

Abstract
Astrocytes send processes to synapses and blood vessels, communicate with other astrocytes through gap junctions and by release of ATP, and thus are an integral component of the neurovascular unit. Electrical field stimulations in brain slices demonstrate an increase in intracellular calcium in astrocyte cell bodies transmitted to perivascular end-feet, followed by a decrease in vascular smooth muscle calcium oscillations and arteriolar dilation. The increase in astrocyte calcium after neuronal activation is mediated, in part, by activation of metabotropic glutamate receptors. Calcium signaling in vitro can also be influenced by adenosine acting on A2B receptors and by epoxyeicosatrienoic acids (EETs) shown to be synthesized in astrocytes. Prostaglandins, EETs, arachidonic acid, and potassium ions are candidate mediators of communication between astrocyte end-feet and vascular smooth muscle. In vivo evidence supports a role for cyclooxygenase-2 metabolites, EETs, adenosine, and neuronally derived nitric oxide in the coupling of increased blood flow to increased neuronal activity. Combined inhibition of the EETs, nitric oxide, and adenosine pathways indicates that signaling is not by parallel, independent pathways. Indirect pharmacological results are consistent with astrocytes acting as intermediaries in neurovascular signaling within the neurovascular unit. For specific stimuli, astrocytes are also capable of transmitting signals to pial arterioles on the brain surface for ensuring adequate inflow pressure to parenchymal feeding arterioles. Therefore, evidence from brain slices and indirect evidence in vivo with pharmacological approaches suggest that astrocytes play a pivotal role in regulating the fundamental physiological response coupling dynamic changes in cerebral blood flow to neuronal synaptic activity. Future work using in vivo imaging and genetic manipulation will be required to provide more direct evidence for a role of astrocytes in neurovascular coupling.
Keywords: adenosine, cerebral blood flow, epoxyeicosatrienoic acid, neuronal activation, nitric oxide, prostaglandin
Astrocytes outnumber neurons in mammalian brain, particularly in higher mammalian species with a thick cortical mantle and an enlarged neuropil containing neuronal and astrocyte processes. Astrocytes serve many functions in brain. Classically, they are known to have high K+ permeability and to be important in regulating extracellular K+ necessary for maintaining neuronal excitability. They also possess several different types of proton and bicarbonate transporters, some of which are linked to carbonic anhydrase, and are considered to play an important role in brain pH homeostasis. Astrocyte processes surround synapses and possess transporters for uptake of various neurotransmitters, including glutamate. However, our view of astrocyte function has grown considerably over the past decade. Astrocytes can communicate with each other through gap junctions. A sufficiently large increase in Ca2+ in an astrocyte is capable of inducing a spreading wave of increased Ca2+ in adjacent astrocytes (23). This Ca2+ wave depends on localized burst release of ATP (11), which acts on purinergic receptors on adjacent astrocytes in the proximity of gap junctions (45). In vitro, Ca2+ waves can spread over hundreds of micrometers in a time frame of a few seconds. This intercellular communication provides a basis for astrocytes to act as a syncytium for possibly modulating neuronal and vascular function. Moreover, specific populations of astrocytes can release neurotransmitters, such as glutamate, and receptor modulators, such as D-serine, thereby exerting an influence on neuronal activity (30). Glutamate release may occur through multiple mechanisms including connexin hemichannels (120). Release of glutamate has been linked to an increase in intracellular Na+ and to the spread of Na+ waves between astrocytes, thereby leading to the propagation of increased glycolysis through adjacent astrocytes to support Na+,K+-ATPase (13). Although astrocytes are electrically nonexcitable cells, they express glutamatergic, GABAergic, purinergic, adrenergic, and serotonergic receptors, among others. Other important functions of astrocytes involve trophic influences in guiding neuronal processes, stabilizing synaptic connections, guiding capillary tube formation during angiogenesis, influencing the phenotypic expression of specific endothelial proteins, and stabilizing the blood-brain barrier (43). Astrocyte physiology is more complex than previously appreciated because these cells play multiple roles in actively modulating neuronal and vascular function.
In considering the role of astrocytes in cerebrovascular regulation, it is important to understand their pivotal relationship among the various cell types in brain. Blood vessels in the brain are surrounded primarily by foot processes that emanate from astrocytes and, to a lesser degree, by pericytes and macrophages. A typical astrocyte can send processes to hundreds of synapses. Most astrocytes extend at least one process that contacts a blood vessel (104). End-feet abutting capillaries and larger vessels express connexin-43 and purinergic P2Y receptors, which together permit Ca2+ increases to be transmitted 60 μm or more along the abluminal side of the vessel wall (104). Thus astrocytes are in a unique position for sensing neuronal activity, integrating that information, and communicating with blood vessels in brain parenchyma. As a rule, neurons do not directly innervate intraparenchymal vascular smooth muscle. However, subpopulations of GABAergic interneurons come into close contact with astrocyte foot processes and can elicit vasodilation, as reviewed elsewhere in the series (42). These neurons might modulate vascular function, in part, through stimulation of nitric oxide (NO) synthase (NOS) activity, release of vasoactive peptides, or an astrocyte signaling mechanism.
ASTROCYTES AS INTERMEDIARY PROCESSORS IN NEUROVASCULAR COUPLING
Brain slice preparations have been used to provide direct evidence for an astrocyte role in neurovascular coupling. With electrical stimulation of cortical slices, Zonta et al. (122) demonstrated increases in intracellular Ca2+ in astrocyte cell bodies and in astrocyte end-feet on blood vessels followed temporally by an increase in diameter of small arterioles. Astrocytes are known to express metabotropic glutamate receptors (mGluRs) of the subtypes that mediate an increase in Ca2+ through coupling to PLC and increased inositol triphosphate. Administration of mGluR antagonists reduced both the evoked increase in astrocytic Ca2+ and the arteriolar dilation, without affecting evoked increases in neuronal Ca2+, whereas administration of a mGluR agonist mimicked the temporal sequence of the increased astrocytic Ca2+ and arteriolar dilation seen with electrical activation. Furthermore, direct stimulation of perivascular astrocytes during membrane patching elicited dilation of adjacent blood vessels. Therefore, synaptic release of glutamate during neuronal activation is believed to act on astrocytic mGluR receptors to increase Ca2+ in astrocytes and elicit dilation in nearby arterioles.
The brain slice preparation, however, has several limitations. The blood vessels have no blood flow or associated shear stress and have no pressure-induced myogenic tone. The response time of increased astrocytic Ca2+ and arteriolar dilation was in the order of 30 s, which is much slower than the in vivo vascular response time of 1–2 s. Part of the slow response time might be explained by the loss of an adequate amount of synaptic connectivity in the slice and by the 33°C temperature used in this study. Others have reported more rapid Ca2+ increases during electrical stimulation in cortical slices at 35–37°C (32). The lack of effect of NOS inhibitors and N-methyl-D-aspartate (NMDA) receptor antagonists on the evoked vascular response (vs. the positive effects of these agents in vivo) suggests incomplete connectivity and signaling processes.
The effect of neuronal activation on arteriolar vasomotion has been studied in hippocampal slices. Using the thromboxane analog U-46619 to increase vascular tone and induce spontaneous vasomotion, Brown et al. (15) found that stimulation of Schaffer collateral input fibers into the hippocampus reduced the frequency of vasomotion. These findings were extended in cortical slices by Filosa et al. (32), who temporally linked the increase in Ca2+ in astrocyte cell body and end-feet with the subsequent suppression of spontaneous periodic Ca2+ waves in neighboring arterioles in the presence of U-46619. Either with electrical activation of neurons or with an agonist of mGluR receptors, a rapid increase in astrocyte Ca2+ was followed by nearly complete blockage of vascular Ca2+ oscillations and of dilation. However, mGluR antagonists did not prevent the increase in astrocyte Ca2+ and the decreased frequency of vascular Ca2+ oscillations. The latter finding suggests that activation of astrocyte mGluR receptors might not be the only mechanism by which astrocyte Ca2+ is increased during neuronal activation. Overall, the findings demonstrate an important influence of increased Ca2+ in astrocytes during activation on dynamic Ca2+ transients in parenchymal arterioles.
In all of the above-cited work with brain slices, astrocyte and vascular responses to electrical field stimulation were abolished by tetrodotoxin, thereby indicating the requirement for neuronal action potentials for the stimulus to be effective. To demonstrate a direct role of astrocyte Ca2+ in the absence of electrical stimulation of neurons or pharmacological activation of astrocytes, Mulligan and MacVicar (74) used flash photolysis of caged Ca2+ in green fluorescent protein-labeled astrocytes to increase Ca2+ in individual astrocytes in brain slices. When the laser intensity was increased sufficiently to induce a Ca2+ wave that spread to the astrocyte end-feet, arteriolar constriction, rather than dilation, unexpectedly occurred. When the induced Ca2+ transient was sufficiently large to spread to many end-feet in the network, the constriction was more extensive and more severe. Application of norepinephrine to the bath produced increases in astrocyte Ca2+, which, if the Ca2+ wave spread to the end-feet, resulted in constriction beginning within 1.2 s of arrival of the Ca2+ wave at the end-feet. The constrictor response to uncaging of Ca2+ was blocked by a phospholipase A2 (PLA2) inhibitor and by an inhibitor of 20-hydroxyeicosatetraenoic acid (20-HETE) synthesis. Because 20-HETE is known to be synthesized in vascular smooth muscle by cytochrome P450 (CYP) 4A and to constrict cerebral arterioles by inhibiting calcium-sensitive potassium (KCa) channels (5, 107), the authors surmise that Ca2+ waves propagating to the end-feet activate PLA2 and mobilize arachidonic acid, which then is transported to the smooth muscle to act as a substrate for CYP ω-hydroxylase formation of 20-HETE. Application of a mGluR agonist to increase astrocytic Ca2+ also produced constriction, in contrast to the dilation seen in other studies (32, 122). These other studies used a NOS inhibitor or U-46619 to increase vascular tone, whereas no agent was used to increase vascular tone when the constrictor response was observed. However, when Mulligan and MacVicar (74) used a NOS inhibitor, the vascular response to the mGluR agonist was converted from a constrictor response to a dilator response. Because NO inhibits 20-HETE synthesis (5, 107), they suggest that release of NO from neurons during neuronal activation interferes with an astrocyte-mediated constrictor response and permits a dilatory response. Alternatively, whether one observes a constrictor or dilator response to increased astrocytic Ca2+ might simply depend on baseline vascular tone. Without increasing baseline vascular tone in a brain slice preparation, dilatory responses might be more difficult to unmask. It is, therefore, important to investigate these mechanisms in vivo with normal myogenic tone and blood flow.
It will also be important to understand how changes in myogenic tone in pathophysiological states alter the evoked vasodilatory response. Decreases in cerebral myogenic tone, as may occur with severe arterial hypotension, carotid stenosis, or intracranial hypertension, would be expected to limit additional vasodilation during neuronal activation (10). Increases in myogenic tone are associated with increases in 20-HETE synthesis, which counteracts the effect of increased Ca2+ on KCa channels and permits depolarization (36). If the pathophysiological state does not impair evoked neuronal NO production during activation, then one would anticipate that the NO would be capable of inhibiting 20-HETE synthesis and permit opening of vascular KCa channels. One situation in which this might not occur is subarachnoid hemorrhage, in which 20-HETE levels are markedly elevated (18) and excess heme may scavenge neuronally released NO.
METABOLIC COUPLING DURING NEURONAL ACTIVATION
Although overall global cerebral blood flow (CBF) normally changes very little during mental tasks, discretely localized increases occur in regions where neuronal networks increase their activity. Neuronal activity results in a regional increase in cerebral oxygen consumption (CMRO2), but the increase is often too small to detect by positron emission tomography (33, 35, 111). However, glucose, the predominant energy substrate in the brain, is consumed at a disproportionately greater rate than is oxygen (34, 66, 95). Regional increases in lactate have been reported during neuronal activation (48, 94), but the excess glucose uptake is not balanced by lactate efflux (66, 67). One hypothesis is that excess glycolysis occurs in astrocytes, leading to formation of lactate, which is then transported from astrocytes to neurons via monocarboxylic acid transporters (89). Lactate could then serve as an alternative energy substrate in neurons during dynamic fluctuations in neuronal activity. However, this hypothesis remains controversial. The endothelium and neurons have abundant glucose transporters, and glucose supply does not normally limit steady-state oxidative phosphorylation unless arterial glucose concentration is reduced by more than 75%. Nevertheless, data derived to support this hypothesis place the astrocyte in an active position to support dynamic alterations in neuronal energy metabolism.
Interestingly, the hemodynamic response is considered to correlate with synaptic activity as well as with neuronal firing rate (59, 64, 73, 82). Monitoring of NADH fluorescence in synapses suggests a rapid increase in dendritic oxidative metabolism followed by increased glycolysis in astrocytes (56). Infusion of lactate in rats increases the blood flow response to activation, whereas infusion of pyruvate decreases the blood flow response (52). A similar effect has been reported in humans (72). Because the lactate-to-pyruvate ratio is proportional to the NADH-to-NAD+ ratio, these results were interpreted as indicating that a reduction of cytosolic NAD+ is a sensor for coupling blood flow to an imbalance between glycolysis and oxidative phosphorylation. Whether the NADH redox state of astrocytes, where glycolysis is purported to be disproportionately augmented during activation, is a key factor in the blood flow coupling mechanism remains to be determined.
CHANGES IN OXYGENATION DURING ACTIVATION
If astrocytes play an important role in neurovascular coupling, it is useful to understand the dynamics of the metabolic and vascular responses. Classical models of blood flow coupling to increased energy metabolism, such as occurs in muscle during exercise, relied on an initial mismatch of flow to metabolism causing release of vasoactive mediators. Blood flow would then increase in this simple negative feedback model to meet increased energy metabolism. During neuronal activation, the percent increase in CBF is approximately proportional to the percent increase in cerebral glucose consumption but surprisingly is disproportionately greater than the percent increase in CMRO2 (33, 34). Consequently, fractional O2 extraction and the concentration of deoxyhemoglobin in the blood decrease during activation. Because deoxyhemoglobin is paramagnetic, a blood-oxygen-dependent signal can be detected by MRI (64). Many studies have made use of high-spatial resolution, functional MRI to map the topology of functional activation in humans during various sensory, motor, and cognitive tasks and to determine the impact of various neurological disorders on these functional activation maps. Because CBF is used as a surrogate for neuronal activation, it is important to understand the mechanisms of this fundamental physiological response that links cerebral hyperemia to neuronal activity.
The cerebrovascular response to neuronal activation in cerebral cortex has been studied in animal models with sensory activation, using visual stimuli, electrical stimulation of the forelimb or sciatic nerve, and mechanical displacement of the whiskers. In all of these sensory activation models, cortical CBF increases after latency of 1–2 s and reaches a steady state by 5–10 s, although an overshoot can also be present if sensory afferents are stimulated with short pulse durations (68, 97). Several experimental studies, but not all studies, reported a transient dip in vascular oxygenation during the first second of activation preceding the increase in CBF, thereby implying a rapid increase in CMRO2 preceding the increase in CBF and increase in the vascular oxygenation (8, 9, 62, 112). Local vasodilation appears to depend on feed-forward signaling mechanisms rather than on a slow feedback mechanism that senses tissue hypoxia. It can be speculated that astrocyte networks will rapidly sense changes in synaptic activity, integrate this information spatially over many synapses, and augment glycolytic metabolism that is coupled to an increase in blood flow.
The physiological importance of the increase in blood flow exceeding the increase in O2 demand remains in question. Steady-state differences in O2 demand among brain regions are met by differences in steady-state CBF and in capillary density. Because all capillaries in the brain are considered to be perfused continuously by red blood cells, capillary recruitment is not thought to be a major mechanism for dynamic increases in tissue oxygenation (neglecting the fact that red blood cell flux is heterogeneous among capillaries and does have the potential to become better matched to spatial heterogeneity in CMRO2 during activation). As a first approximation, a dynamic increase in CMRO2 requires an increase in O2 flux across an essentially constant endothelial surface area driven by the PO2 gradient between intravascular PO2 and mitochondrial PO2 within cells at different distances from the blood vessels. Consequently, an increase in O2 flux across the endothelial membrane requires either an increase in intravascular PO2, a decrease in mitochondrial PO2, or combination of both. When CMRO2 increases, the tissue PO2 profile becomes steeper to provide an increased O2 flux deep into the tissue, as shown schematically in Fig. 1. If CBF and CMRO2 increase by the same percentage, fractional O2 extraction and end-capillary PO2 would be virtually unchanged (ignoring any potential changes in the spatial heterogeneity of red blood cell flux). In this case, tissue PO2 would be lower at all sites, and mitochondria at the greatest distance from a capillary would be at risk of inadequate O2 supply. On the other hand, if CBF increased by a greater percentage than CMRO2, then O2 extraction would decrease, end-capillary PO2 would increase, and mitochondria at the greatest distance from a capillary would be at less risk of inadequate O2 supply. Therefore, the hyperemic response, in which the increase in CBF is disproportionately greater than the increase in CMRO2, appears to be a homeostatic mechanism that mitigates a decrease in mitochondrial PO2. This concept is supported by data showing that the magnitude of the changes in vascular oxygenation during activation is inversely related to baseline oxygenation (62). Models of O2 extraction also indicate that the percent increase in CBF is necessarily greater than the percent increase in CMRO2 when back-diffusion of O2 from parenchyma to capillaries is assumed to be negligible (17, 49). This assumption is equivalent to assuming all of the O2 that crosses the endothelium into the tissue is consumed and, hence, that PO2 at mitochondria located at the greatest distance from a capillary is at the minimum level necessary for basal oxidative phosphorylation. Therefore, a feed-forward vasodilatory mechanism appears to be involved in neurovascular coupling. Astrocytes may participate in this feed-forward mechanism via several potential pathways.
Fig. 1.
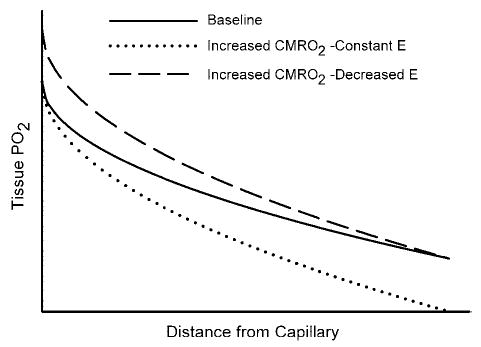
Schematic diagram of tissue PO2 as a function of distance from a capillary at the downstream end of the O2-exchange site. A parabolic PO2 profile is used for illustrative purposes for the simplified case of O2 diffusion into a rectangular slab of tissue of infinite length and a homogeneous consumption of O2 (CMRO2) throughout the parenchyma (solid line at baseline CMRO2). If an increase in CMRO2 results in a proportional increase in blood flow, then there would be no change in fractional O2 extraction (E) and no change in end-capillary PO2. The increase in CMRO2 would then result in a steeper PO2 gradient to maintain the increased O2 flux and in a lower PO2 throughout the tissue, such that mitochondria at the greatest distance from a capillary would be at greater risk of inadequate O2 supply (dotted line). If blood flow increases by a greater percent than the increase in CMRO2, then there would be a decrease in E and an increase in end-capillary PO2. The increase in CMRO2 would still result in a steeper PO2 gradient throughout the tissue, but mitochondria at the greatest distance from a capillary would have less risk of inadequate O2 supply (dashed line).
VASOACTIVE MEDIATORS OF FUNCTIONAL HYPEREMIA
A number of mediators appear to contribute to the complex signaling processes that mediate feed-forward cerebral vasodilation sufficient to decrease O2 extraction in response to neural activity. These include K+, NO, adenosine, prostaglandins, and EETs derived from CYP metabolism of arachidonic acid.
Potassium ions
Evidence for potassium ions is largely indirect and based on the observation that high K+ conductance in perivascular end-feet will act to siphon K+ from synapses to the perivascular space. Conceptually, an increase in interstitial K+ that results from the efflux of K+ out of neurons during increased neuronal activity would be buffered by uptake in synaptic astrocyte processes via inwardly rectifier K+ (Kir) channels (96), and the excess K+ in astrocytes would be preferentially released at foot processes (75, 87). This hypothesis is supported by data from Müller cells in retina (77), but the role for K+ siphoning in other brain regions remains speculative. Cultured astrocytes express KCa channels (39). In situ, rSlo KCa channels are present in astrocyte end-feet, including those abutting the pia mater (93). Moderate increases in perivascular K+ activity arising from astrocyte K+ efflux would then relax arteriolar smooth muscle by acting on vascular Kir channels (31).
NO
The neuronal isoform of NOS is present in a small population of interneurons, some of which are in close proximity to intraparenchymal blood vessels (113). Activation of NMDA receptors on these neurons leads to Ca2+ entry and stimulation of NOS anchored in the vicinity of NMDA receptors by postsynaptic density proteins (21). Because NO can diffuse for considerable distances across neighboring cells, it can produce vasorelaxation of arteriolar smooth muscle. Several lines of evidence support a role for neuronally derived NO in functional hyperemia. A transient burst of NO has been measured within 1 s of neuronal activation and preceding the increase in CBF (16). The neuronal NOS-specific inhibitor, 7-nitroindazole (7-NI) reduces the cortical blood flow response to whisker stimulation by ~50–60% (20, 63). The nonisoform-specific inhibitor Nω-nitro-L-arginine (L-NNA) attenuates functional hyperemia in both wild-type and endothelial NOS null mice (12) but has no effect in neuronal NOS null mice (65).
However, neuronally derived NO is not an essential mediator of the flow response. The attenuating effect of NOS inhibition on the cortical flow response to whisker stimulation is smaller in unanesthetized rats than it is in anesthetized animals (41), and administration of a NOS inhibitor to humans failed to significantly reduce the evoked CBF response in frontal cortex to a learning task (115). Furthermore, neuronal NOS null mice have a normal cortical blood flow response to whisker stimulation, suggesting compensation by other mediators (65). Moreover, inhibition of NOS results in an increase in arteriolar tone and a decrease in baseline CBF. When baseline CBF is restored after NOS inhibition by the use of either a NO donor to clamp the level of NO or a cell-permeant cyclic GMP analog, the CBF response to whisker stimulation is restored (63). These results suggest that the presence of an adequate concentration of NO and cyclic GMP is required for an intact response, but that dynamic fluctuations in NO are not required for mediating the dynamic CBF response. Therefore, NO appears to play more of a role as a modulator, rather than a mediator, of the cortical flow response to activation. As discussed above, in interpreting data from brain slice preparations (74), NO might act to inhibit 20-HETE formation in vascular smooth muscle from PLA2-mobilized arachidonic acid at astrocyte end-feet and thereby permit vasodilation. Regional differences might also be important, in that NO appears to play a more prominent role in cerebellum (119) and thalamus (20, 41).
Adenosine
A role for adenosine in mediating functional hyperemia is supported by evidence that the semiselective adenosine-receptor antagonist theophylline attenuates the increase in CBF during whisker stimulation (Fig. 2) (26) and the vasodilation of extraparenchymal pial arterioles during sciatic nerve stimulation (58). In addition, adenosine deaminase attenuates the CBF response to whisker stimulation (26). Evidence against a major role for adenosine is based on data that show a lack of effect of the semiselective adenosine antagonist caffeine on the cortical flow response to whisker stimulation in unanesthetized rats (41). However, relatively high concentrations of caffeine are required to inhibit the pial arteriolar dilation to adenosine or to sciatic nerve stimulation (71). High concentrations of caffeine probably exert nonspecific effects, such as those on ryanodine receptors, which could offset effects on adenosine receptors.
Fig. 2.
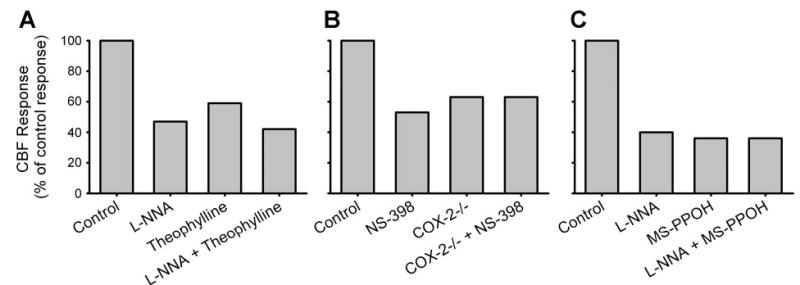
Effect of different inhibitors applied to cortical surface on the cortical blood flow response to sensory activation plotted as a percentage of the control response in each experiment. A: reduction in the response to whisker stimulation with the nitric oxide synthase inhibitor Nω-nitro-L-arginine (L-NNA), the adenosine receptor antagonist theophylline, and combined L-NNA and theophylline [data adapted from Dirnagl et al. (26) and Lindauer et al. (63)]. B: reduction in the response to whisker response with the cyclooxygenase-2 (COX-2) inhibitor N-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulfonamide (NS-398) and in COX-2 null mice (COX-2 −/−) with and without NS-398 [data adapted from Niwa et al. (83)]. C: reduction in response to electrical foreleg stimulation with L-NNA, the epoxygenase inhibitor N-methylsulfonyl-6-(2-propargyloxyphenyl)hexanamide (MS-PPOH), and combined L-NNA and MS-PPOH [data adapted from Peng et al. (91)]. All inhibitors individually reduced the flow response to ~40–60% of the control response. Combining L-NNA with theophylline or MS-PPOH produced either a small or no additional inhibition, suggesting an interaction among these pathways.
Dilation of pial arterioles to exogenous adenosine is mediated primarily by high-affinity A2A receptors and, to a lesser extent, by low-affinity A2B receptors (78, 101). Pial arteriolar dilation to topical glutamate and to sciatic nerve stimulation is attenuated by an A2A antagonist (53, 70). However, preliminary work indicates that the CBF response to whisker stimulation is attenuated by an A2B antagonist, rather than by an A2A antagonist (100), suggesting that the intraparenchymal vascular response is mediated by different adenosine receptors than is the extraparenchymal vascular response.
In addition to direct actions of adenosine on smooth muscle, adenosine can act on astrocytes. Adenosine is derived from the breakdown of ATP in cells and by ecto-ATPase and ecto-5′-nucleotidase (55, 69, 103, 114). ATP is released in synapses as a coneurotransmitter. ATP is also released from astrocytes through connexin hemichannels where ATP can act on adjacent astrocyte P2Y receptors to promote the spread of Ca2+ waves (11, 99, 105). In human astrocytoma cells, ecto-nucleotidase activity colocalizes with sites of ATP release (55). Thus extracellular adenosine concentration is expected to be increased near sites of ATP release. In astrocytes, stimulation of A2B receptors increases cyclic AMP (88), glycogen synthesis (3), and intracellular Ca2+ (92). Adenosine potentiates the increase of Ca2+ waves evoked by ATP through an action largely attributed to A2B receptors (4, 54). In retina, physiological activation with light induces an increase in Ca2+ in Müller glial cells, and the spread of Ca2+ waves is augmented by adenosine primarily through effects on A2B receptors (76). Therefore, A2B receptors are likely to play a role in astrocyte communication with the vasculature. Moreover, purinergic receptor expression is dense along perivascular end-feet (104). If ecto-ATPase and ecto-nucleotidase activity is also present at these sites along the end-feet, localized perivascular adenosine concentration could become elevated to concentrations sufficient to activate low-affinity A2B receptors during stimulation.
Prostaglandins
A role for prostaglandins in functional hyperemia is derived from data showing that the CBF response to whisker stimulation is reduced ~40–50% by a cyclooxygenase-2 (COX-2) inhibitor or by COX-2 gene deletion in mice (Fig. 2) (83). In contrast, a COX-1 inhibitor or COX-1 gene deletion, which attenuated acetylcholine vasodilation, did not impair the CBF response to whisker stimulation (84). Although COX-2 is a highly inducible isoform of COX, it is constitutively expressed in somatosensory cortical neurons, including dendritic and terminal processes adjacent to perivascular astrocyte processes and, in some cases, NOS-positive interneurons (24, 113). In contrast, expression of COX-2 in astrocytes is low. Thus a COX-2 metabolite, such as prostaglandin E2 (PGE2) released from neurons, is postulated to act either directly on vascular smooth muscle, indirectly via perivascular astrocytes, or by an interaction with NOS-containing neurons.
Prostaglandins are also postulated to be released from astrocytes during activation and to mediate vasodilation. Acetylsalicylic acid inhibits vascular dilation during electrical activation in cortical slices (122). Cultured astrocytes release PGE2 when activated by a mGluR agonist (123). Moreover, administration of mGluR antagonists in vivo decreases the CBF response to whisker stimulation in the same manner that they act in slices to reduce astrocyte Ca2+ responses (122). However, the role of an astrocyte-based PGE2 mechanism in vivo is unclear because of the low expression of COX-2 in astrocytes and the lack of dependence of the blood flow response on COX-1.
EETs
Cultured astrocytes from rat express CYP 2C11, which possesses epoxygenase activity (2). Conversion of arachidonic acid to EETs has been described in cultured astrocytes and in brain parenchyma (2, 6, 7). Expression of CYP 2C11 in rat brain colocalizes with glial fibrillary acidic protein-positive astrocytes, including perivascular astrocytes (91). EETs differ from prostaglandins in that they are stored in the phospholipid membrane, including astrocyte membranes, and thus could be mobilized without de novo synthesis (102). Within astrocytes, release of Ca2+ from internal stores induced by thapsigargin causes arachidonic acid to be mobilized followed by formation of EETs and Ca2+ influx (98). Of the four regioisomers of EETs, the 5,6-EET appears to be responsible for enhancing the capacitative Ca2+ influx. Moreover, EETs can promote the opening of iberiotoxin-insensitive KCa channels (39). The resultant efflux of K+ is expected to partially offset the influx of Ca2+ and maintain a hyperpolarized membrane for sustaining Ca2+ entry (38).
In addition to acting intracellularly, EETs can act in a paracrine fashion. For example, EETs serve as an endothelium-derived hyperpolarizing factor in coronary smooth muscle (19). Although a similar paracrine function has not been demonstrated in cerebral endothelium where other hyperpolarizing factors might operate (121), EETs derived from astrocytes may act intercellularly. Addition of glutamate to cultured astrocytes causes release of EETs into the media (1). The release of EETs evoked by a mGluR agonist is reduced by an inhibitor of KCa channels, suggesting a link between hyperpolarization induced by KCa channel opening, sustained Ca2+ influx, and release of EETs by astrocytes (38). In vascular smooth muscle, EETs cause hyperpolarization by opening of KCa channels (2, 37, 44) and dilate cerebral arteries (28, 37, 60). Thus it has been proposed that EETs might serve as an astrocyte-derived vasodilator (44).
In support of this hypothesis, increases in CBF elicited by topical glutamate application to the brain surface or by dialysis of NMDA into striatal tissue are markedly reduced by inhibitors of epoxygenase activity (1, 14). Furthermore, epoxygenase inhibitors applied to the cortical surface reduced the CBF response to whisker stimulation (90) and to electrical stimulation of the forepaw (91). Therefore, an astrocyte-based epoxygenase pathway appears to be critical in the coupling of CBF to neuronal activation in cortex. Consistent with this hypothesis, antagonists of KCa channels also reduce the CBF response to whisker stimulation (40). Further work in vivo is needed using molecular approaches to specifically alter EET signaling and using direct measurements of extracellular EETs to fully understand the role of EETs in neurovascular coupling. Tissue measurements of EETs are complicated by compartmentation of EETs in the membrane phospholipid pool.
Interactions of mediators
The epoxygenase pathway has complex interactions with other mediators of functional hyperemia. In the presence of indomethacin, epoxygenase inhibition is still capable of reducing the CBF response to whisker stimulation, thereby indicating that the actions of the epoxygenase mechanism do not require COX activity (90). On the other hand, the NOS pathway does significantly interact with the epoxygenase pathway. The striatal blood flow response to NMDA was completely blocked by individually inhibiting either NOS or epoxygenase activity (14). Also, no additive effect of combined inhibition was observed with NOS and epoxygenase inhibitors on the cortical CBF response to electrical forelimb stimulation (91). The CBF response was attenuated ~60% with either inhibitor alone or with combined inhibition (Fig. 2). Likewise, an adenosine A2B antagonist combined with an epoxygenase inhibitor, an EET antagonist, or a mGluR antagonist produced no substantial additive inhibition beyond the ~50% inhibition seen with each agent alone during whisker stimulation (100). This observation is consistent with an action of adenosine on astrocyte A2B receptors amplifying Ca2+ stimulation of EETs release. Moreover, NO has been reported to promote capacitative Ca2+ influx in astrocytes (61). Combining a NOS inhibitor with the adenosine antagonist theophylline produced a small additive effect on the CBF response to whisker stimulation (Fig. 2) but did not eliminate the response (26). Therefore, the NOS, adenosine, and EET pathways do not appear to operate by parallel, independent signaling mechanisms, consistent with an interplay at the astrocyte or smooth muscle level.
Indeed, none of the combinations of drug inhibitors and antagonists has been able to completely eliminate the increase in CBF during activation. These findings suggest that other regulatory pathways are recruited when these feed-forward signaling pathways are interrupted. In this regard, it is important to recall that CO2 production is proportional to glucose consumption and that the percent increase in CBF matches the percent increase in glucose consumption. Ordinarily, the match between increased glycolysis and increased CBF results in no measurable change in tissue pH (110). When the feed-forward signaling pathways are interrupted and the CBF response is attenuated, some increase in tissue PCO2 would be anticipated. A consequent decrease in extracellular pH might represent a contingency feedback mechanism for ensuring some degree of vasodilation during activation when other pathways are inoperative.
EXTRAPARENCHYMAL PIAL ARTERIOLES
The discussion thus far has centered on intraparenchymal arterioles and CBF. An adequate parenchymal flow response requires ascending dilation of pial arterioles to maintain intravascular pressure in intraparenchymal feeding arterioles. Mechanisms of ascending vasodilation have not been as well studied in the cerebral circulation as in the skeletal muscle circulation. Pial arterioles receive some innervation from sensory ganglia but not from local cortical neurons. Flow-induced dilation might contribute to ascending dilation of intraparenchymal arterioles, but this mechanism is not thought to be important in pial arterioles where estimated wall shear was unchanged during sciatic nerve stimulation (81). Neuronal NO can also signal pial arterioles. Application of NMDA to the brain surface elicits pial arteriolar dilation that is neuronal NOS dependent (29). Because NOS inhibition reduces pial arteriolar dilation during sciatic nerve stimulation (80), NO derived from neurons might diffuse to surface arterioles and elicit dilation. However, there is some question of whether the diminished dilation is the result of diminished neuronal activation as reflected by a decrease in evoked potentials (79). The pial arteriolar response to sciatic nerve stimulation is reduced by theophylline and by an adenosine A2A antagonist (58, 70). Thus, in contrast to the role of A2B receptors in the intraparenchymal flow response, high-affinity A2A receptors appear to be more important for extraparenchymal arterioles.
Furthermore, NOS-containing neurons are rare in layer 1 of underlying cortex (113). Astrocytes might serve as communicators between cortical NOS neurons and pial arterioles. For example, pial arteriolar dilation to hypercapnia is partially dependent on neuronal NOS activity in rodent brain (51). When superficial astrocytes in the glia limitans are injured after exposure to the gliotoxin L-α-aminoadipic acid, pial arteriolar dilation to hypercapnia becomes attenuated to the same extent as it does with NOS inhibition alone, and NOS inhibition after glial injury has no further effect on hypercapnic dilation (117). Pial arterioles retained normal reactivity to extracellular acidity and to NO donors. Knockdown of connexin-43 had a similar effect as did gliotoxin, suggesting a role for gap-junction communication in the hypercapnic response. In female rats, the glia limitans also plays a role in pial arteriolar dilation to ADP (118), which is not dependent on neuronal NOS. Therefore, the limited amount of currently available information indicates the potential for astrocytes to participate in regulation of extraparenchymal arteriole resistance vessels.
ASTROCYTE FUNCTION IN PATHOPHYSIOLOGICAL STATES
Astrocytes can influence cerebrovascular regulation in pathophysiological states, such as hyperammonemia associated with liver disease. Astrocytes are enriched in glutamine synthetase which forms glutamine from ammonia and glutamate (22, 85). During hyperammonemia, increases in tissue glutamine result in depressed cerebrovascular reactivity to CO2 and acetylcholine due, in part, to glutamine’s interference with NOS activity (46, 47, 57, 86, 108). Astrocyte swelling and increased extracellular K+ activity related to the osmotic effect of glutamine accumulation may also contribute to impaired vascular reactivity (106, 109, 116).
Severe cerebral ischemia results in loss of transcellular ionic gradients and uptake of water. After reperfusion, astrocytes may remain swollen, retain excess K+, and have disturbed intracellular signaling associated with altered Ca2+ stores. The evoked blood flow and glycolytic responses to neuronal activation can remain depressed for hours or days after ischemia despite recovery of basal energy metabolism and evoked potentials (25, 27, 110). The depressed responses may be related to persistent dendritic injury in postsynaptic membranes, persistent astrocyte dysfunction, or impaired signaling in the NOS, COX-2, adenosine, or EETs pathway. The impact of other pathophysiological states on neurovascular coupling is reviewed elsewhere in this review series (50).
SUMMARY OF NEUROVASCULAR SIGNALING
The following hypothesis integrates much of the current evidence for the control of intraparenchymal arterioles in cerebral cortex during neuronal activation (Fig. 3). Glutamate released at excitatory synapses stimulates mGluR on astrocytes and stimulates PLC and inositol triphosphate to release Ca2+ from intracellular stores. Adenosine, formed as a breakdown product of ATP either inside cells or from ecto-ATPase and ecto-5′-nucleotidase acting on ATP released as a coneurotransmitter or by astrocyte connexin hemichannels, acts on astrocyte A2B receptors to amplify the increase in intracellular Ca2+. The increase in Ca2+ activates astrocytic KCa channels, in part by an EET-dependent mechanism, to limit astrocyte depolarization during Ca2+ entry and thereby permit additional Ca2+ influx to amplify the spread of the Ca2+ wave to perivascular end-feet and possibly to neighboring astrocytes.
Fig. 3.
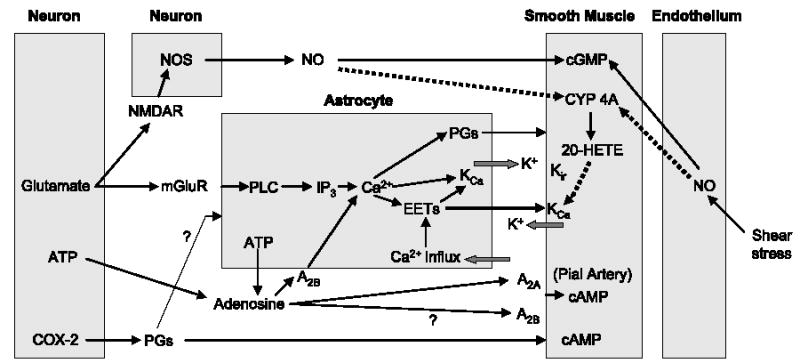
Block diagram illustrating potential signaling pathways of neurovascular coupling and their interactions among neuronal, astrocyte, vascular smooth muscle, and endothelial compartments. Negative control pathways are indicated by dotted lines. cAMP, cyclic AMP; cGMP, cyclic GMP; CYP 4A, cytochrome P450 4A; EETs, epoxyeicosatrienoic acids; 20-HETE, 20-hydroxyeicosatetraenoic acid; IP3, inositol triphosphate; KCa, calcium-sensitive potassium channels; Kir, inward-rectifier potassium channel; mGluR, metabotropic glutamate receptor; NMDAR, N-methyl-D-aspartate receptor; NO, nitric oxide; NOS, nitric oxide synthase; PGs, prostaglandins; PLC, phospholipase C.
The increase in Ca2+ in astrocyte end-feet might have several possible consequences. First, Ca2+ might stimulate PLA2 and mobilize arachidonic acid, which then 1) can be used for synthesis of EETs and for replenishing stored EETs, 2) can be used as a substrate for COX and release of PGE2, or 3) can be transported directly to the smooth muscle and be used as a substrate for 20-HETE synthesis. Second, increased Ca2+ might trigger release of stored EETs from the astrocyte end-feet phospholipid membrane, and the released EETs could then act on smooth muscle KCa channels to produce hyperpolarization and dilation. Third, Ca2+ activation of astrocyte KCa channels will release K+ into the interstitial space, which might be sufficient to hyperpolarize smooth muscle via smooth muscle Kir channels.
Neuronally derived NO may exert several actions. First, increased NO might ensure a sufficient level of smooth muscle cGMP necessary for activation of K+ channels. Second, increased NO will inhibit CYP ω-hydroxylase synthesis of 20-HETE and thereby ensure that 20-HETE does not counteract the opening of vascular KCa channels by EETs or other mediators. Third, NO might help amplify astrocyte Ca2+ influx (61). Furthermore, PGs released from neurons might also act indirectly on astrocyte signaling to evoke vasodilation or act directly on vascular smooth muscle. Lastly, blockage of all of these mediators reduces the magnitude of the vasodilation, but not completely, suggesting a complex interaction among factors mediating activity-induced vasodilation.
In summary, evidence to date implicates an important role of astrocytes in the regulation of the cerebral circulation to physiological stimuli, such as neuronal activation. Astrocytes appear to be capable of monitoring changes in synaptic activity and of spatially integrating this information for signaling microvascular units. However, much of the evidence supporting a role of astrocytes is based on work in vitro. Future studies using more sophisticated molecular approaches and imaging techniques will need to be performed in this expanding field of research to allow better understanding of the role of astrocytes in vivo. Thus many questions remain for future research (Table 1). Future work on the role of astrocytes as a key participant of neurovascular coupling is likely to reveal added complexity of the interrelationships within the neurovascular unit.
Table 1.
Questions for future research on astrocyte function in neurovascular coupling
|
COX, cyclooxygenase; EET, epoxyeicosatrienoic acid; NO, nitric oxide; KCa channels, calcium-sensitive potassium channels.
Acknowledgments
The authors express gratitude to Tzipora Sofare for editorial assistance in preparing this manuscript.
Footnotes
GRANTS
The authors’ work is supported by a grant from the National Heart, Lung, and Blood Institute (HL-59996).
References
- 1.Alkayed NJ, Birks EK, Narayanan J, Petrie KA, Kohler-Cabot AE, Harder DR. Role of P-450 arachidonic acid epoxygenase in the response of cerebral blood flow to glutamate in rats. Stroke. 1997;28:1066–1072. doi: 10.1161/01.str.28.5.1066. [DOI] [PubMed] [Google Scholar]
- 2.Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke. 1996;27:971–979. doi: 10.1161/01.str.27.5.971. [DOI] [PubMed] [Google Scholar]
- 3.Allaman I, Lengacher S, Magistretti PJ, Pellerin L. A2B receptor activation promotes glycogen synthesis in astrocytes through modulation of gene expression. Am J Physiol Cell Physiol. 2003;284:C696–C704. doi: 10.1152/ajpcell.00202.2002. [DOI] [PubMed] [Google Scholar]
- 4.Alloisio S, Cugnoli C, Ferroni S, Nobile M. Differential modulation of ATP-induced calcium signalling by A1 and A2 adenosine receptors in cultured cortical astrocytes. Br J Pharmacol. 2004;141:935–942. doi: 10.1038/sj.bjp.0705707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alonso-Galicia M, Hudetz AG, Shen H, Harder DR, Roman RJ. Contribution of 20-HETE to vasodilator actions of nitric oxide in the cerebral microcirculation. Stroke. 1999;30:2727–2734. doi: 10.1161/01.str.30.12.2727. [DOI] [PubMed] [Google Scholar]
- 6.Amruthesh SC, Boerschel MF, McKinney JS, Willoughby KA, Ellis EF. Metabolism of arachidonic acid to epoxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and prostaglandins in cultured rat hippocampal astrocytes. J Neurochem. 1993;61:150–159. doi: 10.1111/j.1471-4159.1993.tb03550.x. [DOI] [PubMed] [Google Scholar]
- 7.Amruthesh SC, Falck JR, Ellis EF. Brain synthesis and cerebrovascular action of epoxygenase metabolites of arachidonic acid. J Neurochem. 1992;58:503–510. doi: 10.1111/j.1471-4159.1992.tb09749.x. [DOI] [PubMed] [Google Scholar]
- 8.Ances BM. Coupling of changes in cerebral blood flow with neural activity: what must initially dip must come back up. J Cereb Blood Flow Metab. 2004;24:1–6. doi: 10.1097/01.WCB.0000103920.96801.12. [DOI] [PubMed] [Google Scholar]
- 9.Ances BM, Buerk DG, Greenberg JH, Detre JA. Temporal dynamics of the partial pressure of brain tissue oxygen during functional forepaw stimulation in rats. Neurosci Lett. 2001;306:106–110. doi: 10.1016/s0304-3940(01)01868-7. [DOI] [PubMed] [Google Scholar]
- 10.Ances BM, Greenberg JH, Detre JA, Dietrich WD. Acute carotid occlusion alters the activation flow coupling response to forepaw stimulation in a rat model. Stroke. 2000;31:955–960. doi: 10.1161/01.str.31.4.955. [DOI] [PubMed] [Google Scholar]
- 11.Arcuino G, Lin JH, Takano T, Liu C, Jiang L, Gao Q, Kang J, Nedergaard M. Intercellular calcium signaling mediated by point-source burst release of ATP. Proc Natl Acad Sci USA. 2002;99:9840–9845. doi: 10.1073/pnas.152588599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayata C, Ma J, Meng W, Huang P, Moskowitz MA. L-NA-sensitive rCBF augmentation during vibrissal stimulation in type III nitric oxide synthase mutant mice. J Cereb Blood Flow Metab. 1996;16:539–541. doi: 10.1097/00004647-199607000-00002. [DOI] [PubMed] [Google Scholar]
- 13.Bernardinelli Y, Magistretti PJ, Chatton JY. Astrocytes generate Na+-mediated metabolic waves. Proc Natl Acad Sci USA. 2004;101:14937–14942. doi: 10.1073/pnas.0405315101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhardwaj A, Northington FJ, Carhuapoma JR, Falck JR, Harder DR, Traystman RJ, Koehler RC. P-450 epoxygenase and NO synthase inhibitors reduce cerebral blood flow response to N-methyl-D-aspartate. Am J Physiol Heart Circ Physiol. 2000;279:H1616–H1624. doi: 10.1152/ajpheart.2000.279.4.H1616. [DOI] [PubMed] [Google Scholar]
- 15.Brown LA, Key BJ, Lovick TA. Inhibition of vasomotion in hippocampal cerebral arterioles during increases in neuronal activity. Auton Neurosci. 2002;95:137–140. doi: 10.1016/s1566-0702(01)00395-2. [DOI] [PubMed] [Google Scholar]
- 16.Buerk DG, Ances BM, Greenberg JH, Detre JA. Temporal dynamics of brain tissue nitric oxide during functional forepaw stimulation in rats. Neuroimage. 2003;18:1–9. doi: 10.1006/nimg.2002.1314. [DOI] [PubMed] [Google Scholar]
- 17.Buxton RB, Frank LR. A model for the coupling between cerebral blood flow and oxygen metabolism during neural stimulation. J Cereb Blood Flow Metab. 1997;17:64–72. doi: 10.1097/00004647-199701000-00009. [DOI] [PubMed] [Google Scholar]
- 18.Cambj-Sapunar L, Yu M, Harder DR, Roman RJ. Contribution of 5-hydroxytryptamine1B receptors and 20-hydroxyeiscosatetraenoic acid to fall in cerebral blood flow after subarachnoid hemorrhage. Stroke. 2003;34:1269–1275. doi: 10.1161/01.STR.0000065829.45234.69. [DOI] [PubMed] [Google Scholar]
- 19.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 20.Cholet N, Bonvento G, Seylaz J. Effect of neuronal NO synthase inhibition on the cerebral vasodilatory response to somatosensory stimulation. Brain Res. 1996;708:197–200. doi: 10.1016/0006-8993(95)01387-3. [DOI] [PubMed] [Google Scholar]
- 21.Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- 22.Cooper AJ, McDonald JM, Gelbard AS, Gledhill RF, Duffy TE. The metabolic fate of 13N-labeled ammonia in rat brain. J Biol Chem. 1979;254:4982–4992. [PubMed] [Google Scholar]
- 23.Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- 24.Degi R, Bari F, Beasley TC, Thrikawala N, Thore C, Louis TM, Busija DW. Regional distribution of prostaglandin H synthase-2 and neuronal nitric oxide synthase in piglet brain. Pediatr Res. 1998;43:683–689. doi: 10.1203/00006450-199805000-00018. [DOI] [PubMed] [Google Scholar]
- 25.Dietrich WD, Ginsberg MD, Busto R. Effect of transient cerebral ischemia on metabolic activation of a somatosensory circuit. J Cereb Blood Flow Metab. 1986;6:405–413. doi: 10.1038/jcbfm.1986.73. [DOI] [PubMed] [Google Scholar]
- 26.Dirnagl U, Niwa K, Lindauer U, Villringer A. Coupling of cerebral blood flow to neuronal activation: role of adenosine and nitric oxide. Am J Physiol Heart Circ Physiol. 1994;267:H296–H301. doi: 10.1152/ajpheart.1994.267.1.H296. [DOI] [PubMed] [Google Scholar]
- 27.Duckrow RB, LaManna JS, Rosenthal M. Disparate recovery of resting and stimulated oxidative metabolism following transient ischemia. Stroke. 1981;12:677–686. doi: 10.1161/01.str.12.5.677. [DOI] [PubMed] [Google Scholar]
- 28.Ellis EF, Police RJ, Yancey L, McKinney JS, Amruthesh SC. Dilation of cerebral arterioles by cytochrome P-450 metabolites of arachidonic acid. Am J Physiol Heart Circ Physiol. 1990;259:H1171–H1177. doi: 10.1152/ajpheart.1990.259.4.H1171. [DOI] [PubMed] [Google Scholar]
- 29.Faraci FM, Brian JE., Jr 7-Nitroindazole inhibits brain nitric oxide synthase and cerebral vasodilation in response to N-methyl-D-aspartate. Stroke. 1995;26:2172–2176. doi: 10.1161/01.str.26.11.2172. [DOI] [PubMed] [Google Scholar]
- 30.Fellin T, Carmignoto G. Neurone-to-astrocyte signalling in the brain represents a distinct multifunctional unit. J Physiol. 2004;559:3–15. doi: 10.1113/jphysiol.2004.063214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filosa JA, Bonev A, Nelson MT. Potassium ion as the signal in neurovascular coupling (Abstract) FASEB J. 2005;19:A1257. [Google Scholar]
- 32.Filosa JA, Bonev AD, Nelson MT. Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circ Res. 2004;95:e73–e81. doi: 10.1161/01.RES.0000148636.60732.2e. [DOI] [PubMed] [Google Scholar]
- 33.Fox PT, Raichle ME. Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc Natl Acad Sci USA. 1986;83:1140–1144. doi: 10.1073/pnas.83.4.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fox PT, Raichle ME, Mintun MA, Dence C. Nonoxidative glucose consumption during focal physiologic neural activity. Science. 1988;241:462–464. doi: 10.1126/science.3260686. [DOI] [PubMed] [Google Scholar]
- 35.Fujita H, Kuwabara H, Reutens DC, Gjedde A. Oxygen consumption of cerebral cortex fails to increase during continued vibrotactile stimulation. J Cereb Blood Flow Metab. 1999;19:266–271. doi: 10.1097/00004647-199903000-00004. [DOI] [PubMed] [Google Scholar]
- 36.Gebremedhin D, Lange AR, Lowry TF, Taheri MR, Birks EK, Hudetz AG, Narayanan J, Falck JR, Okamoto H, Roman RJ, Nithipatikom K, Campbell WB, Harder DR. Production of 20-HETE and its role in autoregulation of cerebral blood flow. Circ Res. 2000;87:60–65. doi: 10.1161/01.res.87.1.60. [DOI] [PubMed] [Google Scholar]
- 37.Gebremedhin D, Ma YH, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Physiol Heart Circ Physiol. 1992;263:H519–H525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- 38.Gebremedhin D, Narayanan J, Harder DR. Role of astrocytic KCa channel openings in the glutamate receptor mediated release of EETs from rat brain astrocytes (Abstract) FASEB J. 2005;19:A1257. [Google Scholar]
- 39.Gebremedhin D, Yamaura K, Zhang C, Bylund J, Koehler RC, Harder DR. Metabotropic glutamate receptor activation enhances the activities of two types of Ca2+-activated K+ channels in rat hippocampal astrocytes. J Neurosci. 2003;23:1678–1687. doi: 10.1523/JNEUROSCI.23-05-01678.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerrits RJ, Stein EA, Greene AS. Ca2+-activated potassium (KCa) channel inhibition decreases neuronal activity-blood flow coupling. Brain Res. 2002;948:108–116. doi: 10.1016/s0006-8993(02)02957-8. [DOI] [PubMed] [Google Scholar]
- 41.Gotoh J, Kuang TY, Nakao Y, Cohen DM, Melzer P, Itoh Y, Pak H, Pettigrew K, Sokoloff L. Regional differences in mechanisms of cerebral circulatory response to neuronal activation. Am J Physiol Heart Circ Physiol. 2001;280:H821–H829. doi: 10.1152/ajpheart.2001.280.2.H821. [DOI] [PubMed] [Google Scholar]
- 42.Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. doi: 10.1152/japplphysiol.00954.2005. In press. [DOI] [PubMed] [Google Scholar]
- 43.Hansson E, Ronnback L. Glial neuronal signaling in the central nervous system. FASEB J. 2003;17:341–348. doi: 10.1096/fj.02-0429rev. [DOI] [PubMed] [Google Scholar]
- 44.Harder DR, Alkayed NJ, Lange AR, Gebremedhin D, Roman RJ. Functional hyperemia in the brain. Hypothesis for astrocyte-derived vasodilator metabolites. Stroke. 1998;28:229–234. doi: 10.1161/01.str.29.1.229. [DOI] [PubMed] [Google Scholar]
- 45.Haydon PG. Glia: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- 46.Hirata T, Kawaguchi T, Brusilow SW, Traystman RJ, Koehler RC. Preserved hypocapnic pial arteriolar constriction during hyperammonemia by glutamine synthetase inhibition. Am J Physiol Heart Circ Physiol. 1999;276:H456–H463. doi: 10.1152/ajpheart.1999.276.2.H456. [DOI] [PubMed] [Google Scholar]
- 47.Hirata T, Koehler RC, Kawaguchi T, Brusilow SW, Traystman RJ. Impaired pial arteriolar reactivity to hypercapnia during hyperammonemia depends on glutamine synthesis. Stroke. 1996;27:729–736. doi: 10.1161/01.str.27.4.729. [DOI] [PubMed] [Google Scholar]
- 48.Hu Y, Wilson GS. A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J Neurochem. 1997;69:1484–1490. doi: 10.1046/j.1471-4159.1997.69041484.x. [DOI] [PubMed] [Google Scholar]
- 49.Hyder F, Shulman RG, Rothman DL. A model for the regulation of cerebral oxygen delivery. J Appl Physiol. 1998;85:554–564. doi: 10.1152/jappl.1998.85.2.554. [DOI] [PubMed] [Google Scholar]
- 50.Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100:328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- 51.Iadecola C, Zhang F. Permissive and obligatory roles of NO in cerebrovascular responses to hypercapnia and acetylcholine. Am J Physiol Regul Integr Comp Physiol. 1996;271:R990–R1001. doi: 10.1152/ajpregu.1996.271.4.R990. [DOI] [PubMed] [Google Scholar]
- 52.Ido Y, Chang K, Woolsey TA, Williamson JR. NADH: sensor of blood flow need in brain, muscle, and other tissues. FASEB J. 2001;15:1419–1421. doi: 10.1096/fj.00-0652fje. [DOI] [PubMed] [Google Scholar]
- 53.Iliff JJ, D’Ambrosio R, Ngai AC, Winn HR. Adenosine receptors mediate glutamate-evoked arteriolar dilation in the rat cerebral cortex. Am J Physiol Heart Circ Physiol. 2003;284:H1631–H1637. doi: 10.1152/ajpheart.00909.2002. [DOI] [PubMed] [Google Scholar]
- 54.Jimenez AI, Castro E, Mirabet M, Franco R, Delicado EG, Miras-Portugal MT. Potentiation of ATP calcium responses by A2B receptor stimulation and other signals coupled to Gs proteins in type-1 cerebellar astrocytes. Glia. 1999;26:119–128. [PubMed] [Google Scholar]
- 55.Joseph SM, Buchakjian MR, Dubyak GR. Colocalization of ATP release sites and ecto-ATPase activity at the extracellular surface of human astrocytes. J Biol Chem. 2003;278:23331–23342. doi: 10.1074/jbc.M302680200. [DOI] [PubMed] [Google Scholar]
- 56.Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- 57.Kawaguchi T, Brusilow SW, Traystman RJ, Koehler RC. Glutamine-dependent inhibition of pial arteriolar dilation to acetylcholine with and without hyperammonemia in the rat. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1612–R1619. doi: 10.1152/ajpregu.00783.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ko KR, Ngai AC, Winn HR. Role of adenosine in regulation of regional cerebral blood flow in sensory cortex. Am J Physiol Heart Circ Physiol. 1990;259:H1703–H1708. doi: 10.1152/ajpheart.1990.259.6.H1703. [DOI] [PubMed] [Google Scholar]
- 59.Lauritzen M. Reading vascular changes in brain imaging: is dendritic calcium the key? Nat Rev Neurosci. 2005;6:77–85. doi: 10.1038/nrn1589. [DOI] [PubMed] [Google Scholar]
- 60.Leffler CW, Fedinec AL. Newborn piglet cerebral microvascular responses to epoxyeicosatrienoic acids. Am J Physiol Heart Circ Physiol. 1997;273:H333–H338. doi: 10.1152/ajpheart.1997.273.1.H333. [DOI] [PubMed] [Google Scholar]
- 61.Li N, Sul JY, Haydon PG. A calcium-induced calcium influx factor, nitric oxide, modulates the refilling of calcium stores in astrocytes. J Neurosci. 2003;23:10302–10310. doi: 10.1523/JNEUROSCI.23-32-10302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lindauer U, Gethmann J, Kuhl M, Kohl-Bareis M, Dirnagl U. Neuronal activity-induced changes of local cerebral microvascular blood oxygenation in the rat: effect of systemic hyperoxia or hypoxia. Brain Res. 2003;975:135–140. doi: 10.1016/s0006-8993(03)02602-7. [DOI] [PubMed] [Google Scholar]
- 63.Lindauer U, Megow D, Matsuda H, Dirnagl U. Nitric oxide: a modulator, but not a mediator, of neurovascular coupling in rat somatosensory cortex. Am J Physiol Heart Circ Physiol. 1999;277:H799–H811. doi: 10.1152/ajpheart.1999.277.2.H799. [DOI] [PubMed] [Google Scholar]
- 64.Logothetis NK. The underpinnings of the BOLD functional magnetic resonance imaging signal. J Neurosci. 2003;23:3963–3971. doi: 10.1523/JNEUROSCI.23-10-03963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma J, Ayata C, Huang PL, Fishman MC, Moskowitz MA. Regional cerebral blood flow response to vibrissal stimulation in mice lacking type I NOS gene expression. Am J Physiol Heart Circ Physiol. 1996;270:H1085–H1090. doi: 10.1152/ajpheart.1996.270.3.H1085. [DOI] [PubMed] [Google Scholar]
- 66.Madsen PL, Cruz NF, Sokoloff L, Dienel GA. Cerebral oxygen/glucose ratio is low during sensory stimulation and rises above normal during recovery: excess glucose consumption during stimulation is not accounted for by lactate efflux from or accumulation in brain tissue. J Cereb Blood Flow Metab. 1999;19:393–400. doi: 10.1097/00004647-199904000-00005. [DOI] [PubMed] [Google Scholar]
- 67.Madsen PL, Hasselbalch SG, Hagemann LP, Olsen KS, Bulow J, Holm S, Wildschiodtz G, Paulson OB, Lassen NA. Persistent resetting of the cerebral oxygen/glucose uptake ratio by brain activation: evidence obtained with the Kety-Schmidt technique. J Cereb Blood Flow Metab. 1995;15:485–491. doi: 10.1038/jcbfm.1995.60. [DOI] [PubMed] [Google Scholar]
- 68.Martindale J, Berwick J, Martin C, Kong Y, Zheng Y, Mayhew J. Long duration stimuli and nonlinearities in the neural-haemodynamic coupling. J Cereb Blood Flow Metab. 2005;25:651–661. doi: 10.1038/sj.jcbfm.9600060. [DOI] [PubMed] [Google Scholar]
- 69.Melani A, Turchi D, Vannucchi MG, Cipriani S, Gianfriddo M, Pedata F. ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem Int. 2005;47:442–448. doi: 10.1016/j.neuint.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 70.Meno JR, Crum AV, Winn HR. Effect of adenosine receptor blockade on pial arteriolar dilation during sciatic nerve stimulation. Am J Physiol Heart Circ Physiol. 2001;281:H2018–H2027. doi: 10.1152/ajpheart.2001.281.5.H2018. [DOI] [PubMed] [Google Scholar]
- 71.Meno JR, Nguyen TS, Jensen EM, Alexander WG, Groysman L, Kung DK, Ngai AC, Britz GW, Winn HR. Effect of caffeine on cerebral blood flow response to somatosensory stimulation. J Cereb Blood Flow Metab. 2005;25:775–784. doi: 10.1038/sj.jcbfm.9600075. [DOI] [PubMed] [Google Scholar]
- 72.Mintun MA, Vlassenko AG, Rundle MM, Raichle ME. Increased lactate/pyruvate ratio augments blood flow in physiologically activated human brain. Proc Natl Acad Sci USA. 2004;101:659–664. doi: 10.1073/pnas.0307457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mukamel R, Gelbard H, Arieli A, Hasson U, Fried I, Malach R. Coupling between neuronal firing, field potentials, and FMRI in human auditory cortex. Science. 2005;309:951–954. doi: 10.1126/science.1110913. [DOI] [PubMed] [Google Scholar]
- 74.Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- 75.Newman EA. High potassium conductance in astrocyte endfeet. Science. 1986;233:453–454. doi: 10.1126/science.3726539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Newman EA. Calcium increases in retinal glial cells evoked by light-induced neuronal activity. J Neurosci. 2005;25:5502–5510. doi: 10.1523/JNEUROSCI.1354-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Newman EA, Frambach DA. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225:1174–1175. doi: 10.1126/science.6474173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ngai AC, Coyne EF, Meno JR, West GA, Winn HR. Receptor subtypes mediating adenosine-induced dilation of cerebral arterioles. Am J Physiol Heart Circ Physiol. 2001;280:H2329–H2335. doi: 10.1152/ajpheart.2001.280.5.H2329. [DOI] [PubMed] [Google Scholar]
- 79.Ngai AC, Meno JR, Jolley MA, Winn HR. Suppression of somatosensory evoked potentials by nitric oxide synthase inhibition in rats: methodological differences. Neurosci Lett. 1998;245:171–174. doi: 10.1016/s0304-3940(98)00213-4. [DOI] [PubMed] [Google Scholar]
- 80.Ngai AC, Meno JR, Winn HR. L-NNA suppresses cerebrovascular response and evoked potentials during somatosensory stimulation in rats. Am J Physiol Heart Circ Physiol. 1995;269:H1803–H1810. doi: 10.1152/ajpheart.1995.269.5.H1803. [DOI] [PubMed] [Google Scholar]
- 81.Ngai AC, Winn HR. Estimation of shear and flow rates in pial arterioles during somatosensory stimulation. Am J Physiol Heart Circ Physiol. 1996;270:H1712–H1717. doi: 10.1152/ajpheart.1996.270.5.H1712. [DOI] [PubMed] [Google Scholar]
- 82.Niessing J, Ebisch B, Schmidt KE, Niessing M, Singer W, Galuske RA. Hemodynamic signals correlate tightly with synchronized gamma oscillations. Science. 2005;309:948–951. doi: 10.1126/science.1110948. [DOI] [PubMed] [Google Scholar]
- 83.Niwa K, Araki E, Morham SG, Ross ME, Iadecola C. Cyclooxygenase-2 contributes to functional hyperemia in whisker-barrel cortex. J Neurosci. 2000;20:763–770. doi: 10.1523/JNEUROSCI.20-02-00763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res. 2001;88:600–608. doi: 10.1161/01.res.88.6.600. [DOI] [PubMed] [Google Scholar]
- 85.Norenberg MD, Martinez-Hernandez A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979;161:303–310. doi: 10.1016/0006-8993(79)90071-4. [DOI] [PubMed] [Google Scholar]
- 86.Okada T, Watanabe Y, Brusilow SW, Traystman RJ, Koehler RC. Interaction of glutamine and arginine on cerebrovascular reactivity to hypercapnia. Am J Physiol Heart Circ Physiol. 2000;278:H1577–H1584. doi: 10.1152/ajpheart.2000.278.5.H1577. [DOI] [PubMed] [Google Scholar]
- 87.Paulson OB, Newman EA. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science. 1987;237:896–897. doi: 10.1126/science.3616619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peakman MC, Hill SJ. Adenosine A2B-receptor-mediated cyclic AMP accumulation in primary rat astrocytes. Br J Pharmacol. 1994;111:191–198. doi: 10.1111/j.1476-5381.1994.tb14043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pellerin L, Magistretti PJ. Neuroenergetics: calling upon astrocytes to satisfy hungry neurons. Neuroscientist. 2004;10:53–62. doi: 10.1177/1073858403260159. [DOI] [PubMed] [Google Scholar]
- 90.Peng X, Carhuapoma JR, Bhardwaj A, Alkayed NJ, Falck JR, Harder DR, Traystman RJ, Koehler RC. Suppression of cortical functional hyperemia to vibrissal stimulation in the rat by epoxygenase inhibitors. Am J Physiol Heart Circ Physiol. 2002;283:H2029–H2037. doi: 10.1152/ajpheart.01130.2000. [DOI] [PubMed] [Google Scholar]
- 91.Peng X, Zhang C, Alkayed NJ, Harder DR, Koehler RC. Dependency of cortical functional hyperemia to forepaw stimulation on epoxygenase and nitric oxide synthase activities in rats. J Cereb Blood Flow Metab. 2004;24:509–517. doi: 10.1097/00004647-200405000-00004. [DOI] [PubMed] [Google Scholar]
- 92.Pilitsis JG, Kimelberg HK. Adenosine receptor mediated stimulation of intracellular calcium in acutely isolated astrocytes. Brain Res. 1998;798:294–303. doi: 10.1016/s0006-8993(98)00430-2. [DOI] [PubMed] [Google Scholar]
- 93.Price DL, Ludwig JW, Mi H, Schwarz TL, Ellisman MH. Distribution of rSlo Ca2+-activated K+ channels in rat astrocyte perivascular endfeet. Brain Res. 2002;956:183–193. doi: 10.1016/s0006-8993(02)03266-3. [DOI] [PubMed] [Google Scholar]
- 94.Prichard J, Rothman D, Novotny E, Petroff O, Kuwabara T, Avison M, Howseman A, Hanstock C, Shulman R. Lactate rise detected by 1H NMR in human visual cortex during physiologic stimulation. Proc Natl Acad Sci USA. 1991;88:5829–5831. doi: 10.1073/pnas.88.13.5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Raichle ME. Behind the scenes of functional brain imaging: a historical and physiological perspective. Proc Natl Acad Sci USA. 1998;95:765–772. doi: 10.1073/pnas.95.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J Neurophysiol. 1995;73:333–346. doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- 97.Rosengarten B, Lutz H, Hossmann KA. A control system approach for evaluating somatosensory activation by laser-Doppler flow-metry in the rat cortex. J Neurosci Methods. 2003;130:75–81. doi: 10.1016/s0165-0270(03)00209-7. [DOI] [PubMed] [Google Scholar]
- 98.Rzigalinski BA, Willoughby KA, Hoffman SW, Falck JR, Ellis EF. Calcium influx factor, further evidence it is 5,6-epoxyeicosatrienoic acid. J Biol Chem. 1999;274:175–182. doi: 10.1074/jbc.274.1.175. [DOI] [PubMed] [Google Scholar]
- 99.Saez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MV. Connexin-based gap junction hemichannels: gating mechanisms. Biochim Biophys Acta. 2005;1711:215–224. doi: 10.1016/j.bbamem.2005.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shi Y, Harder DR, Koehler RC. Contribution of adenosine A2B receptors and epoxyeicosatrienoic acids to neurovascular coupling during whisker stimulation. Soc Neurosci Abstr. 2004;429.4 [Google Scholar]
- 101.Shin HK, Shin YW, Hong KW. Role of adenosine A2B receptors in vasodilation of rat pial artery and cerebral blood flow autoregulation. Am J Physiol Heart Circ Physiol. 2000;278:H339–H344. doi: 10.1152/ajpheart.2000.278.2.H339. [DOI] [PubMed] [Google Scholar]
- 102.Shivachar AC, Willoughby KA, Ellis EF. Effect of protein kinase C modulators on 14,15-epoxyeicosatrienoic acid incorporation into astroglial phospholipids. J Neurochem. 1995;65:338–346. doi: 10.1046/j.1471-4159.1995.65010338.x. [DOI] [PubMed] [Google Scholar]
- 103.Shukla V, Zimmermann H, Wang L, Kettenmann H, Raab S, Hammer K, Sevigny J, Robson SC, Braun N. Functional expression of the ecto-ATPase NTPDase2 and of nucleotide receptors by neuronal progenitor cells in the adult murine hippocampus. J Neurosci Res. 2005;80:600–610. doi: 10.1002/jnr.20508. [DOI] [PubMed] [Google Scholar]
- 104.Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 106.Sugimoto H, Koehler RC, Wilson DA, Brusilow SW, Traystman RJ. Methionine sulfoximine, a glutamine synthetase inhibitor, attenuates increased extracellular potassium activity during acute hyperammonemia. J Cereb Blood Flow Metab. 1997;17:44–49. doi: 10.1097/00004647-199701000-00006. [DOI] [PubMed] [Google Scholar]
- 107.Sun CW, Falck JR, Okamoto H, Harder DR, Roman RJ. Role of cGMP versus 20-HETE in the vasodilator response to nitric oxide in rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2000;279:H339–H350. doi: 10.1152/ajpheart.2000.279.1.H339. [DOI] [PubMed] [Google Scholar]
- 108.Takahashi H, Koehler RC, Hirata T, Brusilow SW, Traystman RJ. Restoration of cerebrovascular CO2 responsivity by glutamine synthesis inhibition in hyperammonemic rats. Circ Res. 1992;71:1220–1230. doi: 10.1161/01.res.71.5.1220. [DOI] [PubMed] [Google Scholar]
- 109.Tanigami H, Rebel A, Martin LJ, Chen TY, Brusilow SW, Traystman RJ, Koehler RC. Effect of glutamine synthetase inhibition on astrocyte swelling and altered astroglial protein expression during hyperammonemia in rats. Neuroscience. 2005;131:437–449. doi: 10.1016/j.neuroscience.2004.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ueki M, Linn F, Hossmann KA. Functional activation of cerebral blood flow and metabolism before and after global ischemia of rat brain. J Cereb Blood Flow Metab. 1988;8:486–494. doi: 10.1038/jcbfm.1988.89. [DOI] [PubMed] [Google Scholar]
- 111.Vafaee MS, Meyer E, Marrett S, Paus T, Evans AC, Gjedde A. Frequency-dependent changes in cerebral metabolic rate of oxygen during activation of human visual cortex. J Cereb Blood Flow Metab. 1999;19:272–277. doi: 10.1097/00004647-199903000-00005. [DOI] [PubMed] [Google Scholar]
- 112.Vanzetta I, Grinvald A. Increased cortical oxidative metabolism due to sensory stimulation: implications for functional brain imaging. Science. 1999;286:1555–1558. doi: 10.1126/science.286.5444.1555. [DOI] [PubMed] [Google Scholar]
- 113.Wang H, Hitron IM, Iadecola C, Pickel VM. Synaptic and vascular associations of neurons containing cyclooxygenase-2 and nitric oxide synthase in rat somatosensory cortex. Cereb Cortex. 2005;15:1250–1260. doi: 10.1093/cercor/bhi008. [DOI] [PubMed] [Google Scholar]
- 114.Wang TF, Guidotti G. Widespread expression of ecto-apyrase (CD39) in the central nervous system. Brain Res. 1998;790:318–322. doi: 10.1016/s0006-8993(97)01562-x. [DOI] [PubMed] [Google Scholar]
- 115.White RP, Hindley C, Bloomfield PM, Cunningham VJ, Vallance P, Brooks DJ, Markus HS. The effect of the nitric oxide synthase inhibitor L-NMMA on basal CBF and vasoneuronal coupling in man: a PET study. J Cereb Blood Flow Metab. 1999;19:673–678. doi: 10.1097/00004647-199906000-00011. [DOI] [PubMed] [Google Scholar]
- 116.Willard-Mack CL, Koehler RC, Hirata T, Cork LC, Takahashi H, Traystman RJ, Brusilow SW. Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience. 1996;71:589–599. doi: 10.1016/0306-4522(95)00462-9. [DOI] [PubMed] [Google Scholar]
- 117.Xu HL, Koenig HM, Ye S, Feinstein DL, Pelligrino DA. Influence of the glia limitans on pial arteriolar relaxation in the rat. Am J Physiol Heart Circ Physiol. 2004;287:H331–H339. doi: 10.1152/ajpheart.00831.2003. [DOI] [PubMed] [Google Scholar]
- 118.Xu HL, Ye S, Baughman VL, Feinstein DL, Pelligrino DA. The role of the glia limitans in ADP-induced pial arteriolar relaxation in intact and ovariectomized female rats. Am J Physiol Heart Circ Physiol. 2005;288:H382–H388. doi: 10.1152/ajpheart.00727.2004. [DOI] [PubMed] [Google Scholar]
- 119.Yang G, Chen G, Ebner TJ, Iadecola C. Nitric oxide is the predominant mediator of cerebellar hyperemia during somatosensory activation in rats. Am J Physiol Regul Integr Comp Physiol. 1999;277:R1760–R1770. doi: 10.1152/ajpregu.1999.277.6.R1760. [DOI] [PubMed] [Google Scholar]
- 120.Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.You J, Golding EM, Bryan RM., Jr Arachidonic acid metabolites, hydrogen peroxide, and EDHF in cerebral arteries. Am J Physiol Heart Circ Physiol. 2005;289:H1077–H1083. doi: 10.1152/ajpheart.01046.2004. [DOI] [PubMed] [Google Scholar]
- 122.Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
- 123.Zonta M, Sebelin A, Gobbo S, Fellin T, Pozzan T, Carmignoto G. Glutamate-mediated cytosolic calcium oscillations regulate a pulsatile prostaglandin release from cultured rat astrocytes. J Physiol. 2003;553:407–414. doi: 10.1113/jphysiol.2003.046706. [DOI] [PMC free article] [PubMed] [Google Scholar]