

Summary
ClpB is a member of a protein-disaggregating multi-chaperone system in Escherichia coli. The mechanism of protein-folding reactions mediated by ClpB is currently unknown and the functional role of different sequence regions in ClpB is under discussion. We have expressed and purified the full-length ClpB and three truncated variants with the N-terminal, C-terminal, and a double N- and C-terminal deletion. We studied the protein concentration-dependent and ATP-induced oligomerization of ClpB, casein-induced activation of ClpB ATPase, and ClpB-assisted reactivation of denatured firefly luciferase. We found that both the N- and C-terminal truncation of ClpB strongly inhibited its chaperone activity. The reasons for such inhibition were different, however, for the N- and C-terminal truncation. Deletion of the C-terminal domain inhibited the self-association of ClpB, which led to decreased affinity for ATP and to decreased ATPase and chaperone activity of the C-terminally truncated variants. In contrast, deletion of the N-terminal domain did not inhibit the self-association of ClpB and its basal ATPase activity, but decreased the ability of casein to activate ClpB ATPase. These results indicate that the N-terminal region of ClpB may contain a functionally significant protein-binding site, while the main role of the C-terminal region is to support oligomerization of ClpB.
Molecular chaperones and proteases define two mechanisms for the control of structural and functional integrity of proteins inside a cell (1). While chaperones promote protein folding and prevent protein aggregation, proteases eliminate damaged proteins and those that cannot be rescued by chaperones. Among protein families that are involved in such quality control, Clp ATPases (also referred to as Hsp100 proteins) play an essential role as members of both the chaperone and the protease machinery (1, 2). ClpA, ClpX, and ClpY from Escherichia coli associate with peptidase subunits to form ATP-dependent proteases. The role of Clp ATPases in such complexes is to unfold protein substrates and deliver them to the peptidases for degradation (3). In contrast, ClpB from E. coli and Thermus thermophilus as well as Hsp104 from Saccharomyces cerevisiae do not associate with peptidases. Instead, these proteins participate in multi-chaperone systems that efficiently inhibit and reverse protein aggregation (4–7).
ClpA, ClpB, and Hsp104 contain two highly conserved nucleotide-binding domains that are separated by a variable-length middle region and enclosed between less conserved N- and C-terminal regions (see Fig. 1) (2). ClpX and ClpY contain a single nucleotide-binding domain and the C-terminal domain that is similar to that of ClpA, ClpB, or Hsp104 (2). All these proteins express both basal and protein-stimulated ATPase activity. A monomer-hexamer or monomer-dimer-hexamer association equilibrium has been found for ClpA (8), ClpB (9), ClpY (10), and Hsp104 (11). Self-association of Clp ATPases is coupled to nucleotide binding and the hexameric forms are strongly stabilized in the presence of ATP.
Figure 1. Diagram of the ClpB variants produced in this study.
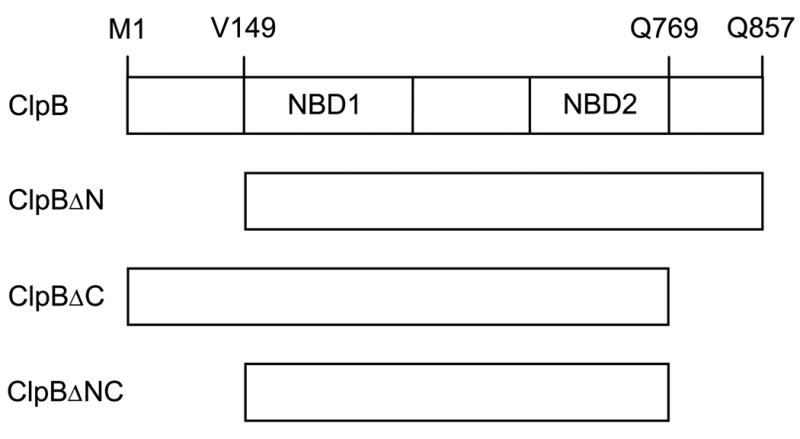
Shown is the full-length ClpB with its two nucleotide-binding domains (NBD1, NBD2) and three truncated forms of ClpB. In ClpBΔN, the amino acids 1 – 148 (the N-terminal domain) have been deleted. In ClpBΔC, the amino acids 770 – 857 (the C-terminal domain) have been deleted. In ClpBΔNC, both the N- and C-terminal domains have been deleted. In ClpBΔN and ClpBΔNC, the first valine residue (corresponding to V149 in the full-length ClpB) has been replaced with methionine.
Since ATP hydrolysis is required for both the protease (12) and the chaperone activity (4, 7) of Clp ATPases, it is evident that the nucleotide-binding domains play an important functional role. The role of the N- and C-terminal regions in Clp ATPases is, however, less clear. The N-terminal sequences of Clp ATPases do not contain any recognized consensus motifs. The C-terminal sequences show a distant similarity to protein-binding PDZ domains (13). Recently, it has been found that isolated C-terminal fragments of ClpA, ClpB, ClpX, and ClpY selectively interact with several proteins (14). It has been postulated that the C-terminal regions in Clp ATPases form “sensor and substrate-discrimination” (SSD) domains that are involved in recognizing protein substrates and guiding them into cavities inside the Clp hexamers. A recently determined crystal structure of ClpY (also known as HslU) does not support, however, the SSD hypothesis (15). In hexameric ClpY, the C-terminal domain of each monomer faces either the outside of an adjacent monomer or the solvent. Thus, the C-terminal domain may not be involved in the transfer of protein substrates into the intra-hexamer cavity.
The ClpB gene contains an internal translation-initiation site and is expressed in vivo as two gene products: a full-length 95-kDa and a truncated 80-kDa polypeptide chain (ClpB95 and ClpB80) (16, 17). The internal initiation site is located near the N-terminus of the first nucleotide-binding domain in ClpB. Thus, the N-terminal region of ClpB95 is not present in ClpB80. It has been found that while both ClpB95 and ClpB80 hydrolyze ATP, the ATPase activity of ClpB95, but not ClpB80, is strongly activated by other proteins (18). This result suggests that not only the C-terminal regions (SSD domains), but also the N-terminal regions in Clp ATPases may be involved in binding protein substrates.
In this study, we focused on the role of the N- and C-terminal domains in ClpB by comparing properties of the full-length and the truncated ClpB forms, in which the terminal regions have been removed. We tested the ClpB variants using four assays that reflect four different aspects of ClpB activity: protein concentration-dependent self-association, ATP-induced self-association, casein-stimulated ATPase activity, and ClpB-assisted protein reactivation. We found that both terminal regions of ClpB are essential for the chaperone activity. However, while the N-terminal region of ClpB is involved in interactions with protein substrates, the C-terminal region supports protein self-association. This result demonstrates a previously unknown function of the PDZ-like SSD domain in ClpB.
Experimental Procedures
Plasmids
The DNA fragments encoding ClpB residues 1-857 (full length ClpB), residues 149–857 (ClpBΔN), residues 1–769 (ClpBΔC), or residues 149–769 (ClpBΔNC) were produced by PCR1 and subcloned between the NdeI and XhoI sites of pET-20b (Novagen). The plasmid pBS-ClpB93 (18) was used as a PCR template. pBS-ClpB93 contains a mutated ClpB DNA sequence without the internal translation initiation site. The cloned DNA fragments contained 3’-end STOP codons to prevent translation of the 6-His C-terminal extension in pET-20b.
Proteins
ClpB and its truncated variants were overexpressed in E. coli strain BL21(DE3)LysS (Novagen) and purified with the procedure similar to that used before to obtain wild type ClpB (9). E. coli cells were grown at 37 °C to A(600 nm) ~ 0.6 in LB broth containing 0.1 mg/ml ampicillin. Protein expression was induced with 0.4 mM isopropyl-β-D-thiogalactoside. Cells were grown at 37 °C for 2 h after induction and were collected by centrifugation. Protein purification steps were performed on ice or at 4 °C. Cell pellets (~10 g) were suspended in buffer A (50 mM Tris-HCl, pH 7.5, 10% glycerol, 1 mM EDTA, 1 mM DTT) and disrupted by sonication. The cell extract was centrifuged at 20,000 g for 45 min. Polyethyleneimine (PEI) was added to the supernatant (final concentration 0.04 % PEI) and the turbid solution was centrifuged at 20,000 g for 60 min. Supernatant was loaded onto a 170-ml Q Sepharose column (Amersham Pharmacia Biotech) after adding 0.1 M KCl. After washing the column with buffer A with 0.1 M KCl at 1 ml/min, proteins were eluted using a 0.1–0.4 M KCl gradient. Collected fractions were analyzed by SDS-PAGE and the fractions containing ClpB were pooled and precipitated in 60% (NH4)2SO4. The precipitate was collected by centrifugation and dissolved in buffer A with 0.1 M KCl. The sample was loaded onto a 70-cm Superdex 200 column (Amersham Pharmacia Biotech) at 0.4 ml/min. Collected fractions were analyzed with SDS-PAGE. Fractions containing >95% pure ClpB were pooled and precipitated with (NH4)2SO4. For further use, the ClpB samples were extensively dialyzed in buffer B (50 mM Tris-HCl, pH 7.5, 0.2 M KCl, 20 mM MgCl2, 10% glycerol, 1 mM EDTA, 1 mM DTT) or in modified buffers (see Figs. 3, 5). Protein concentration was measured using the absorption coefficient A280 = 0.38 cm2/mg (9).
Figure 3. Sedimentation velocity of ClpB and its truncated variants at low and high protein concentration.
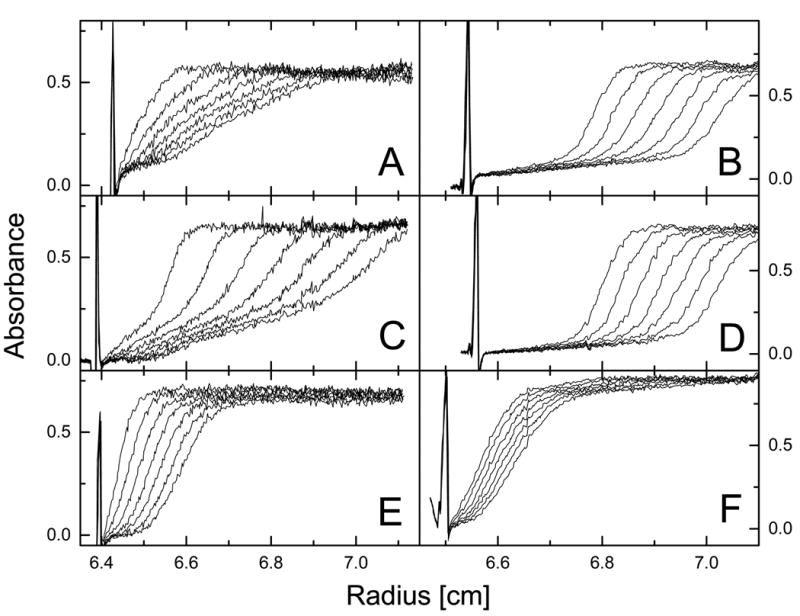
Ultracentrifugation was performed at 50,000 rpm (A, C, E) or 40,000 rpm (B, D, F) at 20 °C. The protein concentration was 0.2 mg/ml (A, C, E) or 4.0 mg/ml (B, D, F) in 50 mM Hepes-KOH, pH 7.5, 0.2 M KCl, 20 mM MgCl2, 1 mM EDTA, 2 mM β-mercaptoethanol. Shown are the series of radial scans of the centrifuge cell at 5 min intervals (A, C, E) or 3.5 min intervals (B, D, F) for the full-length ClpB (A, B), ClpBΔN (C, D), and ClpBΔC (E, F). Protein concentration profiles were measured using absorption at 236 nm (A, C, E) or 290 nm (B, D, F). The direction of sedimentation is to the right and the positions of solution menisci are indicated by the light scattering peaks at ~6.4-6.5 cm.
Figure 5. Gel filtration analysis of ClpB and its truncated variants in the presence of ATP.
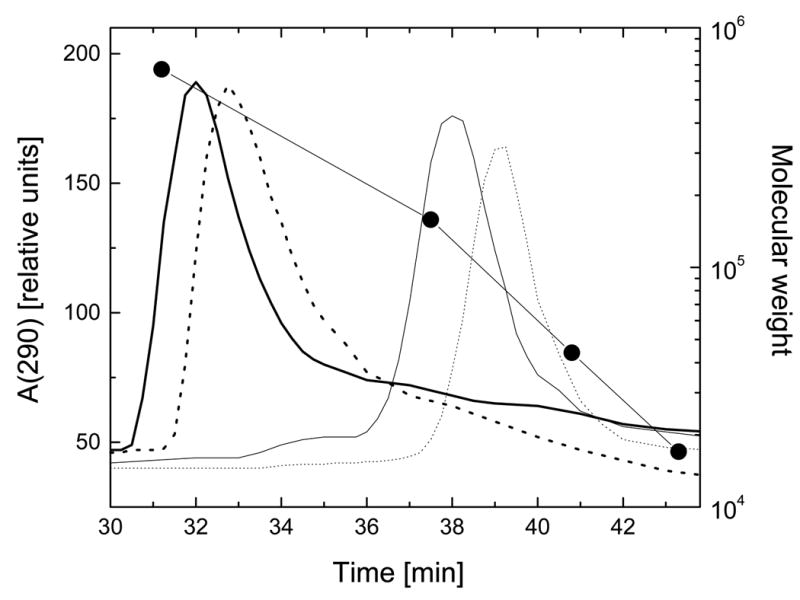
Aliquots of ClpB (thick solid line), ClpBΔN (thick dotted line), ClpBΔC (thin solid line), and ClpBΔNC (thin dotted line) (10 μl of ~2 mg/ml) were injected onto a Superose 6 column. Protein elution profiles were obtained with a 0.06 ml/min flow rate (running buffer: 50 mM Tris/HCl, pH 7.5, 0.2 M KCl, 20 mM MgCl2, 1 mM EDTA, 1 mM DTT, 2 mM ATP) by monitoring absorption at 290 nm. Circles correspond to elution times of thyroglobulin (Mr 670,000), gamma globulin (Mr 158,000), ovalbumin (Mr 44,000), and myoglobin (Mr 17,000).
Circular Dichroism Spectroscopy
CD spectra were measured with a Jasco J-720 spectrometer using a 0.01-cm cylindrical cell. The temperature of the cell was controlled by an external programmable water bath (Fisher Isotemp 1016P). For thermal stability studies, a 0.1-cm water-jacketed cell was used, the temperature was increased step-wise and samples were equilibrated at a given temperature until a steady CD signal was obtained.
Analytical Ultracentrifugation
A Beckman Optima XL-I analytical ultracentrifuge with a four-position AN-Ti rotor was used for sedimentation velocity experiments. Protein solution and a reference buffer were loaded into the right and left sector, respectively, of a double sector 1.2-cm cell with an aluminum centerpiece. Three centrifuge cells containing three protein samples (ClpB, ClpBΔN, and ClpBΔC) were simultaneously analyzed in the centrifugation experiment. After equilibration at 3,000 rpm and 20 °C, the rotor was accelerated to a desired speed and radial scans of protein concentration profiles in the cells were performed. The sedimentation velocity data were analyzed with a time-derivative method (19) using software supplied with the instrument (Beckman). Observed sedimentation coefficients were corrected to s20,w (see Table I) using previously measured values of the density and viscosity of the buffer (8).
Table I. Apparent sedimentation coefficient of ClpB and its truncated forms at low and high protein concentration.
Sedimentation velocity data shown in Fig. 3 were analyzed with the time-derivative method (19). The observed apparent sedimentation coefficients were obtained from the maxima of the apparent sedimentation coefficient distributions (Fig. 4). The observed sedimentation coefficients were corrected to values corresponding to the density and viscosity of water (see Experimental Procedures) and are reported as s20,w in Svedberg units (S). The data for wild type ClpB (0.1 mg/ml and 4.0 mg/ml) are from Ref. 9.
| Protein Sample | s20,w (S) | |
|---|---|---|
| 0.2 mg/ml protein | 4.0 mg/ml protein | |
| ClpB | 4.7 | 16.4 |
| ClpBΔN | 4.5 (minor component)
13.8 (major component) |
14.8 |
| ClpBΔC | 4.2 | 3.9 |
| wt ClpB | 4.4 | 17.1 |
Gel Filtration Chromatography
HPLC gel filtration experiments were performed using Superose 6 PC 3.2/30 column (Amersham Pharmacia Biotech) and a Hewlett Packard 1050 liquid chromatography system with a diode-array UV/Vis absorption detector. Gel filtration standards were obtained from Bio-Rad.
ClpB ATPase Activity
ClpB samples were incubated at 37 °C in the assay buffer (100 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 5 mM ATP, 1 mM EDTA, 1 mM DTT) without or with 0.1 mg/ml κ-casein (Sigma). Inorganic phosphate concentration was determined using the malachite green assay (20, 21).
Titration Calorimetry
Heat of ATP binding to ClpB was measured with a MicroCal MCS isothermal titration calorimeter (MicroCal Inc., Northampton, Massachusetts). The 1.3-ml calorimetric cell was filled with degassed buffer B or with degassed ClpB solution. Aliquots of ATP in buffer B were added with a 250-μl injection syringe. Heat effects of the ATP injections were calculated by integrating the calorimetric output curves using software supplied with the calorimeter.
Chaperone-Assisted Reactivation of Luciferase
Recombinant firefly luciferase (Promega) was diluted into the unfolding buffer (30 mM Hepes-KOH, pH 7.6, 60 mM KCl, 10 mM MgCl2, 1 mM EDTA, 10 mM DTT, 7 M urea) and incubated for 30 min at room temperature. For refolding, the denatured luciferase was rapidly diluted 100-fold into the renaturation buffer (30 mM Hepes-KOH, pH 7.6, 120 mM KCl, 10 mM MgCl2, 5 mM ATP, 1 mM EDTA, 1 mM DTT, 0.1 mg/ml bovine serum albumin) containing ClpB or its variants and DnaK, DnaJ, and GrpE (StressGen Biotechnologies, Victoria, BC, Canada). The activity of luciferase during refolding was determined as described before (4).
Results
Properties of the purified ClpB variants
Fig. 1 shows the postulated domain structure of ClpB and compares four protein variants used in this study. We have purified the full-length 95-kDa ClpB, which corresponds to ClpB95, the N-terminally truncated 80-kDa ClpBΔN, which corresponds to ClpB80, and the C-terminally truncated 85-kDa ClpBΔC. The C-terminal region that has been removed in ClpBΔC is 4 amino acids shorter than the SSD domain from ClpB investigated by Smith et al. (14). We have also purified a doubly-truncated 70-kDa ClpBΔNC with the combined N- and C-terminal deletions. The proteins were >95% pure, as determined by Coomassie blue staining of SDS PAGE gels and their apparent molecular weights corresponded to those predicted from the amino-acid sequence.
We have tested whether the terminal deletions affected the folding of ClpB and its thermodynamic stability. We measured circular dichroism (CD) spectra for ClpB and the truncated variants (Fig. 2A). The CD spectra showed a negative double band at 210–222 nm and a positive band at <200 nm, which indicates that ClpB contains α-helical structures. No apparent difference was observed between the CD spectra of the full-length and truncated ClpB. This result indicates that the average secondary structure (per residue) in all ClpB variants is similar. Thus, terminal truncations in ClpB do not affect secondary structure of the two ATP-binding domains and the middle sequence region (see Fig. 1).
Figure 2. Secondary structure and thermal stability of ClpB and its truncated variants.
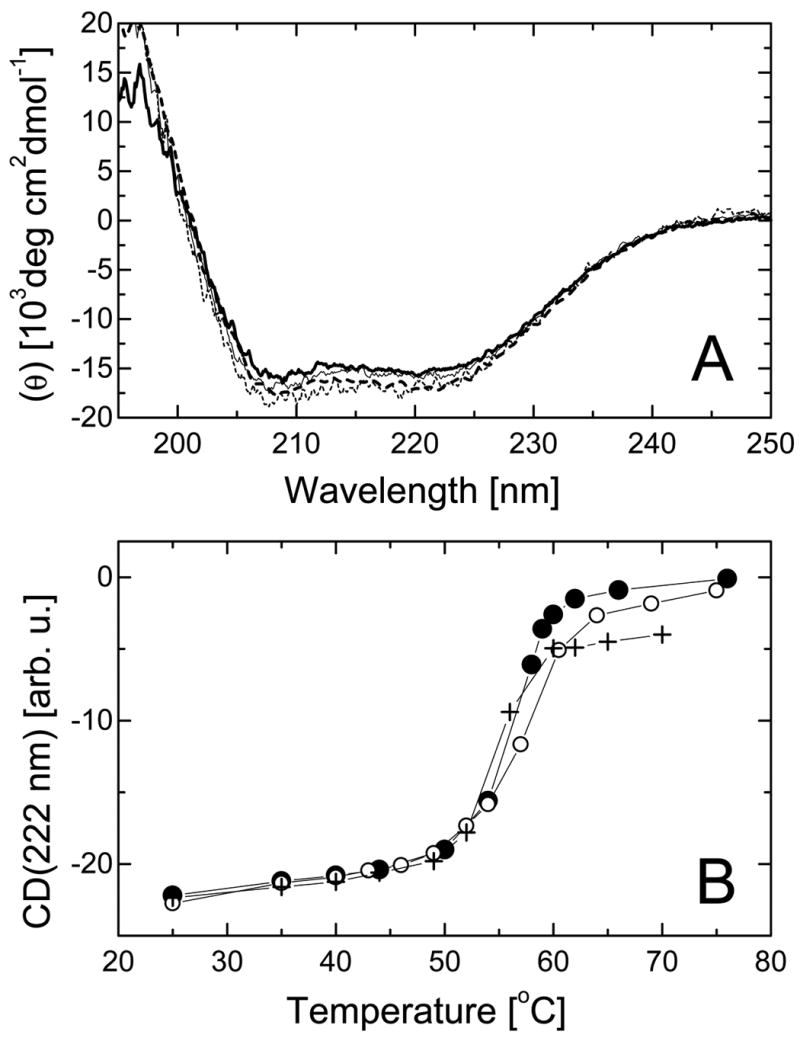
A, Far-UV circular dichroism spectra expressed as mean molar residue ellipticity (θ) of the full-length ClpB (thick solid line), ClpBΔN (thick dashed line), ClpBΔC (thin solid line), and ClpBΔNC (thin dashed line) at 37 °C. B, Temperature-induced changes in the circular dichroism signal at 222 nm for the full-length ClpB (solid circles), ClpBΔN (crosses), and ClpBΔC (open circles).
ClpB contains two Trp residues. W462 is located in the middle sequence region and W543 is within the C-terminal ATP-binding domain (NBD2). The fluorescence spectrum of ClpB showed a single emission maximum at 350 nm and no apparent differences in shape or intensity of the emission band were observed between the full-length ClpB and the truncated variants (not shown). This result indicates that the terminal truncations in ClpB do not produce changes in the environment of Trp residues.
As shown by the temperature dependence of the CD signal (Fig. 2B), ClpB unfolds cooperatively during thermal denaturation with a mid-point temperature of 56 °C. Cooperative transitions with similar mid-point temperatures (± 1 °C) were also observed for the N-terminally and C-terminally truncated ClpB. Cooperative unfolding at 56 °C indicates that the conformation of ClpB is thermodynamically stable at the assay temperatures used in this study. We found that the N- and C-terminal truncations do not destabilize the folded conformation of ClpB. We conclude that the two ATP-binding domains and the middle sequence region form the main folding stability core of ClpB.
Self-association of ClpB
Association equilibria manifest themselves in a protein concentration dependence of the size of a protein. We used sedimentation velocity experiments (Fig. 3) to monitor the difference in apparent size of ClpB between low protein concentration (0.2 mg/ml) and high concentration (4.0 mg/ml). In a sedimentation velocity experiment, protein concentration profiles in a centrifuge cell are measured at different times during centrifugation. The shape of such concentration profiles is related to homogeneity and diffusion properties of species in solution, while the rate of movement of a concentration boundary gives the sedimentation coefficient. At 0.2 mg/ml, both the full-length ClpB and ClpBΔC (Fig. 3A, E) sedimented slower than ClpBΔN (Fig. 3C). However, at 4.0 mg/ml, both the full-length ClpB and ClpBΔN (Fig. 3B, D) sedimented faster than ClpBΔC (Fig. 3F).
Fig. 4 shows apparent distributions of the sedimentation coefficient (g(s*)) for ClpB obtained from the time-derivative analysis (19) of the data in Fig. 3. In Fig. 4, the s* values corresponding to the maxima of g(s*) profiles approximate the observed sedimentation coefficients for molecular species predominating in a protein sample. Single g(s*) maxima were observed for the full-length ClpB and ClpBΔN at high concentration (Fig. 4 A, B, dotted lines) and for ClpBΔC at both low and high concentration (Fig. 4C), which indicates that those protein samples contained a single predominant molecular component. In contrast, the sedimentation data for the full-length ClpB and, more noticeably, ClpBΔN at low concentration (Fig. 4A, B, solid lines) indicated the presence of more components with significantly different sedimentation coefficients. The concentration dependence of g(s*) profiles for ClpB and ClpBΔN is similar to that observed for wild type ClpB (a mixture of ClpB95 and ClpB80). It has been shown that wild type ClpB is monomeric at ~0.1 mg/ml and forms hexamers at 4.0 mg/ml (9). Our results indicate that the population of oligomers of ClpBΔN at 0.2 mg/ml is significantly higher than of the full-length ClpB.
Figure 4. Apparent sedimentation coefficient distributions for ClpB and its truncated variants.
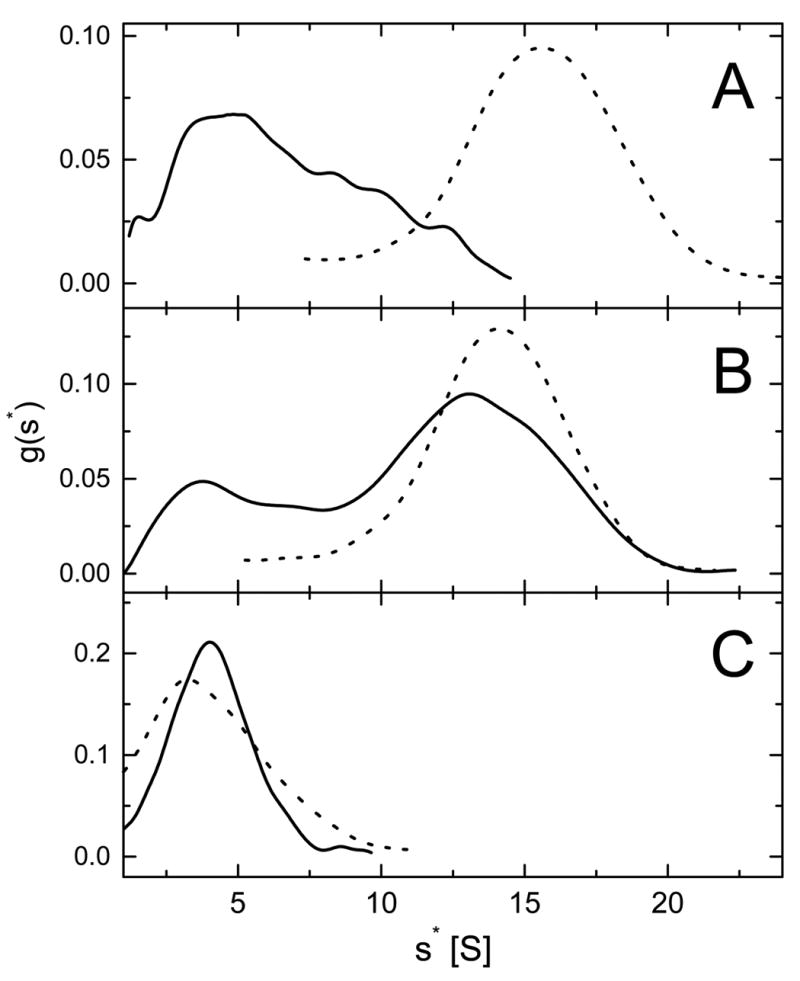
Shown are the results of the time-derivative analysis (19) of four late protein concentration profiles (see Fig. 3) for the full-length ClpB (A), ClpBΔN (B), and ClpBΔC (C) at 0.2 mg/ml (solid lines) and 4.0 mg/ml (dotted lines). The lines show apparent distribution functions g(s*) vs. the sedimentation coefficient s* in Svedberg units (S).
Table I contains the values of sedimentation coefficient of the ClpB variants at low and high protein concentration obtained from the maxima of g(s*). For comparison, the data for wild type ClpB from ref. 9 are also included. At 0.2 mg/ml, the sedimentation coefficients of the dominant component in full-length ClpB, a minor component of ClpBΔN, and ClpBΔC are close to that of the monomeric wild type ClpB. At 4.0 mg/ml, the sedimentation coefficients of the full-length ClpB and ClpBΔN approximate that of the oligomeric wild type ClpB. This result indicates that the full-length ClpB as well as ClpBΔN undergo a concentration dependent self-association. Interestingly, unlike the full-length ClpB, ClpBΔN at low concentration contains a dominant fast sedimenting component 2. This indicates that the association of ClpB is enhanced after removal of the N-terminal domain. In contrast, the sedimentation coefficient of ClpBΔC does not increase upon increasing protein concentration. This result implies that the self-association of ClpB is inhibited after removal of the C-terminal domain.
It has been shown that the associated form of wild type ClpB is stabilized by ATP (9). We used gel filtration chromatography to study self-association of the truncated variants of ClpB in the presence of ATP (protein concentration on the column ~0.1 mg/ml). As shown in Fig. 5, ATP stabilizes hexameric ClpB and ClpBΔN. Monomeric species of the full-length ClpB and ClpBΔN were not found in the presence of ATP (compare to Fig. 4 for 0.2 mg/ml ClpB). However, ATP does not induce self-association of ClpBΔC and ClpBΔNC, which elute as monomers. This result indicates that the association deficiency in the C-terminally truncated variants of ClpB cannot be overcome by ATP binding.
ATPase activity of ClpB
Among the ClpB variants investigated in this work, ClpBΔN displayed the highest rate of ATP hydrolysis (Fig. 6A). The full-length ClpB hydrolyzed ATP with a lower rate than ClpBΔN, while ClpBΔC and ClpBΔNC had no detectable ATPase activity. It is known that Clp ATPases are activated by other proteins (2). We measured the rate of ATP hydrolysis by ClpB in the presence of κ-casein. We found that the ATPase of the full-length ClpB was activated ~20-fold by casein, while that of ClpBΔN was activated only ~5-fold (Fig. 6B). This indicates that the N-terminally truncated ClpB is deficient in transferring the protein binding signal to the ATPase active sites, in agreement with previous results (18). No ATPase activity was detected for ClpBΔC and ClpBΔNC in the presence of casein.
Figure 6. ATP hydrolysis by ClpB and its truncated variants.
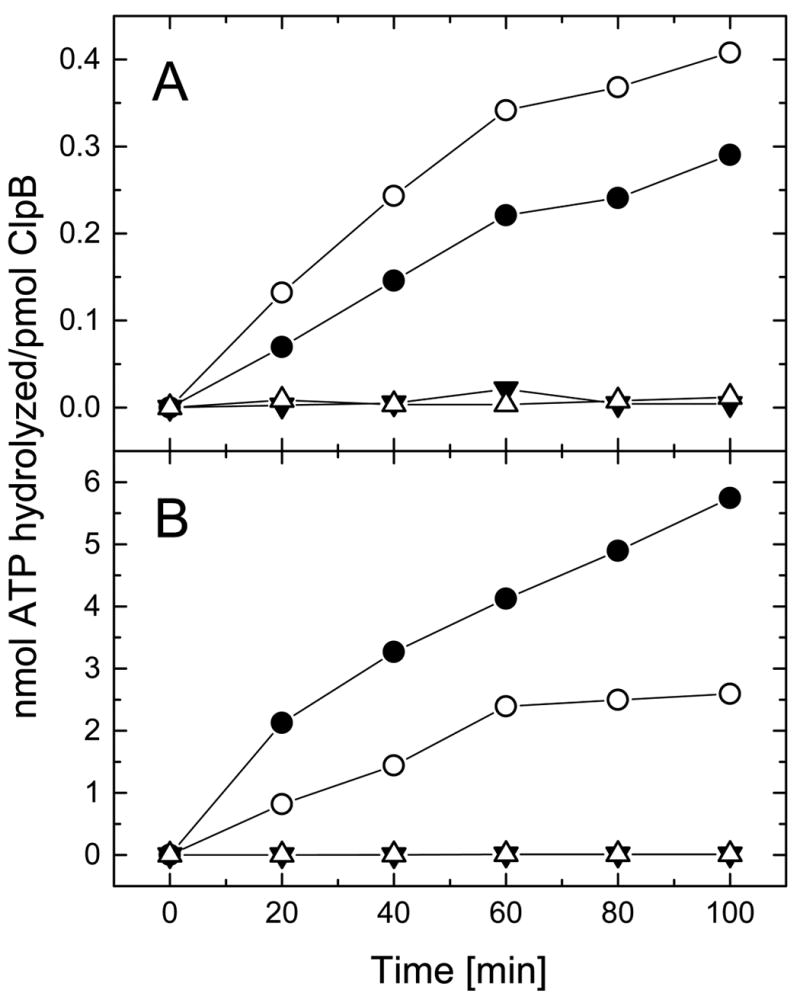
ATPase activity has been measured by incubating the full-length ClpB (filled circles), ClpBΔN (open circles), ClpBΔC (filled triangles), or ClpBΔNC (open triangles) for the indicated time at 37 °C in the assay buffer (see Experimental Procedures). In panel A, 2.5 μg protein was used for each reaction. In panel B, the assay buffer contained 0.1 mg/ml κ-casein and 0.25 μg ClpB or ClpBΔN, or 2.5 μg ClpBΔC or ClpBΔNC. Notice the difference between vertical scales in panels A and B.
We investigated whether the loss of ATPase in ClpBΔC and ClpBΔNC is due to defects in ATP binding to the C-terminally truncated ClpB. Fig. 7 shows calorimetric titrations of ClpB and ClpBΔC with ATP. Injections of ATP into the buffer produced endothermic effects due to the heat of dilution of ATP (Fig. 7A, upper trace). Injections of ATP into solution of the full-length ClpB resulted in a complex calorimetric output due to a combination of ATP binding to ClpB, ATP-induced association of ClpB, and ATP hydrolysis. However, it is evident from the lower trace in Fig. 7A that a strong exothermic reaction became saturated after a few ATP injections and the remaining part of the trace was dominated by the endothermic effect of ATP dilution. A similar calorimetric trace was also measured during titration of ClpBΔN with ATP (not shown). In contrast, injections of ATP into solution of ClpBΔC produced weak exothermic effects that did not show saturation up to 1 mM ATP (Fig. 7A, middle trace, and Fig. 7B). ClpBΔC does not hydrolyze ATP (see Fig. 6), nor does it self-associate in the presence of ATP (see Fig. 5). Thus, heat effects of adding ATP to ClpBΔC can be attributed to protein-nucleotide interactions. We conclude that ClpBΔC binds ATP with a low affinity, most likely with Kd > 1 mM.
Figure 7. Calorimetric titrations of ClpB with ATP.

A, calorimeter output for series of 10-μl injections of buffer B with 5.8 mM ATP into buffer B (upper trace), into 2 mg/ml full-length ClpB (lower trace), or 2 mg/ml ClpBΔC (middle trace) at 30 °C. Positive peaks correspond to endothermic effects and negative – to exothermic effects. The data traces were offset for clarity. B, Cumulative heat effect of titrating ATP into ClpBΔC (from the middle trace in panel A).
Chaperone activity of ClpB
We tested the ClpB variants in a luciferase reactivation assay that had been previously used to detect chaperone activity of ClpB (4). In this assay, no spontaneous recovery of luciferase is observed due to its intense aggregation. Efficient reactivation of luciferase requires ClpB and a group of three other E. coli chaperones: DnaK, DnaJ, and GrpE (4). As shown in Fig. 8, the full-length ClpB efficiently reactivated luciferase with a ~30 min “lag phase”, similar to that observed for wild type ClpB (4). All truncated variants: ClpBΔN, ClpBΔC, and ClpBΔNC were deficient in their chaperone activity. This indicates that both terminal regions of ClpB are essential for its function as a molecular chaperone.
Figure 8. Reactivation of luciferase by ClpB and its truncated variants in the presence of DnaK, DnaJ, and GrpE.
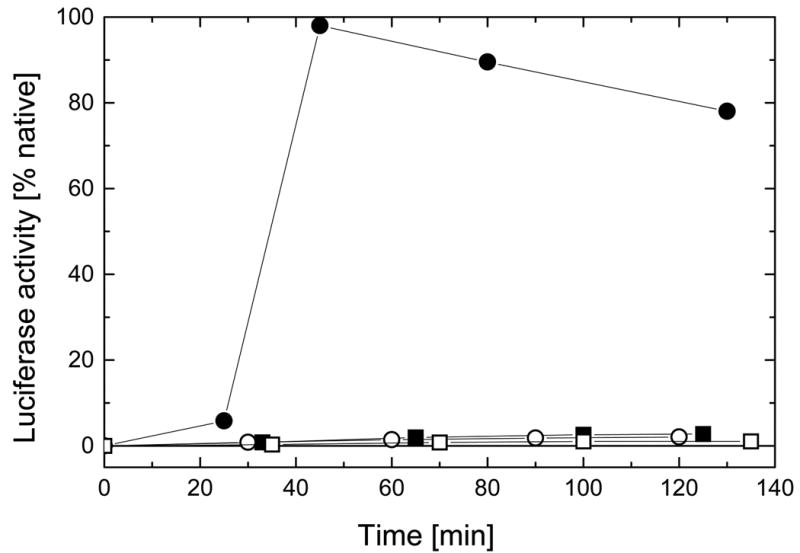
Unfolded luciferase was diluted at room temperature into the refolding buffer (see Experimental Procedures) containing DnaK, DnaJ, GrpE and the full-length ClpB (filled circles), ClpBΔN (open circles), ClpBΔC (filled squares), or ClpBΔNC (open squares). Luciferase activity was measured in aliquots withdrawn after the times indicated. Protein concentrations in the refolding solutions were: 25 nM luciferase, 0.3 μM (hexamer) ClpB or its variants, 1.0 μM DnaK, 1.1 μM DnaJ, 1.2 μM GrpE.
Discussion
We have purified and studied the full-length ClpB and its three terminally truncated variants. We have found full inhibition of the chaperone activity in all truncated ClpB variants. We have also observed inhibition of self-association and ATPase activity in the C-terminally truncated ClpB and inhibition of casein-activated ATPase in the N-terminally truncated ClpB. The N-terminal and C-terminal truncations did not apparently affect the folding and stability of the remaining domains of ClpB. Thus, the observed effects of truncations can be attributed to the roles of the N-terminal and C-terminal parts of ClpB.
As shown in this study for the full-length ClpB and in ref. 9 for wild type protein, there is a reversible equilibrium between monomeric and hexameric ClpB in solution without nucleotides. The most striking structural effect of the C-terminal deletion in ClpB is a complete inhibition of protein oligomerization (see Figs. 3, 4). This result is consistent with position of the C-terminal domain in the crystal structure of ClpY (15). The C-terminal domain of ClpY is located on the outside of the ClpY hexamer and is in contact with the neighboring subunit within the hexamer. Our results indicate that contacts maintained by the C-terminal domain are necessary for stabilization of the ClpB hexamer.
Deletion of the C-terminal and the N-terminal regions of ClpB have opposite effects on protein oligomerization. Compared with the full-length ClpB, the association equilibrium in ClpBΔN is shifted towards the hexamer (Figs. 3, 4). Since both the full-length ClpB (ClpB95) and ClpBΔN (ClpB80) are produced in vivo and form mixed oligomers (9), our results raise a possibility that the association properties of wild type ClpB can be modulated by the relative amounts of ClpB95 and ClpB80.
What is the reason for the observed inhibition of the ATPase activity of ClpB upon removal of the C-terminal domain (see Fig. 6)? Again, the crystal structure of ClpY provides a possible explanation. In the ClpY hexamer, nucleotide molecules are bound at the interfaces between adjacent subunits (15). Our results indicate that nucleotide contacts with both adjacent subunits are necessary for maintaining strong nucleotide binding, because monomeric ClpBΔC binds ATP with low affinity (Fig. 7). In addition, ATP binding to two adjacent ClpB subunits may be necessary to trigger ATP hydrolysis. Since the chaperone function of ClpB requires ATP hydrolysis (4), the final result of the oligomerization deficiency in ClpBΔC and ClpBΔNC is an inhibition of chaperone activity of these proteins (Fig. 8).
Weak binding of ATP to monomeric ClpBΔC indicates that in the monomer-hexamer equilibrium, ATP may bind predominantly to the hexameric ClpB. Such preferential binding of ATP results in a shift of the thermodynamic equilibrium in favor of the hexamer (23). Change in protein association equilibrium manifests itself as the ATP-induced stabilization of hexameric ClpB and ClpBΔN (Fig. 5). Such shifts in coupled equilibria can occur in addition to possible ATP-induced conformational changes within ClpB. Conversely, changes in the relative amounts of ClpB monomers and hexamers can modulate the ability of ClpB to bind and hydrolyze ATP. Enhanced self-association affinity of ClpBΔN, as compared with the full-length ClpB, may be the reason for the elevated basal ATPase activity of ClpBΔN (Fig. 6A).
ClpBΔN displays a much weaker activation of its ATPase in the presence of casein than does the full-length ClpB (Fig. 6B, ref. 18). Since the basal ATPase of ClpB is not inhibited by the N-terminal truncation, we conclude that the N-terminal region of ClpB is involved in either direct interaction with casein or in the coupling between the protein-binding site and the ATPase sites. Defects in protein-binding properties manifest themselves in a decreased chaperone activity of ClpBΔN (Fig. 8). It should be noted, however, that although the ATPase of ClpBΔN does respond to casein to some extent, the chaperone activity of ClpB is completely abolished after the N-terminal truncation. In summary, all terminally truncated variants of ClpB have low chaperone activity, but the reasons for this deficiency are different for the C-terminal and the N-terminal truncation.
Recent publication of the crystal structure of ClpY (HslU) and the ATP-dependent protease ClpYQ (HslU-HslV) gives, for the first time, high-resolution structural information for a Clp ATPase (15). Partial homology between ClpY and ClpB (2) suggests that the C-terminal domain of ClpB and its C-terminal ATP-binding domain (NBD2) may assume conformations similar to analogous regions in ClpY. However, high-resolution information is not available for the N-terminal domain, NBD1, and the middle domain of ClpB. As described above, our experiments in solution support predictions made from the ClpY crystal structure. In particular, we have shown that the C-terminal region of ClpB primarily supports protein self-association. This conclusion is consistent with the crystal structure of ClpY (15) and is different from that of Smith et al. (14), who postulated that the C-terminal fragment forms a substrate-binding domain. Our results indicate that the site, which is essential for chaperone activity and is also responsive to casein binding, is located in the N-terminal region of ClpB. While the N-terminal regions of ClpA and Hsp104 are similar to that of ClpB, other Clp ATPases (ClpX, ClpY) do not contain analogous domains. Thus, ClpX and ClpY may use other sequence motifs to recognize and bind protein substrates.
Unifying biochemical characteristics of Clp ATPases are protein-activated ATPase activity and ATP-induced oligomerization (2). So far, it has not been clear how these two properties are coupled with each other and with the activity of Clp proteins as either ATP-dependent chaperones or ATP-dependent proteases. Our results indicate that the propensity of Clp ATPases to form oligomers is an important feature of these proteins that is necessary for their ATPase activity and the biological function.
Acknowledgments
We thank Dr. Chin Ha Chung (Seoul National University, Korea) for the plasmid pBS-ClpB93. We also thank Dr. Karl J. Kramer (U.S. Grain Marketing Research Laboratory, U.S. Department of Agriculture, Manhattan, Kansas) for making the HPLC system available for our studies. This is contribution 00-468-J from the Kansas Agricultural Experiment Station.
Abbreviations
- DTT
dithiothreitol
- EDTA
ethylenediamine tetraacetic acid
- Hepes
4-(2-hydroxyethyl-)-1-piperazineethanesulfonic acid
- PCR
polymerase chain reaction
- Tris
tris(hydroxymethyl)aminomethane
Footnotes
This work was supported by the National Science Foundation Grant EPS-9550487 with matching funds from the state of Kansas and by the National Institutes of Health Grant GM58626.
It has been shown that the apparent sedimentation coefficients observed when two components are present in solution do not represent the sedimentation coefficients of the pure components (22). This can be the reason for s20,w of the major component in ClpBΔN at 0.2 mg/ml to be less than s20,w of ClpBΔN at 4 mg/ml (Table I).
References
- 1.Wickner S, Maurizi MR, Gottesman S. Science. 1999;286:1888–1893. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- 2.Schirmer EC, Glover JR, Singer MA, Lindquist S. Trends Biochem Sci. 1996;21:289–296. [PubMed] [Google Scholar]
- 3.Weber-Ban EU, Reid BG, Miranker AD, Horwich AL. Nature. 1999;401:90–93. doi: 10.1038/43481. [DOI] [PubMed] [Google Scholar]
- 4.Zolkiewski M. J Biol Chem. 1999;274:28083–28086. doi: 10.1074/jbc.274.40.28083. [DOI] [PubMed] [Google Scholar]
- 5.Goloubinoff P, Mogk A, Ben Zvi AP, Tomoyasu T, Bukau B. Proc Natl Acad Sci U S A. 1999;96:13732–13737. doi: 10.1073/pnas.96.24.13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Motohashi K, Watanabe Y, Yohda M, Yoshida M. Proc Natl Acad Sci U S A. 1999;96:7184–7189. doi: 10.1073/pnas.96.13.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glover JR, Lindquist S. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 8.Maurizi MR, Singh SK, Thompson MW, Kessel M, Ginsburg A. Biochemistry. 1998;37:7778–7786. doi: 10.1021/bi973093e. [DOI] [PubMed] [Google Scholar]
- 9.Zolkiewski M, Kessel M, Ginsburg A, Maurizi MR. Protein Sci. 1999;8:1899–1903. doi: 10.1110/ps.8.9.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kessel M, Wu WF, Gottesman S, Kocsis E, Steven AC, Maurizi MR. FEBS Lett. 1996;398:274–278. doi: 10.1016/s0014-5793(96)01261-6. [DOI] [PubMed] [Google Scholar]
- 11.Parsell DA, Kowal AS, Lindquist S. J Biol Chem. 1994;269:4480–4487. [PubMed] [Google Scholar]
- 12.Gottesman S, Maurizi MR. Microbiol Rev. 1992;56:592–621. doi: 10.1128/mr.56.4.592-621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levchenko I, Smith CK, Walsh NP, Sauer RT, Baker TA. Cell. 1997;91:939–947. doi: 10.1016/s0092-8674(00)80485-7. [DOI] [PubMed] [Google Scholar]
- 14.Smith CK, Baker TA, Sauer RT. Proc Natl Acad Sci U S A. 1999;96:6678–6682. doi: 10.1073/pnas.96.12.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bochtler M, Hartmann C, Song HK, Bourenkov GP, Bartunik HD, Huber R. Nature. 2000;403:800–805. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- 16.Squires CL, Pedersen S, Ross BM, Squires C. J Bacteriol. 1991;173:4254–4262. doi: 10.1128/jb.173.14.4254-4262.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woo KM, Kim KI, Goldberg AL, Ha DB, Chung CH. J Biol Chem. 1992;267:20429–20434. [PubMed] [Google Scholar]
- 18.Park SK, Kim KI, Woo KM, Seol JH, Tanaka K, Ichihara A, Ha DB, Chung CH. J Biol Chem. 1993;268:20170–20174. [PubMed] [Google Scholar]
- 19.Stafford WF., III Anal Biochem. 1992;203:295–301. doi: 10.1016/0003-2697(92)90316-y. [DOI] [PubMed] [Google Scholar]
- 20.Hess HH, Derr JE. Anal Biochem. 1975;63:607–613. doi: 10.1016/0003-2697(75)90388-7. [DOI] [PubMed] [Google Scholar]
- 21.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 22.Stafford WF., III . Modern Analytical Ultracentrifugation. Birkhauser; Boston, Massachusetts: 1994. [Google Scholar]
- 23.Wyman J, Gill SJ. Functional Chemistry of Biological Macromolecules. University Science Books; Mill Valley, California: 1990. Binding and Linkage. [Google Scholar]