

Abstract
Glucocorticoid administration to mice results in a rapid loss of bone mineral density due to an imbalance in osteoblast and osteoclast numbers. Whereas excess glucocorticoids reduce both osteoblast and osteoclast precursors, cancellous osteoclast number surprisingly does not decrease as does osteoblast number, presumably due to the ability of glucocorticoids to promote osteoclast life span. Whether glucocorticoids act directly on osteoclasts in vivo to promote their life span and whether this contributes to the rapid loss of bone with glucocorticoid excess remains unknown. To determine the direct effects of glucocorticoids on osteoclasts in vivo, we expressed 11β-hydroxysteroid dehydrogenase type 2, an enzyme that inactivates glucocorticoids, specifically in the osteoclasts of transgenic mice using the tartrate-resistant acid phosphatase promoter. Bone mass, geometry, and histomorphometry were similar in untreated wild-type and transgenic animals. Glucocorticoid administration for 7 d caused equivalent increases in cancellous osteoblast apoptosis, and equivalent decreases in osteoblasts, osteoid, and bone formation, in wild-type and transgenic mice. In contrast, glucocorticoids stimulated expression of the mRNA for calcitonin receptor, an osteoclast product, in wild-type but not transgenic mice. Consistent with the previous finding that glucocorticoids decrease osteoclast precursors and prolong osteoclast life span, glucocorticoids decreased cancellous osteoclast number in the transgenic mice but not wild-type mice. In accord with this decrease in osteoclast number, the loss of bone density observed in wild-type mice was strikingly prevented in trans-genic mice. These results demonstrate for the first time that the early, rapid loss of bone caused by glucocorticoid excess results from direct actions on osteoclasts.
The adverse effects of glucocorticoid excess on the skeleton are manifested soon after drug administration is initiated (1, 2). Thus, bone mineral density (BMD) decreases by 2–4.5% after just 6 months of glucocorticoid administration to healthy men (3), but subsequently the rate of bone loss declines (2). Considerable evidence indicates that glucocorticoid-induced bone loss occurs in two phases in both humans and mice: an early, rapid phase in which bone mass is lost due to excessive bone resorption and a slower, later phase in which bone is lost due to inadequate bone formation (2, 4).
We previously reported that glucocorticoid administration to mice dramatically reduces osteoblast number and bone formation due to a decrease in osteoblast precursors combined with stimulation of mature osteoblast apoptosis (4). Besides their effects on osteoblasts, glucocorticoid administration suppresses the number of osteoclast precursors. However, osteoclast number does not fall. This maintenance of osteoclast number, despite a reduction in osteoclast precursors, may be due to prolongation of mature osteoclast life span by glucocorticoid-mediated attenuation of osteoclast apoptosis (5). In vitro, glucocorticoids act directly on osteoclasts to attenuate the pro-apoptotic effects of bisphosphonates, and this effect is mediated by the glucocorticoid receptor because it could be reversed by the glucocorticoid receptor antagonist RU486 (5). Nevertheless, it is unknown whether glucocorticoids act directly on osteoclasts in vivo or whether glucocorticoid action on other cells or tissues may contribute to maintenance of osteoclast numbers via altered production of growth factors or cytokines.
In earlier work from our group, we found that osteoblast-specific expression of 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), an enzyme that inactivates glucocorticoids, protected transgenic mice from glucocorticoid-induced apoptosis of osteoblasts and osteocytes and the resultant reduction in the number of cancellous osteoblasts, osteoid production, and bone formation (6). These findings demonstrate that excess glucocorticoids directly affect bone forming cells in vivo.
Using the same strategy to distinguish the direct from indirect effects of glucocorticoids on osteoclasts, in the studies described herein, we overexpressed 11β-HSD2 in mice using the osteoclast-specific murine tartrate-resistant acid phosphatase (TRAP) promoter (7). We report that mice harboring the transgene exhibited decreased cancellous osteoclasts after glucocorticoid administration and were protected from the glucocorticoid-induced early, rapid loss of BMD.
Materials and Methods
Generation of transgenic mice
Constitutive, osteoclastic lineage-specific expression of human 11β-HSD2 was achieved by creating a transgene consisting of the human 11β-HSD2 cDNA placed downstream of the osteoclast-specific murine TRAP gene promoter (7) and upstream from the bovine GH polyadenylation site (Fig. 1A). The construct containing the murine TRAP promoter was kindly provided by Dr. G. D. Roodman (University of Pittsburgh, Pittsburgh, PA). Transgene DNA was excised from the plasmid vector, gel purified two times, and microinjected into fertilized FVB/N eggs at the University of Arkansas for Medical Sciences transgenic mouse facility. Transgenic founders were identified by Southern blotting using a probe consisting of the human 11β-HSD2 cDNA. Transgenic offspring were identified by PCR using the following primer sequences: forward, 5′-CCTCTTGCAGCCTCTCTGAC-3′ and reverse, 5′-CGGC-CGTTACTAGTGGATCT-3′. Transgenic mice were maintained in the FVB/N genetic background by breeding male mice hemizygous for the transgene with wild-type females. All mice used in experiments were hemizygous for the transgene.
Fig. 1.
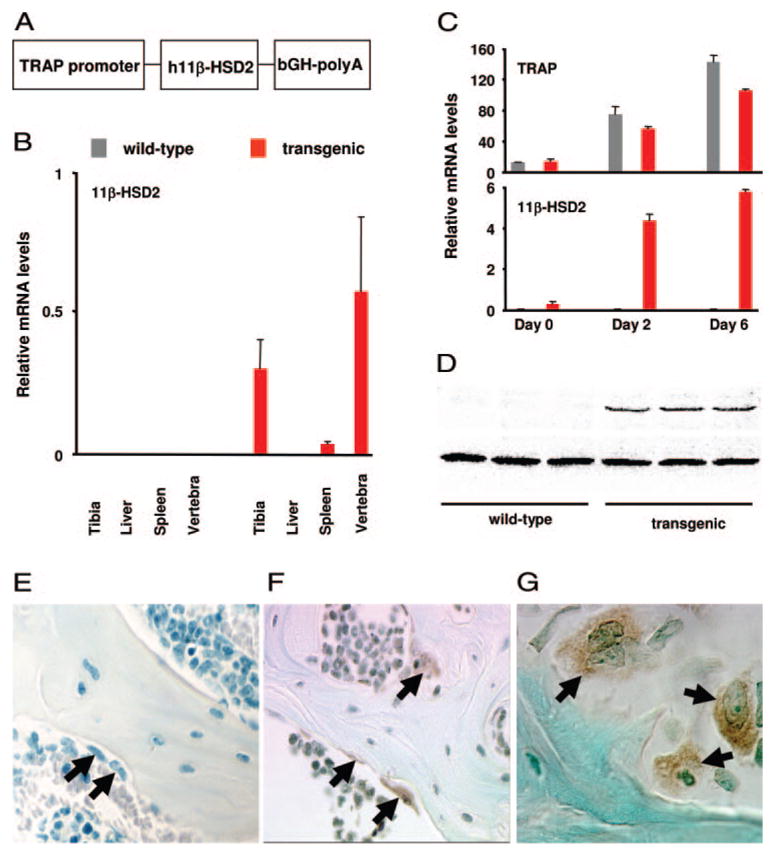
Generation of TRAP-11β-HSD2 transgenic mice. A, The construct used to generate transgenic mice consists of the murine TRAP promoter, the human 11β-HSD2 cDNA, and a bovine GH polyadenylation site (bGH-polyA). B, Total RNA (2 μg), prepared from tibia, liver, spleen, and L6 vertebra of male wild-type or transgenic mice at 4 months of age, was reverse transcribed and subsequently amplified by quantitative RT-PCR for the human 11β-HSD2 and ChoB mRNAs. Values, relative to that of ChoB, are given as the mean ± SD (n = 4). C, Osteoclast progenitors from bone marrow from 6-month-old female wild-type and transgenic mice were individually cultured in presence of RANKL and M-CSF for 0, 2, and 6 d and then analyzed for the expression of murine TRAP and human 11β-HSD2 mRNA by quantitative RT-PCR. Values, relative to that of ChoB, are given as the mean ± SD (n = 3). D, Osteoclast progenitors derived from bone marrow of wild-type and transgenic mice were cultured for 6 d as described above. Cell layer proteins were extracted and subjected to SDS-PAGE followed by immunoblotting. The 41-kDa human 11β-HSD2 bands are shown in the upper panel and the lower panel shows a 30-kDa nonspecific band that was used as a loading control. E–G, Decalcified, paraffin-embedded vertebral sections from 3-month-old wild-type (E) and transgenic (F and G) mice were stained for human 11β-HSD2. Osteoclasts are indicated with arrows. Staining was exclusively in multinucleated osteoclasts from transgenic mice. Magnification is ×200 (E and F) and ×630 (G). Semilunar, scalloped resorption bays, distinctive features of osteoclastic bone resorption, are adjacent to the lower osteoclast (G).
The mice were electronically tagged (Biomedic Data System Inc., Maywood, NJ) and individually kept in plastic cages under standard laboratory conditions with a 12-h light cycle and a constant temperature of 20 C and humidity of 48%. All mice were fed on a standard rodent diet (Formulab Diet 5008; LabDiet, St. Louis, MO) containing 23.5% protein, 6.5% fat, 3.8% fiber, 6.8% ash, 3.3 kcal/g, 3.3 IU/g vitamin D3, 1.0% calcium, and 0.65% phosphorus with water ad libitum. All procedures were approved by the University of Arkansas for Medical Sciences Division of Laboratory and Animal Medicine.
Osteoclast culture
Bone marrow cells were harvested from the femora of untreated wild-type and transgenic mice at 3 or 7 months of age. Cells were suspended in αMEM media with 10% fetal bovine serum (FBS; Hyclone, Logan, UT) and maintained in 75-cm2 flasks for 2 d. Nonadherent cells were collected and plated in 96-well plates (Corning Inc., Corning, NY) at density of 150,000 cells/well in αMEM media supplemented with 10% FBS containing 30 ng/ml receptor activator of nuclear factor-κB ligand (RANKL) and 30 ng/ml macrophage-colony stimulating factor (M-CSF) (R & D Systems, Minneapolis, MN) for up to 6 d, at which point at least 80% of the dish surface was covered by multinucleated osteoclastic cells.
Assessment of gene expression by reverse transcription and real-time PCR
Total RNA from murine tibia, lumbar vertebra (L6), liver, and spleen was extracted using Ultraspec reagent (Biotecx Laboratories, Inc., Houston, TX) following instructions of the manufacturer. RNA was also extracted from osteoclasts formed under culture conditions described above at various times. Equal amounts of RNA (2 μg) from each sample were reverse transcribed using a high-capacity cDNA archive kit (Applied Biosystems, Forster City, CA). The first-strand cDNA was subsequently amplified by real-time PCR using TaqMan universal PCR master mix (Applied Biosystems). The primer and probe sets used were: human 11β-HSD2 forward, 5′-GCTTCAAGACAGAGTCAGTGAGAA-A-3′, reverse 5′-AGGTTGGCCAGCAGCAATT-3′, probe, 5′-FAM-CG-TGGGTCAGTGGGA-NFQ-3′; the mouse housekeeping gene, ribosomal protein S2 (ChoB), forward, 5′-CCCAGGATGGCGACGAT-3′, reverse, 5′-CCGAATGCTGTAATGGCGTAT-3′, probe, 5′-FAM-TCCAGAGCA-GGATCC-NFQ-3′; mouse osteocalcin forward, 5′-GCTGCGCTCTGTC-TCTCTGA-3′, reverse, 5′-TGCTTGGACATGAAGGCTTTG-3′, probe, 5′-FAM-AAGCCCAGCGGCC-NFQ-3′; predesigned primer/probe sets for murine calcitonin receptor, TRAP, RANKL, and osteoprotegerin (OPG) were commercially available (Applied Biosystems), where all primers and probes were synthesized. PCR amplification and detection were carried out on an ABI Prism 7300 real-time PCR system (Applied Biosystems) as follows: denaturation at 95 C for 10 min and 40 cycles of amplification including denaturation at 95 C for 15 sec and annealing/ extension at 60 C for 1 min. Gene expression was quantified using the comparative threshold cycle (Ct) method by subtracting the ChoB Ct value from the Ct values of the gene of interest because amplification efficiencies of these genes were equal.
Assessment of gene expression by Western blotting
Protein was extracted from lumbar vertebra L6 of untreated female wild-type and transgenic mice of 6 months of age by homogenizing the bone in protein lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 2 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, and various protease inhibitors]. Osteoclast progenitors in the nonadherent bone marrow cells from the femora of the same mice were cultured for 6 d in the presence of RANKL and M-CSF, and cell layer protein extracted at d 6 of culture, when at least 80% of the dish surface was covered by multinucleated osteoclastic cells. Protein extracts were quantified using the DC protein assay (Bio-Rad Laboratories, Hercules, CA). Protein samples were boiled for 5 min and 50 μg protein from each extract was fractionated by SDS-PAGE on a 10% gel under reducing conditions. The fractionated proteins were transferred to HyBond poly-vinyl difluoride membranes (Amersham Biosciences Corp., Piscataway, NJ). The blots were blocked in TBS-Tween 20 (TBS-T) buffer [20 mM Tris base, 0.1375 M NaCl, 0.1% Tween 20, 0.01% Thiomerosal (pH 8.0)] containing 5% skim milk for 3 h at room temperature, incubated with rabbit antihuman 11β-HSD2 polyclonal antibody (Alpha Diagnostic International, San Antonio, TX) at 1:1000 dilution in TBS-T overnight at 4 C and then incubated for 3 h at room temperature with bovine antirabbit IgG conjugated with horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:2000 dilution in TBS-T. Signals on the blot were visualized using ECL Advance Western blotting detection kit (Amersham) and scanned in a VersaDoc imaging system (model 3000; BioRad).
Immunohistochemistry
L1–4 from 3-month-old wild-type and transgenic mice were fixed overnight in modified phosphate-buffered formalin (SurgiPath, Richmond IL) and decalcified in 5% EDTA for 1 wk. The vertebrae were then embedded in paraffin and 5-μm longitudinal sections were taken. After paraffin removal and rehydration, antigens were recovered by microwaving the sections in 0.1 M citrate buffer (pH 6.0), for 4 min at 48 C in a H2800 microwave processor (Energy Beam Sciences, Inc., East Granby, CT). The sections were then treated with 0.05% pepsin in 0.1 N HCl, at 37 C for 30 min followed by quenching in 3% H2O2 in methanol for 5 min. Sections were washed twice in PBS and blocked for 20 min in 2.5% normal horse serum. Antihuman 11β-HSD2 antibody (Alpha Diagnostic International) was diluted to 7.5 μg/ml in PBS and incubated with the sections for 1 h at 37 C followed by rinsing and additional incubation for 20 min with biotinylated antirabbit IgG. RTU Elite ABC reagent (VectaStain; Vector Laboratories, Burlingame, CA) was applied for 20 min, washed thoroughly, and developed with diaminobenzidine (Lab Vision, Fremont, CA). Diaminobenzidine staining was enhanced by a 2-min incubation in 0.15% copper sulfate. Sections were counterstained with 2% methyl green for 2 min, dehydrated, and mounted. Sections of human kidney were used as positive controls. The specificity of the anti-11β-HSD2 antibody has been confirmed previously by demonstrating that it stained human kidney tubules and collecting ducts, which express high levels of this protein, but not glomeruli, which do not (6).
Functional assessment of the transgene by osteoclast survival
Osteoclast progenitors in nonadherent bone marrow cell populations were maintained for 3 d under conditions described above. Media were then replaced with fresh αMEM, RANKL, and M-CSF supplemented with dexamethasone or vehicle. In some experiments, 2 × 10−5 M alendronate was also added to the medium, alone or 30 min after dexamethasone. Osteoclast apoptosis was evaluated 18 h after the addition of dexamethasone and/or alendronate by measuring cell layer caspase-3 activity, resulting from cleavage of 7-amino-4-trifluoromethylcoumarin, a fluorogenic substrate (Biomol Research Labs, Plymouth Meeting, PA), as previously described (5). We have shown previously that bisphos-phonates increase active caspase-3 immunostaining, caspase-3 activity, and caspase-9 activity in our osteoclast culture system (5). Under these culture conditions, caspase-3 activity in conditioned medium was negligible. The fluorescent signal generated was detected on a Fluorocount plate reader (Packard Bioscience, Co., Meriden, CT).
BMD Determinations
Starting at 3 months of age, sequential measurements of spinal BMD in live mice were done by dual-energy x-ray absorptiometry on a QDR-2000 Plus densitometer (Hologic, Waltham, MA) using customized software as previously described (8). The measurement was performed at 2- and 4-wk intervals over a 10-wk period.
Evaluation of the basal skeletal phenotype
At 6.5 months of age, the basal skeletal phenotype of the wild-type and transgenic mice was assessed by measuring body weight, bone geometry (femoral length; femoral distal growth plate width; and L5 height, depth and width) and BMD. L1-L4 from wild-type and transgenic mice were fixed and embedded undecalcified in methyl methacrylate and stained for quantitative histological analysis as previously described (4, 5, 9). The histomorphometric examination was done with a computer and digitizer tablet (version 3.00; OsteoMetrics, Decatur, GA) interfaced to a Zeiss Axioscope (Carl Zeiss, Thornwood, NY) with a drawing tube attachment. All measurements were two dimensional, confined to the secondary spongiosa, and made at ×400 magnification (plan-neofluar objective, numerical aperture 0.75). The terminology used is that recommended by the Histomorphometry Nomenclature Committee of the American Society for Bone and Mineral Research (10).
For static measurements, cortical width was measured as previously described (5). The cancellous bone area was expressed as the percentage of tissue area, including bone and marrow as described previously (9). The trabecular width and spacing (the distance between the midpoints of adjacent trabeculae) were measured directly. The osteoblast perimeter was expressed as the number of osteoblasts lining osteoid per millimeter cancellous bone perimeter. The osteoclast perimeter was expressed as the number of osteoclasts per millimeter cancellous bone perimeter.
For dynamic measurements, the rate of mineral apposition was calculated as the mean distance between the midpoints of the two tetracycline labels divided by the interdose duration (5 d). The rates of bone formation was calculated as previously described (9).
Prednisolone administration
At 6.5 months of age, male wild-type and transgenic mice were allocated into four groups with equivalent basal spinal BMD values (13 mice per group) and given sc, slow-release pellets (Innovative Research of America, Sarasota, FL) of prednisolone (1.4 mg/kg·d) as dispensed from a 90-d pellet used to ensure constant prednisolone delivery (4) or placebo for 7 d. The prednisolone dose was based on previous dose-response studies (4). Tetracycline HCl (30 mg/kg body weight) was given ip 6 and 2 d before the animals were killed. Spinal BMD of these mice was determined by dual-energy x-ray absorptiometry at d 0 and 7 of the experiment, L1-L4 were processed for histomorphometry, and L6 was used for analysis of total RNA by RT-PCR as described above.
Statistics
To evaluate treatment effects for the various measurements, ANOVA methods were used with interaction tests included where appropriate. Levene’s test was used to test for homogeneity of variance of a given measurement. The Shapiro-Wilk test was performed to investigate the assumption of normality. When either the homogeneity of variance or normality assumptions were not satisfied, transformations were done or the Kruskal-Wallis nonparametric test was used. Bonferroni methods were used to control the experiment-wise error rate when there were multiple comparisons of interest. All comparisons were defined a priori. P < 0.05 were considered significant.
Results
Generation of transgenic mice expressing 11β-HSD2 in osteoclasts
We generated transgenic mice with a construct consisting of the human 11β-HSD2 inserted downstream from the murine TRAP gene promoter (Fig. 1A), which is expressed in mono- and multinucleated osteoclasts (7). The transgenic mice were generated and maintained in the FVB/N genetic background. Offspring hemizygous for the TRAP-11β-HSD2 transgene were obtained at the expected Mendalian frequency. Real-time PCR analysis of mRNA from tibia, L6, liver, and spleen of 4-month-old male wild-type and trans-genic mice indicated that expression of the transgene was detected only in transgenic mice and in bone but not soft tissues (Fig. 1B). The specificity of the transgene was confirmed by the differentiation-dependent increase in the mRNA levels of the transgene in osteoclasts in vitro (Fig. 1C), the presence of the expected 41-kDa human 11β-HSD2 protein in mature osteoclasts derived from transgenic mice (Fig. 1D), and immunostaining of vertebral bone demonstrating the human 11β-HSD2 protein in osteoclasts of the transgenic mice but not in other cells or the wild-type animals (Fig. 1, E–G). Because osteoclasts constitute only a small fraction of total bone cells, we were unable to detect the 41-kDa human 11β-HSD2 protein band in bone extracts from transgenic mice (data not shown).
Osteoclast-specific expression of 11β-HSD2 transgene did not affect skeletal growth
At 6.5 months of age, body weight, geometry of the femur and L6, BMD, and vertebral cancellous histomorphometry of the wild-type and transgenic mice were indistinguishable (Table 1). Furthermore, serial BMD determinations performed every 2–4 wk during this period showed that both genotypes reached peak BMD at 5.5 months of age (data not shown). There were no gender differences. These findings established that expression of 11β-HSD2 in osteoclasts did not alter skeletal development, peak adult bone mass, or the number of osteoblasts and osteoclasts.
TABLE 1.
Expression of the TRAP-11β-HSD2 transgene does not alter basal bone geometry, mineral density, or vertebral cancellous histomorphometry in 6.5-month-old mice
| Measurement | Wild-type (n = 8) | Transgenic (n = 7) |
|---|---|---|
| Geometry | ||
| Weight (g) | 32.2 ± 4.3 | 35.1 ± 3.4 |
| Vertebral height (mm) | 3.4 ± 0.1 | 3.4 ± 0.1 |
| Vertebral depth (mm) | 1.3 ± 0.1 | 1.4 ± 0.1 |
| Vertebral width (mm) | 2.1 ± 0.4 | 2.3 ± 0.2 |
| BMD (mg/mm2) | 61.1 ± 3.1 | 62.4 ± 4.6 |
| Histomorphometry | ||
| Bone area (%) | 13.1 ± 3.3 | 13.4 ± 1.8 |
| Trabecular width (μm) | 35.2 ± 4.6 | 35.6 ± 2.8 |
| Trabecular spacing (μm) | 241.9 ± 41.2 | 233.5 ± 29.0 |
| Trabecular number (/mm) | 3.7 ± 0.5 | 3.8 ± 0.4 |
| Wall width (μm) | 7.3 ± 0.6 | 6.8 ± 1.2 |
| Osteoid area (%) | 1.6 ± 0.7 | 1.9 ± 0.8 |
| Osteoid width (μm) | 2.4 ± 0.2 | 2.5 ± 0.2 |
| Osteoblast number (mm) | 6.6 ± 1.9 | 7.2 ± 1.8 |
| Osteoclast number (mm) | 1.9 ± 0.3 | 1.7 ± 0.7 |
Values are given as mean ± SD of measurements.
Overexpression of TRAP-11β-HSD2 blocked dexamethasone action on osteoclasts
Basal caspase-3 activity in TRAP-positive osteoclasts obtained from the bone marrow of 3-month-old wild-type and transgenic mice was identical (data not shown). However, as shown in Fig. 2A, in the osteoclasts derived from wild-type mice, treatment with dexamethasone reduced osteoclast ap-optosis as indicated by the 22–26% decrease in caspase-3 activity at the lowest concentrations tested (10−10 and 10−9 M) (P < 0.01). In contrast, in the osteoclasts derived from the transgenic mice, the effects of dexamethasone at 10−10 and 10−9 M were attenuated. With higher doses of dexamethasone, the difference between wild-type and transgenic mice was lost. The lack of effect at the higher doses may be due to overwhelming the capacity of the transgenic 11β-HSD2 enzyme, similar to what occurs in patients receiving high doses of glucocorticoids or with the increased cortisol production that accompanies ectopic production of adrenocor-ticotropic hormone by tumors. In both cases, the human renal 11β-HSD2 may be quenched, resulting in hypertension and hypokalemia (11).
Fig. 2.
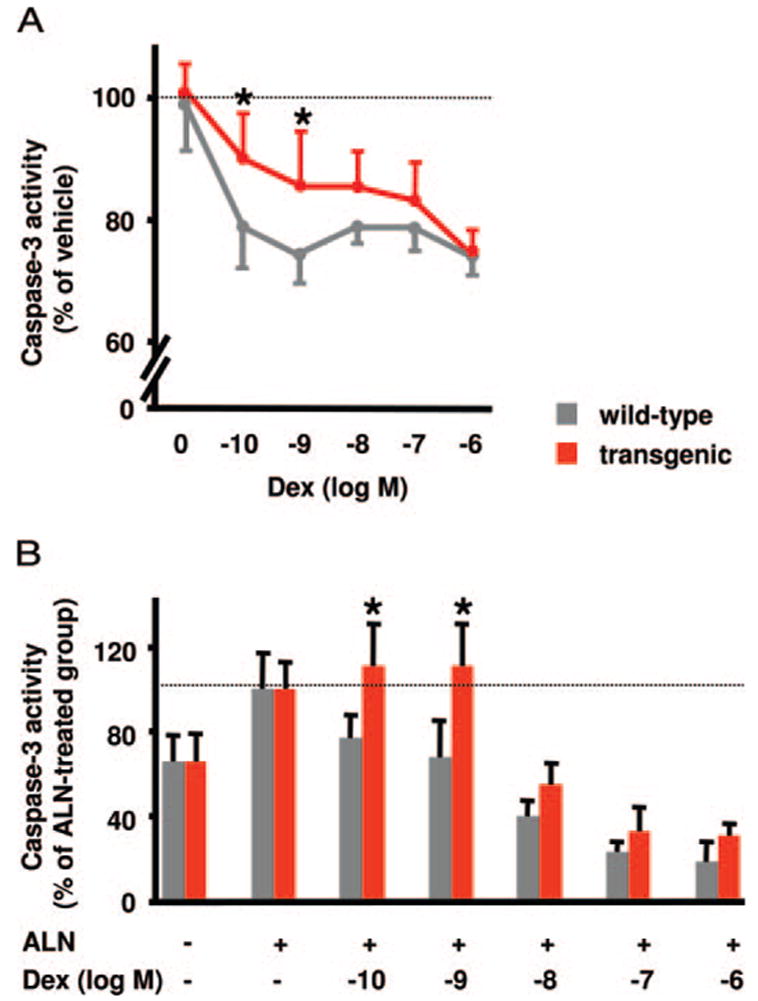
The TRAP-11β-HSD2 transgene blocked dexamethasone (Dex) reduction of basal and alendronate-induced caspase-3 activity in osteoclasts in vitro. Osteoclast progenitors from bone marrow from 3-month-old female wild-type and transgenic mice (A) or 7-month-old male wild-type and transgenic mice (B) were cultured in αMEM supplemented with antibiotics, 10% FBS, 30 ng/ml RANKL, and 30 ng/ml M-CSF for 5 d. During the last 18 h, alendronate (ALN, 2 × 10−5 M), Dex, and ALN or vehicle were added to the medium. Cell layer caspase-3 activity was measured at the end of culture. Values, expressed as percentage of that in the vehicle-treated cultures or ALN-treated cultures, are given as the mean ± SD (n = 6). *, P < 0.05 vs. wild type.
We previously demonstrated that glucocorticoids antagonize bisphosphonate-induced osteoclast apoptosis (5). To determine whether this effect results from direct actions of glucocorticoids on osteoclasts, we examined osteoclast cultures obtained from wild-type and transgenic mice after treatment with alendronate in the presence or absence of dexamethasone. In cultures obtained from 7-month-old wild-type mice, the expected dose-dependent antagonism of alendronate-induced osteoclast apoptosis by dexamethasone was demonstrated (Fig. 2B). In contrast, at the lower concentrations of dexamethasone (10−10 and 10−9 M), the anti-apoptotic effects of the hormone on osteoclasts obtained from the TRAP-11β-HSD2 mice was abrogated. Consistent with the results shown in Fig. 2A, the lowest dose of dexamethasone required to antagonize alendronate-induced caspase-3 activity in TRAP-11β-HSD2 mice was 10−8 M, a dose 100-fold greater than that required in wild-type controls.
Expression of 11β-HSD2 in osteoclasts prevented prednisolone-induced maintenance of vertebral cancellous osteoclasts and loss of spinal BMD
Having established the absence of basal skeletal phenotype, we went on to challenge 6.5-month-old wild-type and transgenic mice with prednisolone administration for 7 d. In wild-type mice, prednisolone administration resulted in a 102% increase in the mRNA level of the calcitonin receptor, an osteoclast-specific gene (12) previously shown to be stimulated by glucocorticoids (13), in vertebrae (P < 0.001, Fig. 3). This effect, however, was prevented in transgenic mice. In contrast, prednisolone caused a practically identical decrease (94 and 96%) in the mRNA levels of osteocalcin, an osteoblast marker (14, 15 c, in both wild-type and transgenic mice (P < 0.001). These results demonstrated that the blockade of glucocorticoid action by the TRAP-11β-HSD2 transgene in vivo was effective and cell type specific. Prednisolone administration did not alter mRNA levels of RANKL and OPG in either genotype (Fig. 3), suggesting that changes in the expression of these mediators of bone resorption (16, 17) were not involved in bone loss during this experiment.
Fig. 3.

The TRAP-11β-HSD2 transgene blocked prednisolone stimulation of calcitonin receptor mRNA expression but not inhibition of osteocalcin. Total RNA was extracted from L6 of 6.5-month-old male mice receiving placebo (plac) or prednisolone (pred) for 7 d. Real-time PCR was performed after reverse transcription. Values, relative to that of ChoB, are given as the mean ± SD (n = 13). *, P < 0.001 vs. placebo.
There were no significant differences in cortical width or cancellous bone area, trabecular separation, trabecular width, and trabecular number between the groups receiving prednisolone (data not shown). As expected, prednisolone caused an increase in the prevalence of osteoblast apoptosis (Fig. 4A) and a decrease in the osteoid perimeter, osteoblast number, and bone formation rate (Fig. 4, B, C, and E) in mice of both genotypes.
Fig. 4.
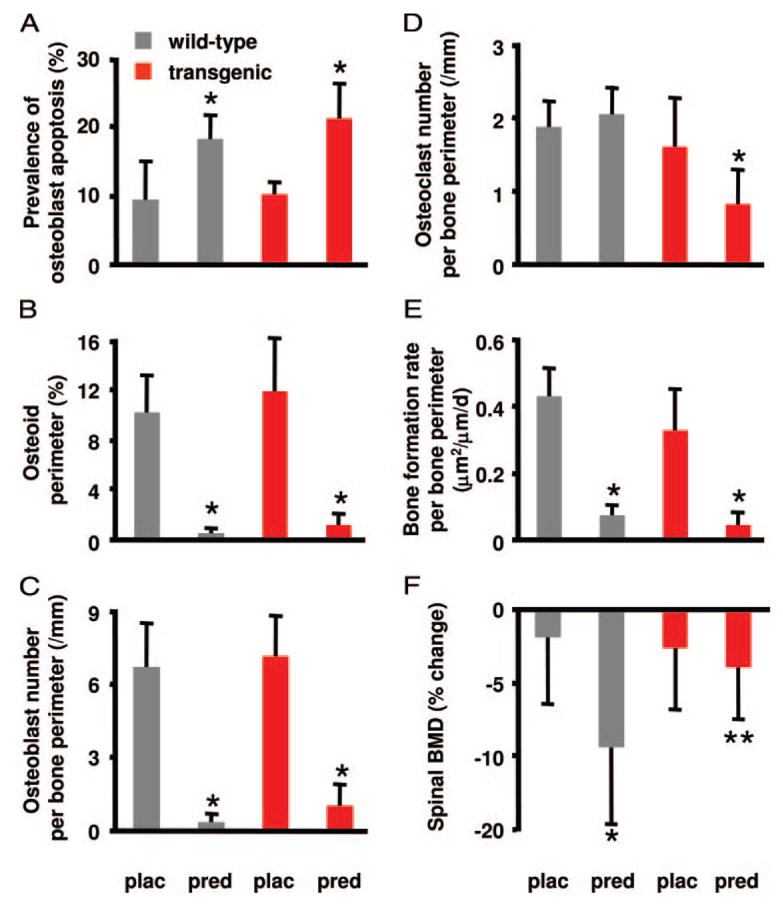
The TRAP-11β-HSD2 trans-gene prevented the loss of BMD after prednisolone administration. Male wild-type and transgenic mice at 6.5 months of age were given placebo (plac) or prednisolone (pred) pellets for 7 d. A, Prevalence of vertebral cancellous os-teoblast apoptosis was evaluated histomorphometrically and values are given as the mean ± SD (n = 7–12). *, P < 0.001 vs. placebo. B, Osteoid perimeter. C, Osteoblast number per bone perimeter. D, Osteoclast number per bone perimeter. E, Bone formation rate per bone perimeter. Values are given as the mean ±SD (n = 7–8). *, P < 0.01 vs. wild type. F, Spinal BMD was measured at beginning and end of the experiment. Values, expressed as percent change from the initial BMD, are given as the mean ± SD (n = 8–12). *, P < 0.001 vs. placebo; **, P < 0.01 vs. wild type.
Consistent with our previous studies (4, 5), prednisolone administration to wild-type mice did not alter osteoclast number (Fig. 4D). In striking contrast, in the transgenic mice in which glucocorticoid action on mature osteoclasts was blocked by expression of 11β-HSD2, prednisolone reduced osteoclast number by 50%. This latter result is most likely due to a loss of the antiapoptotic effect of glucocorticoids on mature osteoclasts such that the prednisolone-mediated reduction in osteoclast precursors now results in decreased osteoclast number. Furthermore and in agreement with the changes in osteoclast number, wild-type mice receiving prednisolone demonstrated a 9.4% decrease in spinal BMD (P < 0.001; Fig. 4F), whereas the loss of BMD was prevented in the transgenic mice.
Discussion
To determine the contribution of direct glucocorticoid action on osteoclasts to the overall adverse effects of a pharmacologic excess of these agents on the skeleton, we blocked the action of glucocorticoids directly on osteoclasts by expressing the enzyme 11β-HSD2 specifically in mature osteoclasts in transgenic mice by use of the TRAP promoter. Glucocorticoid excess induced similar increases in the prevalence of osteoblast apoptosis and decreases in osteoid area, osteoid perimeter, osteoblast number, and bone formation rate in both wild-type and transgenic mice. The seminal observation in these studies is that glucocorticoid administration dramatically reduced the osteoclast number in the transgenic mice but not the wild-type animals.
Glucocorticoid administration causes a rapid decline in bone mass due to a striking remodeling imbalance in the number of osteoblasts and osteoclasts (4). Glucocorticoids inhibit both osteoblastogenesis and osteoclastogenesis, yet cancellous osteoclast numbers are maintained or increased, whereas osteoblasts decrease precipitously (5). We previously obtained evidence that the dichotomous effects on osteoblasts and osteoclasts number may be explained by prolongation of osteoclast survival and curtailment of osteoblast survival. In the results presented in the present report, we obtained additional evidence that this prolongation is due to direct effects of glucocorticoids on osteoclasts. Indeed, consistent with the contention that glucocorticoids prolong osteoclast life span despite a reduction in osteoclast precursors, glucocorticoid administration to TRAP-11β-HSD2 transgenic mice resulted in a reduction in osteoclast number and preservation of bone density. These results provide the first in vivo evidence that glucocorticoids act directly on osteoclasts, and this action is the major cause of glucocorticoid-induced early bone loss. In addition, our findings that basal bone geometric, densitometric, and histomorphometric measurements were similar in both young and adult wild-type and TRAP-11β-HSD2 transgenic mice suggests that endogenous glucocorticoid action in TRAP-expressing osteoclasts is not required for normal skeletal development.
We did not detect significant decreases in the amount of cortical or cancellous bone, by histomorphometry, in the wild-type mice that received prednisolone and lost spinal BMD. However, histomorphometry and BMD measure quite different aspects of bone. The histomorphometric measurements were obtained from one to three longitudinal sections of L1–4, whereas the murine spinal BMD analysis is an integral measurement of all the vertebrae from below the skull to the base of the tail, a much larger skeletal sample. In addition, the coefficient of variation for repeated histomorphometric measurements of microarchitecture is more than 7-fold greater than that for murine BMD (18). The main advantages of histomorphometric measurements lie in quantification of osteoid, bone cells, and the rate of bone formation as demonstrated by the glucocorticoid-induced decrease in osteoid, osteoblasts, and bone formation rate in the wild-type and transgenic mice and the decrease in osteoclasts found only in the TRAP-11β-HSD2 transgenic animals.
We used the 11β-HSD enzyme system as a unique tool to distinguish the direct effects of excess glucocorticoids on bone cells from the indirect effects that occur in almost every other tissue. This system is also a natural prereceptor controller of corticosteroid action (19). Variations in the role of this enzyme system in the metabolism of synthetic glucocorticoids and species-specific differences in substrate specificity do, however, exist as evidenced by observations that dexamethasone is readily metabolized by 11β-HSD2 in rat liver, human kidney, and human osteoblasts but is less effectively inactivated in human lung epithelial cells (20–22). We previously reported that conditional expression of 11β-HSD2 in murine osteocytic cells effectively blocks dexamethasone-induced apoptosis (6). However, it is possible that the 11β-HSD2 system responds to synthetic glucocorticoids in some tissues differently from others and that the results of the in vitro experiments shown here may have been more dramatic if performed with prednisolone rather than dexamethasone (21). Nonetheless, the key observation remains that expressing 11β-HSD2 with the TRAP promoter prevented the action of glucocorticoids on osteoclasts in vivo.
Glucocorticoid administration enhances expression of RANKL and inhibits production of the decoy receptor for RANKL (osteoprotegerin) by stromal cells and osteoblasts in vitro (16). Therefore, changes in these cytokines have been proposed as candidates mediating the proresorptive effects of glucocorticoids on osteoclasts (17). However, we did not detect changes in RANKL and OPG expression in bone obtained from wild-type and transgenic mice after 7 d of prednisolone administration. Our results demonstrating the blockade of glucocorticoid-induced promotion of osteoclast survival and prevention of bone loss in TRAP-11β-HSD2 transgenic mice suggest that direct effects of glucocorticoids on osteoclasts are more important than these mediators in the early, rapid loss of bone mass that follows glucocorticoid administration.
In conclusion, the evidence presented herein demonstrates that osteoclasts are direct targets of glucocorticoid action and that this direct action prolongs the life span of these cells. Moreover, our results indicate that direct action(s) of glucocorticoids on osteoclasts is the major cause of the early, rapid loss of bone in states of glucocorticoid excess.
Acknowledgments
We thank S. Berryhill, T. Chambers, J. A. Crawford, E. A. Hogan, R. S. Shelton, W. W. Webb, and C. Wicker, III for their technical support.
Abbreviations
- BMD
Bone mineral density
- ChoB
ribosomal protein S2
- Ct
threshold cycle
- FBS
fetal bovine serum
- 11β-HSD2
11β-hydrox-ysteroid dehydrogenase type 2
- L
lumbar vertebrae
- M-CSF
macrophage-colony stimulating factor
- OPG
osteoprotegerin
- RANKL
receptor activator of nuclear factor-κB ligand
- TBS-T
TBS-Tween 20
- TRAP
tartrate-resistant acid phosphatase
Footnotes
Disclosure Statement: R.S.W. is a lecturer for Merck and a consultant for Radius. Other authors have nothing to disclose.
Note Added in Proof
While this article was in proofs, Kim et al. (23) reported confirmatory evidence of our seminal findings that glucocorticoid excess prevents osteoclast apoptosis while promoting osteocyte apoptosis (4, 5). However, based on studies with mice in which the glucocorticoid receptor (GR) has been deleted from osteoclasts, Kim et al. have concluded that the adverse effects of these hormones on bone formation are mediated by cells of the osteoclast lineage. These latter findings are in contrast to the evidence shown here that deflection of glucocorticoid action in osteoclasts did not prevent the expected glucocorticoid-induced decrease in osteoblast number, osteoid perimeter, and bone formation or increase in the prevalence of osteoblast apoptosis. Moreover, the findings of Kim et al. are contradicted by previous studies of ours showing that the adverse affects of glucocorticoids on bone formation are the result of direct actions on osteoblasts, as they are abrogated by deflecting the actions of the hormones on this cell type (6). Lack of information on the effect of the osteoclast specific deletion of GR and BMD, osteoblast number, osteoid perimeter, and osteoblast apoptosis precludes us from providing a rational explanation for these seeming discrepancies.
This material is based on work supported by VA Merit Review Grants (to S.C.M. and R.S.W.) from the Office of Research and Development, Department of Veterans Affairs, National Institutes of Health Grants P01-AG13918 and R01-AR46191 (to S.C.M. and R.S.W.) and R01-AR49794 (to C.A.O.), and Tobacco Settlement funds provided by the UAMS College of Medicine.
References
- 1.Reid IR. Pathogenesis and treatment of steroid osteoporosis. Clin Endocrinol (Oxf) 1989;30:83–103. doi: 10.1111/j.1365-2265.1989.tb03730.x. [DOI] [PubMed] [Google Scholar]
- 2.LoCascio V, Bonucci E, Imbimbo B, Ballanti P, Adami S, Milani S, Tartarotti D, DellaRocca C. Bone loss in response to long-term glucocorticoid therapy. Bone Miner. 1990;8:39–51. doi: 10.1016/0169-6009(91)90139-q. [DOI] [PubMed] [Google Scholar]
- 3.Pearce G, Tabensky DA, Delmas PD, Baker HW, Seeman E. Corticosteroid-induced bone loss in men. J Clin Endocrinol Metab. 1998;83:801–806. doi: 10.1210/jcem.83.3.4621. [DOI] [PubMed] [Google Scholar]
- 4.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–282. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinstein RS, Chen JR, Powers CC, Stewart SA, Landes RD, Bellido T, Jilka RL, Parfitt AM, Manolagas SC. Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. J Clin Invest. 2002;109:1041–1048. doi: 10.1172/JCI14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–1841. doi: 10.1210/en.2003-0990. [DOI] [PubMed] [Google Scholar]
- 7.Reddy SV, Hundley JE, Windle JJ, Alcantara O, Linn R, Leach RJ, Boldt DH, Roodman GD. Characterization of the mouse tartrate-resistant acid phosphatase (TRAP) gene promoter. J Bone Miner Res. 1995;10:601–606. doi: 10.1002/jbmr.5650100413. [DOI] [PubMed] [Google Scholar]
- 8.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O’Brien CA, Plotkin L, Fu Q, Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002;298:843–846. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 9.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. The effects of androgen deficiency on murine bone remodeling and bone mineral density are mediated via cells of the osteoblastic lineage. Endocrinology. 1997;138:4013–4021. doi: 10.1210/endo.138.9.5359. [DOI] [PubMed] [Google Scholar]
- 10.Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 11.Torpy DJ, Mullen N, Ilias I, Nieman LK. Association of hypertension and hypokalemia with Cushing’s syndrome caused by ectopic ACTH secretion: a series of 58 cases. Ann NY Acad Sci. 2002;970:134–144. doi: 10.1111/j.1749-6632.2002.tb04419.x. [DOI] [PubMed] [Google Scholar]
- 12.Hattersley G, Chambers TJ. Calcitonin receptors as markers for osteoclastic differentiation: correlation between generation of bone-resorptive cells and cells that express calcitonin receptors in mouse bone marrow cultures. Endocrinology. 1989;125:1606–1612. doi: 10.1210/endo-125-3-1606. [DOI] [PubMed] [Google Scholar]
- 13.Wada S, Akatsu T, Tamura T, Takahashi N, Suda T, Nagata N. Glucocorticoid regulation of calcitonin receptor in mouse osteoclast-like multinucleated cells. J Bone Miner Res. 1994;9:1705–1712. doi: 10.1002/jbmr.5650091106. [DOI] [PubMed] [Google Scholar]
- 14.Frendo JL, Xiao G, Fuchs S, Franceschi RT, Karsenty G, Ducy P. Functional hierarchy between two OSE2 elements in the control of osteocalcin gene expression in vivo. J Biol Chem. 1998;273:30509–30516. doi: 10.1074/jbc.273.46.30509. [DOI] [PubMed] [Google Scholar]
- 15.Aarden EM, Wassenaar AM, Alblas MJ, Nijweide PJ. Immunocytochemical demonstration of extracellular matrix proteins in isolated osteocytes. Histochem Cell Biol. 1996;106:495–501. doi: 10.1007/BF02473312. [DOI] [PubMed] [Google Scholar]
- 16.Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC, Khosla S. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology. 1999;140:4382–4389. doi: 10.1210/endo.140.10.7034. [DOI] [PubMed] [Google Scholar]
- 17.Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res. 2000;15:2–12. doi: 10.1359/jbmr.2000.15.1.2. [DOI] [PubMed] [Google Scholar]
- 18.Weinstein RS, Jia D, Powers CC, Stewart SA, Jilka RL, Parfitt AM, Manolagas SC. The skeletal effects of glucocorticoid excess override those of orchidectomy in mice. Endocrinology. 2004;145:1980–1987. doi: 10.1210/en.2003-1133. [DOI] [PubMed] [Google Scholar]
- 19.Weinstein RS. 2005 Guardian or gate crasher? BoneKEy-OsteoVision 2:6–13 . http://www.bonekey-ibms.org/cgi/content/full/ibmske;2/9/6?ct.
- 20.Best R, Nelson SM, Walker BR. Dexamethasone and 11-dehydrodexa-methasone as tools to investigate the isozymes of 11β-hydroxysteroid dehydrogenase in vitro and in vivo. J Endocrinol. 1997;153:41–48. doi: 10.1677/joe.0.1530041. [DOI] [PubMed] [Google Scholar]
- 21.Garbrecht MR, Schmidt TJ, Krozowski ZS, Snyder JM. 11β-Hydroxysteroid dehydrogenase type 2 and the regulation of surfactant protein A by dexamethasone metabolites. Am J Physiol Endocrinol Metab. 2006;290:E653–E660. doi: 10.1152/ajpendo.00396.2005. [DOI] [PubMed] [Google Scholar]
- 22.Bland R, Worker CA, Noble BS, Eyre LJ, Bujalska IJ, Sheppard MC, Stewart PM, Hewison M. Characterization of 11β-hydroxysteroid dehydrogenase activity and corticosteroid receptor expression in human osteosarcoma cell lines. J Endocrinol. 1999;161:455–464. doi: 10.1677/joe.0.1610455. [DOI] [PubMed] [Google Scholar]
- 23.Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, Ross FP, Teitelbaum SL. Glucocorticoids suppress bone formation via the osteoclast. J Clin Invest. 2006;116:2152–2160. doi: 10.1172/JCI28084. [DOI] [PMC free article] [PubMed] [Google Scholar]