

Abstract
The brain operates through a coordinated interplay of numerous neurons, yet little is known about the collective behaviour of individual neurons embedded in a huge network. We used large-scale optical recordings to address synaptic integration in hundreds of neurons. In hippocampal slice cultures bolus-loaded with Ca2+ fluorophores, we stimulated the Schaffer collaterals and monitored the aggregate presynaptic activity from the stratum radiatum and individual postsynaptic spikes from the CA1 stratum pyramidale. Single neurons responded to varying synaptic inputs with unreliable spikes, but at the population level, the networks stably output a linear sum of synaptic inputs. Nonetheless, the network activity, even though given constant stimuli, varied from trial to trial. This variation emerged through time-varying recruitment of different neuron subsets, which were shaped by correlated background noise. We also mapped the input-frequency preference in spiking activity and found that the majority of CA1 neurons fired in response to a limited range of presynaptic firing rates (20–40 Hz), acting like a band-pass filter, although a few neurons had high pass-like or low pass-like characteristics. This frequency selectivity depended on phasic inhibitory transmission. Thus, our imaging approach enables the linking of single-cell behaviours to their communal dynamics, and we discovered that, even in a relatively simple CA1 circuit, neurons could be engaged in concordant information processing.
Neurons are network elements and do not work alone. Their accurate behaviour or function cannot be estimated only by observing individual neurons, but most physiological studies have focused on single or a few neurons or monosynaptic transmission, the main reason being probably the lack of appropriate experimental procedures to approach network operations. To resolve this dilemma, we adopted a functional imaging technique with multicell loading of calcium fluorophores. This technique was originally introduced by Yuste & Katz (1991) and has recently been put to practical use in vivo (Stosiek et al. 2003; Nimmerjahn et al. 2004; Ohki et al. 2005; Kerr et al. 2005). The remarkable advantages include: (i) simultaneous recordings from 100 to 1000 neurons in a wide area (> 0.1 mm2), (ii) single-cell and single-spike resolution, and (iii) identifiable locations of neurons (including inactive cells during recording periods).
We used this large-scale imaging in order to examine the response of hippocampal CA1 networks to afferent stimulation in vitro. The perspicuous CA1 network architecture can be a minimal model for understanding how single neurons and their synaptic connections combine to execute a coordinated function. In addition, its stereotyped laminar structure allows separating network input and output activities at the level of single neurons. Taking advantage of them, we aimed to examine (i) the input–output (I/O) relationship, (ii) trial-to-trial variation, and (iii) frequency-dependent spike transfer, at the network level.
Methods
Materials
Oregon green BAPTA 1-AM, Pluronic F-127 and sulforhodamine 101 were obtained from Molecular Probes (Eugene, OR, USA). Chromophore EL, sulfinpyrazone, d,l-2-amino-5-phosphonopentanoic acid (AP5), 6-cyano-7-nitroquinoxoxaline-2,3-dione (CNQX), picrotoxin and gabazine (SR-95531) were purchased from Sigma (St Louis, MO, USA). Drugs were dissolved in double-distilled water or DMSO so as to make 1000× stock solutions, except for picrotoxin (100×). The stock solutions were stored at −20°C and diluted immediately before use. They were all bath applied.
Slice preparations
Preliminary data show that, as compared with acutely prepared slices, organotypic cultures are more consistently loaded with calcium indicators and offer improved imaging resolution, presumably because of less cellular debris (data not shown; see Morita et al. 2003). Therefore we decided to use slice cultures, rather than acute slice preparations, in this study, although synaptic connections in organotypic cultures might undergo unnatural remodelling during the incubation period (Robain et al. 1994; Sakaguchi et al. 1994; Gutierrez & Heinemann, 1999).
Hippocampal slice cultures were prepared from postnatal day 7 Wistar/ST rats (SLC, Shizuoka, Japan) as previously described (Yamamoto et al. 1989; Stoppini et al. 1991; Ikegaya, 1999), according to National Institutes of Health guidelines for laboratory animal care and safety. Briefly, rat pups were chilled and decapitated with a small animal guillotine (SN-629, Shinano manufacturing cooperation, Tokyo, Japan). The brains were rapidly removed and cut into horizontal 300-μm-thick slices using a DTK-1500 microslicer (Dosaka, Kyoto, Japan) in aerated, ice-cold Gey's balanced salt solution (Invitrogen, Gaithersburg, MD, USA) supplemented with 25 mm glucose. Entorhino-hippocampal stumps were cultivated on Millicell-CM membranes (Millipore, Bedford, MA, USA) for 7–14 days. Cultures were fed with 1 ml of 50% minimal essential medium, 25% Hanks' balanced salt solution (Invitrogen), 25% horse serum (Cell Culture Laboratory, Cleveland, OH, USA) and antibiotics in a humidified incubator at 37°C in 5% CO2. The medium was changed every 3.5 days.
Ca2+ imaging
Experiments were performed in artificial cerebrospinal fluid (ACSF) consisting of (mm): 127 NaCl, 26 NaHCO3, 1.5 KCl, 1.3 KH2PO4, 1.4 MgSO4, 2.4 CaCl2, and 10 glucose), bubbled with 95% O2 and 5% CO2. Slices were washed three times with ACSF, transferred into a 35-mm dish filled with 2 ml of dye solution, and incubated for 1 h in a humidified incubator at 37°C in 5% CO2. The dye solution is ACSF containing 10 μl of 0.1% Oregon green 488 BAPTA-1AM/DMSO, 2 μl of 10% Pluronic F-127/DMSO, 2 μl of 5% Cremophor EL/DMSO, and 2 μl of 100 mm sulfinpyrazone (Ikegaya et al. 2005). The final concentrations were 0.0005% Oregon green, 0.01% Pluronic F-127, 100 μm sulfinpyrazone, 0.005% Cremophor EL, and 0.8% DMSO.
After being washed, slices were incubated at room temperature for > 30 min, mounted in a recording chamber and perfused with 32°C ACSF at a rate of 1.0–1.5 ml min−1. Incision was made between the CA2 and CA3 regions and between CA1 and the subiculum to reduce recurrent excitation. Images (653 pixels × 492 pixels, 16-bit intensity) were captured at 10 frames s−1 with a CSU10 Nipkow spinning-disk confocal microscope (Yokokawa Electric, Tokyo, Japan), equipped with a Cascade cooled CCD camera (Roper Scientific, Tucson, AZ, USA), a Zeiss AxioSkop2 microscope (Oberkochen, Germany), water-immersion objectives (20×, 0.5 NA, Achroplan, Zeiss), and Metamorph software (Molecular Devices, Union City, CA, USA). Fluorophores were excited with the 488-nm line from an argon–krypton laser (15–20 mW, 641-YB-A01, Melles Griot, Carlsbad, CA, USA) and visualized with a 507-nm long-pass emission filter. Bipolar tungsten electrodes were placed in the CA1 stratum radiatum sufficiently apart from the imaged area to avoid direct stimulation of dendrites of the monitored neurons, and a single pulse or burst train stimuli (50 μs, 60–270 μA) were applied every 30 s to activate Schaffer collateral axons. To minimize photodamage and photobleaching, a laser shutter was opened during the 3–5 s period around the stimulation under the Metamorph Journal control.
Electrophysiological recordings
Patch-clamp recordings were obtained from CA1 pyramidal cells with an Axopatch 700B amplifier (Molecular Devices). For cell-attached and whole-cell recordings, borosilicate glass pipettes (4–9 MΩ) were filled, respectively, with ACSF and internal solution consisting of (mm): 120 potassium gluconate, 20 KCl, 0.1 CaCl2, 10 Hepes, 0.2 EGTA, 3.4 MgATP, and 5 QX-314 (pH 7.2). Signals were low-pass filtered at 1–2 kHz, digitized at 20 kHz and analysed with pCLAMP 9.2 software (Molecular Devices).
Data analysis
Spikes were reconstructed from neurons by using custom-written software in NIH ImageJ (Bethesda, MD, USA) and Microsoft Visual Basic (Redmond, WA, USA), as previously described (Ikegaya et al. 2004). For each cell and each stimulus, the fluorescence change ΔF/F was calculated as:
where F1 is fluorescence intensity at any time point, FBACK is the average background obtained from the whole area of movie at the corresponding time point, F0 is the average baseline of the cell during each prestimulus epoch, and FBACK,0 is the average background baseline of the whole image during each prestimulus epoch. Compensation with FBACK was required because stimulation-evoked presynaptic (probably also dendritic) signals were massively synchronized especially when stimulated at high intensities, contributing to somatic signals as well. Thus, without this compensation, signals of non-spiking cells were contaminated with small Ca2+ transients due to synchronized background signals and occasionally misjudged as spike-triggered transients. In our movies, the total area of the monitored somata constituted only 11.8 ± 1.8% of the whole confocal field, so our ΔF/F measure did not significantly underestimate spike-triggered signals even during synchronous cell activation. Spike-triggered Ca2+ signals were automatically determined as a transient with > 3% amplitude, > 3.5% s−1 maximal first derivative (Δt = 0.7 s) and 1- to 2-frame peak latency. They were then inspected by eye to remove noise detected in error. Spike-related events had fast-rise and slow-decay kinetics (τ > 0.3 s), so they were separable from background or optical noise (Fig. 1B and C). To estimate the total amount of presynaptic inputs, the fluorescence change in the presence of 20 μm CNQX and 50 μm d,l-AP5 was obtained from CA1 stratum radiatum by placing a box (100-μm × 100-μm). This measure might reflect glial Ca2+ waves as well. But the area of glial cells occupied only a small portion (less than 5%) in the entire monitored region. Moreover, glial waves were usually unlocked to electric stimuli, and if there were any, they had much slower kinetics than spike-triggered neural Ca2+ transients. Thus, glial Ca2+ waves contributed only minimally to the amplitude of presynaptic Ca2+ transients, which had a latency of 1 frame. Data were discarded when massive spontaneous activity occurred during imaging.
Figure 1. Reliable reconstruction of action potentials from somatic ΔF/F traces of Ca2+ fluorophores.

A, confocal image of the CA1 pyramidal cell layer of a hippocampal slice bulk-loaded with Oregon green 488 BAPTA-1. B, simultaneous recording of cell-attached extracellular responses and somatic Ca2+ transients with (Spike) and without an action potential (No spike). Stratum radiatum (SR) was stimulated. A portion of the trace (indicated by bar) is magnified in the bottom inset (25 ×). C, left: distribution of the ΔF/F amplitudes in trials with and without spikes. The amplitude was defined as the maximal ΔF/F (%) during the 1 s period after a stimulus. Trials with spikes and without spikes are completely separable. Data of five cells were pooled. Right: relationship between the number of spikes and the amplitude of the resultant ΔF/F increase. Multiple spikes were evoked by repetitive stimulation at 50 Hz, and their actual spike numbers were confirmed with cell-attached patch-clamp recordings. If each pulse in a 50 Hz train did not elicit the corresponding spike, the data were discarded. D, optical signals recorded from the CA1 stratum radiatum (SR), the subregion that contains presynaptic terminals of Schaffer collateral axons, in the presence of 20 μm CNQX and 50 μm d,l-AP5 to block synaptic transmission. Unlike somatic Ca2+ transients, the presynaptic Ca2+ size gradually increased as a function of the electric stimulation intensity.
To estimate the chance level of trial-to-trial variations (Figs 3D and E and 4B), we created spike-matrix surrogates, in which, under the null hypothesis of time- and space-independent processes, we scattered ‘fake’ spikes while maintaining the mean firing probability (Monte Carlo simulation), that is, a procedure in which a spike (‘active bin’) randomly selected from a rasterplot was exchanged with another randomly selected bin, and this was repeated for all active bins. This procedure eliminates a temporal correlation between the spikes without changing the total number of events. For this surrogate, we computed the standard deviation (s.d.) of the summed activity through all cells. This simulation was repeated 100–1000 times to calculate the mean ± s.d. of these s.d. values. In addition, we carried out two other derivatives of this simulation. First, a surrogate rasterplot was created by exchanging individual active bins with either right or left neighbouring bins (Fig. 4C). This procedure collapses temporal correlations between the events but preserves the activity level (i.e. spike probability) of each cell as well as a slow fluctuation of global excitability over time. Second, a surrogate rasterplot was generated by placing vertically clustered bins (3- to 13-cell clusters). This procedure mimics cell-assembly like behaviours (Fig. 4D).
Figure 3. More-than-chance variability of network responses to the same stimuli.

A, representative traces of presynaptic (top) and postsynaptic Ca2+ transients (bottom). Spike and no spikes are shown in black and white, respectively, in the bottom boxes. B, spike responses of 132 cells to 50 successive stimuli. Cell locations are shown in the right-hand map, in which the firing probability of each neuron is indicated in the grey scale. C, number of neurons versus firing probability. D, percentage of activated neurons to the total number of neurons recorded. The broken line (38.7%) shows the mean percentage for all 50 trials. The right-hand two histograms show the distributions (trial-to-trial fluctuations) of the percentage of activated neurons in the original rasterplot and a Monte Carlo-generated surrogate (see text). E, summarized data for all 112 experiments in 28 slices. The across-trial s.d. of the percentage of spiking neurons is plotted against the average firing probability. Each pair of circles indicates an experiment. Open circles, s.d. of real data; filled circles, the Monte Carlo-estimated average s.d. ± s.d. (n = 100 surrogates). Arrow indicates the data shown in panels B–D.
Figure 4. Cell assembly like dynamics can explain trial-to-trial variability: numerical simulation.

A, spike responses of 148 neurons to 50 repetitive constant stimuli. In the 148 × 50 rasterplot, 3159 activities (black bins) were seen. Bottom histogram represents the percentage of active cells to the total at each trial. The s.d. value of the summed activities across trials was 8.8. B, surrogate rasterplot created by Monte-Carlo simulation. By using a random number generator, we scattered 3159 ‘black’ bins in the 148 × 50 blank rasterplot. In this ‘fake’ rasterplot, s.d. was calculated to be 3.7. We repeated this simulation 1000 times and showed the distribution of s.d.s in the right-bottom panel, in which the s.d. of original rasterplot data (s.d. = 8.8) is indicated by the arrow (real data). The real s.d. was consistently higher than s.d.s obtained with 1000 simulations, indicating that the trial-to-trial variation seen in the real net activity cannot be explained by simple stochastic dynamics. C, surrogate rasterplot that was created by exchanging individual activities with either right or left neighbouring bins. This procedure collapses temporal correlations between the events but preserves the activity level (spike probability) of each cell as well as slow fluctuations of global excitability over time. The real s.d. was still higher than expected by this simulation. D, surrogate rasterplot generated by putting vertically clustered bins. This procedure mimics cell-assembly-like behaviours, which was implied by our cluster analyses in Fig. 5. Simulation was carried out by allocating 3-to-13-cell clusters. When the cluster size was larger than 7 cells, the real s.d. was not significantly different from the chance level.
To reveal neuron cliques (Fig. 5B–E), we computed the normalized dot product between all possible pairs of cells <i,j> and constructed the similarity matrix S (Schreiber et al. 2003). If the spike train  (i = 1, …, N) of cell i is represented as a binary vector (1: spike, 0: no spike), the similarity index (type 1) between
(i = 1, …, N) of cell i is represented as a binary vector (1: spike, 0: no spike), the similarity index (type 1) between  and
and  is defined as:
is defined as:
which is equivalent to the cosine of the angle between  and
and  , taking values between 0 and 1. Unless otherwise specified, we used this definition as a similarity index.
, taking values between 0 and 1. Unless otherwise specified, we used this definition as a similarity index.
Figure 5. Cell-assembly-like dynamics underlie trial-to-trial diversity: cluster analysis.

A, the same data as shown in Fig. 4A. B, the rasterplot was converted to a similarity matrix to construct a dendrogram. In this dendrogram, the right-most eight cells had no spikes for any trials. C–D, three examples of neuron groups extracted by the K-means algorithm (C) are shown in the cell map (D). Arrowheads mark trials where all cells in the same neuron group were activated. E–G, cluster dynamics are robust, evidenced by three different similarity indexes (types 1–3). For the definition of these indexes, see Methods. The similarity indexes are all represented in the form of grey-scaled matrices. For each panel, the left-hand matrix is shown in the order of the neuron number (A), the right-hand one in the order assorted with the K-means analysis with 16 clusters.
In addition, we used two other similarity indexes (type 2 and type 3). Because the dot product is normalized by the vector lengths, the type-1 index treats highly active cells and relatively silent cells in the network as equally important entities. To take activity weights into consideration therefore we introduce the similarity index type 2 defined as:
which is a simple dot product, an unnormalized version of the index type 1. It purely reflects how many spikes concur in two cells. In this index, highly active cells tend to be overestimated because these active cells numerically have higher probabilities of coactivation with other cells. We thus used another index (type 3). We digitized a spike train  as a ‘1’ and ‘−1’ binary vector (1: spike, −1: no spike), instead of a ‘1’ and ‘0’ vector, and defined the similarity index type 3 as
as a ‘1’ and ‘−1’ binary vector (1: spike, −1: no spike), instead of a ‘1’ and ‘0’ vector, and defined the similarity index type 3 as 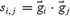 , in which normalization is unnecessary because all vectors have the same length
, in which normalization is unnecessary because all vectors have the same length  . In this index, simultaneous ‘activity’ and ‘silence’ of two cells are equally weighted.
. In this index, simultaneous ‘activity’ and ‘silence’ of two cells are equally weighted.
A hierarchical clustering algorithm was applied to the similarity matrix to construct a dendrogram that assembles all elements into a single tree (Fig. 5B). The matrix was scanned to identify the highest value, and a node was created by joining these two cells. The similarity matrix was updated with this new node replacing the two joined elements. The same process was repeated N − 1 times until only a single element remained. We also manually highlighted neuron groups with the aid of a modified K-means algorithm (Fig. 5C). Cells were randomly assigned to one of K clusters, and the K centroids were determined. Each cell was re-assigned to a cluster to minimize the distance to the cluster centroid, and new centroids were determined. This procedure was iterated until the optimum assignment was attained. Then, the number of clusters (K) was increased with , and the same algorithm was repeated.
To determine whether synaptic inputs and membrane potentials immediately before stimulation influence subsequent spike generation, we compared intracellular traces simultaneously recorded from two cells by computing their Euclidian distance (Fig. 6B). For 30–250 ms fractions of the traces xt and yt before stimulation, the modified Euclidian distance ED was calculated as:
 |
where  and
and  represents the mean membrane potential xt and yt, respectively, between 550 and 30 ms before stimulation, and Δt is the time resolution of whole-cell recordings (Δt = 50 μs).
represents the mean membrane potential xt and yt, respectively, between 550 and 30 ms before stimulation, and Δt is the time resolution of whole-cell recordings (Δt = 50 μs).
Figure 6. Spontaneous synaptic noise is correlated, synchronizing spikes between neurons.

A, representative traces of simultaneous whole-cell recordings from two adjacent neurons. EPSCs (top) and IPSCs (bottom) were isolated by clamping at −90 and 0 mV, respectively. Coincident EPSCs/IPSCs (2 ms jitter) are indicated by broken lines. The right-hand panels are cross-correlograms for EPSC/IPSC relative timings (2 ms bin, n = 4 pairs). B, two neurons responded to an electric stimulus with spikes (S) or no spikes (N). Paired responses to each trial are classified into four types, i.e. S–S, N–N, S–N, and N–S. The modified Euclidian distance between two intracellular traces (from 250 to 30 ms before a stimulus, 40 consecutive trials) was plotted versus these response types (right). *P = 0.031, t38 = 2.25; t test after F-test.
We report the means ± s.d. in all measurements.
Results
Virtually all CA1 neurons in cultured hippocampal slices were loaded with Oregon green 488 BAPTA-1AM (Fig. 1A). To prevent dye leakage, we performed cell-attached recordings of a dye-loaded CA1 neuron and simultaneously measured the fluorescence intensity from the cell body. Each stimulus of the Schaffer collaterals elicited or failed to elicit an action potential in the neuron (Fig. 1B), and the occurrence of a spike was tightly associated with the occurrence of a somatic ΔF/F transient, the amplitude of which was almost constant among trials (Figs 1C left, 2B and 3A) and could be separated from optical noise (Fig. 1C left). This all-or-none nature of somatic ΔF/F signals was not due to the dye saturation because the somatic ΔF/F amplitude increased as the number of spikes involved in a burst increased (Fig. 1C right). Thus, the somatic ΔF/F transients reflect spike outputs, rather than postsynaptic potential, of the observed neurons.
Figure 2. Quasilinear I/O relationship of the CA1 network at the population level.

A, cell map: multineuronal optical recordings from 101 neurons located in CA1 stratum pyramidale. B, presynaptic ΔF/F traces were recorded from the stratum radiatum in the presence of 20 μm CNQX and 50 μm d,l-AP5, whereas postsynaptic ΔF/F traces were recorded from 101 somata in the absence of these antagonists. Stimulus strength was gradually increased from left to right. Postsynaptic activities were converted to a black-and-white rasterplot (right), which further was summed into the bottom histogram showing the percentages of spiking neurons to the total. C, summary of the I/O-transfer relationship of the CA1 network. Top: the average percentages of spiking neurons (i.e. postsynaptic net responses) are plotted against presynaptic activity levels normalized to the maximal, saturated Ca2+ transient size. The stimulus intensity that activated 50% neurons was 123 ± 16 μA (n = 6 slices). Bottom: data were collected from the same slices after exposure to 200 nm and 10 μm gabazine.
We monitored presynaptic activity from CA1 stratum radiatum in the presence of the non-NMDA receptor antagonist CNQX (20 μm) and the NMDA receptor antagonist d,l-AP5 (50 μm) to prevent a possible contamination of postsynaptic activity, e.g. dendritic Ca2+ influx induced by action potential back-propagation (Jaffe et al. 1992; Spruston et al. 1995) or local synaptic activity (Yuste & Denk, 1995). Assuming the uniform distribution of afferent fibres and uniform dye loading, one can consider that ΔF/F is proportional to the total number of presynaptic fibres activated by stimulation, i.e. the sum of presynaptic inputs (Kerr et al. 2005). Indeed, the ΔF/F peak increased in a gradual manner as the stimulus was strengthened (Figs 1D and 2B top). They were almost unchanged for a constant stimulus intensity (Fig. 3A); the coefficient of variance (CV) in the ΔF/F size was only 0.06 ± 0.03 for 50 constant stimuli (n = 3 slices). Therefore, we were able to separate network input and output activities, even though not at the same time, in the same slice.
Quasilinear I/O relationship at the population level
In Fig. 2A and B, spike responses were reconstructed from 101 neurons. As stimulus intensity was increased from 60 to 170 μA, more cells fired spikes. We generated a black-and-white rasterplot that indicated a spike or no spike of each cell at each stimulus intensity (Fig. 2B right) and then collapsed it into an activity histogram, which represents the percentage of activated cells at a given stimulus intensity (Fig. 2B right-bottom).
The averaged I/O relationship is illustrated by plotting the percentage of activated neurons against presynaptic activity levels, i.e. ‘optical fibre volleys’ (Fig. 2C top, n = 6 slices, each 5 trials). The number of spiking cells increased nearly linearly with the sum of presynaptic input. Presynaptic ΔF/F transients (abscissa) and postsynaptic spike responses (ordinate) reached 100% almost at the same point. The presynaptic 100% point was defined as the saturated ΔF/F level obtained at the highest stimulus intensity, whereas postsynaptic 100% indicates the point at which all observed CA1 neurons fired. Therefore, the I/O relationship was almost linear across the entire activity range (see also Kerr et al. 2005).
Gabazine, a GABAA receptor antagonist, abolished the linearity; gabazine reduced the minimal stimulus intensity required to activate all neurons and the threshold intensity at which the network started to react to stimulation (Fig. 2C bottom). It did not affect the relation between stimulus intensities and optical presynaptic fibre volleys (online Supplemental material Fig. 2A). Non-averaged raw data of these experiments are shown in Supplemental Fig. 2B. Similar results were obtained with 50 μm picrotoxin, another GABAA receptor antagonist (data not shown). It is reported that unlike picrotoxin, gabazine at low concentrations (e.g. 200 nm) blocks phasic inhibition without affecting tonic inhibition, but it blocks both phasic and tonic inhibition at higher concentrations (e.g. 10 μm) (Stell & Mody, 2002). We confirmed this with whole-cell patch-clamp recordings (Supplemental Fig. 1). Gabazine-induced collapse of the I/O linearity was found at 200 nm (Fig. 2C bottom). Therefore, phasic inhibition, rather than tonic inhibition, is likely to be responsible for the I/O linearity.
Trial-to-trial variability of postsynaptic neuronal responses
Upon examining the I/O relationship, we noticed that spike responses of individual cells varied from trial to trial (Fig. 3A). An example of trial-to-trial variation of 132 spiking cells during 50 consecutive stimuli at a fixed intensity (120 μA) is shown in Fig. 3B. A few neurons responded faithfully to the repetitive stimuli, whereas others fired much less reliably. Figure 3C illustrates the distribution of the mean firing probability of each cell. Figure 3D left histogram indicates the trial-to-trial fluctuation in the percentage of activated cells, which further was collapsed into the Fig. 3D middle histogram as a distribution map. The percentage of activated cells was 38.7% on average, but it unstably went up and down across trials, with s.d., i.e. the degree of trial-to-trial variation, equal to 8.9%. Thus, the postsynaptic net CV (0.23 = 8.9/38.7) was significantly higher than that of presynaptic ΔF/F (= 0.06 ± 0.03), indicating that presynaptic variations, e.g. stochastic release of neurotransmitter or trial-to-trial variation in the number of presynaptic fibres activated by electric stimulation, if any, cannot fully explain the postsynaptic net variability.
To estimate the chance level of s.d., we performed Monte Carlo simulation, in which we created a surrogate rasterplot with a random number generator by setting the mean firing probability to 38.7%. In this ‘mock’ rasterplot, the s.d. value was 4.1%, smaller than that of the real data (Fig. 3D right). This held true for all 1000 repetitions of simulation. Another example is shown in Fig. 4B. We repeated the same experiments with different stimulus intensities (60–270 μA) and showed the data in Fig. 3E (n = 112 experiments), in which the s.d. values are plotted versus the overall mean firing probability because s.d. depends critically on the firing probability; note that s.d. naturally takes smaller values if the firing probability is extremely high or low. In 83 of 112 experiments, s.d.s (open circles) were significantly higher than chance (linked filled circles).
In the example shown in Fig. 3D, there happened to be a trend for spike probability to decrease over time. Although in most cases, network excitability was invariant over time (for instance, see Fig. 4A), we sometimes observed such slow fluctuations (gradual increase/decrease) in firing probability, like Fig. 3D. It is possible therefore that such gradual changes in global excitability might contribute to more-than-chance variability. We can partially rule out this possibility, based on two following reasons, however. First, the data shown in Fig. 3B were not extraordinarily different form other data in terms of the s.d. value and its chance level (indicated by the arrow in Fig. 3E), but rather they correspond to a case in which the variability was close to the chance level. Second, statistical significance was still confirmed by a non-stationary shuffling in which spike positions were randomly exchanged with either right or left neighbouring bins in the rasterplot (P < 0.001; data not shown); note this procedure preserves the slow fluctuation in global excitability (see Fig. 4C). Thus, the net variability cannot be fully accounted for by simple stochastic behaviours of individual cells or a slow change in net excitability.
There were commonly some trials which produced extremely high or low responses (for instance, Trial no. 16 in Fig. 3B and Trial no. 12 in Fig. 4A). To examine how much such occasional outliers contributed to the measure of variability, we again utilized the non-stationary shuffling (Fig. 4C). This procedure eliminated such extreme trials with preserving the spike probability of each cell as well as slow fluctuations of global excitability over time. s.d. obtained from this simulation was significantly lower than that of the real rasterplot. Therefore, occasional hyperactive or hypoactive trials significantly contributed to trial-to-trial variation, although they seemed unlikely to fully explain the total variation.
We now hypothesize that the net variation must come from certain structured dynamics, e.g. cell-assembly-like dynamics (see Supplemental Movie 1; the stimuli no. 2 and no. 4 invoked a similar cell subpopulation). To address this possibility, we computed the similarity of activation patterns between pairs of neurons. In 148-cell recordings in Fig. 5A, we compared 21 756 (= 148 × (148 − 1)) cell pairs by computing the type-1 similarity indexes (see Methods) and depicted a dendrogram. The configuration of the dendrogram, as expected, implies the existence of neuron subgroup dynamics (Fig. 5B). We next used the K-means algorithm as another cluster analysis. This clustering algorithm is sensitive to the initial cluster assignments and often drops into different local minima so that we can identify indistinct quasi-clusters by reiterating the clustering procedure. In Fig. 5C, we illustrated some examples of neuron groups found in the rasterplot in Fig. 5A. Interestingly, identical neurons often participated in different groups (for instance, neuron no. 55 is involved in Groups no. 1 and no. 2). Therefore, the subgrouping seems to be heterogeneous and flexible in nature. Neurons in the subgroups did not have any specific architecture in network geometry and were dispersed in space (Fig. 5D). To determine whether these cluster dynamics are robust, we computed three types of similarity indexes (types 1–3) and sorted their matrices with the K-means algorithm (see Methods). Clusters were evident in all cases (Fig. 5E–G). Therefore, the cluster dynamics are not mathematical artifacts.
We next questioned whether the cluster dynamics contribute to trial-to-trial variation. We modified the Monte-Carlo method. If the neuron order in a rasterplot, which is arbitrarily assigned, is ignored, cell clusters are conceptually identical to vertically grouped active bins. We thus scattered vertically clustered active bins (3–13 consecutive bins) in a blank rasterplot by keeping the total number of active bins equal to that in the original rasterplot (Fig. 4D). The trial-to-trial variability (i.e. s.d.) seen in this simulation was now similar to that in real data if the cluster size was larger than seven bins. Thus, the subgroup dynamics can increase the net variability to a realistic point.
What shapes the cluster dynamics, then? The most probable cause is spontaneously occurring synaptic activity because we activated the CA1 circuit by applying single-pulse stimulation per trial. To examine whether spontaneous activity is temporally correlated among neurons so as to yield the subgroup dynamics, we performed dual patch-clamp recordings from pairs of CA1 pyramidal neurons. Voltage was clamped at −90 and 0 mV to singularize EPSCs and IPSCs, respectively (Fig. 6A traces). As we expected, the correlograms revealed that spontaneous EPSCs and IPSCs were both synchronized between neurons, IPSCs being more correlated (Fig. 6A). Within a 2 ms jitter, 14.8% EPSCs and 34.0% IPSCs concurred in two neurons (n = 4 pairs).
To determine how much correlated noise contributes to coherent spike dynamics, we monitored stimulation-evoked spikes of two neurons in the current-clamp configuration. Stimulus intensity was adjusted to generate 30–70% of firing probabilities in both neurons. When a spike occurred, its latency relative to stimulation was short and invariant across trials and across cells; thus it reflected a monosynaptic event. Across trials, two neurons variably responded with spikes (S) or without spikes (N). As to response combinations, four types of paired responses existed, i.e. S–S, N–N, S–N, and N–S (for example, S–S denotes that both neurons fired). They can roughly be classified into two groups, i.e. the same behaviours (S–S or N–N) and different behaviours (S–N or N–S). To examine whether these behaviours resulted from the differences in preceding membrane potentials, we compared the Euclidian distance between two intracellular traces in the period from 250 to 30 ms before each stimulus (see Methods). This measurement represents the ‘dissimilarity’ between two membrane potentials immediately before stimulation. The distance was slightly, but significantly, shorter when two neurons displayed the same responses (S–S or N–N), as compared to different responses (S–N or N–S). Thus, two neurons that received similar background noises before stimulation tended to produce the same responses (S–S or N–N).
Band-pass filtering properties in CA1 spike transmission
Firing rates of hippocampal neurons range widely from < 1 Hz to > 100 Hz and play an important role in encoding external information (O'Keefe & Dostrovsky, 1971; Wilson & McNaughton, 1993). The final series of experiments was designed to describe the input frequency-dependent responses. The stimulus intensity was lowered to a subthreshold level, which did not evoke a spike by a single-pulse stimulus, and the Schaffer collaterals were activated by a four-pulse train at various rates (5–200 Hz) in the presence of 50 μm AP5 to avoid induction of synaptic plasticity. We did not discriminate spiking activity across four train pulses because of the insufficient frame rate in our movie (i.e. 10 fps scanning versus 5–200 Hz trains); instead we focused on whether or not at least one spike occurred during a train stimulus. Trains were repeated 10 times, and the firing probabilities were averaged within each neuron to determine which frequency more or less reliably activated the neuron. Data of 93 neurons in a slice (Fig. 7A) are massed across neurons in Fig. 7B. The summed activity displayed a frequency preference. On average, the network responded more reliably in the near-gamma frequency (20–40 Hz) range, acting like a band-pass temporal filter. Individual neurons, however, responded differently to train stimuli. Their filtering properties were expediently categorized into low-pass-like, band-pass-like, and high-pass-like filters by thresholding the mean firing probability at 50% (Fig. 7C). Data from seven slices are summarized in Fig. 7D. Cells that did not show spikes or frequency preference were excluded from data analysis because in these cases, stimulation intensity might not be optimal to produce the filtering properties. Data indicate that 63.9% of neurons showed band-pass-like properties, whereas other cells behaved like a low-pass (9.5%) or high-pass (26.6%) filter in the frequency range tested here. When perfused with 200 nm gabazine, almost all neurons started to fire in the entire frequency range (data not shown). Thus, stimulus intensity was reduced to a point that filtering responses emerged in > 80% neurons. It now turned out that all these neurons had a high-pass-like transfer function (Fig. 7D). The same results were obtained with 50 μm picrotoxin (data not shown).
Figure 7. Frequency-dependent spike transmission.

A, grey-scaled rasterplot depicting the firing probabilities of individual neurons activated at a low stimulus intensity of 4-pulse burst trains in the 5–200 Hz range. B, percentages of activated neurons versus frequencies of bursty stimulation. C, diversity of frequency preferences. Neurons no. 4 (top), no. 10 (middle) and no. 56 (bottom) showed low-pass-like, band-pass-like or high-pass-like profiles, respectively. The filtering threshold was set to 50% (indicated by dashed lines). D, distribution of neurons belonging to each filtering category in the absence (open bars, 631 neurons in 7 slices) and presence of 200 nm gabazine (filled bars, 404 neurons in 4 slices).
Discussion
Trial-to-trial net variation
Central neurons are known to react to repeated presentation of the same stimulus with high variability (Henry et al. 1973; Tomko & Crapper, 1974; Rose, 1979; Softky & Koch, 1993). Evidence is accumulating that this variability results from random ongoing background activity (Arieli et al. 1996; Azouz & Gray, 1999; Kisley & Gerstein, 1999). The variability, however, has long been examined in one or a few neurons and remains to be elucidated in a large cell population. We have shown that trial-to-trial variation seen at the single-cell level does not average out even at the population level and that the variation is higher than would be expected by chance. Interestingly, this non-stationarity seemed to arise from time-varying recruitment of different neuron subpopulations because mimicking cell-assembly-like metadynamics increased the net variability to a point that met with our empirical data.
What brings about the cell-assembly-like dynamics? Axonal electrical coupling and recurrent excitation, both of which are present in CA1 pyramid–pyramid circuits (Deuchars & Thomson, 1996; Draguhn et al. 1998), are possible factors. In addition, we found that background synaptic noise is weakly correlated in space and time. The coherent noise could depolarize (or hyperpolarize) a metapopulation of neurons and yield dynamic subset behaviours. This seems consistent with work by Cobb et al. (1995) who found that single GABAergic interneurons can synchronize the firing of a set of hippocampal pyramidal cells. One direct approach to address our hypothesis is to investigate the effect of gabazine and picrotoxin. Unfortunately, these antagonist extremely enhanced the firing probability even at very weak stimulation intensities, leading the population dynamics to a nearly all-or-none form (see Fig. 2C), and we hence were unable to perform this experiment. Instead, we carried out intracellular recordings from two neurons and tried to compare their spiking behaviours with membrane potential dynamics immediately before stimulation. This idea was based on a report showing that membrane potential fluctuations reliably designate spike timings (Mainen & Sejnowski, 1995). We indeed found that, when two neurons displayed similar kinetics of ongoing fluctuations in membrane potentials, these neurons tended to produce the same responses to a subsequent stimulus. Thus, the correlated background noise is likely to contribute to the dynamics of cell-assembly formation.
Quasilinear I/O relationship
In general, the linearity reasonably helps minimize a loss of information transfer because the graded output covers a wide range of input levels. Our data show that the summed activity of individual neurons is apparently proportional to the overall network input. At the single neuron level, it has already been shown that CA1 dendrites can linearly integrate synaptic inputs (Cash & Yuste, 1999; Gasparini & Magee, 2006). The linear dendritic summation, however, occurs only within individual cells and is not necessarily linked to the net linearity; note firing is an all-or-none event of a cell and its reliability is not determined only by synaptic input levels but is also affected by many other factors, including spike threshold, channel noise, adaptation and refractory period. It is thus surprising that the non-linear units (spikes of single neurons) somehow produce the global linearity. Computational simulation shows that synaptic noise can shape a linear population response (van Vreeswijk & Sompolinsky, 1996). Consistent with this, we found that the linearity was distorted by GABAA receptor antagonists, suggesting the role of inhibitory background noise. Because even a low concentration of gabazine abolished the linearity, phasic inhibition, shown to be correlated between neurons (Fig. 6A), is required for the I/O linearity.
Band-pass filter
GABAA receptor antagonists shifted the band-pass filtering property of CA1 neurons to a high-pass function. Thus, the band-pass filtering depended on inhibitory synaptic transmission, rather than short-term monosynaptic plasticity (Fortune & Rose, 2001) or intrinsic cellular resonance (Pike et al. 2000; Fellous et al. 2001; Izhikevich et al. 2003). In other words, without inhibition, CA1 neurons simply act like integrate-and-fire units, that is, EPSP accumulation is more efficient at higher frequencies. In this case, we do not think that background noise plays a pivotal role. In these experiments, the afferent activation was repeated four times, and therefore polysynaptic inhibition contributes to spike responses. In hippocampal local circuits, inhibition flow dynamically changes depending on the input frequencies (Andersen et al. 1963; Pouille & Scanziani, 2001, 2004; Mori et al. 2004). Such a frequency-dependent switch in GABAergic feedforward/feedback routing may also contribute to the band-pass-like transfer function.
Summary
In this work, we imaged input and output activities from large neuron populations to elucidate how single CA1 neurons execute integrative dynamics at the multicellular level. The main findings are: (1) the network responds nearly in proportion to single-pulse inputs, but non-linearly to multipulse inputs, i.e. in a band-pass manner, and (2) the net responses vary from trial to trial, beyond chance levels. In conclusion, even in simple CA1 circuits, neurons can work in harmony together to make an organized output to the cortex although their individual responses are ostensibly unreliable and stochastic. To extrapolate our results, which were obtained from organotypic cultures, to in vivo brain activity, studies are underway at higher levels of experimental preparations, including the whole hippocampus in vitro and the brain of anaesthetized animals.
Supplementary Material
Acknowledgments
This work was supported in part by a Grant-in-Aid for Science Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (No. 17023015, no. 17650090, and no. 17689004), the Sumitomo Foundation (No. 050038), and KONICA MINOLTA Imaging Science Foundation.
References
- Andersen P, Eccles JC, Loyning Y. Recurrent inhibition in the hippocampus with identification of the inhibitory cell and its synapses. Nature. 1963;198:540–542. doi: 10.1038/198540a0. [DOI] [PubMed] [Google Scholar]
- Arieli A, Sterkin A, Grinvald A, Aertsen A. Dynamics of ongoing activity: explanation of the large variability in evoked cortical responses. Science. 1996;273:1868–1871. doi: 10.1126/science.273.5283.1868. [DOI] [PubMed] [Google Scholar]
- Azouz R, Gray CM. Cellular mechanisms contributing to response variability of cortical neurons in vivo. J Neurosci. 1999;19:2209–2223. doi: 10.1523/JNEUROSCI.19-06-02209.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash S, Yuste R. Linear summation of excitatory inputs by CA1 pyramidal neurons. Neuron. 1999;22:383–394. doi: 10.1016/s0896-6273(00)81098-3. [DOI] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Deuchars J, Thomson AM. CA1 pyramid–pyramid connections in rat hippocampus in vitro: dual intracellular recordings with biocytin filling. Neuroscience. 1996;74:1009–1018. doi: 10.1016/0306-4522(96)00251-5. [DOI] [PubMed] [Google Scholar]
- Draguhn A, Traub RD, Schmitz D, Jefferys JG. Electrical coupling underlies high-frequency oscillations in the hippocampus in vitro. Nature. 1998;394:189–192. doi: 10.1038/28184. [DOI] [PubMed] [Google Scholar]
- Fellous J-M, Houweling AR, Modi RH, Rao RPN, Tiesinga PHE, Sejnowski TJ. Frequency dependence of spike timing reliability in cortical pyramidal cells and interneurons. J Neurophysiol. 2001;85:1782–1787. doi: 10.1152/jn.2001.85.4.1782. [DOI] [PubMed] [Google Scholar]
- Fortune ES, Rose GJ. Short-term synaptic plasticity as a temporal filter. Trends Neurosci. 2001;24:381–385. doi: 10.1016/s0166-2236(00)01835-x. [DOI] [PubMed] [Google Scholar]
- Gasparini S, Magee JC. State-dependent dendritic computation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:2088–2100. doi: 10.1523/JNEUROSCI.4428-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez R, Heinemann U. Synaptic reorganization in explanted cultures of rat hippocampus. Brain Res. 1999;815:304–316. doi: 10.1016/s0006-8993(98)01101-9. [DOI] [PubMed] [Google Scholar]
- Henry GH, Bishop PO, Tupper RM, Dreher B. Orientation specificity and response variability of cells in the striate cortex. Vision Res. 1973;13:1771–1779. doi: 10.1016/0042-6989(73)90094-1. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y. Abnormal targeting of developing hippocampal mossy fibers after epileptiform activities via L-type Ca2+ channel activation in vitro. J Neurosci. 1999;19:802–812. doi: 10.1523/JNEUROSCI.19-02-00802.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegaya Y, Aaron G, Cossart R, Aronov D, Lampl I, Ferster D, Yuste R. Synfire chains and cortical songs: temporal modules of cortical activity. Science. 2004;304:559–564. doi: 10.1126/science.1093173. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y, Le Bon-Jego M, Yuste R. Large-scale imaging of cortical network activity with calcium indicators. Neurosci Res. 2005;52:132–138. doi: 10.1016/j.neures.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Izhikevich EM, Desai NS, Walcott EC, Hoppensteadt FC. Bursts as a unit of neural information: selective communication via resonance. Trends Neurosci. 2003;26:161–167. doi: 10.1016/S0166-2236(03)00034-1. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, Johnston D, Lasser-Ross N, Lisman JE, Miyakawa H, Ross WN. The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature. 1992;357:244–246. doi: 10.1038/357244a0. [DOI] [PubMed] [Google Scholar]
- Kerr JN, Greenberg D, Helmchen F. Imaging input and output of neocortical networks in vivo. Proc Natl Acad Sci U S A. 2005;102:14063–14068. doi: 10.1073/pnas.0506029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisley MA, Gerstein GL. Trial-to-trial variability and state-dependent modulation of auditory-evoked responses in cortex. J Neurosci. 1999;19:10451–10460. doi: 10.1523/JNEUROSCI.19-23-10451.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainen ZF, Sejnowski TJ. Reliability of spike timing in neocortical neurons. Science. 1995;268:1503–1506. doi: 10.1126/science.7770778. [DOI] [PubMed] [Google Scholar]
- Mori M, Abegg MH, Gahwiler BH, Gerber U. A frequency-dependent switch from inhibition to excitation in a hippocampal unitary circuit. Nature. 2004;431:453–456. doi: 10.1038/nature02854. [DOI] [PubMed] [Google Scholar]
- Morita M, Higuchi C, Moto T, Kozuka N, Susuki J, Itofusa R, Yamashita J, Kudo Y. Dual regulation of calcium oscillation in astrocytes by growth factors and proinflammatory cytokines via the mitogen-activated protein kinase cascade. J Neurosci. 2003;23:10944–10952. doi: 10.1523/JNEUROSCI.23-34-10944.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Meth. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- O'Keefe J, Dostrovsky J. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res. 1971;34:171–175. doi: 10.1016/0006-8993(71)90358-1. [DOI] [PubMed] [Google Scholar]
- Ohki K, Chung S, Ch'ng YH, Kara P, Reid RC. Functional imaging with cellular resolution reveals precise micro-architecture in visual cortex. Nature. 2005;433:597–603. doi: 10.1038/nature03274. [DOI] [PubMed] [Google Scholar]
- Pike FG, Goddard RS, Suckling JM, Ganter P, Kasthuri N, Paulsen O. Distinct frequency preferences of different types of rat hippocampal neurones in response to oscillatory input currents. J Physiol. 2000;529:205–213. doi: 10.1111/j.1469-7793.2000.00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Routing of spike series by dynamic circuits in the hippocampus. Nature. 2004;429:717–723. doi: 10.1038/nature02615. [DOI] [PubMed] [Google Scholar]
- Robain O, Barbin G, Billette de Villemeur T, Jardin L, Jahchan T, Ben-Ari Y. Development of mossy fiber synapses in hippocampal slice culture. Dev Brain Res. 1994;80:244–250. doi: 10.1016/0165-3806(94)90109-0. [DOI] [PubMed] [Google Scholar]
- Rose D. An analysis of the variability of unit activity in the cat's visual cortex. Exp Brain Res. 1979;37:595–604. doi: 10.1007/BF00236826. [DOI] [PubMed] [Google Scholar]
- Sakaguchi T, Okada M, Kawasaki K. Sprouting of CA3 pyramidal neurons to the dentate gyrus in rat hippocampal organotypic cultures. Neurosci Res. 1994;20:157–164. doi: 10.1016/0168-0102(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Schreiber S, Fellous J-M, Tiesinga PH, Sejnowski TJ. A new correlation-based measure of spike timing reliability. Neurocomputing. 2003;52–54:925–931. doi: 10.1016/S0925-2312(02)00838-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softky WR, Koch C. The highly irregular firing of cortical cells is inconsistent with temporal integration of random EPSPs. J Neurosci. 1993;13:334–350. doi: 10.1523/JNEUROSCI.13-01-00334.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Schiller Y, Stuart G, Sakmann B. Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science. 1995;268:297–300. doi: 10.1126/science.7716524. [DOI] [PubMed] [Google Scholar]
- Stell BM, Mody I. Receptors with different affinities mediate phasic and tonic GABAA conductances in hippocampal neurons. J Neurosci. 2002;22:RC223. doi: 10.1523/JNEUROSCI.22-10-j0003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Meth. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A. 2003;100:7319–7324. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko GJ, Crapper DR. Neuronal variability: non-stationary responses to identical visual stimuli. Brain Res. 1974;79:405–418. doi: 10.1016/0006-8993(74)90438-7. [DOI] [PubMed] [Google Scholar]
- van Vreeswijk C, Sompolinsky H. Chaos in neuronal networks with balanced excitatory and inhibitory activity. Science. 1996;274:1724–1726. doi: 10.1126/science.274.5293.1724. [DOI] [PubMed] [Google Scholar]
- Wilson MA, McNaughton BL. Dynamics of the hippocampal ensemble code for space. Science. 1993;261:1055–1058. doi: 10.1126/science.8351520. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Kurotani T, Toyama K. Neural connections between the lateral geniculate nucleus and visual cortex in vitro. Science. 1989;245:192–194. doi: 10.1126/science.2749258. [DOI] [PubMed] [Google Scholar]
- Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- Yuste R, Katz LC. Control of postsynaptic Ca2+ influx in developing neocortex by excitatory and inhibitory neurotransmitters. Neuron. 1991;6:333–344. doi: 10.1016/0896-6273(91)90243-s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.