G419A (25%), and only S117N was sensitive to 0.1 mm NAAG (20%). Ampicillin (10 mm) and amoxicillin (5 mm) had no significant inhibitory effect in any group. Gly-Sar influx into non-injected oocytes was not modified by any of the drugs tested (not shown).
Electrophysiology
Substrate selectivity
We measured the currents induced by Gly-Sar and the selected drugs in oocytes voltage clamped at −50 mV. Figure 3A shows a continuous record from a representative experiment, in which a WT hPEPT1-expressing oocyte was exposed to 0.5 mm Gly-Sar or 20 mm cephalexin. Addition of these substrates to the perfusion buffer generated inward currents in expressing oocytes, which were reversed upon replacement with substrate-free buffer.
Figure 3. Substrate-induced inward currents in X. laevis oocytes expressing hPEPT1.

substrate-induced inward currents in X. laevis oocytes expressing hPEPT1. Between 4 and 7 days postinjection with transporter cRNA, oocytes were mounted in a two-microelectrode voltage-clamp system, superfused with Na+buffer at pH 5.0 and held at −50 mV. Currents in response to the addition of Gly-Sar (0.5 mm) or/and the following substrates were recorded: ampicillin (10 mm), amoxicillin (5 mm), cephalexin (20 mm), cefadroxil (2 or 10 mm), d-aminolevulinic acid (d-ALA) (0.5 mm), bestatin (0.5 mm) and N-acetyl-Asp-Glu (NAAG) (0.1 mm). A, individual trace for Gly-Sar and the β-lactam antibiotic cephalexin in WT hPEPT1. B, inward currents evoked by Gly-Sar and the selected compounds in oocytes expressing WT (black bars), S117N (grey bars) or G419A (white bars) hPEPT1. C, effect of the same drugs on inward currents evoked by 0.5 mm Gly-Sar. Results were normalized to the current evoked by 0.5 mm Gly-Sar: 240 ± 14 (WT), 241 ± 22 (S117N) and 232 ± 18 nA (G419A). Data are shown as means ± s.e.m. for at least three oocytes from different donor frogs; in each experiment, the same oocyte was used to test all compounds. *Significant difference between WT and variant hPEPT1s (P > 0.05).
In the absence of substrate, replacement of buffer at pH 7.5 with buffer at pH 5.0 induced an inward current in expressing oocytes that was larger than that observed in non-injected oocytes (∼30 nA at −50 mV). This H+ leak was ∼5% of the current induced by saturating concentration of Gly-Sar. For example, in a WT-expressing oocyte, the H+ leak at −50 mV was 60 nA, and the current evoked by 20 mm Gly-Sar was 600 nA.
Gly-Sar-evoked currents (i) were unaffected by external Na+, and (ii) were significantly attenuated at pH 7.5. For example, currents evoked in a representative WT-expressing oocyte by 1 mm of Gly-Sar in Na+ pH 5.0, Cho pH 5.0, Na+ pH 7.5 and Cho pH 7.5 were 225, 220, 50 and 55 nA, respectively; a similar pH dependence and Na+ independence was found in Gly-Sar currents induced in oocytes expressing S117N and G419A (not shown).
We compared the currents generated by Gly-Sar with those evoked in the same oocyte by each of the different drugs. Experiments were carried out at pH 5.0, the optimum for Gly-Sar transport (Mackenzie et al. 1996a). Results were normalized to the current induced by 0.5 mm Gly-Sar 240 ± 14 (WT), 241 ± 22 (S117N) and 232 ± 18 nA (G419A). All drugs produced inward currents. As shown in Fig. 3B, the currents due to 0.1 mm NAAG, 0.5 mm δ-ALA and 0.5 mm bestatin were ∼14, 25 and 30%, respectively, of that generated by 0.5 mm Gly-Sar, and the currents induced by 20 mm cephalexin and 2 mm cefadroxil were ∼38 and 50%, respectively. Penicillins caused a poor response: the currents evoked by 10 mm ampicillin or 5 mm amoxicillin accounted for less than 20% of the current due to 0.5 mm Gly-Sar. WT and variants displayed an identical profile of substrate selectivity, except that the currents induced by δ-ALA in G419A and S117N were 25 and 40% lower than in WT. At the concentrations used, none of the compounds evoked detectable currents in non-injected oocytes (not shown).
Next, we evaluated the ability of the selected compounds to interact with the currents evoked by Gly-Sar, in the same oocytes as in Fig. 3B. In the representative experiment shown in Fig. 3A, a WT oocyte was superfused with 0.5 mm Gly-Sar, first in the absence, and consecutively in the presence of 20 mm cephalexin. The drug caused an abrupt 30% decrease in the Gly-Sar current. As represented in Fig. 3C, addition of 0.1 mm NAAG, 0.5 mm δ-ALA and 0.5 mm bestatin led to the inhibition of ∼35, 40 and 45%, respectively, of the current induced by 0.5 mm Gly-Sar. Inhibition due to 20 mm cephalexin, 5 mm amoxicillin and 10 mm ampicillin was ∼30, 45 and 55%, respectively. Gly-Sar currents were not inhibited by 10 mm cefadroxil. No significant differences were found among WT and variant hPEPT1s.
Voltage dependence of steady-state currents
We investigated the characteristics of Gly-Sar-evoked currents and compared them with those induced by cefadroxil, as a model for drug transport. Figure 4 shows typical current records in a representative oocyte expressing WT hPEPT1 before (Fig. 4A) and after addition of 0.5 mm (Fig. 4B) or 10 mm (Fig. 4C) Gly-Sar. Stepping the membrane potential of hPEPT1-expressing oocytes from −50 mV to a series of test values (+50 to −150 mV) resulted in the generation of a transient, presteady-state current (ON response), distinct from the fast (τ ∼1 ms) initial spike due to the lipid bilayer capacitance, which relaxed to the steady-state with a time constant τON of 4–12 ms (see for example Fig. 6B). When the membrane potential was returned to the holding value, an equal but opposite transient current was observed (OFF response). hPEPT1 transients were reduced upon addition of Gly-Sar. As shown in Fig. 4, this was particularly evident in the OFF response.
Figure 4. Time course of the total currents measured in a single WT hPEPT1-expressing oocyte in the absence (A) and presence of 0.5 (B) and 10 mm (C) Gly-Sar.

Currents were recorded as the membrane potential was held at −50 mV and stepped to test values (+50 to −150 mV in 20 mV increments, see Methods). For clarity, only the traces for +50, −10, −50, −90 and −150 mV are presented. The pulse protocol is depicted in A (top panel). Dotted traces show current at the holding potential (−50 mV) and emphasize the difference caused by the addition of Gly-Sar. The presteady-state currents associated with hPEPT1 are evident in A, especially for the OFF response, and disappear as the concentration of Gly-Sar is increased (B and C).
At each test membrane potential, net steady-state Gly-Sar and cefadroxil-induced currents were calculated as the difference between the steady-state current in the presence and in the absence of substrate. In the example shown in Fig. 4, steady-state current at −50 mV was 125 nA in the absence of substrate (Fig. 4A), 275 nA in the presence of 0.5 mm Gly-Sar (Fig. 4B), and 550 nA in the presence of 10 mm Gly-Sar (Fig. 4C). Thus, at −50 mV, the net steady-state current due to 0.5 and 10 mm Gly-Sar was 150 and 425 nA. Symbols in Fig. 5A and B show the steady-state currents evoked by increasing concentrations of Gly-Sar (IGS, Fig. 5A) and cefadroxil (ICEF, Fig. 5B) in the same WT-expressing oocyte. For both substrates, the I–V curves approached zero at +50 mV. Currents induced by <2 mm Gly-Sar and all the concentrations of cefadroxil used were inhibited at large negative potentials. The voltage at which the IGS–V relationship saturated shifted in the hyperpolarizing direction as the concentration of dipeptide increased, from −90 mV at 0.1 mm to −130 mV at 2 mm (Fig. 5A). The currents evoked by cefadroxil were inhibited at potentials more negative than −90 mV (Fig. 5B).
Figure 5. Kinetics of Gly-Sar and cefadroxil-evoked currents.

A, voltage dependence of the steady-state currents induced by Gly-Sar in a representative oocyte expressing wild-type hPEPT1. B, voltage dependence of the steady-state currents evoked by cefadroxil in the same oocyte. Substrate-dependent inward currents were measured at pH 5.0 by the two-microelectrode voltage-clamp technique. Symbols are experimental data; continuous lines are the predictions of the model shown in Figure 9 for all the Gly-Sar and cefadroxil concentrations tested; dashed lines in B are the model predictions for higher concentrations of cefadroxil (25–500 mm). C, voltage dependence of the apparent affinity constant for Gly-Sar in oocytes expressing WT (circle), S117N (triangle) and G419A (square) hPEPT1. The kinetic parameters of transport were determined by measuring the steady-state currents evoked, at the test potentials shown, by increasing concentrations of the dipeptide (0.01, 0.1, 0.25, 0.5, 1, 2, 5, 10 and 20 mm) in Na+ buffer and at pH 5.0. Data are expressed as means ± s.e.m. for at least three oocytes from different donor frogs. The continuous line is the model prediction from Fig. 9 over the voltage range −30 to −150 mV; the dashed line is the projection of the model for the voltage range −10 to +50 mV. D, predicted voltage dependence of the apparent affinity constant for cefadroxil (). The curve was obtained by simulation of the model shown in Fig. 9.
Steady-state data were fitted to eqn (1) to estimate the apparent kinetic constants of Gly-Sar transport in WT and variant hPEPT1 (symbols in Fig. 5C and Table 1). Between −30 and −150 mV, (Fig. 5C) and (Table 1) were voltage dependent and decreased with depolarization. In WT-expressing oocytes, decreased from ∼3 mm at −150 mV to ∼1 mm at −30 mV (Fig. 5C), and decreased from ∼1000 (Table 1) to ∼300 nA. No significant differences were found among WT, S117N and G419A in the values for any given voltage (Fig. 5C); at −50 mV, values were (mm): 1.5 ± 0.3, 1.6 ± 0.1 and 1.2 ± 0.1, respectively (Table 1). Furthermore, when normalized to the obtained at −150 mV (Table 1), the − V curves of all variants were identical (not shown). Owing to the low magnitude of the currents measured between +50 to −10 mV, we were unable to estimate the kinetics of Gly-Sar over this voltage range. Due to the limited solubility of cefadroxil at pH 5.0, we could not measure currents at concentrations above 10 mm, and thus could not obtain the transport kinetics of this cephalosporin.
Table 1. Kinetic constants of Gly-Sar and cefadroxil transport.
| K0.5 (mm) | Imax (nA) | |||
|---|---|---|---|---|
|
|
|
|||
| −50 mV | −150 mV | −50 mV | −150 mV | |
| GLY-SAR | ||||
| ″Predicted | 1.2 | 2.7 | 553 | 1349 |
| ″WT | 1.5 ± 0.3 | 3.1 ± 0.2 | 456 ± 15 | 1078 ± 49 |
| ″S117N | 1.6 ± 0.1 | 3.6 ± 0.3 | 545 ± 33 | 1340 ± 75 |
| ″G419A | 1.2 ± 0.1 | 3.2 ± 0.2 | 433 ± 57 | 1041 ± 100 |
| Cefadroxil | ||||
| ″Predicted | 6.2 | 23.5 | 411 | 732 |
Substrate-dependent inward currents were measured at pH 5.0 by means of two-microelectrode voltage-clamp in oocytes expressing wild-type (WT), S117N or G419A hPEPT1. Kinetic parameters for Gly-Sar steady-state currents were determined as described in Fig. 5. Apparent affinity constants (K0.5) and highest maximum currents (Imax) at −50 and −150 mV are shown. Data are expressed as means ± s.e.m. for at least 3 oocytes from different donor frogs. Predicted values for the kinetic parameters of Gly-Sar and cefadroxil transport were obtained from simulation of the model described in Fig. 9
Presteady-state currents: hPEPT1 charge movements
Presteady-state transient currents represent charge translocations associated with voltage-dependent changes in the conformation of a cotransporter as it goes through the transport cycle (Loo et al. 1993). hPEPT1-expressing oocytes exhibited presteady-state currents in response to step changes in membrane potential (Fig. 4). Such transients (i) were not observed in non-injected oocytes (not shown), and (ii) were blocked by the addition of substrates in a concentration-dependent manner (see Fig. 4B and C for Gly-Sar). To isolate the presteady-state currents mediated by hPEPT1, we fitted the total currents to eqn (2). An example of the OFF transient currents observed in a WT oocyte at pH 5.0 is shown in Fig. 6A (continuous lines). Presteady-state currents were significantly reduced at pH 7.5 (see Mackenzie et al. 1996a).
WT and variants followed identical kinetics of presteady-state current relaxation. At pH 5.0, the transient currents for the ON response relaxed with time constants (τON) that ranged from 4 to 12 ms (Fig. 6B). The τON–V curve fitted a Gaussian relationship, with a maximum of ∼10–12 ms at a voltage (Vτmax) of ∼−35 mV (Table 2). The value for was consistent among oocytes, but the values of varied from batch to batch. For example, in WT and variant oocytes from a different donor frog, was ∼12 ms but Vτmax was more positive than +20 mV. In the OFF response, τ was voltage independent at ∼10–12 ms (τON, Table 2).
To calculate the equivalent charge moved (Q), we integrated the presteady-state currents with time. At each test voltage, the charge transfer for the ON and OFF responses (QON and QOFF) was equal; for clarity, only the values of QOFF are shown (Fig. 6C). The charge–voltage (Q–V) curve was fitted to a Boltzmann relation to obtain (i) Qmax, the maximum charge transfer, and (ii) V0.5, the voltage at which half of the maximum charge has moved. At pH 5.0, Qmax was ∼10–12 nC and V0.5 was ∼−25 mV (Table 2). As described for Vτmax, some batch-to-batch variation was observed in V0.5; for example, in the oocytes from a different donor frog, V0.5 was ∼−10 mV. When the pH was increased (i) Qmax decreased significantly, and (ii) V0.5 shifted in the hyperpolarizing direction. In the oocytes shown in Fig. 6, Qmax at pH 7.5 was ∼5 nC and V0.5 was ∼−90 mV. Values of Q, Qmax and V0.5 were similar in WT-, S117N- and G419A-expressing oocytes (Fig. 6C and Table 2).
Effect of substrates on hPEPT1 charge transfer
Figure 7A exemplifies the effect of 1 mm Gly-Sar and 2 mm cefadroxil on the Q–V distribution in the same WT-expressing oocyte from Fig. 6. Addition of either substrate (i) led to a decrease in Qmax, and (ii) caused the shift of V0.5 to more negative potentials. Thus, in the absence and in the presence of 1 mm Gly-Sar or 2 mm cefadroxil, Qmax was 10.4 ± 0.4, 4.3 ± 0.3 and 6.2 ± 0.6 nC, and V0.5 was −27 ± 3, −41 ± 4 and −31 ± 6 mV, respectively, (standard errors of the Boltzmann fits).
The reduction in Qmax (ΔQmax) and the shift in V0.5 (ΔV0.5) induced by Gly-Sar and cefadroxil were dependent upon the external concentration of substrate. Qmax was reduced about 90% by 10 mm Gly-Sar, whereas only 50% of the maximum movable charge disappeared upon addition of 10 mm cefadroxil (Table 2). Accordingly, ΔV0.5 was ∼−45 mV for 10 mm Gly-Sar and ∼−20 mV for 10 mm cefadroxil (Table 2).
We calculated the percentage of decrease in Qmax induced by each concentration of Gly-Sar and cefadroxil according to:
where and represent Qmax in the absence and in the presence of substrate. The Gly-Sar dose–response curve (Fig. 7B) was saturable and followed a simple hyperbolic relationship. For the oocytes shown in Fig. 6, the values of were (mm): 0.6 ± 0.1 (WT, Fig. 7B), 0.7 ± 0.1 (S117N) and 0.8 ± 0.1 (G419A) (standard errors of the fits). At the concentrations of cefadroxil used, the %ΔQmax–[cefadroxil] relationship did not saturate (Fig. 7C).
Ampicillin, amoxicillin, cephalexin, δ-ALA, bestatin and NAAG also modified the kinetics of hPEPT1 charge movement; the effect of these drugs in the same oocytes from Figs 6 and 7 is shown in Fig. 8. In the presence of 0.1 mm NAAG, 0.5 mm δ-ALA and 0.5 mm bestatin, ΔQmax was ∼40–50% (Fig. 8A), and ΔV0.5 was ∼−9, −10 and −13 mV, respectively (Fig. 8B). Upon addition of 20 mm cephalexin, 5 mm amoxicillin or 10 mm ampicillin, Qmax dropped ∼25, 40 and 60%, respectively (Fig. 8A) and V0.5 shifted by ∼−10 mV (Fig. 8B). The effect of all compounds on Qmax and V0.5 was similar in oocytes expressing WT, S117N and G419A hPEPT1 (Fig. 8, Table 2).
Discussion
The combination of an extremely broad substrate specificity and a high transport rate has made the intestinal brush-border peptide transporter a prime candidate for enhancing oral drug delivery. To date, insights into drug transport through human PEPT1 have been limited to competition studies (Daniel, 2004). In the present work, we used tracer uptake and electrophysiological methods to investigate the molecular interactions between hPEPT1, the dipeptide Gly-Sar, and a representative selection of drugs, namely: (i) β-lactam antibiotics such as ampicillin, amoxicillin, cephalexin and cefadroxil; (ii) the antineoplastic immunomodulator bestatin; (iii) δ-ALA, a medium-chain omega-amino fatty acid that has been implicated in porphyrin-based cancer diagnosis and treatment (Rubio-Aliaga & Daniel, 2002); and (iv) the neuropeptide NAAG. Based on comprehensive steady-state and presteady-state analysis, we extended our 6-state kinetic model for hPEPT1 (Mackenzie et al. 1996a) and obtained a complete set of kinetic parameters that account for the global behaviour of the transporter in the presence of neutral substrates. To test the hypothesis that variations in drug response among individuals are caused by alterations in the genes involved in drug absorption (Sadée, 1999), we evaluated the physiological implications of S117N and G419A, the two most frequent single-nucleotide polymorphisms of hPEPT1. Unless otherwise noted, experiments were carried out at pH 5.0, the pH optimum for Gly-Sar transport (Mackenzie et al. 1996a).
Transporter–substrate interactions
Substrate specificity
Gly-Sar transport was electrogenic, pH dependent, Na+ independent, sensitive to membrane potential, and followed saturation kinetics (Figs 2–5), in accordance with previous results (Mackenzie et al. 1996a). The obtained from radiotracer fluxes was identical to those determined from current measurements in the voltage range −30 to −70 mV (1–2 mm, Fig. 5C and Table 1; and Mackenzie et al. 1996a).
Because the radiolabelled drugs are not commercially available, we could not demonstrate directly that they are substrates for hPEPT1. However, we found that ampicillin, amoxicillin, cephalexin, cefadroxil, δ-ALA, bestatin and NAAG all induced inward currents (Fig. 3A). The direct relationship between substrate-dependent currents and transport, as measured by radiotracer uptake, has been confirmed for other carriers, such as the Na+–glucose (SGLT1), the Na+–phosphate (NaPi) and the Na+–Cl−–GABA (GAT1) cotransporters (Mackenzie et al. 1998; Forster et al. 1999; Loo et al. 2000). In one case, however, it should be noted that the generation of currents has proven insufficient to demonstrate actual substrate translocation (Diez-Sampedro et al. 2003).
The currents evoked by all drugs were of lesser magnitude than those generated by equal or lower concentrations of Gly-Sar. Furthermore, the amount of current induced by the antibiotics was comparable to that evoked by concentrations up to 200-fold lower of δ-ALA, bestatin and NAAG (Fig. 3B). The precise interpretation of these results is a challenge, since variations in transport can be due to differences in affinity, in turnover rate, or in both Where there is a lack of detailed kinetics, it is difficult to determine the relative contribution of each factor in each case. Thus, our data allow us only to speculate that the substrate selectivity of hPEPT1 at pH 5.0 is: Gly-Sar > NAAG, δ-ALA, bestatin > cefadroxil, cephalexin > ampicillin, amoxicillin.
The uptake of 5 μm [3H]Gly-Sar was blocked by cefadroxil, cephalexin and δ-ALA, but not by bestatin, ampicillin, amoxicillin or NAAG (Fig. 2B). Because of solubility limitations, we were unable to obtain kinetic data for these drugs. Hillgren and coworkers (Zhang et al. 2004) reported that Gly-Sar uptake into hPEPT1-expressing HeLa cells was inhibited to a significantly higher degree by δ-ALA and bestatin than by cephalexin and cefadroxil. Cephalexin, cefadroxil and ampicillin are low-affinity blockers of Gly-Sar uptake into Caco-2 cells, with inhibition constants between 7 and 14 mm in the pH range 5.0–6.0 (Bretschneider et al. 1999). On the other hand, bestatin and δ-ALA have been characterized as high-affinity substrates (K0.5 ∼0.5 mm) for rat and rabbit PEPT1 (Doring et al. 1998; Terada et al. 2000).
A close look at the physicochemical properties of the different substrates could help explain some of these findings. For example, β-lactams have a tripeptide-like backbone and a more complex three-dimensional structure than Gly-Sar or δ-ALA (see Fig. 3 of Rubio-Aliaga & Daniel, 2002), which may complicate the interactions with the binding site or act on the translocation pathway. On the other hand, it has been suggested that substrate affinity of peptide transporters can be reduced by acetylation of the α-amino group (Wang et al. 1998), which could help justify the relatively low transportability of NAAG. In addition, it has been observed that the transport rate of PEPT1 anionic substrates increases with decreasing pH, which has been attributed to a hypothetical inductive effect of protons at the substrate-binding site (Irie et al. 2005). Thus, it is possible that NAAG, which carries a large anionic charge at acidic pH (−3), requires pH values lower than 5.0 to be transported effectively.
All of the drugs used in this study appear to be transported, as judged by their ability to generate inward currents (Fig. 3B). The currents induced by the drugs in the presence of 0.5 mm Gly-Sar were in most cases less than those due to the same concentration of Gly-Sar alone (Fig. 3C). One possible explanation for this apparent inhibitory effect is that these compounds are transported at a lower turnover rate than Gly-Sar. For example, based on Fig. 2B, it can be roughly estimated that the K0.5 of the β-lactam cephalexin at −50 mV is 10 mm. With this assumption, the inward current shown in Fig. 3B can be fitted to eqn (1) to determine that the Imax of this drug is 15% of that of Gly-Sar. Alternatively, the lack of additive effects of Gly-Sar and drugs on inward H+ transport may indicate interactions between dipetides, drugs and the transporter that result in a decrease of the total rate of H+–solute cotransport by hPEPT1.
Voltage dependence of hPEPT1
Steady-state inward currents evoked by Gly-Sar and the model drug cefadroxil at pH 5.0 were concentration dependent and increased with hyperpolarization. At <2 mm, Gly-Sar currents were inhibited by large hyperpolarizing potentials, and this inhibition was relieved at higher Gly-Sar concentrations (Fig. 5A). In the voltage range −150 to −30 mV, the estimated Gly-Sar apparent affinity constants () and current maxima () decreased as membrane potential became more positive (Fig. 5C and Table 1; and Mackenzie et al. 1996a). For 0.5–10 mm cefadroxil, the currents were inhibited at membrane potentials more negative than −90 mV (Fig. 5B).
In the absence of substrate, hPEPT1 presteady-state currents were generated following step changes in membrane potential (Figs 4A and 6A). These transporter-mediated transients (i) were attenuated at pH 7.5 and upon addition of substrates (Fig. 4B and C), (ii) relaxed with time constants (τ) that (a) in the ON response, followed a Gaussian distribution with membrane potential ( ∼10–12 ms), and (b) in the OFF response, were voltage-independent (τOFF ∼10–12 ms) (Fig. 6B and Table 2), and (iii) represented hPEPT1 charge movements that fitted a Boltzmann relation (eqn (2)) with Qmax ∼10–12 nC (Fig. 6C and Table 2; and Mackenzie et al. 1996a). The midpoints of the τON–V and Q–V distributions, namely Vτmax and V0.5, were similar in oocytes from the same frog, but varied significantly amongst preparations. For example, V0.5 ranged from −20 mV in oocytes from one frog (Table 2) to +30 mV in oocytes from a different one (see also Mackenzie et al. 1996a).
Addition of substrates modified the kinetics of charge distribution, by (i) decreasing Qmax, and (ii) shifting V0.5 in the hyperpolarizing direction (Figs 7 and 8). The ΔQmax and ΔV0.5 values were dependent upon the external concentration of substrate (Figs 7 and 8; Table 2). The ΔQmax and ΔV0.5 caused by the different compounds (Fig. 8) further indicate that they bind with relative affinities: Gly-Sar > NAAG, δ-ALA, bestatin > cefadroxil, cephalexin > ampicillin, amoxicillin.
Transport model for hPEPT1
Description of the model
To gain insights into the mechanism of H+–oligopeptide cotransport, we re-examined and extended our previous kinetic model (Mackenzie et al. 1996a). The goal was to determine if the 6-state kinetic model could account for the observed steady-state and presteady-state kinetics of Gly-Sar and cefadroxil transport by hPEPT1. Previously, we only simulated presteady-state currents in the absence of substrate (Mackenzie et al. 1996a). Here, we have assumed that ligand binding to hPEPT1 is ordered, with H+ binding before the substrate (Fig. 9). Our model assumes that cotransport is a series of conformational changes induced by ligand (H+ and dipeptide) binding and membrane voltage (Fig. 9). In a transport cycle, one H+ binds to the outside-orientated empty transporter [C]′ (state C1) to form the complex [CH]′ (state C2). The substrate-loaded protein [CHS]′ (state C3) undergoes a conformational change (C3 ↔ C4) resulting in H+–dipeptide cotransport. It was necessary to include an additional state to account for the observed inhibition of cotransport by negative voltages at low substrate concentrations (Fig. 5A and B; and Mackenzie et al. 1996a): a second proton binds to the transporter in state C2 to form the complex [CHH]′ (state C7), inaccessible to the substrate.
Starting from the set of rate constants used to describe the presteady-state currents in the absence of substrate (Mackenzie et al. 1996a), we sought an expanded set of rate constants to include substrate binding and translocation for a global fit to our experimental data for H+–dipeptide cotransport and the presteady-state kinetics in the presence of substrates. We also imposed an additional constraint in that the model had to be consistent with the kinetics of reverse H+–oligopeptide transport (Kottra & Daniel, 2001). We obtained a numerical solution for the 16 rate constants and three voltage-dependence parameters that account qualitatively and quantitatively for the steady-state and presteady-state experimental data (see legend to Fig. 9). For the set of constants, we examined (i) the I–V relations of the Gly-Sar and cefadroxil-coupled steady-state currents, (ii) the time course of the presteady-state currents, and (iii) the τ–V, and (iv) the Q–V relationships. The model predictions are compared directly with the experimental data in Figs 5–7 and Tables 1 and 2.
Fit of the model
The predicted steady-state I–V relations for the Gly-Sar and cefadroxil cotransport fit the experimental data over the entire range of substrate concentrations and voltage (Fig. 5A and B). The voltage dependence of Gly-Sar transport (K0.5) between −30 and −150 is well described by the model (Fig. 5C and D, and Table 1), and the same behaviour is predicted for cefadroxil (Fig. 5D). For both substrates, the predicted K0.5 is at a minimum at −50 mV (see values in Table 1) and increases steadily with hyperpolarization and depolarization. In both cases, the voltage-dependent increase in K0.5 can be explained as a reduction in the fraction of transporters in state C2. Hyperpolarization drives carriers in substrate-accessible state C2 to unavailable state C7. On the other hand, depolarizing voltages decrease the rate of proton binding to the transporter (C1 → C2) and increase the rate of dissociation (C2 → C1).
As anticipated, the model provides an accurate description of the presteady-state kinetics of hPEPT1 at pH 5.0 in the absence of substrate (Fig. 6 and Table 2; and Mackenzie et al. 1996a). Thus, the model predicts that step changes in membrane potential generate transient currents that rise rapidly to a peak before decaying to the steady state. Presteady-state currents in the ON and OFF response are equal but opposite in sign; an example of the predicted OFF transients is presented in Fig. 6A. For both ON and OFF responses, two relaxation time constants (τ) are predicted by the model: (i) a fast component, of less than 1 ms, which is beyond the resolution of the voltage clamp (not shown), and (ii) a slower one (τON, τOFF) in the range 3–11 ms (Fig. 6B and Table 2). The predicted values of τON are voltage dependent (Fig. 6B), and the τON–V curve fits a Gaussian relationship, with = 10.8 ms and Vτmax = − 36 mV (Table 2). The predicted time constant for the OFF response (τOFF) is voltage independent at 10.7 ms (Table 2). The Q–V curves (Fig. 6C) fit a Boltzmann equation, with Qmax = 10.6 nC, V0.5 = −27 mV (Table 2) and z = 1. At pH 7.5, the predicted values of Qmax and V0.5 are 6.5 nC and −70 mV, and again this is consistent with the experimental results.
The behaviour of hPEPT1 charge movements as a function of the external concentration of electroneutral substrates such as Gly-Sar and cefadroxil is also predicted by the model. As shown in Fig. 7 and Table 2, the reduction in charge transfer by substrates is simulated both qualitatively and quantitatively. The predicted reductions in Qmax with [Gly-Sar] (Fig. 7B) and [cefadroxil] (Fig. 7C) follow hyperbolic relationships, with 0.6 mm (Gly-Sar) and 2.5 mm (cefadroxil). In addition, the model correctly predicts the shift in V0.5 due to 10 mm Gly-Sar and cefadroxil (Table 2).
The results suggest that the lower transport rate of cefadroxil relative to Gly-Sar is due to (i) a lower affinity and (ii) a lower turnover rate. The values of are one order of magnitude below those of (Fig. 5C and D, and Table 1). At −50 mV, the predicted is 6.2 mm, fivefold higher than (Table 1). The cefadroxil current maxima are expected to be half of those for Gly-Sar (Table 1). The turnover number, calculated as the ratio of Imax at −150 mV to Qmax (Loo et al. 1993), is 130 s−1 for Gly-Sar and 70 s−1 for cefadroxil (Tables 1 and 2).
Interpretation of the model
Our model predicts that, in a transport cycle, at saturating substrate concentration, −50 mV and pH 5.0, the rate limiting step for substrate (Gly-Sar) and drug (cefadroxil) transport by hPEPT1 is the return of the empty transporter from the internal to the external membrane surface (C6 → C1) (Fig. 9). The difference in turnover rate between Gly-Sar and cefadroxil is due to a difference in Imax, the maximal transport rate. Imax is not solely determined by the rate-limiting step, but depends on all the rate constants in the transport cycle (see eqns A37 and A41 of Parent et al. 1992b). Imax is reduced in cefadroxil because of a lower binding rate (see legend to Fig. 9). Even with the reduction in k23 from Gly-Sar to cefadroxil from 105 to 1.25 × 104m−1 s−1, the cefadroxil binding rate k°23 × [cefadroxil] is greater than k61, that is, C6 → C1 remains rate limiting. The shift of V0.5 to more negative values, and the reduction of Qmax with increasing concentrations of substrate and drug are consistent with the model prediction that there is a shift in the distribution of carrier states. In the presence of H+ alone, at pH 5.0 and Vm = −50 mV, the occupancy probabilities are C1, 0.13; C2, 0.59; C3, 0; C4, 0; C5, 0.003; C6, 0.26; C7, 0.02. At a saturating concentration of Gly-Sar (10 mm), the occupancy probabilities are: C1, 0.04; C2, 0.06; C3, 0.09; C4, 0.02; C5, 0.10; C6, 0.69; C7, 0.002. At saturating cefadroxil (100 mm), they are: C1, 0.02; C2, 0.03; C3, 0.2; C4, 0.14; C5, 0.08; C6, 0.53; C7, 0.001. Thus, the negative shift of V0.5 and the reduction in Qmax are due to the shift of the transporter from being predominantly in state C2 in the presence of H+ alone to being in state C6 in the presence of saturating substrate concentrations, at Vm = −50 mV. This contrasts with kinetic models for other transporters, such as the Na+–glucose cotransporter SGLT1 and the Na+–iodide cotransporter NIS. In these, the rate-limiting step seems to be the dissociation of the driving cation into the cytoplasm (C5 ⇌ C6) (Eskandari et al. 1997; Loo et al. 1998).
The set of rate constants for hPEPT1 (Fig. 9) indicate that the system is asymmetric, e.g. in the absence of a driving force provided by the pH gradient (external and internal pH 7.5), the predicted values for inward and outward transport are 6 and 65 mm. This is consistent with the report by Kottra & Daniel (2001) that Gly-l-Gln transport by rabbit PEPT1 was asymmetric: in the absence of a pH gradient, the K0.5 was 0.7 mm in the inward direction and 3.3 mm in the outward direction.
The nature of the interactions (C2 ⇌ C7) between the transporter and the ligands (H+, Gly-Sar and cefadroxil) in the voltage-dependent inhibition of hPEPT1-mediated transport is not clear. The data indicate that there is a competition between protons and substrates. Our model for this competition assumes that the inhibition at low pH and large negative potentials is due to the binding of a second proton to the transporter to form state C7, after the first H+-binding site is occupied. Our simulations (not shown) indicate that the substrate and/or drug can not bind to the transporter in state C7, otherwise high Gly-Sar concentrations would increase rather than relieve the inhibition by protons (see for example Fig. 5A). While the strength of the interaction, represented by the pseudo-rate constant , is independent of substrate concentration, it is determined by the nature of the substrate per se. We estimate to be 5 × 105m−1 s−1 for Gly-Sar, and 3 × 106m−1 s−1 for cefadroxil. The higher value of for cefadroxil simply reflects the relatively poorer ability of the cephalosporin to overcome the inhibitory effects of protons, due perhaps to the lower affinity of this drug.
Recently, Inui and coworkers (Irie et al. 2005) proposed a model for the transport of charged and neutral substrates by hPEPT1. In their model, two protons bind first (CoHH), and then anionic substrates can bind to the two-proton-bound transporter (Fig. 3 of Irie et al. 2005). Without further assumptions, it is not clear how their model can account for the inhibition of the electroneutral Gly-Sar transport at low pH and negative membrane voltages. Our simulations indicate that additional constraints include the requirement that the binding of the second proton does not involve a charge movement, and neutral substrates can not bind to the two-proton bound transporter. With these additional assumptions, our model is equivalent to that proposed by Irie et al. (2005) for neutral substrates.
The inhibition of substrate transport at low substrate concentrations and large negative membrane voltages appears to be a common observation in cation-driven cotransporters. Besides hPEPT1, it has been observed in the plant H+–hexose cotransporter (STP1) cloned from Arabidopsis thaliana (Boorer et al. 1994) and the rat Na+–Cl−–GABA transporter rGAT1 (Soragna et al. 2005). The molecular structural basis for the inhibition observed in GAT1 is suggested in the recently obtained crystal structure of LeuTAa, a bacterial homologue of GAT1: it is found that, at the first Na+-binding site, the Na+ interacts with the leucine substrate (Yamashita et al. 2005).
Testing the pharmacogenetics hypothesis
According to this hypothesis, the interindividual variability in drug response is due to sequence variations in genes affecting drug disposition (Sadée, 1999). To test this hypothesis for hPEPT1, we investigated the cellular phenotype of the two common variants, S117N (25%) and G419A (8%) (Fig. 1).
S117N and G419A maintained the essential features of hPEPT1 function: (i) the levels of protein expression in Xenopus oocytes, (ii) the acid stimulation of radiolabelled Gly-Sar uptake (Fig. 2A), and (iii) the kinetic characteristics of Gly-Sar transport (Fig. 5C and Table 1). No significant differences were found among WT and variants with regards to the characteristics of voltage-dependent charge movement, such as (i) effect of pH, (ii) τON–V curves (Fig. 6B and Table 2), (iii) values of τOFF (Table 2), and (iv) Q–V relationships (Fig. 6C and Table 2). Both variants also shared similar characteristics with the WT with respect to their drug interactions, such as (i) transport of amoxicillin, ampicillin, cephalexin, cefadroxil, bestatin and NAAG (Fig. 3B), (ii) blocking effects of most drugs on Gly-Sar-evoked currents (Fig. 3C), (iii) inhibition of radiolabelled Gly-Sar uptake by cephalexin and cefadroxil (Fig. 2B), and (iv) effect of all compounds on presteady-state charge distribution, as judged by (a) reduction in Qmax, and (b) hyperpolarization of V0.5 (Fig. 8 and Table 2). Some subtle differences were observed, e.g. the apparent affinity of δ-ALA transport was significantly lower in the variants than in the WT, as evaluated by inhibition of radiolabelled Gly-Sar influx (Fig. 2B) and induction of currents (Fig. 3B).
Our results indicate that, under normal physiological conditions, S117N and G419A are functionally neutral. These findings are supported by more limited studies of the variants expressed in HeLa, Cos7 and CHO cells (Zhang et al. 2004; Anderle et al. 2006).
In summary, ampicillin, amoxicillin, cephalexin, cefadroxil, δ-ALA, bestatin and NAAG are substrates for hPEPT1, as indicated by their ability to generate inward currents. The currents induced by the drugs in the presence of Gly-Sar are equal or less than those due to Gly-Sar alone, indicating that the drugs are transported at a lower turnover rate than the dipeptide and/or that substrates and drugs interact to decrease the total rate of transport by hPEPT1. All compounds modified the kinetics of hPEPT1 charge distribution, by leading to concentration-dependent decrease in Qmax and hyperpolarization of V0.5. Steady-state and presteady-state data suggest that the substrate specificity of hPEPT1 at pH 5.0 is Gly-Sar > NAAG, δ-ALA, bestatin > cefadroxil, cephalexin > ampicillin, amoxicillin. This indicates that the presence of natural peptides at physiological concentrations in the gut may reduce the oral availability of these drugs. The essential kinetic and drug recognition features of the WT are retained by the common hPEPT1 variants S117N and G419A. To explain the global behaviour of hPEPT1 in the presence of neutral substrates, we extended our previous 6-state kinetic model and obtained a comprehensive set of parameters that fit our steady-state and presteady-state data qualitatively and quantitatively. Our model suggests that (i) dipeptides and hPEPT1 drug substrates are transported by the same mechanism, (ii) the rate-limiting step in the hPEPT1 transport cycle is the reorientation of the empty carrier within the membrane, and (iii) variations in the rate of drug cotransport are due to differences in affinity and turnover rate.
Acknowledgments
We wish to thank Teresa Ku for the preparation and management of oocytes, and the UCSF Pharmacogenetics Core Facility for the hPEPT1 variants Ser117Asn and Gly419Ala in pGEM. This work was supported by National Institutes of Health Grants DK19567 and GM61390 (to E.M.W.). Monica Sala-Rabanal was supported by a Postdoctoral Scholarship from the Ministerio de Educación y Ciencia (Government of Spain).
References
- Anderle P, Nielsen CU, Pinsonneault J, Krog PL, Brodin B, Sadee W. Genetic Variants of the human dipeptide transporter PEPT1. J Pharmacol Exp Ther. 2006;316:636–646. doi: 10.1124/jpet.105.094615. [DOI] [PubMed] [Google Scholar]
- Boll M, Markovich D, Weber WM, Korte H, Daniel H, Murer H. Expression cloning of a cDNA from rabbit small intestine related to proton-coupled transport of peptides, β-lactam antibiotics and ACE-inhibitors. Pflugers Arch. 1994;429:146–149. doi: 10.1007/BF02584043. [DOI] [PubMed] [Google Scholar]
- Boorer KJ, Loo DDF, Wright EM. Steady-state and presteady-state kinetics of the H+–hexose cotransporter (STP1) from Arabidopsis thaliana expressed in Xenopus oocytes. J Biol Chem. 1994;269:20417–20424. [PubMed] [Google Scholar]
- Bretschneider B, Brandsch M, Neubert R. Intestinal transport of β-lactam antibiotics: analysis of the affinity at the H+–peptide symporter (PEPT1), the uptake into Caco-2 cell monolayers and the transepithelial flux. Pharm Res. 1999;16:55–61. doi: 10.1023/a:1018814627484. [DOI] [PubMed] [Google Scholar]
- Daniel H. Molecular and integrative physiology of intestinal peptide transport. Annu Rev Physiol. 2004;66:361–384. doi: 10.1146/annurev.physiol.66.032102.144149. [DOI] [PubMed] [Google Scholar]
- Daniel H, Kottra G. The proton oligopeptide cotransporter family SLC15 in physiology and pharmacology. Pflugers Arch. 2004;447:610–618. doi: 10.1007/s00424-003-1101-4. [DOI] [PubMed] [Google Scholar]
- Diez-Sampedro A, Hirayama BA, Osswald C, Gorboulev V, Baumgarten K, Volk C, Wright EM, Koepsell H. A glucose sensor hiding in a family of transporters. Proc Natl Acad Sci U S A. 2003;100:11753–11758. doi: 10.1073/pnas.1733027100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doring F, Walter J, Will J, Focking M, Boll M, Amasheh S, Clauss W, Daniel H. Delta-aminolevulinic acid transport by intestinal and renal peptide transporters and its physiological and clinical implications. J Clin Invest. 1998;101:2761–2767. doi: 10.1172/JCI1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandari S, Loo DDF, Dai G, Levy O, Wright EM, Carrasco N. Thyroid Na+–I− symporter. Mechanism, stoichiometry and specificity. J Biol Chem. 1997;272:27230–27238. doi: 10.1074/jbc.272.43.27230. [DOI] [PubMed] [Google Scholar]
- Fei YJ, Kanai Y, Nussberger S, Ganapathy V, Leibach FH, Romero MF, Singh SK, Boron WF, Hediger MA. Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature. 1994;368:563–566. doi: 10.1038/368563a0. [DOI] [PubMed] [Google Scholar]
- Forster IC, Loo DDF, Eskandari S. Stoichiometry and Na+ binding cooperativity of rat and flounder renal type II Na+–Pi cotransporters. Am J Physiol. 1999;276:F644–F649. doi: 10.1152/ajprenal.1999.276.4.F644. [DOI] [PubMed] [Google Scholar]
- Ganapathy ME, Brandsch M, Prasad PD, Ganapathy V, Leibach FH. Differential recognition of β-lactam antibiotics by intestinal and renal peptide transporters, PEPT 1 and PEPT 2. J Biol Chem. 1995;270:25672–25677. doi: 10.1074/jbc.270.43.25672. [DOI] [PubMed] [Google Scholar]
- Ganapathy ME, Huang W, Wang H, Ganapathy V, Leibach FH. Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem Biophys Res Commun. 1998;246:470–475. doi: 10.1006/bbrc.1998.8628. [DOI] [PubMed] [Google Scholar]
- Ganapathy ME, Prasad PD, Mackenzie B, Ganapathy V, Leibach FH. Interaction of anionic cephalosporins with the intestinal and renal peptide transporters PEPT 1 and PEPT 2. Biochim Biophys Acta. 1997;1324:296–308. doi: 10.1016/s0005-2736(96)00234-9. [DOI] [PubMed] [Google Scholar]
- Hediger MA, Ikeda T, Coady M, Gundersen CB, Wright EM. Expression of size-selected mRNA encoding the intestinal Na+–glucose cotransporter in Xenopus laevis oocytes. Proc Natl Acad Sci U S A. 1987;84:2634–2637. doi: 10.1073/pnas.84.9.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Subramanian P, Mosberg HI, Amidon GL. Use of the peptide carrier system to improve the intestinal absorption of 1-α-methyldopa: carrier kinetics, intestinal permeabilities, and in vitro hydrolysis of dipeptidyl derivatives of 1-α-methyldopa. Pharm Res. 1989;6:66–70. doi: 10.1023/a:1015855820488. [DOI] [PubMed] [Google Scholar]
- Inui K, Tomita Y, Katsura T, Okano T, Takano M, Hori R. H+-coupled active transport of bestatin via the dipeptide transport system in rabbit intestinal brush-border membranes. J Pharmacol Exp Ther. 1992;260:482–486. [PubMed] [Google Scholar]
- Irie M, Terada T, Katsura T, Matsuoka S, Inui K. Computational modelling of H+-coupled peptide transport via human PEPT1. J Physiol. 2005;565:429–439. doi: 10.1113/jphysiol.2005.084582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottra G, Daniel H. Bidirectional electrogenic transport of peptides by the proton-coupled carrier PEPT1 in Xenopus laevis oocytes: its asymmetry and symmetry. J Physiol. 2001;536:495–503. doi: 10.1111/j.1469-7793.2001.0495c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Fei YJ, Prasad PD, Ramamoorthy S, Han H, Yang-Feng TL, Hediger MA, Ganapathy V, Leibach FH. Human intestinal H+–peptide cotransporter. Cloning, functional expression, and chromosomal localization. J Biol Chem. 1995;270:6456–6463. doi: 10.1074/jbc.270.12.6456. [DOI] [PubMed] [Google Scholar]
- Loo DDF, Eskandari S, Boorer KJ, Sarkar HK, Wright EM. Role of Cl− in electrogenic Na+-coupled cotransporters GAT1 and SGLT1. J Biol Chem. 2000;275:37414–37422. doi: 10.1074/jbc.M007241200. [DOI] [PubMed] [Google Scholar]
- Loo DDF, Hazama A, Supplisson S, Turk E, Wright EM. Relaxation kinetics of the Na+–glucose cotransporter. Proc Natl Acad Sci U S A. 1993;90:5767–5771. doi: 10.1073/pnas.90.12.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo DDF, Hirayama BA, Gallardo EM, Lam JT, Turk E, Wright EM. Conformational changes couple Na+ and glucose transport. Proc Natl Acad Sci U S A. 1998;95:7789–7794. doi: 10.1073/pnas.95.13.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie B, Loo DDF, Fei Y, Liu WJ, Ganapathy V, Leibach FH, Wright EM. Mechanisms of the human intestinal H+-coupled oligopeptide transporter hPEPT1. J Biol Chem. 1996a;271:5430–5437. doi: 10.1074/jbc.271.10.5430. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Loo DDF, Panayotova-Heiermann M, Wright EM. Biophysical characteristics of the pig kidney Na+–glucose cotransporter SGLT2 reveal a common mechanism for SGLT1 and SGLT2. J Biol Chem. 1996b;271:32678–32683. doi: 10.1074/jbc.271.51.32678. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Loo DDF, Wright EM. Relationships between Na+–glucose cotransporter (SGLT1) currents and fluxes. J Membr Biol. 1998;162:101–106. doi: 10.1007/s002329900347. [DOI] [PubMed] [Google Scholar]
- Parent L, Supplisson S, Loo DDF, Wright EM. Electrogenic properties of the cloned Na+–glucose cotransporter. I. Voltage-clamp studies. J Membr Biol. 1992a;125:49–62. doi: 10.1007/BF00235797. [DOI] [PubMed] [Google Scholar]
- Parent L, Supplisson S, Loo DDF, Wright EM. Electrogenic properties of the cloned Na+–glucose cotransporter: II. A transport model under nonrapid equilibrium conditions. J Membr Biol. 1992b;125:63–79. doi: 10.1007/BF00235798. [DOI] [PubMed] [Google Scholar]
- Rubio-Aliaga I, Daniel H. Mammalian peptide transporters as targets for drug delivery. Trends Pharmacol Sci. 2002;23:434–440. doi: 10.1016/s0165-6147(02)02072-2. [DOI] [PubMed] [Google Scholar]
- Sadée W. Pharmacogenomics. BMJ. 1999;319:1286–1289. doi: 10.1136/bmj.319.7220.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soragna A, Bossi E, Giovannardi S, Pisani R, Peres A. Relations between substrate affinities and charge equilibration rates in the rat GABA cotransporter GAT1. J Physiol. 2005;562:333–345. doi: 10.1113/jphysiol.2004.076703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple CS, Boyd CA. Proton-coupled oligopeptide transport by rat renal cortical brush border membrane vesicles: a functional analysis using ACE inhibitors to determine the isoform of the transporter. Biochim Biophys Acta. 1998;1373:277–281. doi: 10.1016/s0005-2736(98)00093-5. [DOI] [PubMed] [Google Scholar]
- Terada T, Saito H, Mukai M, Inui K. Recognition of β-lactam antibiotics by rat peptide transporters, PEPT1 and PEPT2, in LLC-PK1 cells. Am J Physiol. 1997;273:F706–F711. doi: 10.1152/ajprenal.1997.273.5.F706. [DOI] [PubMed] [Google Scholar]
- Terada T, Sawada K, Irie M, Saito H, Hashimoto Y, Inui K. Structural requirements for determining the substrate affinity of peptide transporters PEPT1 and PEPT2. Pflugers Arch. 2000;440:679–684. doi: 10.1007/s004240000339. [DOI] [PubMed] [Google Scholar]
- Wang H, Fei YJ, Ganapathy V, Leibach FH. Electrophysiological characteristics of the proton-coupled peptide transporter PEPT2 cloned from rat brain. Am J Physiol. 1998;275:C967–C975. doi: 10.1152/ajpcell.1998.275.4.C967. [DOI] [PubMed] [Google Scholar]
- Wenzel U, Gebert I, Weintraut H, Weber WM, Clauss W, Daniel H. Transport characteristics of differently charged cephalosporin antibiotics in oocytes expressing the cloned intestinal peptide transporter PepT1 and in human intestinal Caco-2 cells. J Pharmacol Exp Ther. 1996;277:831–839. [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+–Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Zhang EY, Fu DJ, Pak YA, Stewart T, Mukhopadhyay N, Wrighton SA, Hillgren KM. Genetic polymorphisms in human proton-dependent dipeptide transporter PEPT1: implications for the functional role of Pro586. J Pharmacol Exp Ther. 2004;310:437–445. doi: 10.1124/jpet.104.065912. [DOI] [PubMed] [Google Scholar]
- Zhu T, Chen XZ, Steel A, Hediger MA, Smith DE. Differential recognition of ACE inhibitors in Xenopus laevis oocytes expressing rat PEPT1 and PEPT2. Pharm Res. 2000;17:526–532. doi: 10.1023/a:1007556630189. [DOI] [PubMed] [Google Scholar]