

Abstract
D13 protein trimers, which form an external lattice providing curvature to the membrane of vaccinia virus immature virions, are the target of the drug rifampicin. We obtained 63 rifampicin-resistant mutants following random PCR mutagenesis of the D13L gene and 5 that arose spontaneously. Sequencing indicated that 26 mutants contained a single, unique, amino acid substitution whereas others contained 2 or more. The single mutations, including 6 previously identified, mapped to 24 different amino acids that were distributed over the length of the protein with the majority clustered between amino acids 17 to 33, 222 to 243 and 480 to 488. Two or three different substitutions occurred in six of the 24 amino acids. Representative mutant viruses of each cluster replicated to wild type levels in the absence of rifampicin and nearly two logs higher than wild type in the presence of drug. The large number and fitness of the mutations are remarkable in view of the extreme sequence conservation (99 to 100% amino acid identity amongst all orthopoxviruses). Clustering of mutations could suggest the presence of a rifampicin-binding pocket comprised of discontinuous regions of D13.
Keywords: poxvirus, drug-resistant mutants, rifampicin
Introduction
Poxviruses are large, enveloped DNA viruses that replicate and assemble in the cytoplasm of infected cells. Crescent-shaped membranes and spherical immature virions (IVs) are the first viral forms that can be recognized by electron microscopy in cells infected with vaccinia virus (VACV) and other poxviruses (Dales and Siminovitch, 1961; de Harven and Yohn, 1966). The curvature of the crescent and IV membranes is determined by an external lattice comprised of trimers of a 63-kDa protein encoded by the D13L (VACWR118) gene (Heuser, 2005; Szajner et al., 2005). During the subsequent stage of morphogenesis, the IV condenses into the infectious brick-shaped intracellular mature virion (MV) that no longer displays the lattice, indicating that D13 acts as a temporary scaffold. D13 is conserved in all poxviruses and a segment has a low but significant sequence identity with capsid proteins of certain other large DNA viruses (Iyer, Aravind, and Koonin, 2001), leading to the idea that poxvirus morphogenesis may recapitulate evolution from an ancestral icosahedral virus to a more complex form (Szajner et al., 2005).
The external lattice, originally thought to be a spicule layer, does not form on the surface of viral membranes when cells are infected with VACV in the presence of the drug rifampicin (Grimley et al., 1970; Moss et al., 1969; Nagayama, Pogo, and Dales, 1970), a semisynthetic antibiotic with a molecular weight of 823 derived from Streptomyces mediterranei. Instead, irregular viral membranes surrounding masses of electron dense material are observed. Although rifampicin binds to the β subunit of the DNA-dependent RNA polymerase of bacteria and inhibits transcription, the drug has little or no effect on transcription of vaccinia virus, which encodes its own rifampicin-insensitive RNA polymerase (Moss, Katz, and Rosenblum, 1969). Instead, there is considerable evidence that the D13 scaffold protein is the target of rifampicin: VACV mutations that confer resistance to rifampicin map exclusively to the D13L gene (Baldick and Moss, 1987; McNulty-Kowalczyk and Paoletti, 1993; Tartaglia, Piccini, and Paoletti, 1986); repression of D13L expression blocks virus assembly, resulting in the formation of irregular membranes that closely resemble those formed in the presence of rifampicin (Zhang and Moss, 1992); and D13 accumulates in large cytoplasmic masses in the presence of rifampicin, preventing association with crescent and IV membranes (Miner and Hruby, 1989; Mohandas and Dales, 1995; Sodeik et al., 1994).
In the present study, random PCR mutagenesis was used to generate a large library of D13 mutations that confer resistance to rifampicin. The number and fitness of the mutations are remarkable in view of the sequence conservation of the protein (99 to 100% identity in orthopoxviruses). Most of the mutations clustered within short segments at the N- and C-termini and also in the central region of D13, suggesting a putative rifampicin-binding pocket comprised of discontinuous regions of D13.
Results
Isolation of rifampicin-resistant mutants with missense substitutions in D13
Random PCR mutagenesis was performed under conditions intended to introduce 0 to 3 nucleotide changes into each copy of the D13L gene. The DNA was then transfected into BS-C-1 cells that had been infected with VACV strain WR. After harvesting the infected cells, lysates were applied to fresh monolayers in the presence of rifampicin (100 μg/ml). Virus was picked from the drug-resistant plaques and subjected to a second round of plaque purification in the presence of the same concentration of rifampicin. Stocks of each rifampicin-resistant virus were then prepared and the D13L gene of each was sequenced. Of 63 rifampicin-resistant isolates, 25 had single missense mutations in the D13L gene and 22 of these were unique. The other rifampicin-resistant mutants had two or more substitutions in the D13L gene. In addition, we isolated five spontaneous rifampicin-resistant mutants after one amplification passage of individual virus plaques; each had a single amino acid substitution in the D13L gene and four of these were unique relative to the ones obtained by PCR mutagenesis. The locations of the 26 new single mutations in addition to 6 previously identified spontaneous mutations differing from the above are shown in Fig. 1. The 32 mutations occurred in 24 amino acids (6 of the amino acids had 2 or 3 different substitutions), which were distributed between amino acids 17 to 94, 175 to 314, and 429 to 488. Within these regions, clustering of mutations occurred between amino acids 17 to 33, 222 to 243 and 480 to 488.
Fig. 1.
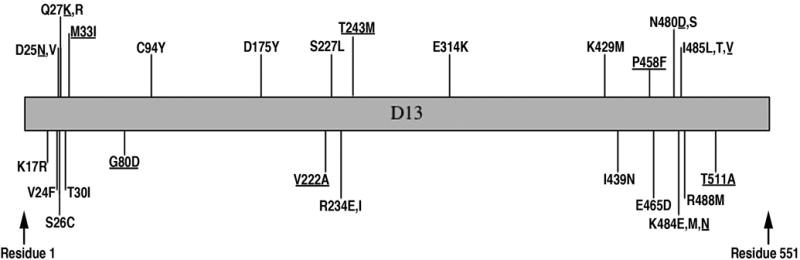
Amino acid substitutions that confer resistance to rifampicin. The position number of each amino acid conferring rifampicin-resistance is shown. The original amino acid is to the left of the number and the substitution(s) on the right. Spontaneous mutations are underlined; others were obtained after random PCR mutagenesis of the D13L gene.
Demonstration that the D13 mutations confer rifampicin-resistance
The occurrence of D13 mutations in many of the rifampicin-resistant VACV mutants could have been coincidental, since the infected cells were transfected with DNA containing the randomly mutagenized D13 gene. To determine whether the D13 amino acid substitutions confer rifampicin-resistance, we made specific mutations in plasmid pJAG9.4, which contains the wild type D13 gene and approximately 200 bp of flanking sequence on each side. Cells were infected with VACV strain WR and transfected with either non-mutagenized or mutagenized pJAG9.4. After 24 h, the cells were harvested and the number of rifampicin-resistant mutants was determined by plaque assay. Data from four replicate transfections are shown in Fig. 2. The number of resistant mutants obtained was significantly higher after transfection with each of the mutated D13 genes than with the wild type gene, confirming the relationship of D13 amino acid substitutions with rifampicin-resistance. The difference in frequency of rifampicin-resistant mutants did not correlate with position of the gene and could have been related to the DNA preparations or other factors.
Fig. 2.

Frequency of rifampicin-resistant mutants following transfection of D13L DNA with site-specific mutations. Plasmid pJAG9.4 containing wild type D13L gene and flanking DNA or the same plasmid with site-specific mutations, corresponding to those found in rifampicin-resistant mutants generated by random PCR mutagenesis, were transfected into BS-C-1 cells infected with VACV WR. After 24 h, the titer of rifampicin-resistant virus in each transfected culture was determined by plaque assay. The relative numbers of rifampicin-resistant plaques were determined by dividing the number detected after transfection of each mutated plasmid by the number obtained by transfection of the wild type gene in the same experiment. Bars represent standard error (N=4).
Plaque morphology of mutant viruses
In the absence of rifampicin, the plaque sizes of most mutants were similar to each other and to wild type virus. Two exceptions were R234E and N480S; after 48 h the plaques were ~75% of the size of wild type virus plaques. In the presence of 100 μg per ml of rifampicin, the wild type virus made no plaques whereas the numbers of plaques of the mutants were unaffected. However, the plaques of the mutant viruses were smaller in the presence of rifampicin than in its absence and varied slightly in size. Plaques of wild type virus and representative mutants mapping to different locations in the D13 open reading frame are shown in Fig. 3.
Fig. 3.
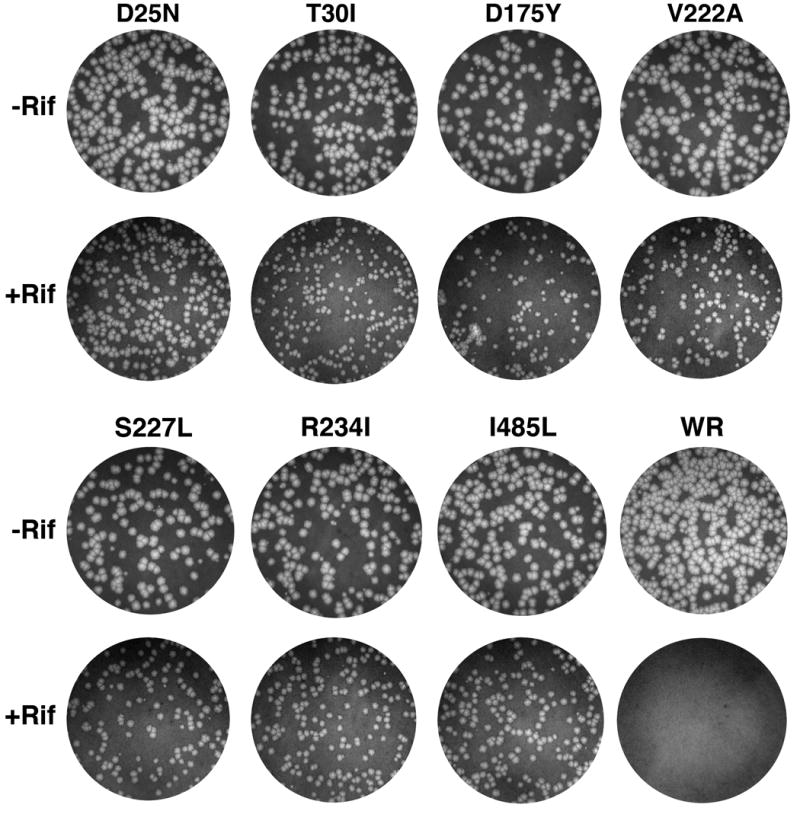
Plaque morphology of rifampicin-resistant mutants. Plaques that formed in 2 days on BS-C-1 cell monolayers in 6-well plates by representative rifampicin-resistant mutants and wild type VACV in the presence (+Rif) and absence (−Rif) of 100 μg per ml of rifampicin were visualized by staining with crystal violet and photographed. Mutants are designated by the codon substitution in the D13L gene. Wild type VACV strain WR is designated WR.
Virus growth in the presence and absence of rifampicin
One-step growth experiments with 3 plaque-forming units (PFU) of virus per cell were carried out to assess the effects of the drug on virus yield. In each case, the mutant replicated approximately 1.5 logs higher than the wild type virus in the presence of rifampicin (Fig. 4). The 21 h yields of seven mutants and wild type virus in the presence and absence of rifampicin were determined in triplicate as shown in Fig. 5A. The difference in virus yield between the wild type and mutant viruses was not statistically significant in the absence of rifampicin, whereas in the presence of drug the difference between the wild type virus and each of the mutants was highly significant (p<0.005). The wild type virus was inhibited by 98.7% under the latter conditions. We also followed the growth of viruses when cells were infected with 0.33 PFU of virus per cell. Even at this lower multiplicity of infection, where a growth defect of mutant viruses might be more apparent, the yields were similar in the absence of rifampicin (Fig. 5B). Moreover, the yields of mutant viruses in the presence and absence of rifampicin were very similar, whereas growth of wild type virus was inhibited by 98.3% (Fig. 5B).
Fig. 4.

One-step virus growth in the presence and absence of rifampicin. BS-C-1 cells were infected with 3 PFU per cell of wild type VACV WR or mutants designated by the codon substitution in the D13L gene. After the 1 h adsorption, the cells were incubated for 0, 8, 21 or 44 h in the absence (uninterrupted line) or presence (dashed line) of 100 μg of rifampicin per ml. Virus titers were determined by plaque assay in the absence of rifampicin.
Fig. 5.
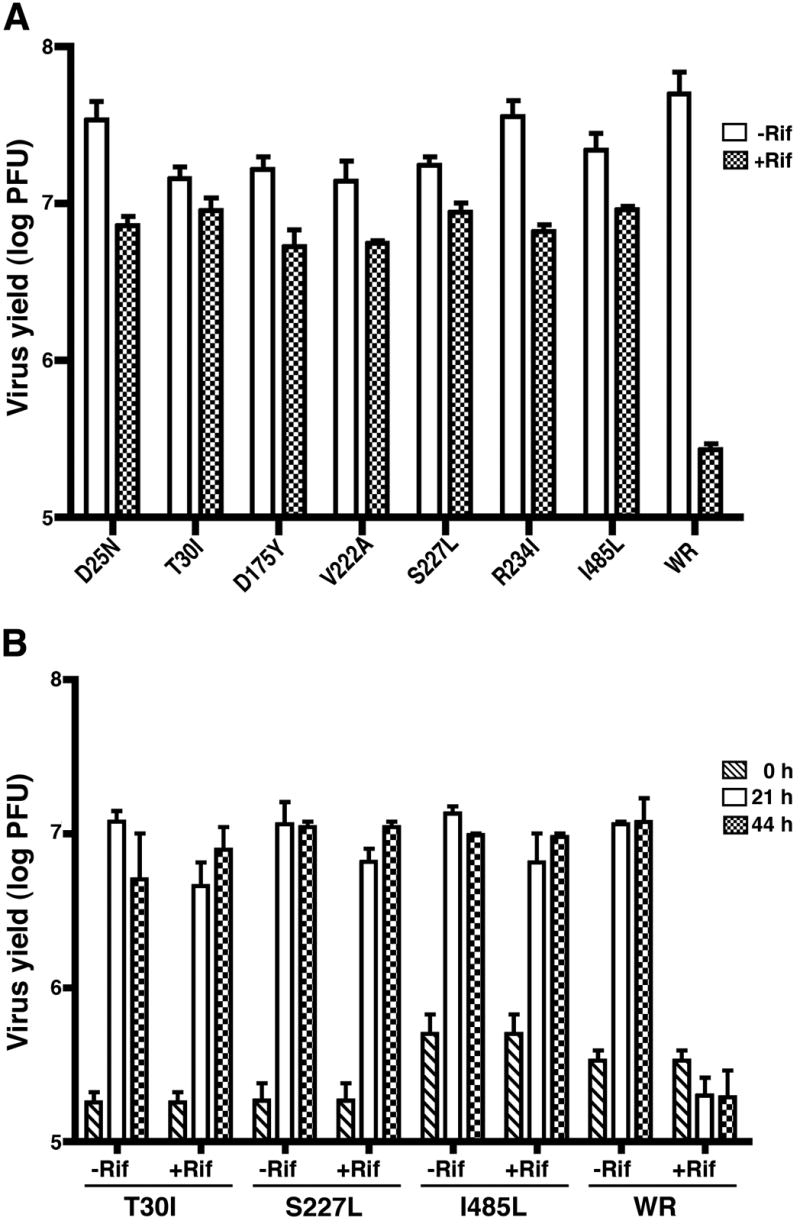
Virus yields in the presence or absence of rifampicin. (A) BS-C-1 cells were infected in triplicate with 3 PFU per cell of mutant or wild type VACV WR in the absence (Rif−) or presence (Rif+) of 100 μg of rifampicin per ml as described in the legend to Fig. 4. After 21 h, virus yields were determined by plaque assay in the absence of rifampicin. Standard error bars are shown. (B) Cells were infected in duplicate as described in panel A except that the multiplicity of infection was 0.33 PFU per cell and virus yields were determined at 0, 21 and 44 h. Abbreviations for mutant and wild type viruses are the same as in Fig. 4.
Discussion
Our interest in the D13 protein of VACV stemmed from its intriguing function as an essential component of the temporary scaffold specifying the curvature of IVs, role as a target for the drug rifampicin, and high conservation among poxviruses. The 99 to 100% sequence identity of D13 amongst all orthopoxvirus species suggested that the protein might not tolerate many mutations without loss of function. The ability to select rifampicin-resistant mutants provided us with an opportunity to more thoroughly map the sites of rifampicin-resistance, while allowing us to determine the location of amino changes with minimal effects on replication. Our strategy was to transfect randomly PCR mutagenized D13L DNA into cells infected with wild type VACV and isolate plaques that formed in the presence of rifampicin. In this manner we isolated 63 drug-resistant mutants and identified the codon changes by DNA sequencing. Of these, 25 had single base changes that encoded amino acid substitutions in D13L and 22 were unique. Each of the 22 mutations was shown to mediate drug-resistance. We also isolated five spontaneous rifampicin-resistant mutants and found that four of these were unique with respect to the others. Together with the six spontaneous mutants reported previously, there are now a total of 32 changes in 24 amino acids of D13 known to confer rifampicin-resistance. Seven of the 24 amino acids cluster near the N-terminus (cluster 1, between amino acids 17 and 33), four near the center of the protein (cluster 2, between amino acids 222 and 243), and four near the C-terminus (cluster 3, between amino acids 480 and 488). These sites were also notable for the occurrence of multiple substitutions at the same amino acid. The remaining eight amino acids were distributed on the fringes of the clusters. Many of the mutations were non-conservative i.e. from a non-charged to a charged amino acid or vice versa or a small to a bulky amino acid or vice versa. However, a few changes were conservative such as K17R, E465D and I485L. In view of the extreme conservation of orthopoxvirus D13 homologs, we were surprised by the fitness of the mutants. In the absence of rifampicin, the yields of representative mutants from each of the clusters were similar to those of wild type virus. Their unimpaired replication accounts for the ease of isolation of spontaneous rifampicin-resistance mutants, which we detected after one passage of individual plaques. Although wild type VACV (strain WR) does not make plaques in the presence of 100 μg per ml of rifampicin, the resistant mutants made the same number of slightly smaller plaques in the presence of drug. It is difficult to know whether some of the size reduction is due to non-specific cellular effects of the drug or to a weak effect on D13. Under one step growth conditions in the presence of rifampicin, the yields of representative viruses mapping to different regions of D13 were 1.5 to 2 logs higher than that of wild type virus.
Rifampicin is a bacteriocidal antibiotic that is used to treat tuberculosis and leprosy as well as other infections. It interferes with transcription by binding to bacterial RNA polymerase. Rifampicin-resistance of Escherichia coli mutants map to the center of the rpoB gene, affecting only a limited number of amino acids that are thought to be present in a binding pocket (Jin and Gross, 1988). The concentration of rifampicin needed to inhibit vaccinia virus replication is higher than that of bacteria and the drug has not found clinical utility as an antiviral agent. Although direct binding of rifampicin to D13 has not been reported, that fact that all spontaneous VACV rifampicin-resistant mutants map to D13 makes this likely. In contrast to E. coli rpoB, D13 mutations that confer rifampicin-resistance form clusters at the two ends of the protein as well as in the central region. Although speculative, we suggest that these discontinuous regions of D13 form a rifampicin-binding pocket. A segment of D13, between amino acids 71 to 195, has a limited but statistically significant similarity with predicted structural elements of the capsid proteins of iridoviruses and phycodnaviruses (Iyer, Aravind, and Koonin, 2001). The C-terminal jelly roll-like domain of the capsid protein of the latter viruses is totally absent from D13 and is replaced by a predicted β-strand-rich domain without detectable structural similarity to a domain of any other protein. Unfortunately, few of the rifampicin-resistance mutations map to the homology region and therefore the full interpretation of our data must await structural analysis of D13. Based on electron microscopic images of D13, we doubt that rifampicin interferes with trimer formation and more likely prevents interaction of D13 with a viral membrane protein (Szajner et al., 2005).
Materials and Methods
PCR mutagenesis
PCR mutagenesis of the D13L gene in plasmid pJAG9.4 (a gift of P. Szajner) was carried out using the GeneMorph Random Mutagenesis Kit (Stratagene) according to the manufacturers direction to achieve 0 to 3 mutations per kbp. For mutagenesis of the entire D13L gene, the primer pair used was 5′ CTATGAAGATAAGGAAAAGGTC 3′ and 5′ TAGCTCCGTATTCCAGACT 3′. To enrich for mutations downstream of the N-terminal cluster, the oligonucleotide 5′ ATATATGCCGCAATATATTTCTC 5′ was used in place of the first primer. DNA was purified using the QIAquick PCR Purification kit (Qiagen).
Isolation and sequencing of rifampicin-resistant mutants
BS-C-1 cells in individual wells of a 6-well plate were infected at a multiplicity of 1 PFU per cell and 1 h later were transfected with 0.5 to 1.0 μg of PCR product described above. Each well received DNA from an independent PCR reaction. After 21 h, the cells were harvested and lysed by freezing and thawing. Stock solutions of 10 mg/ml of rifampicin (Sigma-Aldrich St. Louis MO) were prepared in DMSO and stored at -20°C or -80°C for use at a 100-fold dilution. Dilutions of the lysates were incubated with BS-C-1 cells for 1 h. The plates were washed and covered with a 1% agarose (low melting point, Invitrogen) overlay containing 100 μg of rifampicin per ml at 37°C for 2 days. Virus from individual plaques was then subjected to a second round of plaque purification under the same conditions. The cloned rifampicin-resistant viruses were amplified in a 24-well plate and the DNA isolated with a QIAamp DNA blood mini kit (Qiagen). Amplicons, prepared from each DNA preparation, were sequenced using an ABI PRISM 3100 Genetic Analyzer.
To isolate spontaneous rifampicin-resistant mutants, 5 plaques of wild type VACV WR were amplified separately in a 6-well plate and harvested. Dilutions of virus from the single round of amplification were used to infect 6-well plates of BS-C-1 cells covered by an agarose overlay containing 100 μg of rifampicin per ml. Virus from individual plaques was subjected to a second round of plaque purification. The cloned rifampicin-resistance virus was then amplified and the DNA sequenced as above.
Verification of rifampicin-resistance mutations by site-specific mutagenesis
The Quickchange site directed mutagenesis kit (Stratagene) was used to introduce specific mutations into the D13L gene in pJAG9-4. BS-C-1 cells were infected with 109 PFU of VACV WR. After 1 h, the cells were transfected with 1 μg of plasmid. The cells were harvested approximately 26 h later and disrupted by freezing and thawing. Dilutions of the lysate were then titered in the presence of 100 μg of rifampicin per ml and plaques were visualized by staining the plates with crystal violet (0.1% in 20% ethanol) and washing with water.
Determination of virus yields in the presence and absence of rifampicin
BS-C-1 cell monolayers in 6-well plates were infected with 0.2 ml of virus at a multiplicity of 0.33 or 3 PFU per cell. After 1 h at 37°C, the plates were washed and 2 ml of medium containing 2.5% bovine serum with or without 100 μg per ml of rifampicin was added. The cells were harvested at various times and lysed by freezing and thawing three times and sonicating. Virus dilutions were adsorbed to BS-C-1 cells for 1 h, covered with an overlay of 0.5% methyl cellulose (Sigma-Aldrich) in minimum essential medium with Earle’s salts (Quality Biologicals) and incubated for 2 days at 37°C. Virus plaques were visualized by staining with crystal violet.
Acknowledgments
We thank Norman Cooper for providing cells, Andrea Weisberg for help preparing figures, and Wolf Resch for statistical analysis and modeling the D13 protein. The work was supported by NIAID intramural research funds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baldick CJ, Moss B. Resistance of vaccinia virus to rifampicin conferred by a single nucleotide substitution near the predicted NH2 terminus of a gene encoding an Mr 62,000 polypeptide. Virology. 1987;156:138–145. doi: 10.1016/0042-6822(87)90444-2. [DOI] [PubMed] [Google Scholar]
- Dales S, Siminovitch L. The development of vaccinia virus in Earle's L strain cells as examined by electron microscopy. J Biophys Biochem Cytol. 1961;10:475–503. doi: 10.1083/jcb.10.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Harven E, Yohn DS. The fine structure of monkey tumor poxvirus. Cancer Res. 1966;26:995–1008. [PubMed] [Google Scholar]
- Grimley PM, Rosenblum EN, Mims SJ, Moss B. Interruption by rifampin of an early stage in vaccinia virus morphogenesis: Accumulation of membranes which are precursors of virus envelopes. J Virol. 1970;6:519–533. doi: 10.1128/jvi.6.4.519-533.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J. Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic "honeycomb" surface coat. J Cell Biol. 2005;169:269–283. doi: 10.1083/jcb.200412169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Aravind L, Koonin EV. Common origin of four diverse families of large eukaryotic DNA viruses. J Virol. 2001;75:11720–11734. doi: 10.1128/JVI.75.23.11720-11734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin DJ, Gross CA. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol. 1988;202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- McNulty-Kowalczyk A, Paoletti E. Mutations in ORF D13L and other genetic loci alter the rifampicin phenotype of vaccinia virus. Virology. 1993;194:638–646. doi: 10.1006/viro.1993.1303. [DOI] [PubMed] [Google Scholar]
- Miner JN, Hruby DE. Rifampicin prevents virosome localization of L65, an essential vaccinia virus polypeptide. Virology. 1989;170:227–237. doi: 10.1016/0042-6822(89)90370-x. [DOI] [PubMed] [Google Scholar]
- Mohandas AR, Dales S. Involvement of spicules in the formation of vaccinia virus envelopes elucidated by a conditional lethal mutant. Virology. 1995;214:494–502. doi: 10.1006/viro.1995.0060. [DOI] [PubMed] [Google Scholar]
- Moss B, Katz E, Rosenblum EN. Vaccinia virus directed RNA and protein synthesis in the presence of rifampicin. Biochem Biophys Res Commun. 1969;36:858–865. doi: 10.1016/0006-291x(69)90688-3. [DOI] [PubMed] [Google Scholar]
- Moss B, Rosenblum EN, Katz E, Grimley PM. Rifampicin: A specific inhibitor of vaccinia virus assembly. Nature. 1969;224:1280–1284. doi: 10.1038/2241280a0. [DOI] [PubMed] [Google Scholar]
- Nagayama A, Pogo BGT, Dales S. Biogenesis of vaccinia: separation of early stages from maturation by means of rifampicin. Virology. 1970;40:1039–1051. doi: 10.1016/0042-6822(70)90150-9. [DOI] [PubMed] [Google Scholar]
- Sodeik B, Griffiths G, Ericsson M, Moss B, Doms RW. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J Virol. 1994;68:1103–1114. doi: 10.1128/jvi.68.2.1103-1114.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szajner P, Weisberg AS, Lebowitz J, Heuser J, Moss B. External scaffold of spherical immature poxvirus particles is made of protein trimers, forming a honeycomb lattice. J Cell Biol. 2005;170:971–981. doi: 10.1083/jcb.200504026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia J, Piccini A, Paoletti E. Vaccinia virus rifampicin-resistance locus specifies a late 63,000 Da gene product. Virology. 1986;150:45–54. doi: 10.1016/0042-6822(86)90264-3. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Moss B. Immature viral envelope formation is interrupted at the same stage by lac operator-mediated repression of the vaccinia virus D13L gene and by the drug rifampicin. Virology. 1992;187:643–653. doi: 10.1016/0042-6822(92)90467-4. [DOI] [PubMed] [Google Scholar]