

Abstract
EtOH (ethanol) damages the hippocampus, a brain region that is involved in learning and memory processes. The mechanisms responsible for this effect of EtOH are not fully understood. We recently demonstrated that acute EtOH exposure potently stimulates oscillatory activity driven by the excitatory actions of GABA in the CA3 region of the neonatal rat hippocampus. This activity can be recorded during the growth spurt period as giant depolarizing potentials (GDPs). Here, we characterized the effects of prolonged EtOH exposure on GDPs. In the first study, we prepared hippocampal coronal slices from neonatal rats and exposed these to control artificial cerebrospinal fluid (ACSF) or ACSF plus 50 mM EtOH for 3–4 hr. We then performed whole-cell patch-clamp electrophysiological recordings from CA3 pyramidal neurons, which revealed that tolerance to the GDP stimulating effects of EtOH did not occur after continuous exposure. In the second study, we exposed neonatal rats to air or air plus 1.9 g/dL EtOH in vapor chambers for 4 hours/day for 1 or 3 days (neonatal peak blood EtOH concentration = 40–45 mM). We then performed slice electrophysiological studies 24 hours after the end of EtOH exposure and found that there was no statistically significant difference in the acute effect of 50 mM EtOH on GDP frequency in samples from neonates exposed to air or air plus EtOH. These findings indicate that EtOH persistently stimulates network-driven oscillatory activity in the developing hippocampus. We propose that the lack of adaptive response to continuous EtOH exposure could make immature neuronal networks particularly vulnerable to the actions of this agent.
Keywords: chronic, GABA, excitatory, depolarizing, network, fetal
Numerous animal and human studies have demonstrated that EtOH (ethanol) exposure damages the developing hippocampus, leading to deficits in learning and memory (Berman & Hannigan, 2000; Bonthius et al., 2001a; Bonthius et al., 2001b; Mattson et al., 2001; Hamilton et al., 2003). These deficits are a consequence of structural and functional hippocampal abnormalities that persist into adulthood (Sutherland et al., 1997; Krahl et al., 1999; Berman & Hannigan, 2000; Richardson et al., 2002; Savage et al., 2002). Multiple candidate mechanisms could be responsible for these effects of EtOH, including alterations in cellular energetics, gene expression, cell-cell interactions, cell signaling pathways and synaptic transmission (Goodlett et al., 2005). However, the precise contribution of these mechanisms to EtOH-induced hippocampal damage has yet to be determined.
Developing neuronal circuits in many brain regions exhibit spontaneous correlated activity that is thought to play a central role in synaptic refinement (Yuste, 1997; Penn & Shatz, 1999). In the hippocampus, these oscillations are driven by the excitatory actions of GABAA receptors; these receptors have excitatory actions in immature neurons because these cells have a relatively high intracellular concentration of Cl− (Ben-Ari, 2002). In the CA3 hippocampal region, these oscillations are denoted as giant depolarizing potentials (GDPs). In rats, these events are present during the first 8–12 days of postnatal life (Ben-Ari, 2002; Owens & Kriegstein, 2002). GDPs trigger transient elevations in intracellular Ca2+ via activation of voltage-gated Ca2+ channels that may play a role in synapse maturation and dendritic growth (Leinekugel et al., 1997; Berridge, 1998; Ben-Ari, 2002; Groc et al., 2002; Owens & Kriegstein, 2002; Lauri et al., 2003; Mantelas et al., 2003; Colin-Le Brun et al., 2004; Fiszman & Schousboe, 2004). Thus, alterations in these network driven oscillatory events could negatively impact hippocampal neuronal development.
We recently reported that acute EtOH exposure potently excites immature neuronal networks by increasing GDP frequency in the CA3 region of the neonatal hippocampus (Galindo et al., 2005). This effect was driven by an increase in the excitatory actions of GABAA receptors. In this study, we further characterized the EtOH modulation of GDPs by investigating whether tolerance develops to this effect.
Experimental Procedures
Tissue preparation and solutions
Unless indicated, all chemicals were from Sigma-RBI-Fluka (St. Louis, MO). Animal procedures were approved by the Institutional Animal Care and Use Committee of the University of New Mexico Health Sciences Center and conformed to National Institutes of Health guidelines. Experiments were performed in 400 μm-thick coronal hippocampal slices that were prepared from neonatal Sprague-Dawley rats, as previously described (Galindo et al., 2005). Artificial cerebrospinal fluid (ACSF) contained (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2 CaCl2 and 10 glucose, equilibrated with 95%O2/5%CO2. Slices were allowed to recover for ≥ 80 min at room temperature prior to any experimental procedure.
Prolonged exposure of slices to EtOH
Slices from a single animal were rapidly hemisected in freshly oxygenated ACSF. Subsequently, the left and right sides from each slice were randomly segregated into chambers containing either ACSF alone or ACSF plus 50 mM EtOH at 32–33 °C (Fig. 1). During the incubation procedure, solutions were continuously bubbled with a mixture of 95 % O2 and 5 % CO2. The slices were sequentially placed into the recording chamber starting from the most ventral region, alternating between control and contralateral EtOH-exposed sister slices. Only a single cell was recorded per slice. Recordings were obtained from slices incubated in control or EtOH solution for a period of 3 to 4 hrs, which was chosen to model a single episode of EtOH exposure during the third trimester of human pregnancy. Slices were withdrawn from EtOH in the recording chamber. The recording chamber was perfused with ACSF or ACSF + EtOH at a rate of 2–3 ml/min at 32–33 °C.
Fig. 1. In Vitro EtOH Exposure Paradigm.
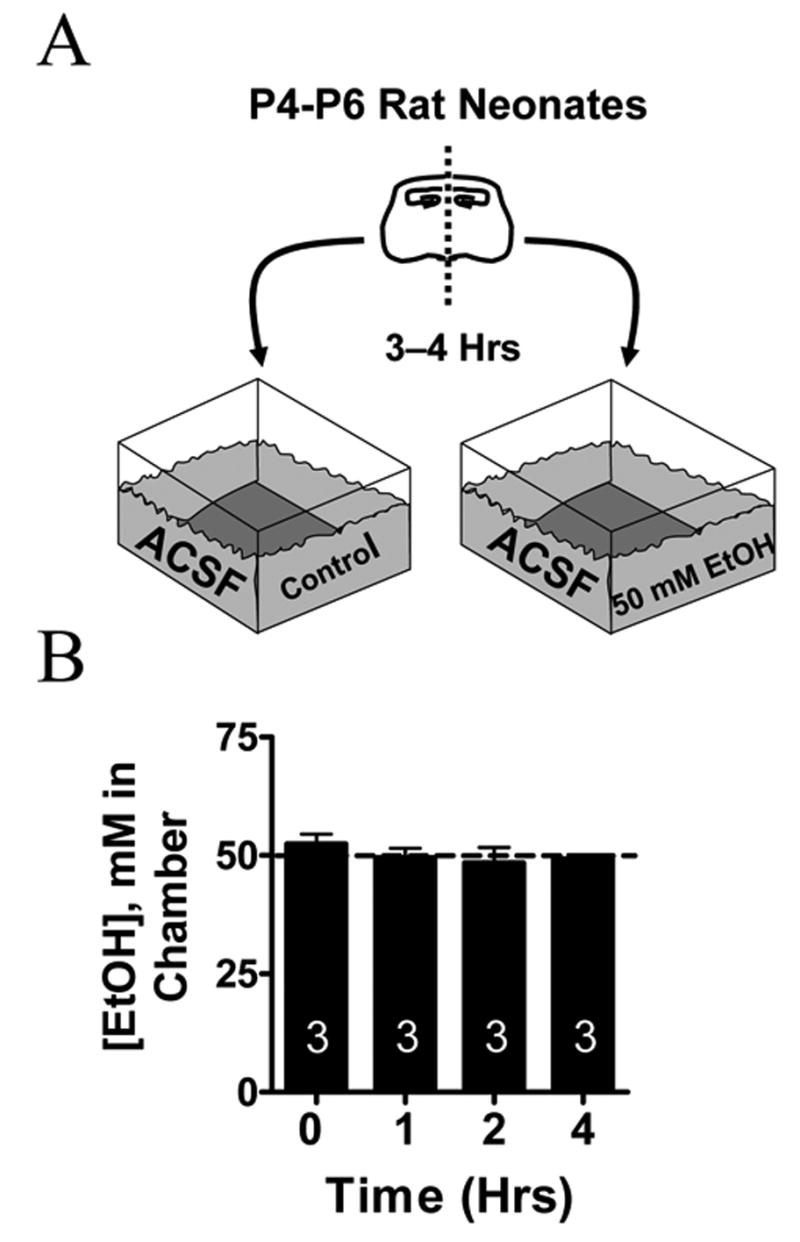
(A) Scheme illustrating the procedure used to expose neonatal (P4–P6) hippocampal slices to 50 mM EtOH for 3–4 hrs. Sister slices were segregated into a chamber that contained continuously oxygenated ACSF solution with or without EtOH. Left and right sister slices were randomly assigned to each chamber. All slices were allowed to equilibrate for a period greater than 80 min prior to the start of the experiment. (B) EtOH concentrations in the ACSF do not significantly change as a function of time. Numbers in each bar represent the number of experiments.
Electrophysiological recordings
Whole-cell voltage-clamp and current-clamp electrophysiological recordings were performed under infrared-differential interference contrast microscopy with an Axopatch 200B amplifier (Axon Laboratories, Union City, CA). Microelectrodes had resistances of 4–6 MΩ. We recorded GDPs in the whole-cell mode from CA3 neurons using the following internal solution (in mM): 140 CsCl, 2 MgCl2, 1 CaCl2, 10 EGTA, 10 HEPES (pH 7.3), 4 Na2-ATP and 2 QX-314 (Tocris-Cookson, Ellisville, MO). Pharmacologically isolated GABAA-mediated spontaneous postsynaptic currents (GABAA-sPSCs) were recorded utilizing the above internal solution in ACSF containing 10 μM NBQX and 100 μM DL-AP5. Voltage-clamp experiments were performed at a holding potential of −60 mV. Access resistances were between 15–25 M; if access resistance changed more than 20%, the recording was discarded. Data were acquired with pClamp7 (Axon Laboratories) and analyzed with Minis Analysis program (Synaptosoft, Decatur, GA). The effect of acute EtOH exposure was quantified with respect to the average of control and washout responses. For the GABAA-sPSC recordings, the Kolmogorov-Smirnov test (KS test) was used initially to test for significant differences between treatments in individual cells; a p 0.01 was considered to be statistically significant. Statistical analyses of pooled data were performed by unpaired or paired t test; a p 0.05 was considered to be statistically significant. Data are presented as mean ± SEM.
Exposure of neonatal rats to EtOH vapor
We exposed neonatal rat pups and their respective mothers to EtOH between postnatal days 3 to 6 in inhalation chambers essentially as previously described (Heaton et al., 2000). This paradigm consists of exposing neonatal pups and rat dams to EtOH vapor by placing animals in transparent sealed chambers (La Jolla Research Inc, La Jolla, CA) connected to an intake hose that continuously delivers EtOH vapor by via a heating flask that receives a constant drip of 95% liquid EtOH controlled by a regulating peristaltic pump. The EtOH vapor is continuously removed by an exhaust hose connected to a vacuum line. Litters were culled to 10 pups and exposed to air or air plus EtOH. All experiments were started at the same time of the day (10:00 am) and the EtOH vapor in the chamber was monitored every hour. This paradigm was designed in an attempt to model single or repeated (i.e. three) daily maternal drinking sessions during the third trimester equivalent of human pregnancy producing blood alcohol levels approximately 2 to 3-times higher than the legal intoxication limit.
Determination of EtOH levels
The concentrations of EtOH in ACSF and plasma were determined by a standard alcohol dehydrogenase-based assay. A standard EtOH concentration curve was generated utilizing either ACSF or plasma from non-EtOH exposed neonates. Samples were exposed to an enzyme reaction assay involving the reduction of NAD+ (β-NAD, free acid, grade I, Roche, Indianapolis, IN) and oxidation of EtOH by alcohol dehydrogenase (Roche). NAD+ absorbance was read at a wavelength of 340 nm.
Results
Prolonged EtOH exposure of immature hippocampal slices results in a sustained increase in GDP and GABAA-SPSC frequency in pyramidal neurons
Sister hippocampal slices were incubated in ACSF alone or ACSF plus 50 mM EtOH for 3–4 hr (Fig. 1A). Solutions were continuously bubbled with 95% O2 plus 5% CO2. A control experiment demonstrated that the concentration of EtOH did not significantly change over the course of 4 hr under these experimental conditions (Fig. 1B). No gross changes in neuronal morphology were qualitatively appreciable under infrared-differential interference contrast microscopy after prolonged exposure to this concentration of EtOH.
We initially recorded GDPs from control and EtOH exposed slices. The first group of slices was incubated for 3–4 hr in ACSF and then transferred to the recording chamber. A stable baseline recording of GDP activity was obtained in ACSF (average frequency = 0.27 ± 0.01 Hz; average duration = 615 ± 44 ms; average amplitude = 488 ± 122 pA; n = 11). Then, slices were exposed to 50 mM EtOH for 7 min. In agreement with our previous report (Galindo et al., 2005), we found that acute EtOH exposure reversibly increases GDP frequency (31 ± 5 %; n = 11; Fig. 2A, B) without affecting the amplitude (−5.2 ± 3 %; n = 11) or duration (−4.2 ± 3 %; n = 11) of these events.
Fig. 2. Prolonged EtOH Exposure Results in a Persistent Elevation in GDP Frequency.
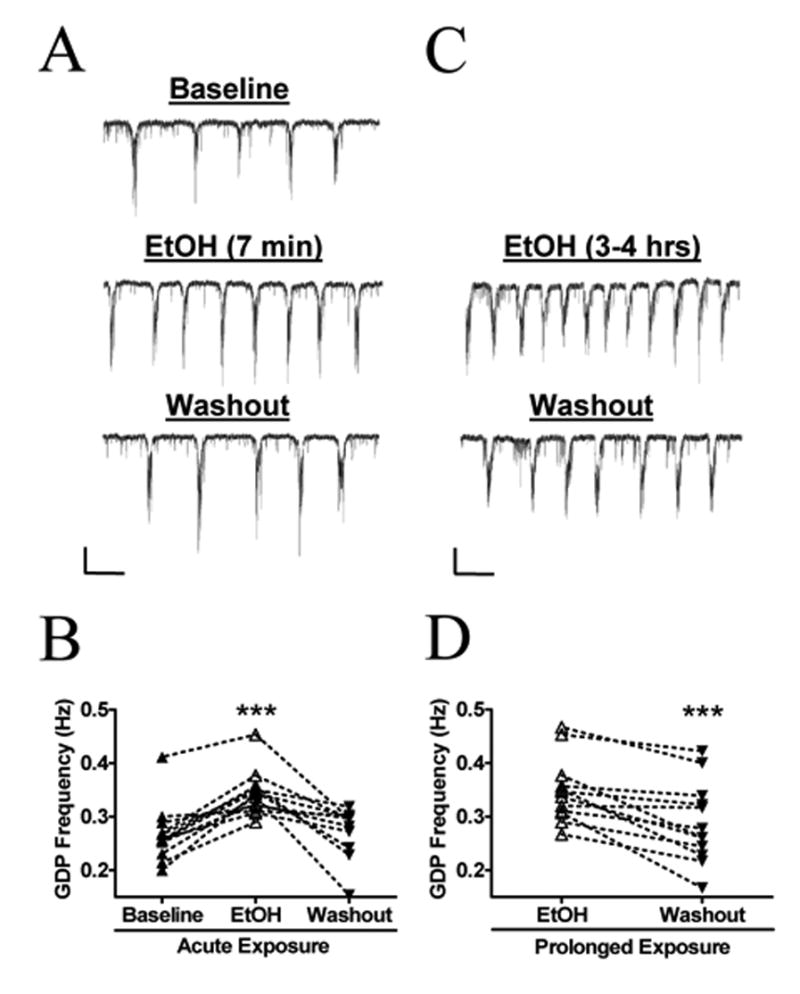
(A) Sample trace demonstrating a reversible increase in GDP frequency in CA3 pyramidal neurons from control slices acutely treated with EtOH for 7 minutes. (B) Summary of the effect of acute ETOH exposure on GDP frequency (n = 11). ***p < 0.001 by repeated measures ANOVA followed by Tukey’s posthoc test. (C) Sample traces from the sister slice of 2A treated with 50 mM EtOH for a period of 3 to 4 hrs. The frequency of GDPs decreases following EtOH washout. (D) Summary of the effect prolonged EtOH exposure on GDP frequency (n = 12). The basal GDP frequency during prolonged ETOH exposure was significantly higher than the baseline frequency of acutely exposed slices (0.35 ± 0.02 Hz vs 0.26 ± 0.01 Hz; <0.01 by unpaired t-test). The percent change in GDP frequency between acute-ETOH and prolonged-EtOH slices was not statistically different by unpaired t-test (31 ± 5 % vs. 25 ± 7 %). ***p < 0.001 by paired t-test. Scale bar for both traces = 100 pA/4 s.
We then assessed whether this effect persisted after continuous EtOH exposure for several hours. Because it is difficult to record from neurons in the whole-cell patch-clamp configuration for prolonged periods, we determined the effect of EtOH by calculating the change in GDP frequency between EtOH exposure and washout. Sister slices from those used for the acute EtOH exposure experiments described above were continuously incubated in ASCF plus 50 mM EtOH for 3–4 hr. Slices were then transferred to the recording chamber, which was perfused with ASCF plus 50 mM EtOH. This procedure prevented the initiation of EtOH withdrawal. A stable baseline recording of GDP activity was obtained in ACSF plus EtOH (average frequency = 0.35 ± 0.01 Hz; n = 12). Then, the perfusion media was switched to ACSF alone (washout). Under this condition, we found that GDP frequency changed by 25 ± 7 % (Fig. 2C, D; n = 12). As expected, basal GDP frequency during prolonged ETOH exposure was significantly higher than the baseline frequency of acutely exposed slices (0.35 ± 0.02 Hz vs 0.26 ± 0.01 Hz; p <0.01 by unpaired t-test; Fig. 2B, D). Furthermore, the change in GDP frequency between EtOH and washout was not significantly different from the effect of acute EtOH by unpaired t-test. Neither the amplitude (−2.6 ± 4 %; n = 12) nor the duration (−0.5 ± 2 %; n = 12) of these events changed upon EtOH washout.
As previously reported, the EtOH-induced elevation of GDP frequency is associated with an increase in GABAA-sPSCs frequency (Galindo et al., 2005). In agreement with that report, we found that acute EtOH exposure reversibly increased GABAA-sPSCs frequency (36 ± 5 %; n = 5; KS test p < 0.01 in all cells; Fig. 3A, B) without affecting the amplitude (8.8 ± 10 %; n = 5) or half-width (4.2 ± 4.6 %; n = 5) of these events. In slices incubated for 3–4 hours with 50 mM EtOH, GABAA-sPSCs frequency changed by 54 ± 13 % upon washout (n = 5; KS test p < 0.01 in all cells; Fig. 3C, D). Unpaired t-test revealed that this value was not significantly different from the change induced by acute EtOH exposure in sister slices. The amplitude (−4.2 ± 6.3 %; n = 5) of these events did not change upon EtOH washout. There was a small, but significant (p <0.05 by one-sample t-test) increase in the half-width of GABAA-sPSCs following washout (12 ± 3 %; n = 5).
Fig. 3. Prolonged EtOH Exposure Results in a Sustained Increase in Action Potential-Dependent GABAA-sPSCs.
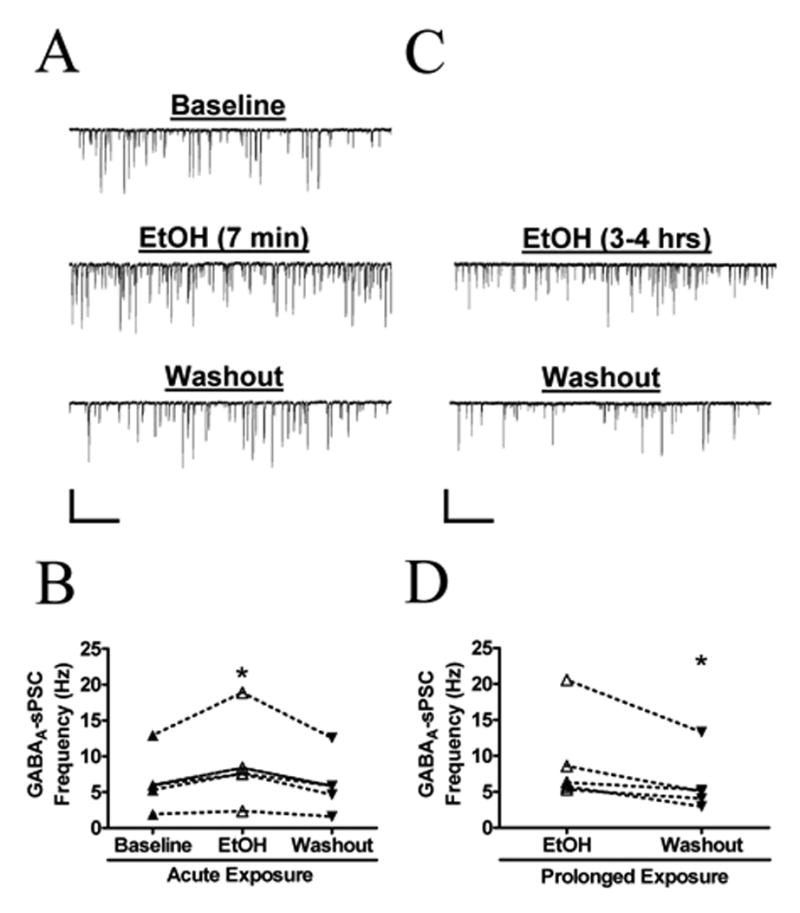
(A) Sample trace demonstrating the reversible increase in GABAA-sPSC frequency during acute, 7-minute, application of 50 mM EtOH to neonatal CA3 pyramidal neurons. (B) Summary of the effect of acute ETOH exposure on GABAA-sPSC frequency (n = 5). *p < 0.05 by repeated measures ANOVA followed by Tukey’s posthoc test. (C) Sample recording demonstrating the decrease in GABAA-sPSC frequency following washout from prolonged (3 to 4 hr) exposure to EtOH (50 mM). Scale bar for both traces = 200 pA/2 s. (D) Summary of the effect prolonged EtOH exposure on GABAA-sPSC frequency (n = 5). The percent change in GDP frequency between acute-ETOH and prolonged-EtOH slices was not statistically different by unpaired t-test (36 ± 5 % vs. 54 ± 13 %). *p < 0.05 by paired t-test.
EtOH exposure in vivo does not induce tolerance to the acute effect of EtOH on GDP frequency
We exposed neonatal rats along with their respective mothers to air alone or air plus EtOH vapor for 4 hours per day. Blood EtOH concentrations increased linearly with respect to the concentration of EtOH in the chamber (Fig. 4A). Interestingly, in 4 neonates, we detected blood EtOH concentrations near 90 mM (400 mg/dL), which would have expected to be lethal. However, breathing patterns in these neonates were comparable to those of controls and significant motor behavior was still appreciable in these animals. These qualitative observations are in general agreement with reports indicating that young rats are more resistant to the acute intoxicating effects of EtOH (Hollstedt et al., 1980; Fang et al., 1997; Silveri & Spear, 1998). In two of the mothers, we were able to measure blood EtOH concentrations and these were either undetectable or substantially lower than those found in the neonatal blood (Fig. 4A). A control experiment in a litter that was exposed to a chamber EtOH concentration of 1.5 g/dL revealed that neonatal blood EtOH concentrations peaked at 5 hr (i.e. 1 hr after termination of exposure) and returned to baseline by 9 hrs (Fig. 4B). For experiments, animals were exposed to chamber concentrations of 1.9 g/dL for 4 hr/day in an attempt to reach the concentrations used in vitro (50 mM; 230 mg/dL). Two animals from each litter were sacrificed after either a 1- or 3-day exposure to this regimen. This procedure was repeated in 3 different litters. As shown in Table 1, exposure to ~1.9 g/dL of EtOH concentrations in vapor chambers produced blood EtOH concentrations between 40–45 mM (184–207 mg/dL) in P4 and P6 neonates (measured immediately after the end of the 4 hr exposure period). Importantly, there was no statistically significant difference in the weights of control and EtOH exposed animals.
Fig. 4. Characterization of the In Vivo EtOH-Exposure Paradigm.
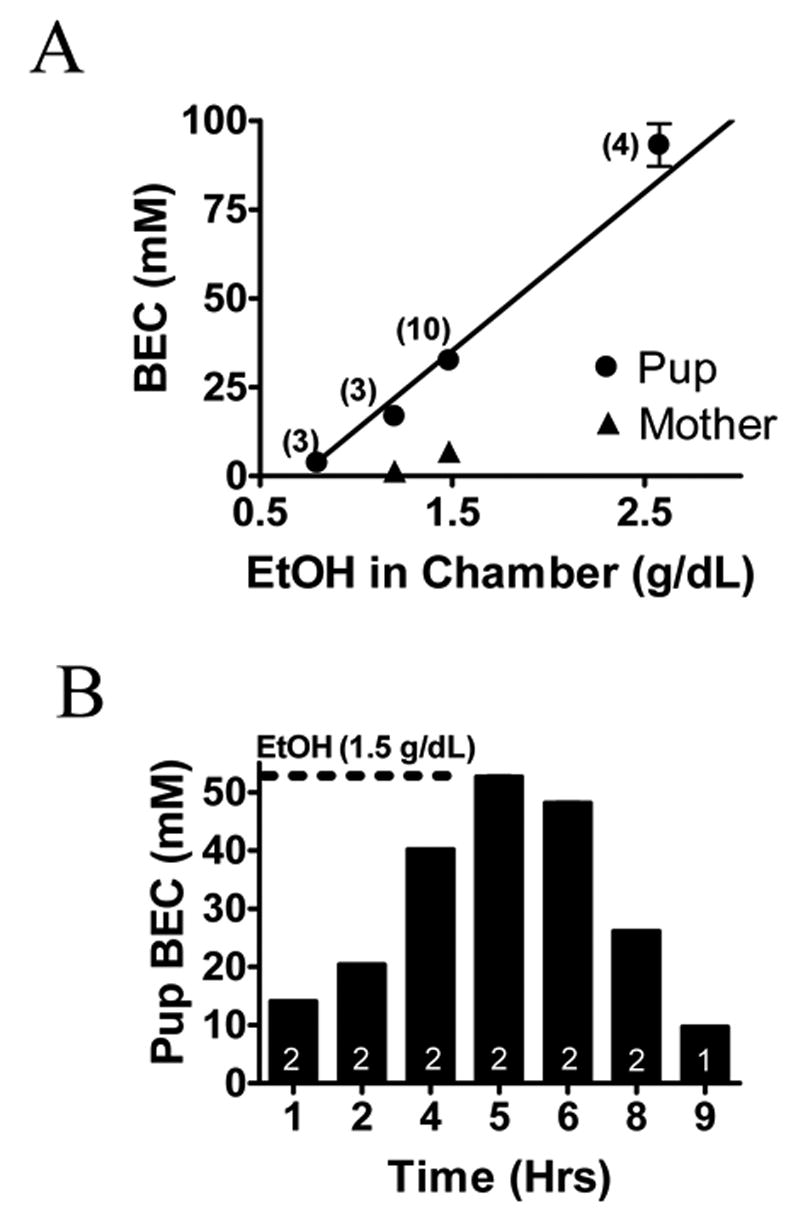
(A) Linear regression plot demonstrating increases in blood EtOH concentration (BEC) of neonatal rats of ages P4 and P5 and mothers as a function of EtOH concentration in the chamber. BEC measurements were obtained immediately following a 4-hr drug-exposure period. Notice the low BECs in the mother of the pups exposed to 1.0 and 1.5 g/dL of EtOH. As a point of reference, the legal intoxication limit in the U.S. is 17.4 mM = 80 mg/dL. (B) Histogram showing BEC in neonatal rat pups exposed to 1.5 g/dL of EtOH in the vapor chamber as a function of time. BEC gradually increased reaching a peak BEC at 1 hr after termination of EtOH exposure.
Table 1.
Characteristics of Rat Pups Exposed to Control or EtOH-containing Vapor Chamber
| Control 1-day Exposure | Ethanol 1-day Exposure | Control 3-day Exposure | Ethanol 3-Day Exposure | |
|---|---|---|---|---|
| No. of litters used | 3 | 3 | 3 | 3 |
| No. of animals used for recordings | 6 | 6 | 6 | 6 |
| Age of recording | P4 | P4 | P6 | P6 |
| [EtOH] in Vapor Chamber (g/dL) | 0 | 1.87 ± 0.13 | 0 | 1.99 ± 0.06 |
| BEC (mM) at t = 4 hrs | 0 | 41.64 ± 4.52 | 0 | 44.61 ± 0.93 |
| BEC (mg/dL) at t = 4 hrs | 0 | 191 ± 20 | 0 | 205 ± 4 |
| Weight (g) | 8.35 ± 1.37 | 8.04 ± 1.10 | 11.36 ± 1.95 | 10.91 ± 1.99 |
Neonatal rat pups 4–6 days old from 3 different litters were exposed for 1 to 3 days to a vapor chamber with circulating air (control) or ~1.9 g/dL of vaporized EtOH. No statistical differences were found by unpaired t-test when comparing the EtOH concentration in the vapor chamber or BEC between 1-day and 3 day exposed animals. There were also no differences in pup weight among control and EtOH-treated groups in the 1-day and 3-day experiments. All data are expressed as mean ± SEM.
We then measured the acute effects of EtOH on slices from animals exposed to air or air plus ~1.9 g/dL of EtOH vapor. In slices prepared 24 hrs after neonates were exposed to 4 hr/day of air for 1 day, acute exposure to 50 mM EtOH increased GDP frequency by 41 ± 10 % (Fig. 5A, B; n = 6). In slices prepared 24 hrs after neonates were exposed to 4 hr/day of air plus EtOH for 1 day, acute exposure to 50 mM EtOH increased GDP frequency to a similar extent (Fig. 5C, D; 59 ± 21 %; n = 6; not significantly different from air group by unpaired t-test). We quantified baseline GDP frequency in slices prepared 24 hr after 1 day vapor chamber exposure and found that it was not significantly different between the air (0.25 ± 0.02 Hz; n = 6) and air plus EtOH (0.18 ± 0.04 Hz; n = 6) groups.
Fig. 5. The Sensitivity of GDPS to acute EtOH exposure is not affected by 1-day in vivo EtOH exposure in vapor chambers.
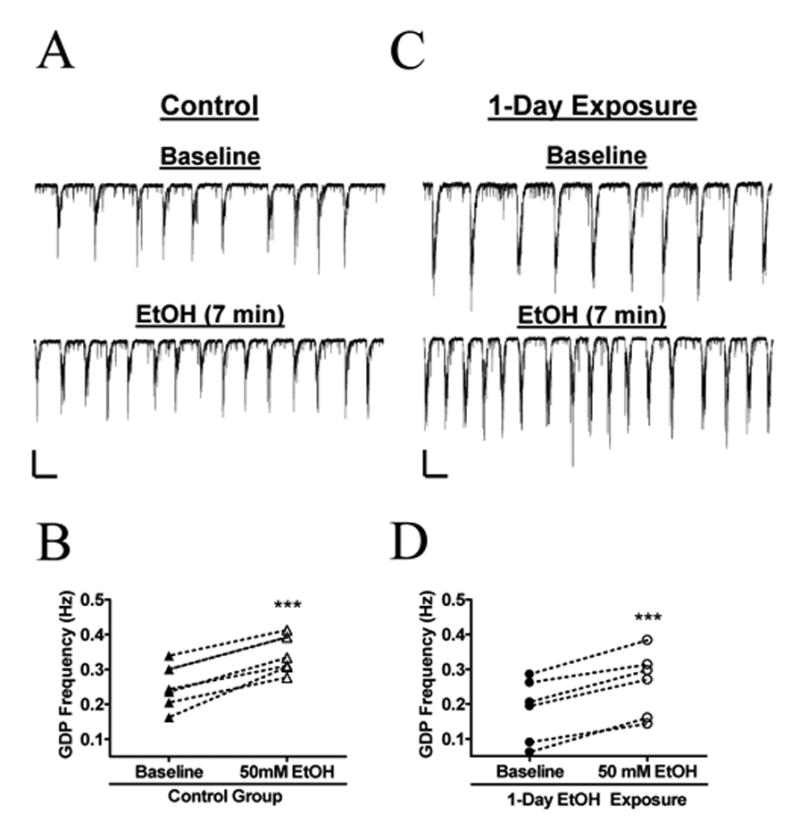
(A) Sample trace demonstrating an increase in GDP frequency during acute, 7-minute, application of 50 mM EtOH to a slice prepared 24 hr after a single 4 hr exposure to air in the vapor chamber. (B) Summary on the effect of acute EtOH on air-exposed rat pups ( n = 6). (C) Sample recording showing an increase in GDP frequency during acute EtOH exposure in slices prepared 24 hr after a single 4 hr exposure to 1.9 g/dL of EtOH in the vapor chamber. (D) Summary of the effect of acute EtOH on 1-day EtOH exposed animals (n = 6). ***p < 0.001 by paired t-test. Scale bars for both set of traces = 200 pA/2 s.
In slices prepared 24 hrs after neonates were exposed to 4 hr of air/day for 3 days, acute 50 mM EtOH increased GDP frequency by 74 ± 26 % (Fig. 6A, B; n = 8). In slices prepared 24 hrs after neonates were exposed to 4 hr/day of air plus EtOH for 3 days, acute 50 mM EtOH increased GDP frequency to a similar extent (Fig. 6C, D; 62 ± 20 %; n = 7; not significantly different from air group by unpaired t-test). We quantified baseline GDP frequency in slices prepared 24 hr after 3 day vapor chamber exposure and found that it was not significantly different between the air (0.14 ± 0.03 Hz; n = 8) and air plus EtOH (0.16 ± 0.03 Hz; n = 8) groups.
Fig. 6. The Sensitivity of GDPS to acute EtOH exposure is not affected by 3-day in vivo EtOH exposure in vapor chambers.
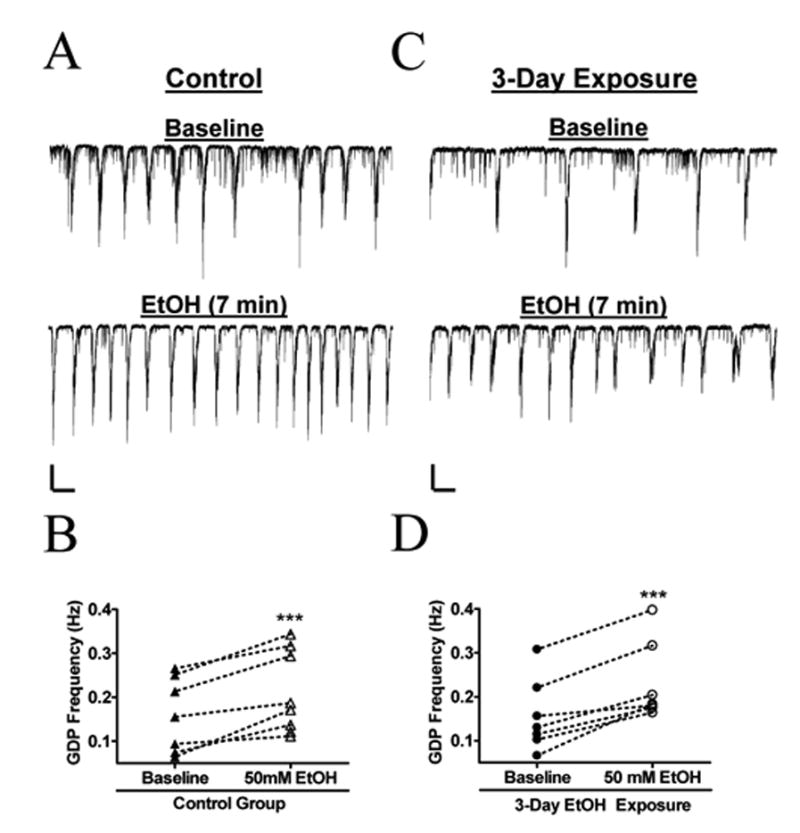
(A) Sample trace demonstrating an increase in GDP frequency during acute, 7-minute, application of 50 mM EtOH to a slice prepared 24 hr after 4 hr x 3 days exposure to air in the vapor chamber. (B) Summary on the effect of acute EtOH on air-exposed rat pups (n = 8). (C) Sample recording showing an increase in GDP frequency during acute application of 50 mM EtOH to a slice prepared 24 hr after 4 hr x 3 days exposure to 1.9 g/dL of EtOH in the vapor chambers. (D) Summary of the effect of acute EtOH on 3-day EtOH exposed animals ( n = 7). ***p < 0.001 by paired t-test. Scale bars for both set of traces = 200 pA/2 s.
Discussion
The main finding of this study is that GDPs do not develop tolerance to the modulatory effect of EtOH in CA3 pyramidal neurons from neonatal rats. We initially examined whether exposure to EtOH for a relatively short period of time was associated with tolerance development. Exposure of hippocampal slices to 50 mM EtOH for 3–4 hr followed by washout produced the same percent change in GDP frequency than that produced by acute (7 min) exposure to the same EtOH concentration. A similar effect was observed with GABAA-sPSCs, which in part drive GDP activity. These findings suggest that that during continuous EtOH exposure (i.e. over a few hours), GABAA receptor-driven excitatory activity in immature neuronal networks remains persistently increased; i.e. acute tolerance does not develop. In contrast to our findings, Allan and Harris (Allan & Harris, 1987) detected tolerance development to acute EtOH modulation of GABAA receptor-mediated 36Cl− flux in mouse cerebellar microsacs 60 min after in vivo injection of EtOH. A similar finding was recently reported by Wallace et al. (2006) with PKCε +/+ mice but not PKCε −/− mice. Thus, acute EtOH tolerance can be observed under some experimental conditions; i.e. when assaying activation of GABAA receptors by exogenous agonist in preparations from cerebella of mature mice but not when studying network activity driven by synaptically activated GABAA receptors in preparations from hippocampi of immature rats.
We also investigated whether tolerance developed after more prolonged exposure to EtOH. One- or three-day EtOH exposure of neonates via inhalation chambers did not change acute EtOH sensitivity of GDPs to EtOH. These findings are in general agreement with those of recent studies where EtOH modulation of GABAA receptor function was evaluated using electrophysiological techniques in acute slices from animals exposed to EtOH in vivo. Kang et al. (1998) studied GABAA receptor-mediated function in rats that were given 60 doses of 6 g/kg EtOH every 24 h by gastric intubation, with repeated intoxicating and withdrawal episodes. Rats treated with this paradigm develop a kindling-like persistent increase in withdrawal hyperexcitability. Interestingly, this paradigm of chronic intermittent EtOH exposure did not result in tolerance development of GABAA receptor-mediated inhibitory postsynaptic potentials in the hippocampal CA1 region. Instead, sensitization to the acute modulatory effects of 60 mM EtOH was detected. More recently, Liang et al. (2006) demonstrated that this gain of synaptic GABAA receptor sensitivity to acute EtOH exposure is associated with a loss of extrasynaptic GABAA receptor responsiveness to EtOH. Lack of tolerance development to the acute effects of EtOH on GABAergic synaptic transmission was also observed in slices from monkeys that self-administered EtOH daily for 18 months (Weiner & Valenzuela, 2006). Specifically, acute EtOH exposure increased evoked inhibitory postsynaptic currents in dentate granule cells to a similar extent in slices from control and chronically EtOH exposed monkeys. The issue of tolerance development of GABAergic responses was also addressed in a study with amygdala slices from rats exposed to EtOH for 2 weeks via inhalation chambers (Roberto et al., 2004). It was found that acute EtOH (5–66 mM) significantly enhanced evoked inhibitory postsynaptic responses equally in naive and chronically EtOH exposed rats, indicating that tolerance did not develop to the acute modulatory effects of EtOH. Taken together with our results, these findings suggest that GABAergic synapses in the developing and mature central nervous systems do not become tolerant to the acute modulatory effects of EtOH after long-term exposure to this drug.
Another finding of our study is that one- or three-day EtOH exposure of neonates via inhalation chambers did not significantly change basal GDP frequency, suggesting a lack of development of adaptive changes on excitatory GABAergic transmission in neonatal CA3 pyramidal neurons. This finding is not surprising given the relatively short duration of our exposure paradigm and the relatively low EtOH concentrations used in our experiments. Studies have typically found compensatory adaptations after relatively long periods of exposure to relatively high levels of EtOH. For instance, chronic intermittent EtOH exposure (6 g/kg) for 120 days produced a long-lasting decrease in GABAergic activity in hippocampal slices (Kang et al., 1996; Cagetti et al., 2003). In agreement with that study, electrophysiological studies revealed a decrease in GABA release probability in dentate granule cells from monkeys that administered EtOH for 18 months (BEC = 5–235mg/dL) (Weiner & Valenzuela, 2006). Unlike the hippocampus, exposure to BEC = 150–200 mg/dL for 2 weeks increased GABA release in the amygdala of rats (Roberto et al., 2004). Consequently, future experiments should assess whether longer exposure of neonates to EtOH results in compensatory alterations in excitatory network activity.
In conclusion, EtOH persistently changes correlated neuronal network activity driven by the excitatory actions of GABA in hippocampal neurons from neonatal rats. These effects of EtOH are likely to impair normal synaptic maturation. Moreover, Nunez et al (Nunez et al., 2003a; 2003b) recently demonstrated that activation of voltage-gated Ca2+ channels by GABAA-receptor mediated depolarization triggers apoptosis in the neonatal hippocampus. Consequently, these effects of EtOH could also alter neuronal survival. Given that GDPs have been detected in non-human primates during the third trimester of pregnancy, it is possible that similar effects occur in humans. Our data imply that network driven oscillatory activity does not adapt to continuous and/or repeated EtOH exposure, making developing synapses particularly susceptible to the deleterious effects of this widely abused substance.
Acknowledgments
Supported by National Institutes of Health grants AA12684 and AA15614. Rafael Galindo was supported by funds from the UNM-SOM MD/PhD program and minority supplement AA 12684-S1. We thank D. Partridge, D. Savage, M. Johnson and A. Azimi for helpful discussions.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionate
- BEC
blood ethanol concentration
- BMI
(−)-bicuculline methiodide
- DL-AP5
DL-2-amino-5-phosphonovaleric acid
- EtOH
ethanol
- GABA
γ-aminobutyric acid
- GDP
giant depolarizing potential
- PSC
postsynaptic current
- sPSC
spontaneous PSC
- mPSC
miniature PSC
- NBQX
2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide
- NMDA
N-methyl-D-aspartate
- P
postnatal day
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allan AM, Harris RA. Acute and chronic ethanol treatments alter GABA receptor-operated chloride channels. Pharmacol Biochem Behav. 1987;27:665–670. doi: 10.1016/0091-3057(87)90192-4. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Berman RF, Hannigan JH. Effects of prenatal alcohol exposure on the hippocampus: spatial behavior, electrophysiology, and neuroanatomy. Hippocampus. 2000;10:94–110. doi: 10.1002/(SICI)1098-1063(2000)10:1<94::AID-HIPO11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bonthius DJ, Pantazis NJ, Karacay B, Bonthius NE, Taggard Da, Lothman EW. Alcohol exposure during the brain growth spurt promotes hippocampal seizures, rapid kindling, and spreading depression. Alcohol Clin Exp Res. 2001a;25:734–745. [PubMed] [Google Scholar]
- Bonthius DJ, Woodhouse J, Bonthius NE, Taggard DA, Lothman EW. Reduced seizure threshold and hippocampal cell loss in rats exposed to alcohol during the brain growth spurt. Alcohol Clin Exp Res. 2001b;25:70–82. [PubMed] [Google Scholar]
- Cagetti E, Liang J, Spigelman I, Olsen RW. Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Mol Pharmacol. 2003;63:53–64. doi: 10.1124/mol.63.1.53. [DOI] [PubMed] [Google Scholar]
- Colin-Le Brun I, Ferrand N, Caillard O, Tosetti P, Ben-Ari Y, Gaiarsa JL. Spontaneous synaptic activity is required for the formation of functional GABAergic synapses in the developing rat hippocampus. J Physiol. 2004;559:129–139. doi: 10.1113/jphysiol.2004.065060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z, Ionescu P, Gong D, Kendig J, Harris A, Eger EI., 2nd Maturation decreases ethanol minimum alveolar anesthetic concentration in mice as previously demonstrated in rats: there is no species difference. Anesth Analg. 1997;85:160–163. doi: 10.1097/00000539-199707000-00029. [DOI] [PubMed] [Google Scholar]
- Fiszman ML, Schousboe A. Role of calcium and kinases on the neurotrophic effect induced by gamma-aminobutyric acid. J Neurosci Res. 2004;76:435–441. doi: 10.1002/jnr.20062. [DOI] [PubMed] [Google Scholar]
- Galindo R, Zamudio PA, Valenzuela CF. Alcohol is a potent stimulant of immature neuronal networks: implications for fetal alcohol spectrum disorder. J Neurochem. 2005;94:1500–1511. doi: 10.1111/j.1471-4159.2005.03294.x. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Horn KH, Zhou FC. Alcohol teratogenesis: mechanisms of damage and strategies for intervention. Exp Biol Med (Maywood) 2005;230:394–406. doi: 10.1177/15353702-0323006-07. [DOI] [PubMed] [Google Scholar]
- Groc L, Petanjek Z, Gustafsson B, Ben-Ari Y, Hanse E, Khazipov R. In vivo blockade of neural activity alters dendritic development of neonatal CA1 pyramidal cells. Eur J Neurosci. 2002;16:1931–1938. doi: 10.1046/j.1460-9568.2002.02264.x. [DOI] [PubMed] [Google Scholar]
- Hamilton DA, Kodituwakku P, Sutherland RJ, Savage DD. Children with Fetal Alcohol Syndrome are impaired at place learning but not cued-navigation in a virtual Morris water task. Behav Brain Res. 2003;143:85–94. doi: 10.1016/s0166-4328(03)00028-7. [DOI] [PubMed] [Google Scholar]
- Heaton MB, Mitchell JJ, Paiva M, Walker DW. Ethanol-induced alterations in the expression of neurotrophic factors in the developing rat central nervous system. Brain Res Dev Brain Res. 2000;121:97–107. doi: 10.1016/s0165-3806(00)00032-8. [DOI] [PubMed] [Google Scholar]
- Hollstedt C, Olsson O, Rydberg U. Effects of ethanol on the developing rat. II. Coordination as measured by the tilting-plane test. Med Biol. 1980;58:164–168. [PubMed] [Google Scholar]
- Kang M, Spigelman I, Sapp DW, Olsen RW. Persistent reduction of GABA(A) receptor-mediated inhibition in rat hippocampus after chronic intermittent ethanol treatment. Brain Res. 1996;709:221–228. doi: 10.1016/0006-8993(95)01274-5. [DOI] [PubMed] [Google Scholar]
- Kang MH, Spigelman I, Olsen RW. Alteration in the sensitivity of GABA(A) receptors to allosteric modulatory drugs in rat hippocampus after chronic intermittent ethanol treatment. Alcohol Clin Exp Res. 1998;22:2165–2173. [PubMed] [Google Scholar]
- Krahl SE, Berman RF, Hannigan JH. Electrophysiology of hippocampal CA1 neurons after prenatal ethanol exposure. Alcohol. 1999;17:125–131. doi: 10.1016/s0741-8329(98)00043-3. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Lamsa K, Pavlov I, Riekki R, Johnson BE, Molnar E, Rauvala H, Taira T. Activity blockade increases the number of functional synapses in the hippocampus of newborn rats. Mol Cell Neurosci. 2003;22:107–117. doi: 10.1016/s1044-7431(02)00012-x. [DOI] [PubMed] [Google Scholar]
- Leinekugel X, Medina I, Khalilov I, Ben-Ari Y, Khazipov R. Ca2+ oscillations mediated by the synergistic excitatory actions of GABA(A) and NMDA receptors in the neonatal hippocampus. Neuron. 1997;18:243–255. doi: 10.1016/s0896-6273(00)80265-2. [DOI] [PubMed] [Google Scholar]
- Liang J, Zhang N, Cagetti E, Houser CR, Olsen RW, Spigelman I. Chronic intermittent ethanol-induced switch of ethanol actions from extrasynaptic to synaptic hippocampal GABAA receptors. J Neurosci. 2006;26:1749–1758. doi: 10.1523/JNEUROSCI.4702-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantelas A, Stamatakis A, Kazanis I, Philippidis H, Stylianopoulou F. Control of neuronal nitric oxide synthase and brain-derived neurotrophic factor levels by GABA-A receptors in the developing rat cortex. Brain Res Dev Brain Res. 2003;145:185–195. doi: 10.1016/j.devbrainres.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Schoenfeld AM, Riley EP. Teratogenic effects of alcohol on brain and behavior. Alcohol Res Health. 2001;25:185–191. [PMC free article] [PubMed] [Google Scholar]
- Nunez JL, Alt JJ, McCarthy MM. A new model for prenatal brain damage. I. GABAA receptor activation induces cell death in developing rat hippocampus. Exp Neurol. 2003a;181:258–269. doi: 10.3201/eid0906.030118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez JL, Alt JJ, McCarthy MM. A novel model for prenatal brain damage. II. Long-term deficits in hippocampal cell number and hippocampal-dependent behavior following neonatal GABAA receptor activation. Exp Neurol. 2003b;181:270–280. doi: 10.3201/eid0906.020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR. Is there more to GABA than synaptic inhibition? Nat Rev Neurosci. 2002;3:715–727. doi: 10.1038/nrn919. [DOI] [PubMed] [Google Scholar]
- Penn AA, Shatz CJ. Brain waves and brain wiring: the role of endogenous and sensory-driven neural activity in development. Pediatr Res. 1999;45:447–458. doi: 10.1203/00006450-199904010-00001. [DOI] [PubMed] [Google Scholar]
- Richardson DP, Byrnes ML, Brien JF, Reynolds JN, Dringenberg HC. Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. Eur J Neurosci. 2002;16:1593–1598. doi: 10.1046/j.1460-9568.2002.02214.x. [DOI] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci. 2004;24:10159–10166. doi: 10.1523/JNEUROSCI.3004-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DD, Becher M, de la Torre AJ, Sutherland RJ. Dose-dependent effects of prenatal ethanol exposure on synaptic plasticity and learning in mature offspring. Alcohol Clin Exp Res. 2002;26:1752–1758. doi: 10.1097/01.ALC.0000038265.52107.20. [DOI] [PubMed] [Google Scholar]
- Silveri MM, Spear LP. Decreased sensitivity to the hypnotic effects of ethanol early in ontogeny. Alcohol Clin Exp Res. 1998;22:670–676. doi: 10.1111/j.1530-0277.1998.tb04310.x. [DOI] [PubMed] [Google Scholar]
- Sutherland RJ, McDonald RJ, Savage DD. Prenatal exposure to moderate levels of ethanol can have long-lasting effects on hippocampal synaptic plasticity in adult offspring. Hippocampus. 1997;7:232–238. doi: 10.1002/(SICI)1098-1063(1997)7:2<232::AID-HIPO9>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Wallace MJ, Newton PM, Oyasu M, McMahon T, Chou WH, Connolly J, Messing RO. Acute Functional Tolerance to Ethanol Mediated by Protein Kinase C varepsilon. Neuropsychopharmacology. 2006 doi: 10.1038/sj.npp.1301059. advance online publication 15 March 2006. [DOI] [PubMed] [Google Scholar]
- Weiner JL, Valenzuela CF. Ethanol modulation of GABAergic transmission: The view from the slice. Pharmacol Ther. 2006;111:533–54. doi: 10.1016/j.pharmthera.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Yuste R. Introduction: spontaneous activity in the developing central nervous system. Semin Cell Dev Biol. 1997;8:1–4. doi: 10.1006/scdb.1996.0114. [DOI] [PubMed] [Google Scholar]