

Abstract
Thiamine (vitamin B1) deficiency (TD) causes region selective neuronal loss in the brain; it has been used to model neurodegeneration that accompanies mild impairment of oxidative metabolism. The mechanisms for TD-induced neurodegeneration remain incompletely elucidated. Inhibition of protein glycosylation, perturbation of calcium homeostasis and reduction of disulfide bonds provoke the accumulation of unfolded proteins in the endoplasmic reticulum (ER), and cause ER stress. Recently, ER stress has been implicated in a number of neurodegenerative models. We demonstrated here that TD up-regulated several markers of ER stress, such as GRP78, GADD153/Chop, phosphorylation of eIF2α and cleavage of caspase-12 in the cerebellum and the thalamus of mice. Furthermore, ultrastructural analysis by electron microscopic study revealed an abnormality in ER structure. To establish an in vitro model of TD in neurons, we treated cultured cerebellar granule neurons (CGNs) with amprolium, a potent inhibitor of thiamine transport. Exposure to amprolium caused apoptosis and the generation of reactive oxygen species in CGNs. Similar to the observation in vivo, TD up-regulated markers for ER stress. Treatment of a selective inhibitor of caspase-12 significantly alleviated amprolium-induced death of CGNs. Thus, ER stress may play a role in TD-induced brain damage.
Keywords: cell death, cerebellum, neurodegeneration, nutrition, vitamin B1
Introduction
Thiamine (vitamin B1) deficiency (TD) induces regionally selective neuronal death in the brains of humans and animals; these regions include the thalamus, midbrain, brainstem and cerebellum (Victor et al., 1989; Baker et al., 1999). TD-induced neuronal loss accompanies a mild and chronic impairment of oxidative metabolism as well as inflammatory responses and glial activation (Ke and Gibson, 2004). Selective cell death, inflammation, glial activation and abnormalities in oxidative metabolism are common in many aging-related neurodegenerative diseases, such as Alzheimer's disease (AD)(Gibson and Zhang, 2001), Parkinson's disease (PD)(Schwab et al., 1996) and progressive supranuclear palsy (Park et al., 2001). The neurological disorder that is most clearly associated with TD in humans is Wernicke-Korsakoff syndrome (WKS), which is characterized by severe memory loss, cholinergic deficits and selective cell death in specific brain regions (Victor et al., 1989; Todd and Butterworth, 1999; Calingasan and Gibson, 2000; Ke et al., 2003). These features of the TD model make it amenable to investigate the cellular mechanisms of neurodegeneration.
The mechanisms for aging-related neurodegeneration remain incompletely understood. Recently, endoplasmic reticulum (ER) stress has been implicated in various neurodegenerative processes, such as brain ischemia (Tajiri et al., 2004; Hayashi et al., 2003; 2004), AD (Katayama et al., 2004), PD (Silva et al., 2005; Smith et al., 2005), Huntington's disease (HD) (Hirabayashi et al., 2001) and amyotrophic lateral sclerosis (Turner and Atkin, 2006). The ER is an important organelle involved in posttranslational protein processing and transport. Approximately one third of all cellular proteins are translocated into the lumen of the ER where posttranslational modification, folding and oligomerization occur. The ER is also the site for the biosynthesis of steroids, cholesterol and other lipids. A number of cellular stress conditions, such as perturbed calcium homeostasis or redox status, elevated secretory protein synthesis rates, altered glycosylation levels, and cholesterol overloading, can interfere with oxidative protein folding. This can subsequently lead to the accumulation of unfolded or misfolded proteins in the ER lumen and activate compensatory mechanism, which has been referred to as ER stress response or unfolded protein response (UPR)(Kaufman, 1999; Ron, 2002; Shen et al., 2004; Xu et al., 2005). Several sensors of ER stress have been identified. These include pancreatic ER kinase (PERK), the kinase encoded by the inositol requiring (IRE) 1 gene and activating transcription factor (ATF) 6. PERK phosphorylates the α subunit of the eukaryotic initiation factor 2 (eIF2α), which attenuates the initiation of translation in response to ER stress. The activation of IRE1 and ATF6 signaling promotes the expression of ER-localized chaperones, such as GRP78 and GRP94, which facilitate the restoration of proper protein folding within the ER (Kaufman, 1999; Ron, 2002; Shen et al., 2004; Xu et al., 2005). These protective responses result in an overall decrease in translation, enhanced protein degradation and increased levels of ER chaperones, which consequently increase the protein folding capacity of the ER. However, sustained ER stress ultimately leads to the apoptotic death of the cell (Rutkowski and Kaufman, 2004; Xu et al., 2005).
The cause for TD-induced neuronal damage remains unclear. Several potential mechanisms have been proposed; these include mitochondrial dysfunction (Park et al., 2000; Singleton and Martin, 2001), impairment of oxidative metabolism (Ke and Gibson, 2004; Gibson et al., 2005) and acidosis (Hakim and Pappius, 1983; Pannunzio et al., 2000). Since oxidative stress may cause ER stress (Hayashi et al., 2003; Xue et al., 2005) and TD induces oxidative stress, we hypothesized that TD may cause ER stress which contributes to TD-induced brain damage. With both in vivo and in vitro approaches, the current study was designed to determine whether TD induced ER stress in neurons.
Material and Methods
Animals and reagents
Sprague-Dawley rats and C57BL/6J mice were obtained from Shanghai Laboratory Animal Co. Ltd (Shanghai, China). The procedure for animal surgery was performed in accordance with the Guideline of Animal Care and Use Committee of the Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences (SIBS), Chinese Academy of Sciences (CAS). 5-(and-6-)-chloromethyl-2′,7′-dichlorodi-hydrofluorescein diacetate acetyl ester (CM-H2DCFDA) was obtained from Molecular Probes (Eugene, OR, USA). A specific inhibitor for caspase-12 (Z-ATAD-FMK) was purchased from R&D Systems (Cat #:1079-100, Minneapolis, MN, U.S.A). Other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO, USA), unless otherwise mentioned. Anti-GADD153 antibody was obtained from Santa Cruz Biotech (Santa Cruz, CA, USA). Anti-caspase-12 antibody was obtained from Calbiochem (La Jolla, CA, USA). Anti-α-tubulin antibody was purchased from Sigma Chemical Co. Anti-GRP78 antibody was obtained from StressGen Bioreagents (Ann Arbor, Michigan, USA). All other antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA).
Cultures of cerebellar granule neurons
Thirty 6- or 7-day-old Sprague-Dawley rat pups were used in the study. Cultures of cerebellar granule neurons (CGNs) were generated using a previously described method with a slight modification (Chen et al., 2004). Briefly, the rat pups were decapitated under deep anesthesia and cerebella removed. The cerebella were minced with a sterile razor blade and suspended in 10 ml of trypsin solution (0.025%) at 37°C. After incubation for 15 min, an equal volume of a solution containing DNAse (130 Kunitz units/ml) and trypsin inhibitor (0.75 mg/ml) was added, and the tissue was sedimented by a brief (5 s) centrifugation. The tissue was dissociated by trituration, and the cell suspension was mixed with 4% bovine serum albumin and centrifuged. The cell pellet was re-suspended in Neurobasal/B27 medium containing B27 (2%), KCl (25 mM), glutamine (1 mM), penicillin (100 units/ml) and streptomycin (100 μg/ml). Cells were plated into poly-d-lysine (50 μg/ml)-coated cell culture wells or dishes, and maintained at 37°C in a humidified environment containing 5% CO2.
Induction of thiamine deficiency
The animal TD model has been previously described (Ke et al., 2003). Briefly, adult male C57BL/6J mice (20-25 g) were housed in a controlled environment (one mouse/cage at 23°C and 53% humidity). The animals were fed with either a control diet or a thiamine deficient diet (ICN Nutrition Biomedicals, Cleveland, OH, USA) ad libitum. TD animals also received a daily intraperitoneal injection of a thiamine antagonist, pyrithiamine hydrobromide (5 μg/10g body weight, Sigma Chemical Co.), while control animals were injected with saline. Pyrithiamine is a potent inhibitor of thiamine pyrophosphokinase and blocks the synthesis of thiamine diphosphate (TDP). A total of one hundred twenty animals were used for immunoblotting analysis. There were fifteen animals per treatment group and the brains from these animals were combined to generate three independent pooled tissue samples (five animals per pooled sample, n = 3). The brain tissues were combined to minimize the variation among individual animals. The statistical analyses (One-way ANOVA) were carried out on samples from three independent pools of brains.
TD in cerebellar granule neurons (CGNs) was induced by the treatment of amprolium. Amprolium is a competitive inhibitor of thiamine transport and effectively depletes intracellular thiamine (Bettendorff et al., 1995; Park et al., 2000). After cultured in Neurobasal/B27 medium for 7 days, CGNs were treated with amprolium (0, 0.5, 1 or 1.5 mM) for indicated times.
Sample preparation
After treatment, the mice were killed by decapitation and the cerebella and thalamus were immediately dissected from TD and control mice. Animals were anaesthetized by intraperitoneal injection of chloral hydrate (350 mg/kg). The thalamus was dissected according to the method of Glowinski and Iversen (1966). The tissues were frozen in liquid nitrogen and stored at −80°C for later analysis. Proteins were extracted with a previously described method (Li et al. 2002). Briefly, tissues were homogenized in an ice-cold lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1 mM PMSF, 0.5% NP-40, 0.25% SDS, 5 μg/ml leupeptin, and 5 μg/ml aprotinin. Homogenates were centrifuged at 20,800 g for 30 min at 4°C and the supernatant fraction was collected.
For protein extraction in cultured neurons, the CGNs were lysed in an ice-cold lysis buffer that contained 5 mM EDTA, 1% NP-40, 10 mg/ml PMSF, 10 μg/ml leupeptin and 100 mM sodium orthovanadate in PBS, and centrifuged at 20,800 g for 10 min. The supernatant was designated as the cytoplasmic fraction. The pellets were sonicated in a nuclear extraction buffer (20 mM Tris-HCl, pH7.5, 1% SDS, 5 mM EGTA, 0.5% Triton X-100, 150 mM NaCl, 1 mM DTT, 10 μg/ml leupeptin and 1 mM Pephabloc SC) and centrifuged at 20,800 g for 10 min. The supernatant was collected and designated as the nuclear fraction.
Immunoblotting
The procedure for immunoblotting has been previously described (Li et al., 2002). Aliquots of cytoplasmic or nuclear proteins (50 μg) were loaded into the lanes of a sodium dodecyl sulfate (SDS) polyacrylamide gel. The proteins were separated by electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk or 5% BSA (for detection of phosphorylation) in 0.01 M PBS (pH 7.4) and 0.05% Tween-20 (TPBS) at room temperature for 1 h. Subsequently, the membrane was incubated with primary antibodies directed against target proteins overnight at 4°C. The final dilutions for primary antibodies were: GRP78, 1:10,000; GADD153, 1:500; p-eIF2α, 1:500; caspase-12, 1:1000. After two quick washes in TPBS, the membranes were incubated with secondary antibodies conjugated to horseradish peroxidase (Amersham, Arlington Heights, IL, USA) diluted at 1:5000 in TPBS for 1 h. The immuno-complexes were detected by the enhanced chemiluminescence method (Amersham). The density of immunoblotting was quantified with the software of Quantity One (Bio-Rad Laboratories, Hercules, CA, USA).
Cell viability and nuclear morphology
Cell viability was determined and quantified by MTT assay as previously described (Chen et al., 2004). For examining nuclear morphology, neurons were cultured on cover slips, fixed with 4% paraformaldehyde in phosphate buffer saline (PBS) for 20 min at room temperature, and then incubated with a DNA dye, 4,6-diamidino-2-phenylindole (DAPI, 1 μg/ml in PBS) for 30 min. The staining was visualized under a fluorescent microscope that was excited at a wavelength of 400 nm and measured at 455 nm.
Measurement of reactive oxygen species accumulation
Intracellular reactive oxygen species (ROS) levels were measured using the fluorescent dye H2DCFDA staining method. Once incorporated into cells, H2DCFDA is converted into a nonfluorescent polar derivative (H2DCF) by cellular esterases. H2DCF is rapidly oxidized to the highly fluorescent 2,7-dichlorofluorescein (DCF) by intracellular ROS, mainly H2O2 (LeBel et al., 1992). Therefore, the intensity of the DCF signal reflects the quantity levels of intracellular ROS. Cultured CGNs were plated in 6-well plates and treated with amprolium for the indicated times. After treatment, the cells were incubated with phenol-red-free medium (opti-MEM) (Invitrogen, Carlsbad, California, USA) containing CM-H2DCFDA (10 μM) in the dark for 30 min at 37°C. Intracellular ROS levels (DCF signals) were measured with a flow cytometer (FACSAria™, BD Biosciences, San Jose, CA, USA) at λex of 488 nm and λem of 520 nm. Data acquisition and processing was performed using the Cellquest program (BD Biosciences, San Jose, CA.USA).
Immunofluorescent staining
CGNs were cultured in cover slips and treated with amprolium (1.5 mM) for indicated times. Immunocytofluorescent staining of GADD153 and GRP78 was performed as previously described (Ayoub et al., 2005). After incubated with respective primary antibodies overnight at 4°C (1:200 for anti-GADD153; 1:5000 for anti-GRP78), cultures were treated with a fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit IgG secondary antibody (1:500 dilution in PBS; Vector Laboratories, Burlingame, CA, USA). Nuclei were labeled with DAPI (1 μg/ml in PBS). Images of fluorescence were acquired using the Zeiss LSM 510 META confocal laser-scanning microscope (Carl Zeiss, Jena, Germany). The same settings for filters, pinhole size, stack size, and resolution were used for all captured images.
Transmission electron microscopy
The sections of the thalamus were serially fixed with 2% glutaraldehyde in PBS for 2 h and then with 2% OsO4 for 2 h, dehydrated in a graded series of alcohol and then flat-embedded in Epon 812. Thin sections were prepared using a LKB Ultrotome Nova ultramicrotome (LKB, Broma, Sweden), and stained with uranyl acetate followed by lead citrate. The sections were then examined under a JEOL JEM1230 electron microscope (JEOL, Tokyo, Japan).
Statistical analysis
The alterations in the expression of markers for ER stress were analyzed by one-way ANOVA. Differences in which the p value was less than 0.05 were considered statistically significant. In cases where significant differences were detected, specific post hoc comparisons between treatment groups were examined with Student-Newman Keuls tests or Dunnett's T3 tests. The analyses were performed using SPSS software (SPSS, Chicago, IL, USA).
Results
Animal TD model
The effect of TD on ER stress was first evaluated in mice. TD was evident by weight loss; we have shown that the onset of weight loss can predict the severity of TD-induced pathological changes in the brain (Ke et al., 2003; 2006). We examined the expression of several markers for ER stress in the cerebellum following TD. Glucose regulated protein (GRP) 78 is an ER chaperone protein. GRP78 is usually induced during the unfolded protein response (UPR), which is required to alleviate ER stress. The TD-induced alterations in GRP78 were analyzed by one-way ANOVA with the duration of treatment as a variable. We demonstrated a significant increase in GRP78 expression during TD treatment [F(7,16)=13.16, p < 0.05] (Fig. 1). An initial increase in GRP78 expression was observed on day 6 following TD (p < 0.05); the up-regulation reached the highest level (5.5-fold increase over controls) on day 7 after TD (TD7). The levels of GRP78 sharply decreased after TD7. We further examined the effect of TD on other markers of ER stress, such as growth arrest and DNA-damage inducible protein (GADD153) or C/EBP-homologous protein (Chop), the α subunit of the eukaryotic initiation factor 2 (eIF2α) and caspase-12. One-way ANOVA was also employed to evaluate the statistically significant alterations. In general, the pattern of TD-induced alterations in the expression of GADD153 [F(7,16)=16.33, p < 0.05] and phosphorylated eIF2α [F(7,16)=13.38, p < 0.05] was similar to that of GRP78.
Fig. 1.
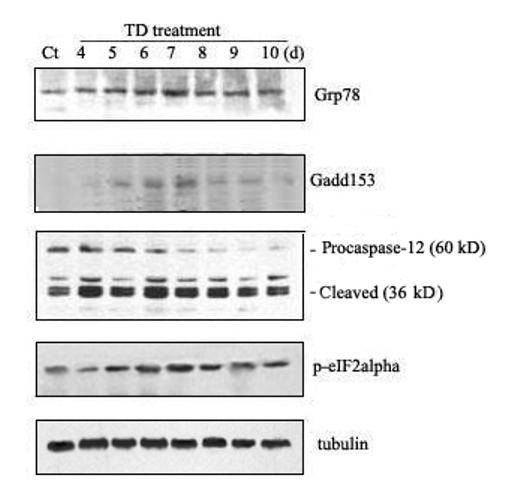
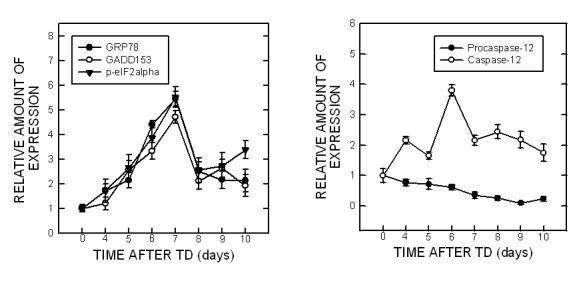
TD-induced ER stress in the cerebellum. A. The mice were divided into control and TD groups (n = 5 for each group). TD was induced in mice as described under the Materials and Methods. At specified times after TD, the cerebellum was removed. For each independent experiment, brain samples collected from five animals at a specified time point were mixed. The expressions of GRP78, GADD153, phospho-eIF2α, and cleaved caspase-12 were determined by immunoblotting. Each data point was the mean of three independent experiments. B. The relative amounts of GRP78, GADD153, phospho-eIF2α and pro-/cleaved-caspase-12 were measured microdensitometrically and normalized to the expression of tubulin.
TD-induced caspase-12 activation was indicated by the cleavage of pro-caspase-12. As shown in Fig. 1, a significant increase in cleaved (active) capases-12 (42 and 35 kDa) was observed after TD [F(7,16)=26.79, p < 0.05]. The cleaved form of caspase-12 increased by 2.8 folds on day 4 after TD (p < 0.05); it reached maximal levels (4.6-fold increase) on day 6 after TD. However, the decrease in pro-caspase-12 (∼60 kDa) was not statistically significant. It was noted that the cleaved form of caspase-12 was present in the controls, indicating a constitutive activation of caspase-12 in the cerebellum (Fig. 1).
To determine whether TD induces ER stress in other brain areas, we examined the effect of TD on the thalamus, which is one of the brain regions most susceptible to TD. Similar to the results observed in the cerebellum, TD significantly increased the expression of GRP78 [F(7,16)=41.97, p < 0.05], GADD153 [F(7,16)=102.96, p < 0.05] and p-eIF2α [F(7,16)=178.81, p < 0.05] (Fig. 2). Unlike in cerebellum, however, there was not significant alteration in caspase-12 following TD in the thalamus (data not shown).
Fig. 2.
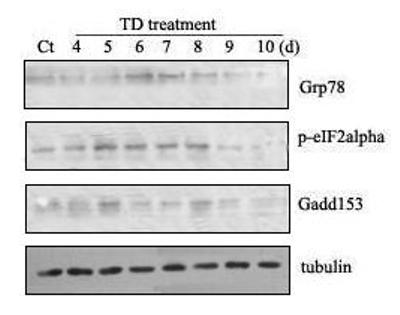
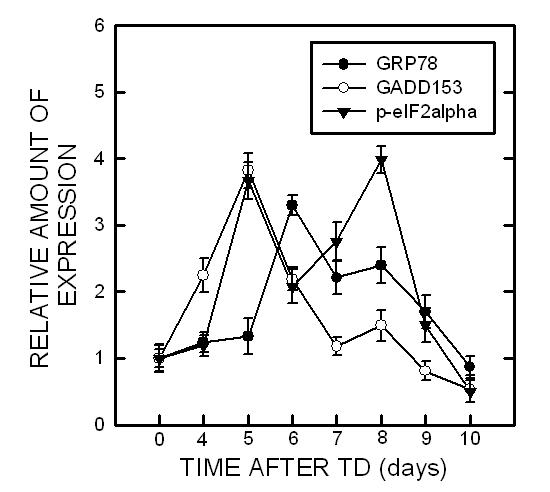
TD-induced ER stress in the thalamus. Notations are as Fig. 1.
Ultrastructure of the ER was analyzed with transmission electron microscopy. ER morphology was altered in the thalamus of TD mice. Swelling ERs accompanied by an enlargement of lumen were frequently observed. Deposition of vesicles with medium electron density in the ER lumen was sometimes spotted (Fig. 3). In addition, the rough ER displayed an irregular packing pattern in the thalamus of TD mice.
Fig. 3.
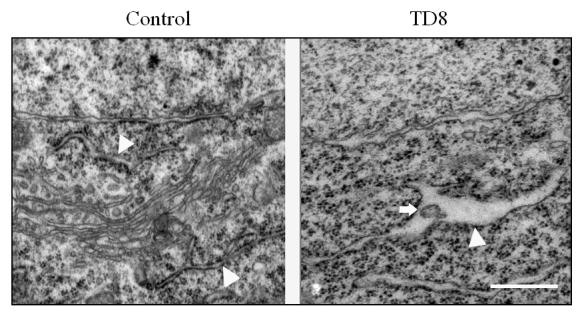
TD-induced alterations in the ultrastructure of the ER in neurons. After TD for 8 days, the neurons in the submedial thalamus nuclei (SmTN) of mice were processed and examined under a transmission electron microscope as described under the Materials and Methods. Arrow heads indicate ER; arrow indicates an inclusion of medium electron density in the ER lumen. Bar = 455 nm
In vitro TD model
Amprolium has been previously shown to effectively deplete intracellular thiamine (Bettendorff et al., 1995; Park et al., 2000). The effect of amprolium on the viability of cultured CGNs was examined by MTT assay. Amprolium induced a dose- and duration-dependent decrease in the viability of cultured CGNs (Fig. 4A). At 0.5, 1, and 1.5 mM, amprolium reduced cell viability by 15%, 22% and 30% respectively after 5 days exposure, by 33%, 43% and 51%, respectively following 7 days exposure. Furthermore, DAPI staining revealed condensation of chromatin and breakdown of the nucleus in amprolium-treated cultures, which is characteristic of apoptotic cells (Fig.4B). We next sought to determine whether TD induced oxidative stress in this cell culture model. As shown in Fig. 5, treatment with amprolium significantly increased the production of ROS in cultured CGNs. The amprolium-induced ROS production occurred as early as one day exposure to amprolium. Therefore, treating CGNs with amprolium provides a relevant in vitro neuronal model of TD.
Fig. 4.
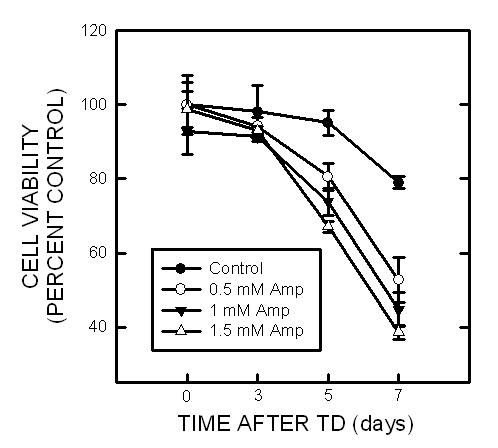
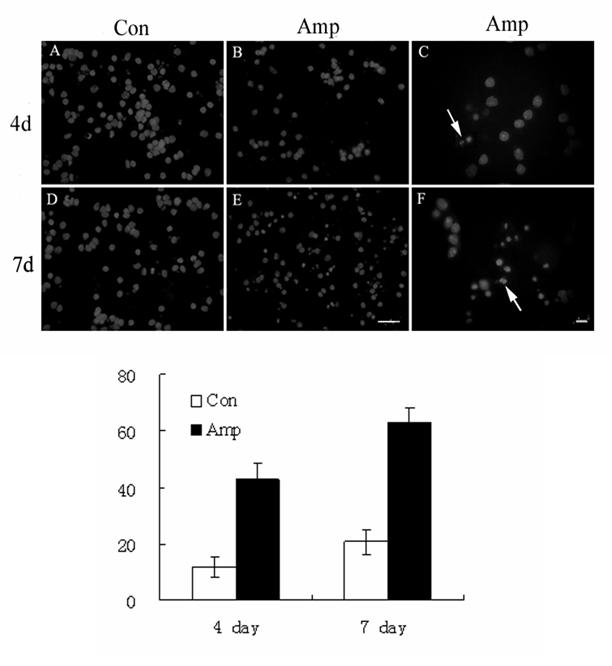
Effect of TD on the viability of cultured CGNs. A. CGNs were treated with amprolium (Amp; 0, 0.5, 1, or 1.5 mM) for specified times, and the viability of cells was determined by MTT. Each data point represents the mean of five independent experiments. B. CGNs were treated with amprolium (Amp; 0 or 1.5 mM) for 4 or 7 days. Cultures were stained with DAPI for visualizing nuclear morphology (top panel); higher magnification shows condensed or fragmented nuclei following TD treatment (arrows), bar = 20 μm. Cells with condensed or fragmented nuclei were counted (bottom panel). Y-axis indicates the percentage of cells with condensed or fragmented nuclei. Each data point represents the mean of three independent experiments.
Fig. 5.
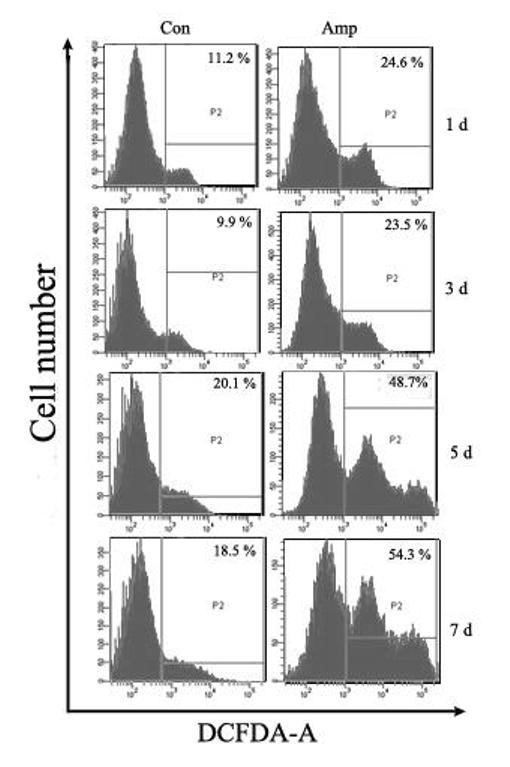
TD-induced ROS production in cultured CGNs. CGNs were exposed to amprolium (Amp; 0 or 1.5 mM) for specified times. After the exposure, cells were labeled with CM-H2DCFDA (10 μM) as described under the Materials and Methods. The percentage of ROS producing cells (positive for DCF fluorescence) was determined by flow cytometry. Each data point represents the mean of three independent experiments.
We then determined whether amprolium induced ER stress in cultured CGNs. TD-induced alterations in the expression of markers for ER stress were analyzed with a quantitative immunoblotting method. At each time point, the control was designated as 1 and TD-induced alterations were normalized to the controls of matched time-points. As shown in Fig. 6, treatment with amprolium significantly increased the expression of GRP78 [F(6,14)=33.59, p < 0.05], GADD153 [F(6,14)=36.33, p < 0.05], p-eIF2α [F(6,14)=213.10, p < 0.05], and the cleavage of pro-caspase-12 [F(6,14)=12.29, p < 0.05] in cultured CGNs. For example, treatment of amprolium (1.5 mM) for 12 and 24 h increased GRP78 expression by 3.6 folds and 4.2 folds, respectively (p < 0.05) (Fig. 6). The profile of amprolium-induced up-regulation of GADD153 expression, eIF2α phosphorylation and caspase-12 activation was similar to that of GRP78. It was particularly noted that a dramatic increase in nuclear GADD153 was observed following exposure to amprolium for 24 h.
Fig. 6.
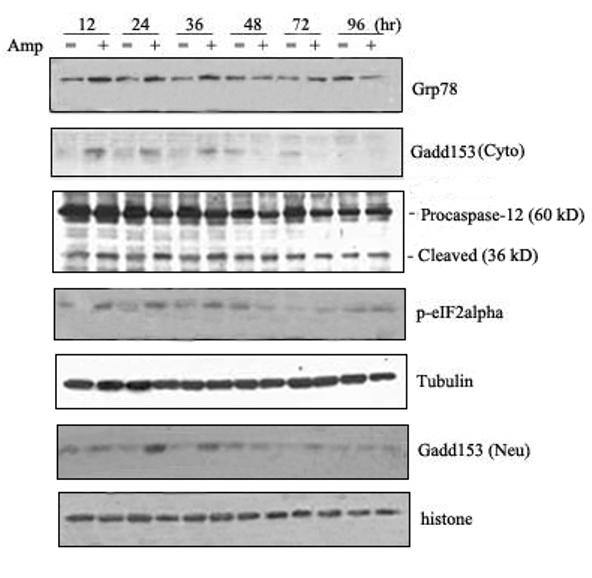
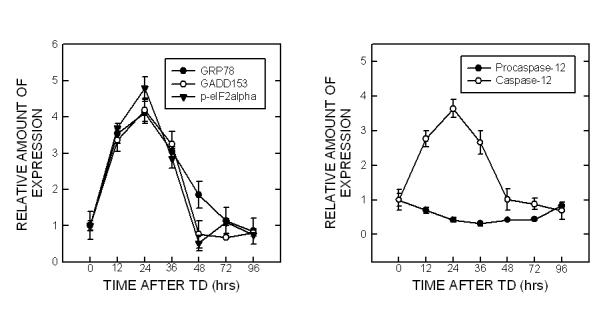
TD-induced ER stress in primary cultures of CGNs. A. CGNs were treated with amprolium (Amp; 0 or 1.5 mM) for specified times. The expression of GRP78, cytoplasmic/ nuclear GADD153, phospho-eIF2α and caspase-12 was determined with immunoblotting. The expression of tubulin and histone H1o served as an internal loading control for cytoplasmic and nuclear protein, respectively. B. The relative amounts of these proteins were measured microdensitometrically and normalized to the expression of tubulin. TD-induced alterations were controls of matched time points. Each data point represents the mean of three independent experiments.
Amprolium-induced alteration in GRP78 and GADD153 was visualized through immunofluorescent staining. Untreated CGNs displayed weak GRP78 immunoreactivity in the cytoplasm (Fig. 7A). Treatment with amprolium (1.5 mM and 12 h) dramatically increased GRP78 immunoreactivity in the cytoplasm. Weak GADD153 immunoreactivity with predominantly perinuclear distribution was observed in control CGNs; amprolium not only increased the intensity of GADD153 immunoactivity, but also induced its nuclear translocation after 24 h of exposure (Fig. 7B). Thus, the results obtained from immunofluorescent images were consistent with that of immunoblots.
Fig. 7.
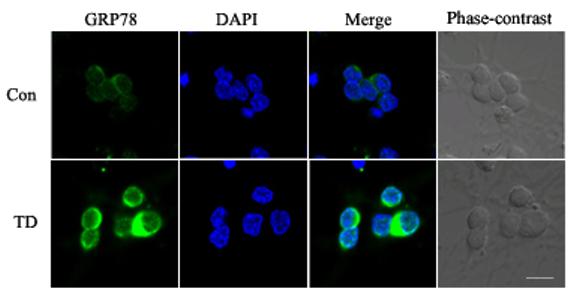
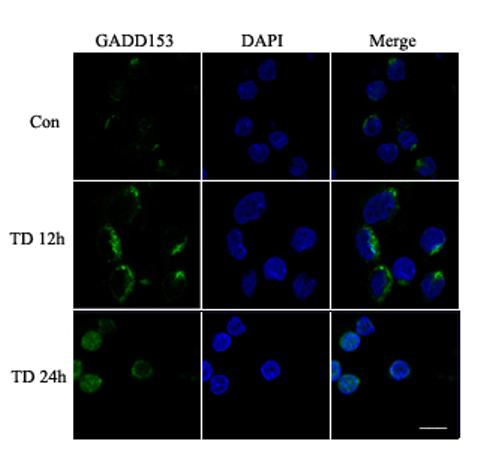
Effect of TD on the expression and localization of GRP78 and GADD153 in cultured CGNs. A. After treating with amprolium (Amp, 0 or 1.5 mM) for 24 h, the expression of GRP78 (green) was examined with immunocytochemistry as described under the Materials and Methods. The nuclei (blue) were visualized with DAPI staining. B. After treating with amprolium (Amp; 0 or 1.5 mM) for 12 and 24 h, the expression of GADD153 (green) was examined with immunocytochemistry. DAPI staining was performed to visualize nuclei (blue). Bar = 10 μm.
Activation of caspase-12 has been suggested to be responsible for ER stress-associated cell death under certain circumstances (Szegedi et al., 2003). Since TD activated caspase-12, we sought to determine whether a selective caspase-12 inhibitor can alleviate TD-induced neuronal death. Z-ATAD-FMK is a synthetic peptide that irreversibly inhibits capase-12. As shown in Fig. 8, treatment with Z-ATAD-FMK significantly mitigated amprolium-induced death of cultured CGNs.
Fig. 8.
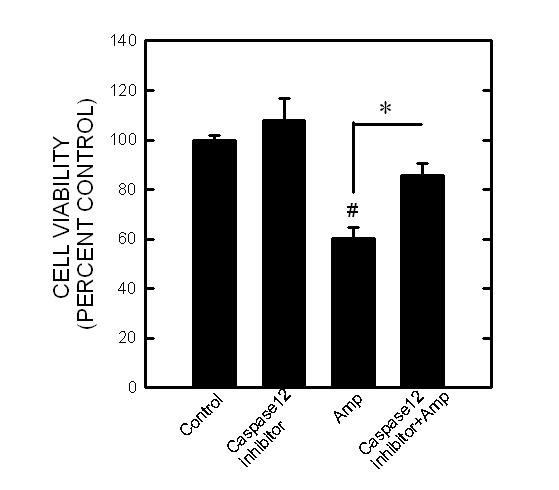
. Effect of caspase-12 inhibitor on the viability of CGNs. CGNs were pretreated with a selective caspase-12 inhibitor (0 or 2 μM) for 30 min and exposed to amprolium (Amp; 0 or 1.5 mM) for 5 days. The viability of cells was determined by MTT assay. Each data point represents the mean of four independent experiments. # p < 0.05, statistically significant difference from untreated control groups. * p < 0.05, statistically significant difference from TD-treated groups.
Discussion
This is the first report showing that TD causes ER stress in neurons. We demonstrate that TD up-regulates various markers for ER stress in vivo and in vitro. Furthermore, our results indicate that blocking the activation of caspase-12, an ER-anchored caspase, alleviates TD-induced neuronal death in vitro. Our findings suggest that ER stress may underlie TD-induced damage to the CNS.
Excessive accumulation of unfolded or misfolded proteins in the ER lumen triggers ER stress response or UPR. ER stress response is an adaptive cellular response which involves (1) cell cycle arrest; (2) attenuation of protein synthesis in order to prevent further protein aggregation and accumulation in the ER; (3) induction of ER-localized chaperone proteins and folding catalysts, such as GRP78/BiP, GRP94, calnexin, and calreticulin; and (4) activation of ER-associated protein degradation that eliminates unwanted aggregates (Kaufman, 1999; Ron, 2002; Szegezdi et al., 2003; Xu et al., 2005). Sustained ER stress ultimately leads to the apoptotic death of the cell. Many neurodegenerative disorders, including Alzheimer's, Huntington's or Parkinson's diseases, are associated with the pathological accumulation of protein aggregates, such as amyloid beta, Huntingtin or α-synuclein (Forloni et al., 2002; Shastry, 2003). An increasing body of evidence suggests that ER stress may be involved in the pathogenesis of these neurodegenerative disorders (Holtz and O'Malley, 2003; Ryu et al., 2002; Chen et al., 2004; Hoozemans et al., 2005; Katayama et al., 2004; Silva et al., 2005).
TD is a classic animal model of selective neurodegeneration. It is believed that chronic animal oxidative abnormalities, glial activation and inflammation caused by TD lead to this selective damage to the CNS. TD has been used to model some aging-related neurodegenerative disorders since they share some common neuropathological features and selective cell death (Schwab et al., 1996; Griffin et al., 1998). TD-induced ER stress in neurons provides a new insight into the cellular mechanism of TD-induced damage. Furthermore, our study offers additional evidence indicating that the pathogenesis of TD and other neurodegenerative disorders may share some common mechanisms. Thus, TD is a useful model for investigating neurodegenerative disorders.
The thalamus is a well-known target of TD. TD induces oxidative stress, neuronal death and the activation of microglial and endothelial cells in the thalamus (Ke and Gibson, 2004). Neuronal death and other neuropathological alterations occur on approximately day 8 after TD in a defined area of the thalamus, called submedial thalamus nuclei (SmTN) in mice (Calingasan et al., 1999; Ke et al., 2003). Neurons in SmTN are completely lost on day 10 following TD. Our results indicate that TD-induced ER stress in the thalamus is evident on day 5 after TD. Therefore, ER stress precedes neuropathological alterations.
The cerebellum is also sensitive to TD in vitro and in vivo (Pannunzio et al., 2000; Mulholland et al., 2005; Mulholland, 2006). Cerebellar degeneration associated with a loss of Purkinje cells as well as the shrinkage of the molecular and granule cell layers is observed in animal TD models (Mulholland, 2006). In humans, TD is a critical factor in the etiology of Wernicke-Korsakoff syndrome (WKS). Cerebellar pathology in WKS includes severe loss of Purkinje cells in the cerebellar vermis and a decrease in the volume of the molecular layer (Baker et al., 1999). Cultured cerebellar granule neurons offer an excellent in vitro model system to investigate the cellular/molecular events following TD. Our study and work from others indicate that TD causes impaired energy metabolism, production of ROS and enhanced cell death in CGNs (Pannunzio et al., 2000); these alterations are consistent with the observations in vivo. Similar to that in the thalamus, TD-induced up-regulation of markers for ER stress well precedes cerebellar neuropathological alterations in vitro and in vivo. Taken together, sustained ER stress caused by TD may be a mechanism underlying TD damage. The most commonly acquired degenerative condition affecting the cerebellum is chronic alcoholism (Maschke et al., 2005; Mulholland et al., 2005). Chronic alcohol abuse may cause TD (Victor et al, 1989; Martin et al., 2003). It would be interesting to determine whether chronic alcohol exposure also induces ER stress in the cerebellum and whether ER-stress contributes to alcohol-induced damage. These studies would provide additional insight into the cellular mechanisms of alcoholism.
The mechanisms for TD-induced ER stress are currently unknown. Although not entirely consistent, it has been shown that ROS can trigger UPR and ER stress response (Hayashi et al., 2003; Xue et al., 2005). Studies using animal models and cultured neurons have established that TD induces oxidative stress (Langlais et al., 1997; Calingasan et al., 1999; Pannunzio et al., 2000). Our study verifies that TD induces ROS in cultured CGNs (Fig. 5). Therefore, it is possible that TD-produced ROS causes ER stress.
ER-anchored caspase-12 is usually activated during ER stress response and is believed to play a central role in ER stress-induced cell death (Szegedi et al., 2003). Caspase-12 activation is involved in neuronal apoptosis in various model systems (Nakagawa et al., 2000; Chan et al., 2002; Chen and Gao, 2002; Szegezdi et al., 2003). In particular, a recent study demonstrates that caspase-12 activation is associated with ER stress-induced degeneration of Purkinje cells (Kyuhou et al., 2006). Our results reveal that TD promotes caspase-12 cleavage, indicating caspase-12 activation. Moreover, blockage of caspase-12 activation provides significant protection against TD-induced cell death, suggesting that caspase-12 plays a pivotal role in TD-induced damage. In addition to caspase-12 activation, we show that TD induces up-regulation and nuclear translocation of GADD153/Chop. GADD153 is a transcription factor and has been shown to play a critical role in ER-stress-induced cell cycle arrest and cell death (Oyadomari and Mori, 2004). Recently, death receptor 5 (DR5/TRAIL-R2), an apoptosis-inducing membrane receptor, has been identified as a target of GADD153 (Shiraishi et al., 2005). GADD153 is implicated in mediating neurodegeneration in animal PD and AD models (Ghribi et al., 2004; Silva et al., 2005). It remains to be determined whether GADD153 activation also contributes to TD-induced neuronal death.
Acknowledgement
We would like to thank Dr. He Xu for assistance in the statistical analyses and Mr. Chun Feng for assistance in confocal laser-scanning microscopy. We also want to thank Ms. Kimberly A. Bower for reading this manuscript. This research was supported by grants from the National Natural Science Foundation of China (30470544, 30471452 and 30570580) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars that is sponsored by State Education Ministry. Dr. ZJ. Ke was also supported by One Hundred Talents Program of Chinese Academy of Sciences. Dr. X. Wang was also supported by China Postdoctoral Science Foundation and Shanghai Postdoctoral Scientific Program.
Abbreviations
- AD
Alzheimer's disease
- CGNs
cerebellar granule neurons
- HD
Huntington's disease
- PD
Parkinson's disease
- ROS
reactive oxygen species
- SmTN
submedial thalamus nuclei
- TD
thiamine deficiency
- WKS
Wernicke-Korsakoff syndrome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Ayoub AE, Cai TQ, Kaplan RA, Luo J. Developmental expression of matrix metalloproteinases 2 and 9 and their potential role in the histogenesis of the cerebellar cortex. J Comp Neurol. 2005;481:403–415. doi: 10.1002/cne.20375. [DOI] [PubMed] [Google Scholar]
- Baker KG, Harding AJ, Halliday GM, Kril JJ, Harper CG. Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke's encephalopathy. Neuroscience. 1999;91:429–438. doi: 10.1016/s0306-4522(98)90664-9. [DOI] [PubMed] [Google Scholar]
- Bettendorff L, Goessens G, Sluse F, Wins P, Bureau M, Laschet J, Grisar T. Thiamine deficiency in cultured neuroblastoma cells: effect on mitochondrial function and peripheral benzodiazepine receptors. J Neurochem. 1995;64:2013–2021. doi: 10.1046/j.1471-4159.1995.64052013.x. [DOI] [PubMed] [Google Scholar]
- Calingasan NY, Chun WJ, Park LC, Uchida K, Gibson GE. Oxidative stress is associated with region-specific neuronal death during thiamine deficiency. J Neuropathol Exp Neurol. 1999;58:946–958. doi: 10.1097/00005072-199909000-00005. [DOI] [PubMed] [Google Scholar]
- Calingasan NY, Gibson GE. Vascular endothelium is a site of free radical production and inflammation in areas of neuronal loss in thiamine-deficient brain. Ann NY Acad Sci. 2000;903:353–356. doi: 10.1111/j.1749-6632.2000.tb06386.x. [DOI] [PubMed] [Google Scholar]
- Chan SL, Culmsee C, Haughey N, Klapper W, Mattson MP. Presenilin-1 mutations sensitize neurons to DNA damage-induced death by a mechanism involving perturbed calcium homeostasis and activation of calpains and caspase-12. Neurobiol Dis. 2002;11:2–19. doi: 10.1006/nbdi.2002.0542. [DOI] [PubMed] [Google Scholar]
- Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004;18:1162–1164. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- Chen L, Gao X. Neuronal apoptosis induced by endoplasmic reticulum stress. Neurochem Res. 2002;27:891–898. doi: 10.1023/a:1020387414086. [DOI] [PubMed] [Google Scholar]
- Forloni G, Terreni L, Bertani I, Fogliarino S, Invernizzi R, Assini A, Ribizzi G, Negro A, Calabrese E, Volonte MA, Mariani C, Franceschi M, Tabaton M, Bertoli A. Protein misfolding in Alzheimer's and Parkinson's disease: genetics and molecular mechanisms. Neurobiol Aging. 2002;23:957–976. doi: 10.1016/s0197-4580(02)00076-3. [DOI] [PubMed] [Google Scholar]
- Ghribi O, Herman MM, Pramoonjago P, Spaulding NK, Savory J. GDNF regulates the A beta-induced endoplasmic reticulum stress response in rabbit hippocampus by inhibiting the activation of gadd 153 and the JNK and ERK kinases. Neurobiol Dis. 2004;16:417–427. doi: 10.1016/j.nbd.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Blass JP, Beal MF, Bunik V. The alpha-ketoglutarate-dehydrogenase complex: a mediator between mitochondria and oxidative stress in neurodegeneration. Mol Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Zhang H. Abnormalities in oxidative processes in non-neuronal tissues from patients with Alzheimer's disease. J Alzheimers Dis. 2001;3:329–338. doi: 10.3233/jad-2001-3308. [DOI] [PubMed] [Google Scholar]
- Glowinski J, Iversen LL. Regional studies of catecholamines in the rat brain. I. The disposition of [3H] norepinephrine, [3H] dopamine and [3H]dopa in various regions of the brain. J Neurochem. 1966;13:655–669. doi: 10.1111/j.1471-4159.1966.tb09873.x. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer's disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim AM, Pappius HM. Sequence of metabolic, clinical, and histological events in experimental thiamine deficiency. Ann Neurol. 1983;13:365–375. doi: 10.1002/ana.410130403. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Oxidative injury to the endoplasmic reticulum in mouse brains after transient focal ischemia. Neurobiol Dis. 2004;15:229–239. doi: 10.1016/j.nbd.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Nishi T, Maier CM, Kinouchi H, Chan PH. Oxidative damage to the endoplasmic reticulum is implicated in ischemic neuronal cell death. J Cereb Blood Flow Metab. 2003;23:1117–1128. doi: 10.1097/01.WCB.0000089600.87125.AD. [DOI] [PubMed] [Google Scholar]
- Hirabayashi M, Inoue K, Tanaka K, Nakadate K, Ohsawa Y, Kamei Y, Popiel AH, Sinohara A, Iwamatsu A, Kimura Y, Uchiyama Y, Hori S, Kakizuka A. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 2001;8:977–984. doi: 10.1038/sj.cdd.4400907. [DOI] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–19377. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W. The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol (Berl) 2005;110:165–172. doi: 10.1007/s00401-005-1038-0. [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer's disease. J Chem Neuroanat. 2004;28:67–78. doi: 10.1016/j.jchemneu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Gene Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Ke ZJ, DeGiorgio LA, Volpe BT, Gibson GE. Reversal of thiamine deficiency-induced neurodegeneration. J Neuropathol Exp Neurol. 2003;62:195–207. doi: 10.1093/jnen/62.2.195. [DOI] [PubMed] [Google Scholar]
- Ke ZJ, Gibson GE. Selective response of various brain cell types during neurodegeneration induced by mild impairment of oxidative metabolism. Neurochem Int. 2004;45:361–369. doi: 10.1016/j.neuint.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Ke ZJ, Bowen WM, Gibson GE. Inflammatory mechanisms mediate microglial activation but not neurodegeneration in response to mild impairment of oxidative metabolism. Neurochem Intl. 2006. 2006 doi: 10.1016/j.neuint.2006.04.011. in press. [DOI] [PubMed] [Google Scholar]
- Kyuhou S, Kato N, Gemba H. Emergence of endoplasmic reticulum stress and activated microglia in Purkinje cell degeneration mice. Neurosci Lett. 2006;396:91–96. doi: 10.1016/j.neulet.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Anderson G, Guo SX, Bondy SC. Increased cerebral free radical production during thiamine deficiency. Metab Brain Dis. 1997;12:137–143. [PubMed] [Google Scholar]
- LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- Li Z, Miller MW, Luo J. Effects of prenatal exposure to ethanol on the cyclin-dependent kinase system in the developing rat cerebellum. Brain Res Dev Brain Res. 2002;139:237–245. doi: 10.1016/s0165-3806(02)00573-4. [DOI] [PubMed] [Google Scholar]
- Martin PR, Singleton CK, Hiller-Sturmhofel S. The role of thiamine deficiency in alcoholic brain disease. Alcohol Res Health. 2003;27:134–142. [PMC free article] [PubMed] [Google Scholar]
- Maschke M, Weber J, Bonnet U, Dimitrova A, Bohrenkamper J, Sturm S, Muller BW, Gastpar M, Diener HC, Forsting M, Timmann D. Vermal atrophy of alcoholics correlate with serum thiamine levels but not with dentate iron concentrations as estimated by MRI. J Neurol. 2005;252:704–711. doi: 10.1007/s00415-005-0722-2. [DOI] [PubMed] [Google Scholar]
- Mulholland PJ. Susceptibility of the cerebellum to thiamine deficiency. Cerebellum. 2006;5:55–63. doi: 10.1080/14734220600551707. [DOI] [PubMed] [Google Scholar]
- Mulholland PJ, Self RL, Stepanyan TD, Little HJ, Littleton JM, Prendergast MA. Thiamine deficiency in the pathogenesis of chronic ethanol-associated cerebellar damage in vitro. Neuroscience. 2005;135:1129–1139. doi: 10.1016/j.neuroscience.2005.06.077. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Pannunzio P, Hazell AS, Pannunzio M, Rao KV, Butterworth RF. Thiamine deficiency results in metabolic acidosis and energy failure in cerebellar granule cells: an in vitro model for the study of cell death mechanisms in Wernicke's encephalopathy. J Neurosci Res. 2000;62:286–292. doi: 10.1002/1097-4547(20001015)62:2<286::AID-JNR13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Park LC, Albers DS, Xu H, Lindsay JG, Beal MF, Gibson GE. Mitochondrial impairment in the cerebellum of the patients with progressive supranuclear palsy. J Neurosci Res. 2001;66:1028–1034. doi: 10.1002/jnr.10062. [DOI] [PubMed] [Google Scholar]
- Park LC, Calingasan NY, Uchida K, Zhang H, Gibson GE. Metabolic impairment elicits brain cell type-selective changes in oxidative stress and cell death in culture. J Neurochem. 2000;74:114–124. doi: 10.1046/j.1471-4159.2000.0740114.x. [DOI] [PubMed] [Google Scholar]
- Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–1388. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, Steele JC, McGeer PL. Neurofibrillary tangles of Guam parkinson-dementia are associated with reactive microglia and complement proteins. Brain Res. 1996;707:196–205. doi: 10.1016/0006-8993(95)01257-5. [DOI] [PubMed] [Google Scholar]
- Shastry BS. Neurodegenerative disorders of protein aggregation. Neurochem Int. 2003;43:1–7. doi: 10.1016/s0197-0186(02)00196-1. [DOI] [PubMed] [Google Scholar]
- Shen X, Zhang K, Kaufman RJ. The unfolded protein response--a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat. 2004;28:79–92. doi: 10.1016/j.jchemneu.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Shiraishi T, Yoshida T, Nakata S, Horinaka M, Wakada M, Mizutani Y, Miki T, Sakai T. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res. 2005;65:6364–6370. doi: 10.1158/0008-5472.CAN-05-0312. [DOI] [PubMed] [Google Scholar]
- Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, Kholodilov N, Greene LA, Burke RE. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. 2005;95:974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton CK, Martin PR. Molecular mechanisms of thiamine utilization. Curr Mol Med. 2001;1:197–207. doi: 10.2174/1566524013363870. [DOI] [PubMed] [Google Scholar]
- Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann NY Acad Sci. 2003;1010:186–194. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- Todd K, Butterworth RF. Mechanisms of selective neuronal cell death due to thiamine deficiency. Ann NY Acad Sci. 1999;893:404–411. doi: 10.1111/j.1749-6632.1999.tb07866.x. [DOI] [PubMed] [Google Scholar]
- Turner BJ, Atkin JD. ER stress and UPR in familial amyotrophic lateral sclerosis. Curr Mol Med. 2006;6:79–86. doi: 10.2174/156652406775574550. [DOI] [PubMed] [Google Scholar]
- Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff syndrome and related neurological disorders due to alcoholism and malnutrition. Second Edition F. A. Davis Company; Philadelphia, PA: 1989. [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Piao JH, Nakajima A, Sakon-Komazawa S, Kojima Y, Mori K, Yagita H, Okumura K, Harding H, Nakano H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem. 2005;280:33917–33925. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]