

Abstract
The coding sequence at the boundaries of exons flanking nuclear introns shows some degree of conservation. To the extent that such sequences might be recognized by the splicing machinery, this conservation may be a derived result of evolution for efficient splicing. Alternatively, such conserved sequences might be remnants of proto-splice sites, which might have existed early in eukaryotic genes and served as the targets for the insertion of introns, as has been proposed by the introns-late theory. The distribution of intron phases, the position of the intron within a codon, is biased with an over-representation of phase 0 introns. Could any distribution of proto-splice sites account for today’s intron phase distribution? Here, we examine the dicodon usage in six model organisms, based on current sequences in the GenBank database, and predict the phase distribution that would be expected if introns had been inserted into proto-splice sites. However, these predictions differ between the various model organisms and disagree with the observed intron phase distributions. Thus, we reject the hypothesis that introns are inserted into hypothetical proto-splice sites. Finally, we analyze the sequences around the splice sites of introns in all six of the species to show that the actual conservation of sequence in exon regions near introns is very small and differs considerably between these species, which is inconsistent with a general proto-splice sites model.
Keywords: intron–exon structure, dicodon usage
The significance of any conservation of DNA sequences near the exon–intron boundaries is an open question. Within the introns, there is very high conservation at and near the boundaries: The GT..AG rule is obeyed very well, with a minor exception, the AT..AC signal, in a small class of nuclear introns (1–2). Within the exon, various groups have conjectured that coding sequences near the boundaries also are conserved, such as the hypothesis of a (C/A)AG|G conservation in mammalian genes and an (A/G)|N conservation in Saccharomyces cerevisiae (the symbol “|” stands for the intron positions) (3–4).
There are two alternative scenarios to account for the origin of conserved exon sequences. One is that the conserved sequence is a splicing signal at the exon boundary that has evolved as a result of natural selection for efficient splicing because the small nuclear RNAs in the splicing apparatus necessarily interact with some of the exon sequence. One clear case of such pairing has been identified in S. cerevisiae (5–6). Alternatively, conserved exon sequences might be remnants of early sequences in the coding regions that served as recognition sites for the insertion of introns, as proposed by Dibb and Newman (7), who called such consensus sequences “proto-splice” sites. Such proto-splice sites have been used as a conceptual basis for introns-late theories (8–10).
Some authors (11–13) have shown that the distribution of intron phases is significantly biased toward the phase 0 introns. Although these authors have argued that this biased distribution was most likely to be a consequence of exon shuffling, an alternative hypothesis would be that the biased intron-phase distribution is a consequence of intron insertion into nonrandomly distributed proto-splice sites.
In this study, we tested such models of proto-splice site insertion by examining their predictions for the intron-phase distribution. We will show that the distribution of hypothetical proto-splice sites in the genomes of six model organisms fails to explain the actual distribution of intron phases. Furthermore, taking the extensive sequence comparisons now available, we can show that most of the conserved information is confined within the intron and that the conserved information content within the exon is very small, and different in different organisms, suggesting only a fragile basis for any proto-splice site model.
METHODS
General Approach.
To analyze the distribution of proto-splice sequences, we analyzed the dicodon distribution in the coding sequence of intron-containing genes. We examined the true dicodon distribution rather than simply using the codon frequencies because the correlation between adjacent codons may affect the distribution of proto-splice sites that cross codons. We calculated the correlation of dicodon frequencies by information analysis and calculated the distribution of various hypothetical proto-splice sites. We then compared such distributions with the actual intron phase distribution.
Finally, to assess the validity of a proto-splice site analysis, which is based on sequence conservation within exons, we determined the distribution of sequences of both exons and introns and calculated the information content of each position, summarized by the logo analysis of Schneider et al. (14).
Exon Databases and Calculation of Intron Phase Proportions.
We chose six model species, which have many sequenced genes and contain representatives of the major eukaryotic lineages. These organisms are Homo sapiens, Drosophila melanogaster, Caenorhabditis elegans, Saccharomyces cerevisiae, Schizosaccharomyces pombe, and Arabidopsis thaliana.
We used computing methods similar to those of Long et al. (12) to develop intron–exon databases of the six model organisms from GenBank database release 96. We deleted the highly redundant genes, such as the Ig superfamily in humans or the Adh (alcohol dehydrogenase) sequences created for population genetics study in Drosophila. We purged the CDS databases (a CDS is the composed coding sequence for an intron-containing gene) to a criterion of 80% to remove duplicates and closely related genes by using the gbpurge program (12). (We also purged each database further to a criterion of 20% with gbpurge and found that the proportions of intron phases and the other properties that we computed in various species did not change or changed insignificantly.) The C. elegans database contains many cosmid sequences that are analyzed by prediction by the genefinder computer program. We purged these hypothetical genes from the C. elegans database for these calculations.
Proto-Splice Sites.
Based on the previous analysis of conservation of coding sequences around the splicing sites (1, 4, 7), we chose four candidates for proto-splice sites: G|G, AG|G, AG|GT, and (C/A)AG|R (where the bar symbol indicates the site of the intron and R is purine A or G). [(C/A)AG|R was proposed by Dibb and Newman (7).]
We used the information content measure of Schneider to evaluate the importance of any sequence conservation (14–15). The amount of information at each nucleotide site i is calculated by
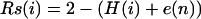 |
where H(i) = −Σfj(i)log2fj(i), fj(i) is the frequency of the base j at position i. Here e(n) = 3/(2ln(2)n) is a correction for sample size (approximate calculation), where n is the number of introns. When there is no conservation, Rs(i) = 0; when frequency of a single base reaches 100% (maximum conservation), Rs(i) = 2.
Dicodon Correlation.
We generated CDS databases for the six model species. We calculated the frequencies of the 64 × 64 dicodons from the CDS databases. We analyzed the frequencies of the dicodon types (stop codon)N4N5N6 as a control for two possible errors: errors caused by any irregularity in the feature tables in GenBank from which the CDS databases were developed and errors arising by the inclusion of pseudogenes. These errors lead to non-zero frequencies of the dicodons of these types and were all removed.
We analyzed the information content in the dicodon sequences (in-frame hexamers) (16) by using the formula
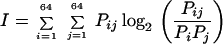 |
where Pij is the frequency of the dicodon i and j; Pi and Pj are codon frequencies of i and j. When all codon pairs i and j are completely independent, Pij = Pi.Pj, and the information I is equal to 0. On the contrary, if one codon (i or j) completely determines the other (j or i), the information I will reach a maximum of six.
We then wrote a computer program to scan every dicodon for proto-splice sites in each phase. To avoid any repeated counting of proto-splice sites that have length equal to or shorter than 3, we counted the dicodons that contained the site only at the 5′ codons and the site across the codon. (Counting 3′ codons yields the same results.) If a dicodon sequence contains more than one proto-splice site, we counted its frequency for each site.
RESULTS
The Six Model Species Show Different Patterns of Intron Phases.
Table 1 shows the distribution of intron phases for the six species. All of the species, except yeast because of a small sample size, showed a significant deviation from an equal–probable distribution (one-third) of intron phases and a preference for phase 0 introns. Furthermore, the species differed. Phase 0 introns ranged from 56% in Arabidopsis to 39% in S. cerevisiae, and phase 1 introns ranged from 36% in humans to 23% in Arabidopsis. This variation in the proportions of intron phases was not consistent with the model that proto-splice sites were used for targeting in the early stages of eukaryote evolution because this model would predict similar distributions of intron phase across the eukaryotic organisms. However, one might assume that the frequencies of proto-splice sites could have evolved differently in the different lineages. Can the distribution of proto-splice sites explain these peculiar distributions of intron phases?
Table 1.
Proportions of three intron phases
| Species | Intron phases, %
|
Intron number | Gene number | P | ||
|---|---|---|---|---|---|---|
| 0 | 1 | 2 | ||||
| A. thaliana | 56 | 23 | 21 | 1342 | 300 | 2.93 × 10−68 |
| S. cerevisiae | 39 | 35 | 26 | 163 | 160 | 0.11 |
| S. pombe | 45 | 29 | 26 | 796 | 364 | 1.46 × 10−11 |
| C. elegans | 47 | 29 | 24 | 1096 | 195 | 1.23 × 10−21 |
| D. melanogaster | 46 | 31 | 23 | 1145 | 385 | 4.73 × 10−21 |
| H. sapiens | 44 | 36 | 20 | 7743 | 1301 | 1.99 × 10−151 |
Prediction of Intron Phases Based on Proto-Splice Sites and Dicodon Usage.
Fig. 1 shows, as an example, the dicodon frequency of human genes by using the 80% purged database. (The dicodon frequencies of the other five species are available on request.) First, we analyzed the correlation between codons by calculating an information content by using a measure that ranges from 0 to 6. Table 2 shows that there were correlations between adjacent codons. However, the correlations differed by up to 2-fold. D. melanogaster and humans had the highest correlations (I = 0.14), and S. pombe had the lowest (I = 0.07).
Figure 1.

Human dicodon table (106).
Table 2.
Dicodon correlation (I)
| A. thaliana | 0.13 |
| S. serevisiae | 0.13 |
| S. pombe | 0.07 |
| C. elegans | 0.13 |
| D. melanogaster | 0.14 |
| H. sapiens | 0.14 |
We then calculated the expected frequencies of intron phase that would be determined by four hypothetical proto-splice sites [G|G, AG|G, AG|GT, and (C/A)AG|R] by using the dicodon frequencies for the six species. Table 3 lists these different patterns and compares them to the observed phases; the P values are given below each dicodon for all species.
Table 3.
Intron phase distribution (%) predicted from dicodon frequency
| Proto-splice site intron phase | G/G
|
AG/G
|
AG/GT
|
(C/A)AG/R
|
Observed
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 2 | |
| A. thaliana | 36 | 43 | 21 | 41 | 37 | 22 | 36 | 48 | 16 | 41 | 42 | 17 | 56 | 23 | 21 |
| P values | 3 × 10−60 | 3 × 10−32 | 1 × 10−75 | 2 × 10−44 | |||||||||||
| S. cerevisiae | 40 | 42 | 18 | 48 | 34 | 18 | 31 | 59 | 10 | 39 | 36 | 25 | 39 | 35 | 26 |
| P values | 0.02 | 0.01 | 6 × 10−14 | 0.95 | |||||||||||
| S. pombe | 40 | 41 | 19 | 47 | 36 | 17 | 35 | 52 | 13 | 32 | 52 | 16 | 45 | 29 | 26 |
| P values | 2 × 10−12 | 2 × 10−11 | 1 × 10−45 | 3 × 10−38 | |||||||||||
| C. elegans | 38 | 50 | 12 | 51 | 44 | 5 | 57 | 38 | 5 | 42 | 45 | 13 | 47 | 29 | 24 |
| P values | 2 × 10−55 | 7 × 10−185 | 10 × 10−182 | 8 × 10−38 | |||||||||||
| D. melanogaster | 55 | 32 | 13 | 75 | 14 | 12 | 77 | 16 | 7 | 71 | 19 | 10 | 46 | 31 | 23 |
| P values | 1 × 10−23 | 6 × 10−105 | 1 × 10−157 | 2 × 10−83 | |||||||||||
| H. sapiens | 46 | 32 | 23 | 57 | 26 | 17 | 59 | 27 | 14 | 50 | 35 | 15 | 44 | 36 | 20 |
| P values | 3 × 10−17 | 4 × 10−124 | 2 × 10−158 | 2 × 10−41 | |||||||||||
The predicted intron phase frequencies were not consistent with the observed proportions of intron phases for many of the species. For example, for A. thaliana, no proto-splice sites or combinations of sites gave intron phase frequencies close to observation because the observed fraction of phase 0 introns was too high. Among 24 comparisons, only the S. cerevisiae phase pattern was very similar to the (C/A)AG|R one; we think this single case is a random match to the 163 introns of S. cerevisiae. All of the other comparisons showed a significant difference between the observed and expected proportions in χ2 tests.
Sequence Conservation at the Exon Side of the Splice Sites.
We analyzed the distribution of 10 bases on both the intron and exon sides of each splice junction in our databases. The logos in Fig. 2 show the sequence conservation at the splice sites for the six species. Here, the total height of the stacked letters at each position is the total amount of information at that position, and the heights of individual letters reflect the proportion of the nucleotides.
Figure 2.

Information content at each position in the 10 bases flanking the exon intron boundary and the intron–exon boundary. The total height at each position is given by the information content at that position. The height of each letter is proportional to the fraction that base is of the total at each position.
Fig. 2 shows that the information content is very uneven between exons and introns in all six species. The amount of information in the exons is very small; more than 90% of the information is contained within the intron. For the limited conservation within the exon sequences in S. cerevisiae, Long et al. (5) have argued that conservation may reflect a molecular role of pairing with the U5 small nuclear RNA rather than a signal for the insertion of introns.
The consensus sequences in the exon regions flanking introns varied among the six species. Taking at least 40% of the total to be the criterion for a consensus nucleotide, we found the sequence A60G75|G54T42 for A. Arabidopsis; A45A55A45N|N for S. cerevisiae; A42A44A47G55|N for S. pombe; A54G70|G41 for D. melanogaster; A40A54G65|N for C. elegans; and A61G81|G56 for H. sapiens. (The subscripts represent percentages of the consensus nucleotides.)
The limited and variable conservation in the exon sequences of different organisms suggests differential local requirements for the splicing processes and does not support the conception of a proto-splice site sequence preexisting in ancestor mRNAs that did not contain introns.
DISCUSSION
By analyzing dicodon frequencies from six model species (A. thaliana, S. cerevisiae, S. pombe, D. melanogaster, C. elegans, and H. sapiens), we have shown that the four candidate hypothetical proto-splice sites [G|G, AG|G, AG|GT, and (C/A)AG|R] cannot explain the actual intron phase distributions across all the species. For G|G, AG|G, and AG|GT sites, there were no similar patterns in the pattern of the three phases in the data. The (C/A)AG|R in S. cerevisiae and H. sapiens showed patterns similar to the intron phase distribution, but the actual differences between the prediction and the expectation were very significant for H. sapiens, for which there were a lot of data. For S. cerevisiae, for which there were only 163 introns in our purged database drawn from the entire genome, we think this coincidence was not significant but was a random match.
In general, the predictions of the proto-splice models change drastically with the species. This model, the most general model of the introns-late theory, fails to account for the uneven distribution of intron phases.
We extended the information content conservation analysis of Stephens and Schneider (15) to these six model species and observed only a very low information content in the coding regions that flank the splice sites. This finding weakens the argument for proto-splice sites.
To date, the only clear examples of the insertion of introns are the spliceosomal introns in the U2 and U6 small nuclear RNA genes in certain yeast species (17–19). However, the coding sequences flanking these introns are random (20). Hence, the example of real insertion of introns does not suggest any proto-splice sites.
A direct model that fits the data of intron phase distribution is the introns-early theory (21–23). The excess of phase 0 introns would be a consequence of the mini-gene nature of primordial exons and the use of exon shuffling. Moreover, the excess of symmetric exons in modern as well as ancient conserved genes supports an important role for exon shuffling both later and also in the early evolution of genes before the divergence of prokaryotes and eukaryotes.
Acknowledgments
We thank T. Schneider for valuable discussions on information analysis and S. Mount for the discussion about the relationship between splicing signals and intron phases. S.J.deS. was supported by the PEW–Latin American Program and Fundacao de Amparo a Pesquisa do Estado de Sao Paulo.
References
- 1.Mount S M. Nucleic Acids Res. 1982;10:459–472. doi: 10.1093/nar/10.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall S L, Padgett R A. Science. 1996;271:1690–1691. doi: 10.1126/science.271.5256.1716. [DOI] [PubMed] [Google Scholar]
- 3.Horowitz D S, Krainer A R. Trends Genet. 1994;10:100–106. doi: 10.1016/0168-9525(94)90233-x. [DOI] [PubMed] [Google Scholar]
- 4.Csank C, Taylor F M, Martindale D W. Nucleic Acids Res. 1990;18:5133–5141. doi: 10.1093/nar/18.17.5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long, M., de Souza, S. J. & Gilbert, W. (1997) Cell, in press.
- 6.Newman A J, Norman C. Cell. 1992;68:743–754. doi: 10.1016/0092-8674(92)90149-7. [DOI] [PubMed] [Google Scholar]
- 7.Dibb N J, Newman A J. EMBO J. 1989;8:2015–2022. doi: 10.1002/j.1460-2075.1989.tb03609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer J D, Logsdon J M. Curr Opin Genet Dev. 1991;1:470–477. doi: 10.1016/s0959-437x(05)80194-7. [DOI] [PubMed] [Google Scholar]
- 9.Logsdon J M, Jr, Tyshenko M G, Dixon C, Jafari J D, Walker V K, Palmer J D. Proc Natl Acad Sci USA. 1995;92:8507–8511. doi: 10.1073/pnas.92.18.8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long, M. & Stoltzfus, A. (1997) HMS Beagle: A BioMedNet Publication. Issue 1 (Feb. 1). Available at http://hmsbeagle.com. Accessed November 24, 1997.
- 11.Fedorov A, Suboch G, Bujakov M, Fedorova L. Nucleic Acids Res. 1992;20:2553–2557. doi: 10.1093/nar/20.10.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long M, Rosenberg C, Gilbert W. Proc Natl Acad Sci USA. 1995;92:12495–12499. doi: 10.1073/pnas.92.26.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomita M, Shimuzu N, Brutlag S. Mol Biol Evol. 1996;13:11–15. doi: 10.1093/oxfordjournals.molbev.a025687. [DOI] [PubMed] [Google Scholar]
- 14.Schneider T D, Stormo G D, Gold L, Ehrenfeucht A. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- 15.Stephens R M, Schneider T D. J Mol Biol. 1992;228:1124–1136. doi: 10.1016/0022-2836(92)90320-j. [DOI] [PubMed] [Google Scholar]
- 16.Farber R, Lapeds A, Sirotkin K. J Mol Biol. 1992;226:471–479. doi: 10.1016/0022-2836(92)90961-i. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi Y, Urushiyama S, Tani T, Ohshima Y. Mol Cell Biol. 1991;5:1022–1231. doi: 10.1128/mcb.13.9.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tani T, Ohshima Y. Genes Dev. 1991;5:1022–1031. doi: 10.1101/gad.5.6.1022. [DOI] [PubMed] [Google Scholar]
- 19.Tani T, Ohshima Y. Nature (London) 1989;337:87–90. doi: 10.1038/337087a0. [DOI] [PubMed] [Google Scholar]
- 20.Long M, de Souza S J, Gilbert W. Curr Opin Genet Dev. 1995;5:774–778. doi: 10.1016/0959-437x(95)80010-3. [DOI] [PubMed] [Google Scholar]
- 21.Doolittle W F. Nature (London) 1978;272:581–582. [Google Scholar]
- 22.Gilbert W. Nature (London) 1978;271:501. doi: 10.1038/271501a0. [DOI] [PubMed] [Google Scholar]
- 23.Gilbert W. Cold Spring Harbor Symp Quant Biol. 1987;52:901–905. doi: 10.1101/sqb.1987.052.01.098. [DOI] [PubMed] [Google Scholar]