

Abstract
Cardiovascular disease is the leading cause of death in people with type 2 diabetes and is linked to insulin resistance even in the absence of diabetes. Here we show that mice with combined deficiency of the insulin receptor and insulin-like growth factor 1 (IGF-1) receptor in cardiac and skeletal muscle develop early-onset dilated cardiomyopathy and die from heart failure within the first month of life despite having a normal glucose homeostasis. Mice lacking the insulin receptor show impaired cardiac performance at 6 months, and mice lacking the insulin receptor plus one Igf1r allele have slightly increased mortality. By contrast, mice lacking the IGF-1 receptor or the IGF-1 receptor plus one Ir allele appear normal. Morphological characterization and oligonucleotide array analysis of gene expression demonstrate that prior to development of these physiological defects, mice with combined deficiency of both insulin and IGF-1 receptors have a coordinated down-regulation of genes encoding components of the electron transport chain and mitochondrial fatty acid beta-oxidation pathways and altered expression of contractile proteins. Thus, while neither the insulin receptor nor IGF-1 receptor in muscle is critical for glucose homeostasis during the first month of life, signaling from these receptors, particularly the insulin receptor, is required for normal cardiac metabolism and function.
Type 2 diabetes is associated with a significant increase in the risk of cardiovascular disease (CVD), and CVD is the leading cause of death in people with type 2 diabetes (24). Important mediators of increased CVD risk are dyslipidemia and hyperglycemia, and treatment of these factors significantly decreases the risk of CVD events (10, 36). Increasing evidence indicates that insulin resistance, a major factor in the pathogenesis of type 2 diabetes, is associated with increased risk of CVD even in the absence of hyperglycemia (18, 51). Most of the cardiac morbidity and mortality in diabetic patients relates to atherosclerotic disease, but there may also be an increased risk of heart failure independent of vascular pathology associated with diabetes (15, 40). The direct role of insulin signaling in cardiomyocytes has been investigated in transgenic mouse models. Mice with a cardiac muscle-specific deletion of the insulin receptor (CIRKO) and mice lacking the insulin receptor (IR) in both cardiac and skeletal muscle (MIRKO) have a small heart phenotype but show only modest reductions in cardiac function (4, 22). MIRKO mice that have deletion of the insulin receptor in both skeletal muscle and cardiac muscle also exhibit dyslipidemia, another aspect of the metabolic syndrome and a risk factor for CVD (8). One interpretation of the preserved cardiac function in CIRKO and MIRKO mice is that signaling from the insulin receptor is not critical for cardiac development and function. Another possibility is that other mechanisms activating the same signaling pathway may compensate for the absence of the IR.
The IR and insulin-like growth factor 1 (IGF-1) receptor (IGF-1R) are structurally similar. Both are found at the cell surface as α2β2 heterotetramers with transmembrane ligand-binding α-subunits and intracellular, tyrosine kinase-containing β-subunits. Furthermore, both receptors, once activated, can phosphorylate and/or interact with the same intracellular protein substrates, including members of the insulin receptor substrate (IRS) family, and Src homology and collagen domain protein (Shc). IR and IGF-1R also activate many of the same downstream signaling molecules, such as phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (5, 44).
IGF-1R signaling, like IR signaling, in cardiac and skeletal muscle has been suggested to play key roles in the growth, development, and differentiation of these organs as well as in the regulation of whole-body metabolism (16, 17, 32, 42, 43). To investigate a possible functional overlap of the two receptors in cardiac and skeletal muscle tissue, we generated muscle-specific IR and IGF-1R double-knockout (MI2RKO) mice. We report here that while disruption of either IR or IGF-1R alone in cardiac and skeletal muscle has no effect on mortality in the mouse, the combined lack of both receptors results in early-onset dilated cardiomyopathy and death from heart failure within the first month of life. Thus, some level of IR or IGF-1R signaling is required for normal cardiac development and function and based on combinatorial knockouts, the IR seems more critical than the IGF-1R. Oligonucleotide array experiments and morphological analysis demonstrate that changes in the cardiac muscle contractile apparatus, the electron transport chain (ETC), and the mitochondrial fatty acid beta-oxidation (MFABO) pathways in MI2RKO, and to a lesser extent, in MIRKO hearts may be responsible for the development of heart failure in animals lacking IR and IGF-1R signaling in the heart.
MATERIALS AND METHODS
Animals and genotyping.
MCK-Cre, Irlox, and Igf1rlox mice were generated as previously described (8, 19). In the initial breeding protocol, mice doubly heterozygous for floxed alleles of the Ir and Igf1r genes (Irlox/wt Igf1rlox/wt [wt indicates wild type] [DLoxHet]) were crossed with DLoxHet MCK-Cre mice, which in addition to being heterozygous for Irlox and Igf1rlox, were heterozygous for MCK-Cre, i.e., a transgene for the Cre recombinase under the control of the muscle-specific muscle creatine kinase (MCK) promoter. Because of the low expected frequency of double- and single-knockout progeny (3.1% each based on a Mendelian distribution) from this protocol, two lines that had higher expected frequencies of study group animals were established as follows. For line A, Irlox/lox Igf1rlox/wt mice were crossed with Irlox/lox Igf1rlox/wt MCK-Cre mice to produce Irlox/lox Igf1rlox/lox double Lox control (DLox) mice (expected frequency, 12.5%), Irlox/lox MCK-Cre (muscle-specific IR knockout [MIRKO]; expected frequency, 12.5%) mice, Irlox/lox Igf1rlox/wt MCK-Cre (MIRKO/muscle-specific IGF-1R [MIGF1R] heterozygous [MIRKO/+]; expected frequency, 25%) mice, and Irlox/lox Igf1rlox/lox MCK-Cre (MI2RKO; expected frequency, 12.5%) mice. For line B, Irlox/wt Igf1rlox/lox mice were crossed with Irlox/wt Igf1rlox/lox MCK-Cre mice to produce Irlox/lox Igf1rlox/lox control mice (DLox mice; expected frequency, 12.5%), Igf1rlox/lox MCK-Cre (muscle-specific IGF-1R knockout [MIGF1RKO]; expected frequency, 12.5%) mice, Irlox/wt Igf1rlox/lox MCK-Cre (MIGF1RKO/mouse-specific IR [MIR] heterozygous [MIGF1RKO/+]; expected frequency, 25%) mice, and Irlox/lox Igf1rlox/lox MCK-Cre (MI2RKO; expected frequency, 12.5%) mice. The final strains were on a mixed background of C57BL/6J, 129Sv, and FVB. Accordingly, all experiments were performed with littermate controls as the best genetic match to the study group. Mice were housed in pathogen-free facilities on a 12-h light, 12-h dark cycle and had free access to food (Mouse Diet 9F; PMI Nutrition International) and water. Genotyping was performed by PCR analysis of genomic DNA extracted from tail snips. Each mouse was genotyped for MCK-Cre, Irlox, and Igf1rlox. The sense and antisense primers for the genes were as follows: for the MCK-Cre transgene, sense (5′-AGA TGA CCT TGA ACT GCT GG-3′) and antisense (5′-CGC CGC ATA ACC AGT GAA AC-3′) primers; for the Irlox allele, sense (5′-GAT GTG CAC CCC ATG TCT G-3′) and antisense (5′-CTG AAT AGC TGA GAC CAC AG-3′); and for the Igf1rlox allele, sense (5′-ATG AAT GCT GGT GAG GGT TGT CTT-3′) and antisense (5′-ATC TTG GAG TGG TGG GTC TGT TTC-3′). All procedures involving animals were approved by the Animal Care Committee of the Joslin Diabetes Center.
Analytical procedures.
Blood glucose levels were measured from whole venous blood using an automatic glucometer (Glucometer Elite; Bayer, Tarrytown, NY). Plasma insulin levels were determined by an enzyme-linked immunosorbent assay using mouse insulin as a standard (Crystal Chem, Downers Grove, IL). Prior to the glucose tolerance tests, 2.5-week-old mice were fasted for 3 hours, and 4- and 6-month-old animals were fasted overnight. At time zero, glucose was injected intraperitoneally (2 g/kg body weight), and blood glucose levels were determined from samples drawn at the indicated time points. Insulin tolerance tests were performed on random-fed mice by intraperitoneal injection of human insulin (Humulin; Eli Lilly, Indianapolis, IN) using a dose of 0.75 U/kg body weight for 2.5-week-old mice and a dose of 1.25 U/kg body weight for 4- and 6-month-old mice, respectively. Blood glucose levels were determined from samples drawn immediately before and at the indicated time points after injection of insulin.
Light microscopy and EM.
For light microscopy, male mice were anesthetized at postnatal day 8 (P8) and postnatal day 20 (P20). The heart was quickly removed and fixed in Bouin's solution. Sections (5 μm) perpendicular to the long axis of cardiomyocytes were prepared for hematoxylin and eosin and trichrome staining. Cell size was determined by delineating cell borders with fluorescein-tagged wheat germ agglutinin staining (Sigma) and measuring the cell circumference using commercially available software (SigmaPro). For electron microscopy (EM), hearts from anesthetized mice were quickly removed and fixed with a 2.5% solution of glutaraldehyde in a 0.1 M phosphate buffer, pH 7.4. After the samples were postfixed in 2% osmium tetroxide, they were dehydrated in ethanol, cleared with propylene oxide, and embedded in propylene oxide and Araldite 502 epoxy resin (1:1). One-micron sections were stained with methylene blue and analyzed to determine the regions of proper orientation. Blocks were then trimmed for thin sectioning. Thin sections were cut on an LKB Nova ultramicrotome and were stained with uranyl acetate and lead citrate before being photographed on a Philips 301 transmission electron microscope.
Echocardiography.
Transthoracic echocardiography on nonanesthetized animals was conducted as described previously (26) using an Acuson Sequoia C-256 echocardiograph machine and a 15-MHz probe. In brief, the heart was imaged in the two-dimensional parasternal short-axis view, and an M-mode echocardiogram of the midventricle was recorded at the level of papillary muscles. The heart rate, posterior wall thickness, and end-diastolic and end-systolic internal dimensions of the left ventricle (LV) were measured from the M-mode image. LV fractional shortening was defined as the end-diastolic dimension minus the end-systolic dimension normalized for the end-diastolic dimension and was used as an index of cardiac contractile function.
Immunoprecipitation and immunoblot analysis.
Western blot analysis was performed using a protocol modified from the method of Brüning et al. (8). In brief, mice were anesthetized by injection of pentobarbital and then injected with either saline, 3 IU of insulin, or 1 mg/kg recombinant human IGF-1 (PeproTech, Rocky Hill, NJ) via the inferior vena cava. After 5 min, the liver, heart, and skeletal muscle were removed and immediately frozen in liquid nitrogen. For preparation of protein extracts, tissues were homogenized in homogenization buffer (25 mM Tris-HCl, pH 7.4, 10 mM Na3VO4, 100 mM NaF, 50 mM Na4P2O7, 10 mM EGTA, 10 mM EDTA, protease inhibitor cocktail [P8340; Sigma, St. Louis, MO], 1% NP-40) using a Polytron homogenizer. Particulate matter was removed by two rounds of centrifugation (both at 4°C), once on a Jouan CR312 centrifuge (3,000 rpm, 10 min) and once on a Beckman Ti70 centrifuge (55,000 rpm, 1 h). Immunoprecipitation and immunoblotting were performed essentially as previously described (25) except that adsorbed proteins were released from protein A beads by incubation in sodium dodecyl sulfate sample buffer with 100 mM dithiothreitol at 95°C for 10 min. For immunoprecipitation and immunoblotting of the IR, we used an antibody against the C-terminal sequence of the IR β-subunit (JD433) that we produced, whereas for immunoprecipitation of IGF-1R, we used an antibody against the N terminus of the IGF-1R β-subunit (catalog no. sc-713; Santa Cruz Biotechnology, Santa Cruz, CA). For immunoblotting of phosphotyrosine, we used anti-phosphotyrosine, clone 4G10 (Upstate, Lake Placid, NY). For immunoblotting of phospho-Akt and total Akt, we used antibodies against phospho-Ser473 Akt and the carboxy terminus of Akt (catalog no. 9271 and 9272, respectively; Cell Signaling, Beverly, MA).
Microarray analysis.
Hearts were quickly removed from male mice and snap-frozen in liquid nitrogen on postnatal days 8 and 20. Total RNA was extracted from the heart muscle by homogenizing in TRIzol. Total RNA was purified using the RNeasy kit (QIAGEN) and used for cRNA synthesis (54). Fifteen micrograms of cRNA was hybridized to Affymetrix MG430A 2.0 chips. Intensity values were quantitated by using MAS 5.0 software (Affymetrix). Array values were normalized for overall intensity using linear regression and then normalized to a median of 1,500. To avoid loss of information regarding genes expressed at low levels, the data were not further filtered before analysis. Differences between groups were evaluated by the t test with unequal variance. GenMAPP and MAPPFINDER (12) were used to integrate expression data with known pathways, utilizing all probe sets with P < 0.05 for the MI2RKO, MIRKO, or MIGF1RKO versus DLox comparison. The pathways with the highest scores for each genotype versus DLox were further analyzed. For each gene with at least one probe set meeting the P < 0.05 criterion, the mean ratio of all probe sets (even those not meeting statistical significance criteria) is reported. This procedure had the effect of limiting the effect of outlier probe sets but may underestimate the true changes in gene expression. All primary array data are available at the Diabetes Genome Anatomy Project (DGAP) website (www.diabetesgenome.org).
Quantitative reverse transcriptase PCR (RT-PCR).
Total RNA isolated from day 8 hearts (1 μg) was used as the template for cDNA synthesis using the RT-for-PCR kit and random hexamer primers (BD Biosciences). Quantitative PCR was performed using the ABI Prism 7000 instrument and software and a SYBR green reaction mixture (Applied Biosystems) according to the manufacturer's instructions. All reactions yielded products with a single dissociation peak and a single band on ethidium-stained agarose gels. PCRs using as the template the product of mock cDNA synthesis reactions lacking reverse transcriptase yielded values more than 10 threshold cycles (CT) greater than reactions using cDNA template. Signals were normalized to the expression of TATA-binding protein (TBP), which was not expressed differently in the four strains studied according to the microarray data. Arbitrary units were calculated as follows: 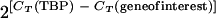 . The amplification primers were chosen from PrimerBank (http://pga.mgh.harvard.edu/primerbank) (53) and were as follows: TBP (TCTACCGTGAATCTTGGCTGT/CTGGCTCATAGCTCTTGGCTC), the fetal form (beta) of myosin heavy chain (β-MHC) (TTCATCCGAATCCATTTTGGGG/GCATAATCGTAGGGGTTGTTGG), Cycs (ATCAGGGTATCCTCTCCCCAG/CCAAATCTCCACGGTCTGTTC), Ndufa3 (ATGGCCGGGAGAATCTCTG/AGGGGCTAATCATGGGCATAAT), Cox4i1 (ATTGGCAAGAGAGCCATTTCTAC/CACGCCGATCAGCGTAAG T), Atp5b (AATCCCTCATCGAACTGGACG/GGTTCATCCTGCCAGAGACTA), Slc25a20 (GACGAGCCGAAACCCATCAG/AGTCGGACCTTGACCGTGT), Cpt2 (TCCCAATGCCGTTCTCAAAAT/CAGCACAGCATCGTACCCA), Pecr (CTCCGCCATACAGTGCAACAT/CAGGTTGGTTTCTATCACAGCA), and Hadha (TGCATTTGCCGCAGCTTTAC/GTTGGCCCAGATTTCGTTCA).
. The amplification primers were chosen from PrimerBank (http://pga.mgh.harvard.edu/primerbank) (53) and were as follows: TBP (TCTACCGTGAATCTTGGCTGT/CTGGCTCATAGCTCTTGGCTC), the fetal form (beta) of myosin heavy chain (β-MHC) (TTCATCCGAATCCATTTTGGGG/GCATAATCGTAGGGGTTGTTGG), Cycs (ATCAGGGTATCCTCTCCCCAG/CCAAATCTCCACGGTCTGTTC), Ndufa3 (ATGGCCGGGAGAATCTCTG/AGGGGCTAATCATGGGCATAAT), Cox4i1 (ATTGGCAAGAGAGCCATTTCTAC/CACGCCGATCAGCGTAAG T), Atp5b (AATCCCTCATCGAACTGGACG/GGTTCATCCTGCCAGAGACTA), Slc25a20 (GACGAGCCGAAACCCATCAG/AGTCGGACCTTGACCGTGT), Cpt2 (TCCCAATGCCGTTCTCAAAAT/CAGCACAGCATCGTACCCA), Pecr (CTCCGCCATACAGTGCAACAT/CAGGTTGGTTTCTATCACAGCA), and Hadha (TGCATTTGCCGCAGCTTTAC/GTTGGCCCAGATTTCGTTCA).
Statistics.
Values are expressed as means ± standard errors of the means. Data were subjected to statistical analysis using Student's t test (unequal variance) with differences between means considered significant for P values of <0.05.
RESULTS
Generation of MI2RKO mice.
To obtain muscle-specific disruption of Ir and/or Igf1r, we used the Cre-loxP gene recombination approach with the Cre recombinase under the control of the MCK promoter, which we have previously shown will restrict the deletion of a LoxP flanked gene to cardiac and skeletal muscle (8). Using this approach, six groups of mice were generated for study: control double Lox mice, muscle-specific IR knockout mice, muscle-specific IGF-1R knockout mice, MIRKO/MIGF1R heterozygous mice, MIGF1RKO/MIR heterozygous mice, and muscle-specific IR/IGF-1R double knockout mice. To validate the tissue-specific knockout of IR and/or IGF-1R, we analyzed tyrosine phosphorylation in vivo in the liver, heart, and skeletal muscle of DLox, MIRKO, MIGF1RKO, and MI2RKO mice in response to exogenous insulin or IGF-1. In the liver, insulin stimulation resulted in increased IR tyrosine phosphorylation in all genotypes, indicating normal IR signaling (Fig. 1a). Furthermore, liver IR protein levels were comparable in all four genotypes. In heart and skeletal muscle, IR phosphorylation in response to insulin was markedly reduced (>90%) in MIRKO and MI2RKO mice than in DLox and MIGF1RKO mice. While insulin-stimulated tyrosine phosphorylation of IR was barely detectable, some IR protein (15 to 30% of DLox) was found in MI2RKO whole-muscle protein extract, especially the heart. This most likely represents IR expressed in noncardiomyocyte cell types within the heart (4) and appears to be greater in MI2RKO heart lysate than in MIRKO heart lysate, suggesting that the combined lack of IR and IGF-1R in the cardiomyocyte, which leads to cardiac failure (see below), results in an increase in the expression of IR in surrounding noncardiomyocyte cells and/or an increase in the number of noncardiomyocyte cells within the heart itself. As expected, IGF-1R tyrosine phosphorylation in response to IGF-1 was markedly reduced in cardiac and skeletal muscle tissue of MIGF1RKO and MI2RKO mice than in DLox and MIRKO mice (Fig. 1b).
FIG. 1.

In vivo IR and IGF-1R signaling deficiency in muscle tissue of MI2RKO mice. Phosphotyrosine (pY) immunoblot analysis of (a) IR or (b) IGF-1R immunoprecipitates of tissue protein extracts from mice injected with saline (−) or with insulin or IGF-1 (+).
MI2RKO mice die within the first month of life.
Mice of all genotypes were born at the expected frequency (the frequency expected if the genotype had a Mendelian distribution) and grew normally for the first 2 weeks of life (data not shown). However, during the third week of life, MI2RKO growth rate slowed, and at postnatal day 20, MI2RKO mice weighed 15 to 20% less than control mice (Table 1). Furthermore, starting as early as P16, some MI2RKO mice appeared to be gasping for air and were less active. Once an individual MI2RKO mouse entered this stage, it would typically die within the next 2 days, with death being spontaneous or precipitated by even the stress of routine handling. In a study group of 69 mice, all 15 MI2RKO mice died between P17 and P27, whereas there were no deaths among 54 DLox, MIRKO, MIGF1RKO, MIRKO/+, and MIGF1RKO/+ pups during this time period (Fig. 2). In a second study group, again all MI2RKO mice died between P17 and P27. In addition, 3 mice out of a total of 13 MIRKO/+ mice in this group died within the first 6 months of life, including 1 mouse that showed symptoms similar to those described for MI2RKO mice beginning at 3 months of age. No deaths were observed in the two study groups for DLox, MIRKO, MIGF1RKO, and MIGF1RKO/+ mice up to 6 months of age. Furthermore, there was no statistical difference in body weight between DLox, MIRKO, MIGF1RKO, MIRKO/+, and MIGF1RKO/+ male mice between 1 and 6 months of age (data not shown). Thus, deficiency of both IR and IGF-1R signaling in muscle results in early death. One Ir allele in muscle is sufficient for survival and normal growth in the absence of IGF-1R, whereas loss of both Ir alleles and one Igf1r allele in the MIRKO/+ mice is sufficient for normal growth but appears to be associated with some increased early death, i.e., prior to 6 months of age.
TABLE 1.
MI2RKO male mice have small hearts at postnatal days 8 and 20 and small cardiomyocytes at postnatal day 20
| Timea | Mice | Heart wt (mg)b | Body wt (g)b | HW/BW (%)b | Cell circumference (μm)c |
|---|---|---|---|---|---|
| P8 | DLox | 27.6 ± 0.8 (13) | 4.4 ± 0.2 (13) | 0.64 ± 0.02 (13) | 26.5 ± 1.1 (7) |
| MI2RKO | 23.5 ± 1.1d (10) | 4.5 ± 0.2 (10) | 0.54 ± 0.03d (10) | 26.8 ± 1.2 (5) | |
| P20 | DLox | 68.6 ± 2.5 (14) | 9.9 ± 0.3 (22) | 0.67 ± 0.03 (9) | 49.7 ± 0.6 (4) |
| MIRKO | 57.2 ± 2.6d,g (6) | 10.3 ± 0.7 (12) | 0.49 ± 0.01e,g (4) | 49.4 ± 0.4 (4) | |
| MIGF1RKO | 79.3 ± 2.7d (11) | 10.4 ± 0.3 (15) | 0.75 ± 0.03 (9) | 49.0 ± 1.2 (4) | |
| MI2RKO | 50.5 ± 2.3e,g (11) | 8.3 ± 0.2e,g,h (15) | 0.60 ± 0.04f,h (7) | 39.7 ± 0.9e (4) |
P8, postnatal day 8; P20, postnatal days 19 to 21.
Values are means ± standard errors of the means. The number of mice is shown in parentheses.
Values are means ± standard errors of the means. The number of sections is shown in parentheses. At least 100 cell outlines per heart were measured for volume estimation.
Significantly different from value obtained with DLox mice (P < 0.01).
Significantly different from value obtained with DLox mice (P < 0.001).
Significantly different from value obtained with MIGF1RKO mice (P < 0.01).
Significantly different from value obtained with MIGF1RKO mice (P < 0.001).
Significantly different from value obtained with MIRKO mice (P < 0.05).
FIG. 2.

MI2RKO mice die within 4 weeks of birth. A group of 69 mice including all study group genotypes were monitored closely for 4 weeks after birth. All 15 MI2RKO mice died within this time period.
MI2RKO mice have normal glucose homeostasis.
Despite the lack of insulin and IGF-1 receptors in muscle, at 2 weeks of age, MI2RKO mice had slightly lower random-fed blood glucose levels compared with DLox control mice, and plasma insulin levels were normal (Fig. 3a and b). Furthermore, 2- to 3 week-old MI2RKO mice showed normal responses in glucose and insulin tolerance tests (Fig. 3c and d) despite nearly abolished insulin- and IGF-1-stimulated Akt phosphorylation (Fig. 3e), indicating that in young mice, muscle IR and IGF-1R signaling are not required for normal glucose homeostasis. Consistent with this observation, mice with all of the intermediate genotypes, MIRKO, MIGF1RKO, MIRKO/+, and MIGF1RKO/+, also had normal blood glucose and plasma insulin levels at 2 weeks of age (Fig. 3a and b). Moreover, no statistically significant differences were observed in MIRKO, MIGF1RKO, MIRKO/+, or MIGF1RKO/+ mice compared with DLox control mice up to 6 months of age in fed and fasted blood glucose and fasted plasma insulin levels or in glucose and insulin tolerance tests. There were also no significant differences in fed plasma insulin levels between the groups, although there was large variation within groups (Fig. 4a to e).
FIG. 3.

MI2RKO mice have normal glucose homeostasis. (a) Blood glucose and (b) plasma insulin levels were measured in 2-week-old (2 wks), random-fed male mice (four to nine mice per group). The value that was significantly different (P < 0.05) from the value for DLox mice is indicated by an asterisk. (c) Glucose tolerance tests were performed on 2.5-week-old DLox and MI2RKO male mice (three to five mice per group). (d) Insulin tolerance tests were performed on 2.5-week-old DLox and MI2RKO male mice (three mice per group). (e) Phospho-Akt (pAkt) (phospho-Ser473) and total Akt immunoblot analysis of whole-skeletal-muscle protein extracts from mice stimulated with saline (−), insulin (Ins), or IGF-1 (IGF1).
FIG. 4.

Mice with combinatorial knockouts have normal glucose homeostasis. (a) Blood glucose levels in random-fed and fasted 6-month-old male DLox, MIRKO, MIGF1RKO, MIRKO/+, and MIGF1RKO/+ mice (7 to 10 mice per group). (b and c) Plasma insulin levels were measured in 6-month-old random-fed mice (b) or mice fasted overnight (c) (six to nine mice per group). (d) Glucose tolerance tests were performed on 4- and 6-month-old DLox, MIRKO, MIGF1RKO, MIRKO/+, and MIGF1RKO/+ male mice (five to seven mice per group). (e) Insulin tolerance tests were performed on mice of the same genotypes and at the same ages as in panel d (seven to nine mice per group).
MI2RKO mice have small hearts and reduced cardiomyocyte size.
Necropsy of mice sacrificed at day P8 and P20 revealed significant reductions in MI2RKO heart weight of approximately 15% and 25%, respectively, compared to DLox control mice (Table 1). When heart weight was expressed relative to body weight (as a ratio of heart weight to body weight [HW/BW]), MI2RKO animals also had a significantly reduced HW/BW ratio at 8 days of age. However, by 20 days of age, MI2RKO mice had reduced body weight compared to all other genotypes, such that the HW/BW ratio was not statistically different from that of DLox control mice. Consistent with previous reports (4, 22), MIRKO mice also had smaller hearts than the control mice did, both in terms of absolute weight and HW/BW ratio, but they were significantly larger than those of MI2RKO mice. In contrast, the hearts from MIGF1RKO mice were slightly heavier than the hearts from control mice.
To determine whether the reduced heart weight was due to reduction in cell size or cell number, we estimated cell size in DLox and MI2RKO mice at day 8 and in all genotypes at day 20 by staining heart muscle sections with wheat germ agglutinin to allow accurate measurement of cell circumference. There was no significant difference in the circumference of cardiomyocytes from MIRKO, MIGFR1KO, and DLox hearts at day 20, but cardiomyocytes from the hearts of MI2RKO animals were significantly reduced in size. This is similar to the finding in cardiac muscle-specific insulin receptor knockout mice at 12 weeks of age (4). The fact that our MIRKO mice did not show any reduction in cardiomyocyte size may be explained by the much younger age of the MIRKO mice in the current study. Indeed, differences in cardiomyocyte size between wild-type and MI2RKO hearts were not apparent at postnatal day 8 (Table 1) consistent with an age-dependent development of this clinical phenotype. The finding that the cell size phenotype in young mice is exaggerated in MI2RKO mice compared to MIRKO mice is consistent with a role for both IR and IGF-1R in cardiomyocyte growth.
Prior studies have implicated the IR/IGF-1R-PI3K pathway and the downstream target Akt in the regulation of cardiac growth (see Discussion). In DLox, MIRKO, and MIGF1RKO mice, intravenous injection of both insulin and IGF-1 caused activation of Akt as measured by Akt phosphorylation (Fig. 5a). and Fig. 5b). Despite this defect in in vivo heart function, there was no compensatory increase in the heart rate. The echocardiographic data also revealed a thinning of the interventricular septum and the posterior wall of the left ventricle in MI2RKO mice, consistent with MI2RKO mice developing dilated cardiomyopathy. This conclusion was supported by histological analysis showing a marked thinning of the ventricular walls and enlarged ventricles in MI2RKO hearts (Fig. 5b and c). By contrast, knockout of either receptor alone in MIRKO and MIGF1RKO mice had no effect on echocardiographic parameters at P17.
FIG. 5.

MI2RKO mice develop dilated cardiomyopathy. (a) Phospho-Akt (pAkt) (phospho-Ser473) and total Akt immunoblot analysis of whole-heart protein extracts from mice stimulated with saline (—), insulin (Ins), or IGF-1 (IGF1). (b) Longitudinal sections of hearts removed from 20-day-old DLox, MIRKO, MIGF1RKO, and MI2RKO mice (left panels). Representative echocardiograms from 17-day-old male mice (right panels). (c) Transverse sections of hearts in 20-day-old DLox and MI2RKO mice. The right ventricle (RV) and left ventricle (LV) are indicated.
To investigate whether mice with partial deficiency in IR/IGF-1R signaling would develop reduced cardiac function with age, 6-month-old DLox, MIRKO, MIGF1RKO, MIRKO/+, and MIGF1RKO/+ male mice were subjected to echocardiographic analysis. Consistent with data from CIRKO mice (4), MIRKO mice showed a 16% reduction in left ventricular fractional shortening versus DLox mice, although this did not reach statistical significance (data not shown). Additional knockout of one allele of the Igf1r in MIRKO/+ mice resulted in an even greater reduction (23% [P < 0.05]) in left ventricular fractional shortening, indicating a more severe heart defect and providing a possible explanation for the small number of premature deaths observed among MIRKO/+ mice. By contrast, MIGF1RKO and MIGF1RKO/+ mice had normal echocardiographic parameters. Thus, complete knockout of IR and IGF-1R signaling in muscle results in dilated cardiomyopathy and death from heart failure. One Ir allele was sufficient for normal cardiac function even in the absence of IGF-1R. Mice lacking IR in cardiac muscle had reduced cardiac function, but one Igf1r allele was sufficient to prevent heart failure in most animals.
MI2RKO hearts have ultrastructural abnormalities.
Histological analysis of MI2RKO hearts at postnatal day 20 revealed normal gross tissue architecture without increased interstitial fibrosis on hematoxylin and eosin and trichrome staining (data not shown). Electron microscopy, however, revealed irregular and disrupted sarcomeric Z and M lines, as well as increased mitochondria with central crowding in MI2RKO hearts relative to wild-type control hearts (Fig. 6). The mitochondria in MI2RKO hearts also appeared less electron dense than those from wild-type control hearts.
FIG. 6.

Altered morphology of cardiac muscle of MI2RKO hearts at the ultrastructural level. The Z and M lines of MI2RKO hearts are disrupted, and the number of mitochondria is increased. Arrows indicate Z lines in DLox (Lox) and MI2RKO (DKO) myofibrils. Electron microscopic images were obtained from longitudinal sections of cardiac muscle from DLox and MI2RKO mice at postnatal day 20 and processed as described in Materials and Methods. Scale bars, 1 μm.
Altered gene expression in MI2RKO hearts.
To further investigate the cause of the abnormalities in cardiac function in MI2RKO hearts, we performed microarray gene expression analysis. RNA was isolated from the hearts of three to six animals of each genotype at both postnatal days 8 and 20 and was used for microarray analysis. Of 22,602 probe sets represented on the array (Affymetrix MG403A 2.0 chips), 529 were differentially expressed (nominal P < 0.01) between DLox and MI2RKO hearts as early as postnatal day 8. Similar numbers of genes were changed in the hearts of MIRKO and MIGF1RKO mice at day 8, with approximately equal numbers of genes up-regulated and down-regulated (Fig. 7). By day 20, far more genes were significantly changed in the MI2RKO hearts than in the hearts of the two single knockout strains, likely in response to the developing heart failure. All of the primary data are available on the Diabetes Genome Anatomy Project (DGAP) website (www.diabetesgenome.org), and thus, we focus here on those alterations that are most likely contributory to the cardiac failure.
FIG. 7.

Number of probe sets detecting altered gene expression (P < 0.01). Heart failure is accompanied by a large shift in gene expression patterns. This graph indicates the number of probe sets up- and down-regulated in the hearts of MIRKO, MIGF1RKO, and MI2RKO mice at postnatal days 8 and 20. The Affymetrix chip contained 22,602 probe sets. Between three and six hearts were analyzed, each with an individual RNA preparation and chip hybridization, for each genotype and time point.
In agreement with the observed ultrastructural abnormalities in the contractile apparatus of MI2RKO hearts, by day 20, expression of 10 genes encoding sarcomeric proteins were changed by more than 1.5-fold (Table 3). Interestingly, all were up-regulated from 1.6- to 54.3-fold in MI2RKO hearts compared to DLox hearts. Of these genes, seven are structural components of the Z disk (41), suggesting that changes in the stoichiometry of these proteins might contribute to the disorganization of Z lines seen by EM. One of these proteins, cardiac ankyrin repeat protein (CARP) (up-regulated eightfold), is found both in the nucleus and at the Z disk and has a role in regulation of other structural genes of the sarcomere (3, 20, 57). By contrast, the expression of these genes was not substantially altered in MIGF1RKO hearts at day 20, consistent with the normal cardiac function in this strain. Τmp2, CARP, alpha-actin 1 (Acta1), and myomesin 2 were also up-regulated by at least twofold in MIRKO hearts compared to DLox hearts at day 20. These changes are consistent with the nonsignificant reduction in cardiac function (left ventricular fractional shortening) in MIRKO hearts at day 20 and 6 months (Table 2).
TABLE 3.
Genes of the contractile apparatus are up-regulated in MI2RKO hearts at postnatal day 20a
| Contractile apparatus proteinb | Mean ratio of expressionc at:
|
|||||
|---|---|---|---|---|---|---|
| P8
|
P20
|
|||||
| MI2RKO | MIRKO | MIGF1RKO | MI2RKO | MIRKO | MIGF1RKO | |
| β-MHC | 4.32 ± 0.52 | 54.31 ± 5.20 | ||||
| {5.74 ± 0.92d | 1.88 ± 0.53 | 0.99 ± 0.24 | ||||
| Tnni1 | 22.55 ± 5.52 | |||||
| Tpm2 | 1.44 ± 0.10 | 0.91 ± 0.04 | 1.50 ± 0.11 | 8.63 ± 1.16 | 2.26 ± 0.24 | 1.24 ± 0.15 |
| CARP | 0.96 ± 0.06 | 7.96 ± 0.31 | 2.12 ± 0.19 | 1.31 ± 0.12 | ||
| Acta1 | 3.92 ± 0.71 | 2.65 ± 0.28 | ||||
| Flnc | 2.81 ± 0.41 | |||||
| Myom2 | 0.68 ± 0.04 | 2.82 ± 0.24 | 2.16 ± 0.23 | |||
| Actn4 | 1.75 ± 0.09 | |||||
| Smpx | 1.55 ± 0.18 | |||||
| Des | 0.77 ± 0.07 | 0.78 ± 0.05 | 1.57 ± 0.17 | |||
Genes encoding proteins associated with the contractile apparatus were selected if the mean change in expression (n-fold change) by microarray for any pairwise comparison of MI2RKO, MIRKO, or MIGF1RKO mice versus the mean change in expression for DLox mice for all probe sets at either time point was >1.5.
Genes encoding proteins directly associated with the Z disk are shown in bold type. β-MHC, myosin heavy chain, beta isoform; Tnni1, troponin, slow skeletal, 1; Tpm2, tropomyosin 2; CARP, cardiac ankyrin repeat protein; Acta1, alpha-actin 1; Flnc, filamin C; Myom2, myomesin 2; Actn4, alpha-actinin 4; Smpx, small muscle protein, X-linked; Des, desmin.
The mean ratio of expression of the indicated mice versus that of DLox mice for all probe sets specific to the genes encoding the listed proteins is reported. Values are means ± standard errors of the means. Cells are blank when no probe sets met the significance criterion. The lower values for β-MHC at P8 indicated by the bracket represent quantitative real-time PCR data, for which mean ratios versus that of DLox mice are reported regardless of significance.
Significantly different from value obtained with DLox mice (P < 0.01).
TABLE 2.
Echocardiographic analysis of in vivo heart function in 17-day-old male micea
| Mice (n) | IVS-D (mm) | IVS-S (mm) | LV-D (mm) | LV-S (mm) | PW-D (mm) | PW-S (mm) | LVFS (%) | HR (no. of contractions min−1) |
|---|---|---|---|---|---|---|---|---|
| DLox (8) | 0.40 ± 0.00 | 0.75 ± 0.04 | 3.09 ± 0.06 | 1.26 ± 0.08 | 0.40 ± 0.00 | 0.76 ± 0.04 | 59.1 ± 2.3 | 728 ± 11 |
| MIRKO (3) | 0.37 ± 0.03 | 0.70 ± 0.06 | 3.07 ± 0.09 | 1.40 ± 0.15 | 0.40 ± 0.00 | 0.73 ± 0.03 | 54.8 ± 3.2 | 700 ± 20 |
| MIGF1RKO (2) | 0.40 ± 0.00 | 0.75 ± 0.05 | 3.20 ± 0.10 | 1.10 ± 0.20 | 0.40 ± 0.00 | 0.75 ± 0.05 | 66.0 ± 5.6 | 705 ± 15 |
| MI2RKO (6) | 0.28 ± 0.02b | 0.48 ± 0.02b | 3.30 ± 0.07b | 1.98 ± 0.07b | 0.30 ± 0.03b | 0.50 ± 0.03b | 39.4 ± 2.1b | 645 ± 15b |
Values are means ± standard errors of the means. IVS-D, diastolic interventricular septum wall thickness; IVS-S, systolic interventricular septum wall thickness; LV-D, diastolic left ventricle diameter; LV-S, systolic left ventricle diameter; PW-D, diastolic left ventricle posterior wall thickness; PW-S, systolic left ventricle posterior wall thickness; LVFS, left ventricular fractional shortening; HR, heart rate.
Significantly different from value obtained with DLox mice (P < 0.05).
The most up-regulated gene was the fetal form (beta) of myosin heavy chain, with expression 54-fold greater in MI2RKO mice than in DLox mice at day 20. β-MHC was also up-regulated by 4.3-fold at day 8 and was the only gene of the contractile apparatus with greatly altered expression at the early time point. Quantitative RT-PCR confirmed an up-regulation of β-MHC (5.7-fold [P < 0.01]) at day 8 (Table 3, lower values indicated by the bracket). The expression of the adult form of myosin heavy chain, α-MHC, was not significantly altered at day 8 or 20 compared to DLox. A shift from the adult (alpha, fast) to fetal (beta, slow) isoform of MHC has been observed in failing hearts, and it has been suggested that this isotype switch might cause further deterioration in cardiac function (35). Near complete experimental replacement of α-MHC with β-MHC in transgenic mice results in a modest defect in LV shortening, but no signs of LV failure even with chronic exercise (23, 50). Therefore, the approximately fivefold increase in β-MHC seen in day 8 MI2RKO mice cannot explain subsequent development of heart failure and likely represents a response to early myocardial dysfunction that is not clinically apparent.
Expression of electron transport and mitochondrial fatty acid beta-oxidation genes is reduced in MI2RKO hearts.
In order to identify changes in gene expression that might cause the heart failure phenotype, rather than be secondary to cardiac failure, we chose to focus further on gene expression changes at day 8, when all strains appeared healthy and were growing normally. At this point, a total of 2,501 genes were altered in expression in MI2RKO hearts versus DLox hearts at the P < 0.05 level, but of these, only 53 genes were changed by more than 2.5-fold with a nominal P value of <0.01; the maximal change was 9-fold (data not shown). Using MAPPFINDER and GenMAPP (12) to integrate gene expression data with known pathways for all probe sets with nominal P values of <0.05, the top ranked map annotator and pathway profiler (MAPP) terms for the comparison between MI2RKO and DLox hearts were electron transport chain and mitochondrial fatty acid beta-oxidation genes with Z scores of 4.3 and 3.1, respectively. Of 27 genes included in the ETC MAPP (complexes I through IV, F1Fo ATPase, and cytochrome c) meeting the P < 0.05 significance level, 27 of 27 were down-regulated, although as has been observed in other situations with insulin resistance, the magnitude of decrease was modest (mean of 19%) (Table 4). Similarly, of MFABO genes with P < 0.05, all eight genes were down-regulated by a mean of 22% (Table 5).
TABLE 4.
Genes of the electron transport chain are down-regulated in MI2RKO and MIRKO hearts
| Electron transport chain proteina | Mean ratio of expressionb
|
||
|---|---|---|---|
| MI2RKO | MIRKO | MIGF1RKO | |
| Cytochrome c | |||
| Cycs | 0.84 ± 0.04 | ||
| {0.79 ± 0.07c | 1.04 ± 0.04 | 1.07 ± 0.04 | |
| Cyct | 0.55 ± 0.13 | ||
| Complex I (NADH dehydrogenase [ubiquinone]) | |||
| Ndufa3 | 0.72 ± 0.04 | 0.72 ± 0.04 | |
| {0.73 ± 0.03d | 0.93 ± 0.07 | 0.99 ± 0.06 | |
| Ndufa6 | 0.84 ± 0.04 | ||
| Ndufa7 | 0.83 ± 0.02 | 0.86 ± 0.03 | |
| Ndufb4 | 0.84 ± 0.04 | 0.90 ± 0.06 | |
| Ndufb9 | 0.80 ± 0.03 | 0.76 ± 0.03 | 0.80 ± 0.04 |
| Ndufb2 | 0.82 ± 0.03 | ||
| Ndufa4 | 0.73 ± 0.06 | ||
| Ndufa1 | 0.73 ± 0.06 | 0.75 ± 0.04 | 0.68 ± 0.03 |
| Ndufv2 | 0.79 ± 0.03 | 0.87 ± 0.03 | |
| Ndufa9 | 0.79 ± 0.03 | 0.81 ± 0.03 | 0.77 ± 0.05 |
| Complex II (succinate dehydrogenase) | |||
| Sdhc | 1.13 ± 0.12 | 1.08 ± 0.16 | |
| Sdhd | 1.25 ± 0.15 | ||
| Complex III (ubiquinol-cytochrome c) reductase | |||
| Uqcrc1 | 0.98 ± 0.06 | 1.00 ± 0.11 | |
| Uqcrc2 | 0.93 ± 0.07 | 1.00 ± 0.11 | |
| Uqcrb | 0.87 ± 0.08 | ||
| 1500040F11Riken | 0.71 ± 0.07 | 0.71 ± 0.05 | 0.69 ± 0.08 |
| Complex IV (cytochrome c oxidase) | |||
| Cox4i1 | 0.77 ± 0.05 | 0.73 ± 0.04 | |
| {0.82 ± 0.08e | 0.94 ± 0.07 | 0.88 ± 0.09 | |
| Cox5a | 1.02 ± 0.10 | ||
| Cox5b | 0.75 ± 0.04 | 0.77 ± 0.05 | 0.75 ± 0.04 |
| Cox6a2 | 0.77 ± 0.04 | 0.60 ± 0.05 | |
| Cox7a2 | 0.86 ± 0.12 | 0.86 ± 0.04 | |
| Cox8b | 0.75 ± 0.06 | 0.73 ± 0.06 | |
| Cox8a | 0.71 ± 0.07 | 0.85 ± 0.17 | |
| Complex V (ATP synthase) | |||
| Atp5a1 | 0.94 ± 0.08 | ||
| Atp5b | 0.83 ± 0.03 | 0.82 ± 0.05 | |
| {0.85 ± 0.05f | 0.87 ± 0.05e | 0.97 ± 0.08 | |
| Atp5c1 | 0.95 ± 0.07 | ||
| Atp5d | 0.83 ± 0.03 | 0.84 ± 0.06 | 0.90 ± 0.08 |
| Atp5e | 0.82 ± 0.04 | ||
| Atp5j | 0.83 ± 0.04 | ||
| Atp5o | 0.96 ± 0.06 | 1.16 ± 0.15 | |
| Atp5g1 | 0.85 ± 0.04 | 0.82 ± 0.06 | |
| Atp5g2 | 0.99 ± 0.10 | ||
| Atp5g3 | 0.79 ± 0.04 | 0.77 ± 0.05 | 0.78 ± 0.05 |
| Atp5h | 0.80 ± 0.03 | 0.84 ± 0.02 | 0.80 ± 0.02 |
| Atp5k | 0.67 ± 0.06 | ||
| Atp5j2 | 0.85 ± 0.03 | 0.85 ± 0.03 | |
| Atp5l | 0.80 ± 0.04 | ||
| Atp5s | 0.82 ± 0.06 | 0.84 ± 0.04 | |
| Atpi (inhibitor) | 1.37 ± 0.15 | ||
| No. of genes significantly decreased (microarray data)/total no. of genes | 27/27 | 26/30 | 13/17 |
All genes encoding electron transport chain proteins for which at least one probe set had a P value of <0.05 for one of the pairwise comparisons of MI2RKO, MIRKO, or MIGF1RKO heart versus DLox heart are listed.
For each individual pairwise comparison, the mean ratio of expression of the indicated mice versus that of DLox mice for all probe sets specific to genes encoding the listed proteins is reported. Cells are blank when no probe set met the significance criterion. The lower set of values indicated by each set of brackets is quantitative real-time PCR data, for which mean ratios versus that of DLox are reported regardless of significance.
Significantly different from value obtained with DLox mice (P < 0.05).
Significantly different from value obtained with DLox mice (P < 0.0005).
Significantly different from value obtained with DLox mice (P = 0.08).
Significantly different from value obtained with DLox mice (P = 0.07).
TABLE 5.
Genes of mitochondrial fatty acid beta-oxidation are down-regulated in MI2RKO hearts
| Fatty acid beta-oxidation proteina | Enzyme | Mean ratio of expressionb
|
||
|---|---|---|---|---|
| MI2RKO | MIRKO | MIGF1RKO | ||
| Facl2 | Fatty acid-CoA ligase,g long chain, 2 | 0.90 ± 0.03 | 1.09 ± 0.05 | 1.05 ± 0.05 |
| Cpt1a | Carnitine palmitoyltransferase 1a | 1.25 ± 0.14 | ||
| Slc25a20 | Carnitine/acylcarnitine translocase | 0.85 ± 0.08 | ||
| {0.80 ± 0.07f | 1.06 ± 0.11 | 1.12 ± 0.10 | ||
| Cpt2 | Carnitine palmitoyltransferase 2 | 0.80 ± 0.12 | 1.31 ± 0.12 | |
| {0.60 ± 0.05e | 0.84 ± 0.05c | 1.03 ± 0.08 | ||
| Acadm | Acyl-CoA dehydrogenase, medium chain | 0.77 ± 0.04 | ||
| Acads | Acyl-CoA dehydrogenase, short chain | 0.65 ± 0.04 | 0.73 ± 0.05 | |
| Pecr | Peroxisomal trans-2-enoyl-CoA reductase | 0.65 ± 0.04 | ||
| {0.99 ± 0.10 | 0.92 ± 0.09 | 1.05 ± 0.13 | ||
| Hadha | Trifunctional enzyme alpha subunit | 0.80 ± 0.04 | ||
| {0.77 ± 0.05d | 0.96 ± 0.05 | 0.97 ± 0.05 | ||
| Scp2 | Sterol carrier protein 2 | 0.81 ± 0.06 | 0.85 ± 0.03 | |
| No. of genes significantly decreased (microarray data)/total no. of genes | 8/8 | 0/1 | 2/5 | |
The genes encoding all proteins for which at least one probe set on the microarray had a P value of <0.05 for one of the pairwise comparisons of MI2RKO, MIRKO, or MIGF1RKO heart versus DLox heart are listed.
For each individual pairwise comparison, the mean ratio of expression of the indicated mice versus DLox mice for all probe sets specific to the genes encoding the listed proteins is reported. Cells are blank when no probe set met the significance criterion. The lower set of values in brackets are quantitative real-time PCR data, for which mean ratios versus DLox are reported regardless of significance.
Significantly different from value obtained with DLox mice (P < 0.05).
Significantly different from value obtained with DLox mice (P < 0.005).
Significantly different from value obtained with DLox mice (P < 0.0005).
Significantly different from value obtained with DLox mice (P = 0.08).
CoA, coenzyme A.
The top ranked MAPP terms for the comparison between MIRKO and DLox hearts were also ETC genes (Z = 6.6), and of the 30 ETC genes with P < 0.05 for the MIRKO versus DLox comparison, 26 were down-regulated by a mean of 19% (Table 3). By contrast, in the MIRKO mouse heart, none of the genes involved in MFABO were significantly decreased (Table 5).
MFABO was one of the two top ranked MAPP terms for the MIGF1RKO versus DLox heart comparison (Z = 2.22) with ribosomal proteins being the other (Z = 3.18). However, of the five genes on the MFABO MAPP that met the P < 0.05 criterion for this comparison, two were down-regulated, while the others were up-regulated (Table 5), in contrast to the consistent pattern of down-regulation seen in MI2RKO hearts. The ETC MAPP achieved a Z score of only 1.66 for the MIGF1RKO versus DLox heart comparison, with 13 of 17 genes with P < 0.05 down-regulated (Table 4). Inspection of the ribosomal protein MAPP revealed 16 genes up-regulated by a mean of 22% and 2 genes down-regulated by a mean of 16%.
These data demonstrate that there is coordinate down-regulation of ETC genes in MIRKO and MI2RKO hearts (Z scores of 4.3 and 6.6, respectively) but that ETC genes were not greatly overrepresented among genes with altered expression in MIGF1RKO hearts (Z score of 1.66). This suggests that the coordinate decrease in ETC genes in MI2RKO and MIRKO hearts is due to their common defect, deletion of the insulin receptor. A similar coordinated down-regulation is found in the MFABO pathway of MI2RKO hearts, and this is unique to the double mutant hearts.
We chose four genes of the ETC pathway and four genes of the MFABO pathway for measurement of gene expression by quantitative RT-PCR experiments (Tables 4 and 5, lower values indicated by each set of brackets). Significantly reduced expression in MI2RKO versus DLox hearts was confirmed for cytochrome c (21% decrease) and Ndufa3 (complex I, 27% decrease), and reductions in Cox4i1 (complex IV, 18% decrease) and Atp5b (complex V, 15% decrease) just missed statistical significance (P = 0.08 and P = 0.07, respectively). In the MFABO pathway, significantly reduced expression in MI2RKO versus DLox hearts was confirmed for Cpt2 (40% reduction [P < 0.0005]) and Hadha (23% decrease [P < 0.005]), while a decrease in Slc25a20 expression (20% [P = 0.08]) narrowly missed significance. Quantitative PCR did not confirm a reduction in Pecr expression in MI2RKO hearts.
Taken together, the gene expression data suggest a coordinated down-regulation of ETC genes in MI2RKO and MIRKO hearts and a coordinated down-regulation of MFABO genes in MI2RKO hearts. These alterations in genes required for energy generation correspond to alterations in cardiac function: MIRKO hearts exhibit mild defects in contractility at 6 months, while MI2RKO hearts fail by 3 weeks of age. This suggests a model in which lack of insulin signaling down-regulates genes of the ETC pathway and decreases ATP production capacity in MIRKO hearts, leading to decreased myocardial contractile force, but not overt heart failure. We hypothesize that the additional down-regulation of MFABO genes required for utilization of the hearts favored fuel, fatty acids, in MI2RKO hearts decreases the input of acyl coenzyme A into the Krebs cycle. This second hit decreases production of ATP below a sustainable threshold during a period of rapid cardiac growth, contributing to the development of heart failure.
DISCUSSION
Despite the homology between the IR and IGF-1R and their intracellular signaling pathways, disruption of the Ir and Igf1r genes in mice at the whole-body level produces very different phenotypes (1, 21, 28), indicating that IR signaling and IGF-1R signaling are physiologically distinct. This functional in vivo differentiation could be due to a number of factors, including tissue-specific differences in the relative expression of the two receptors, differences in signaling pathways activated by each receptor, modulation of IGF-1 activity by specific binding proteins, and differences in the rate of ligand dissociation and receptor internalization (5). Since a combined lack of both receptors at the whole-body level results in perinatal lethality (29), we have studied the overlap between these two pathways in muscle by creating a double tissue-specific knockout. We find that while disruption of IGF-1R alone in muscle has no effect on mortality in the mouse and disruption of IR plus one Igf1r allele, MIRKO/+, leads to a modest increase in mortality at 6 months, the combined lack of both receptors results in early-onset dilated cardiomyopathy and death from heart failure within the first month of life. Thus, some level of IR or IGF-1R signaling is required for heart development and function, and on the basis of combinatorial knockout data, the IR seems more critical than the IGF-1R.
Prior studies have implicated IR/IGF-1R signaling in the regulation of cardiac development, growth, and function. Mice with a cardiac muscle-specific disruption of IR (CIRKO) present with a small heart phenotype due to a decrease in cardiomyocyte size (4). Deletion of both IR and IGF-1R in the heart causes a more severe cell growth defect such that MI2RKO hearts are even smaller than those of the MIRKO mouse and already have reduced cell size by day 20, whereas the cell size is not changed in MIRKO heart at day 20. Conversely, overexpression of IGF-1R specifically in the mouse heart produces cardiac hypertrophy and an increase in cardiomyocyte size (32). This effect on cardiomyocyte growth seems to be mediated via the PI3K pathway. Thus, cardiac hypertrophy is also observed in mice in which this pathway is stimulated by cardiac muscle-specific expression of constitutively active forms of PI3K (47) and in animals in which the phosphatase PTEN (phosphatase and tensin homologue deleted on chromosome 10) has been knocked out (11). PTEN inhibits PI3K signaling by dephosphorylating phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)P3], the product of PI3K activity. Conversely, cardiac hypotrophy is observed in mice in which this pathway is down-regulated by cardiac muscle-specific expression of a dominant-negative form of PI3K (47) or by muscle-specific deletion of 3′-phosphoinositide-dependent protein kinase 1 (mPDK-1−/−). Interestingly, the mPDK-1−/− mouse presents with a phenotype very similar to the one described here for MI2RKO mice, including enlarged ventricles, a thinning of the heart walls, reduced in vivo cardiac function by echocardiography, and early death, although mPDK-1−/− mice live longer than MI2RKO mice (5 to 11 weeks versus 3 to 4 weeks) (34). These observations suggest that an early event in causing the death of the MI2RKO mouse is a failure to activate the PI3K pathway and the downstream effector PDK-1, although the more severe phonotype of MI2RKO mice may be due to actions of the insulin and/or IGF-1 receptor not medicated through PDK-1. PDK-1 is an important regulator of several downstream kinases, including protein kinase C, p70S6K, and protein kinase B/Akt. Cardiac muscle-specific expression of Akt results in cardiomyocyte hypertrophy (9, 48) and rescues the small heart phenotype of CIRKO mice (49), indicating that Akt plays a role in regulating heart growth. Consistent with this hypothesis, Akt activation in response to insulin or IGF-1 was markedly reduced in MI2RKO hearts.
In addition to effects on cardiac muscle growth, IGF-1 has been shown to have a role in response to increased myocardial loading. Rats subjected to swimming exercise increase expression of IGF-1 in heart tissue (45), and IGF-1 has a rapid, calcium-dependent, positive inotropic effect on human heart muscle isolated from failing hearts that is PI3 kinase pathway dependent (52). These findings suggest that increased IGF-1 signaling is part of the normal response to increased loading and that increased IGF-1 signaling can improve compromised cardiac function. Thus, intact IGF-1 signaling may be important in limiting the degree of cardiac dysfunction in CIRKO and MIRKO hearts that lack only the IR.
Mechanistically, the double insulin-IGF-1 signaling defect appears to produce both structural and functional abnormalities in the heart. The cells of MI2RKO hearts have severe abnormalities in the organization of the sarcomere, including disorganization of the Z and M lines. This is accompanied by changes in the expression level of several genes encoding structural components of the sarcomere, including up-regulation of the fetal form of myosin heavy chain, as well as multiple structural components of the Z disk. It is possible that this results in altered stoichiometry of sarcomeric Z-disk components and the ultrastructural disorganization observed.
Increased expression of β-MHC and CARP has been associated with pressure overload heart failure (2, 35, 56), and high-level overexpression of β-MHC reduces cardiac contractility (7, 23). However, only β-MHC was overexpressed in MI2RKO hearts prior to the onset of overt heart failure, and the level of overexpression at day 8 is not as high as that which is associated with heart failure based on overexpression studies of otherwise normal hearts (23, 50). Therefore, it is possible that alterations in sarcomeric gene expression are a response to cardiac dysfunction, rather than its cause.
Gene array analysis also revealed coordinated down-regulation of electron transport chain gene expression in MI2RKO hearts as early as day 8. A similar, but somewhat less severe, change is also observed in MIRKO, but not MIGF1RKO, hearts. This is consistent with the fact that in contrast to MIGF1RKO mice, MIRKO mice do develop nonsignificant, but consistent, defects in left ventricular shortening at 20 days and 6 months, suggesting that the down-regulation of ETC genes may have consequences for cardiac function. This is also consistent with the phenotype of the cardiac muscle-specific deletion of the insulin receptor (CIRKO), which also had mildly increased LV systolic dimension, decreased posterior wall thickness, and decreased fractional shortening as the mouse ages (4).
Deletion of both the IR and IGF-1R was required for a coordinated down-regulation of mitochondrial fatty acid β-oxidation genes, and this was observed as early as day 8. Fatty acids are the primary fuel of the adult heart, and thus, a defect in MFABO might be poorly tolerated, especially in the context of a defect in the electron transport chain. Although the magnitude of the gene expression changes detected by microarray and quantitative RT-PCR analysis was modest (maximum 40% reduction [Tables 4 and 5]), many genes in both pathways were decreased. This is similar to the modest, but coordinated, changes in gene expression observed in skeletal muscle in insulin-deficient diabetic mice and type 2 diabetic humans (33, 37). According to metabolic control theory, modest changes in multiple sites of control may lead to large changes in flux through pathways required for ATP production (7). This suggests that the changes identified here could have a significant effect in a tissue that is highly dependent on a steady supply of ATP, such as the heart. Ultrastructural analysis of MI2RKO hearts revealed increased numbers of mitochondria that appear less electron dense than in control animals. This is consistent with a proliferative response to lack of available ATP, which would be the expected consequence of a combined defect in the electron transport chain and mitochondrial fatty acid β-oxidation. Together these combined defects are likely to be important contributors to the cardiomyopathy phenotype of MI2RKO mice.
Diabetic patients and lean, insulin-resistant offspring of diabetic parents have decreased rates of insulin-stimulated ATP synthesis and increased intramyocellular lipid concentrations (30, 38, 39). Consistent with this finding, electron transport and mitochondrial fatty acid beta-oxidation genes are down-regulated in the skeletal muscles of patients with diabetes mellitus (DM) or those with a family history of DM (33, 37). The levels of PGC1α and NRF, transcription factors involved in the activation of electron transport and mitochondrial fatty acid beta-oxidation genes were found to be down-regulated in DM patients (33, 37). We examined expression of genes that regulate transcription of nucleus-encoded mitochondrial genes and genes in the MFABO pathway, including NRF-1, PGC1α, PGC1β (PERC), TFAM, MEF isotypes, YY1, CREM, CREB1, CREB3, PPARα, ERRα, and SIN3. There was equivocal evidence for a modest increase in MEF2c in MI2RKO hearts (two of five probe sets showed 30% and 50% increases, while the other three probe sets showed no change). None of the other genes were differentially regulated in MI2RKO hearts. Therefore, we did not find evidence that changes in the levels of these transcription factor that could serve as a proximate cause of the observed changes in expression of ETC and MFABO genes. In previous studies from our laboratory of adipose tissue taken from the fat-specific IR knockout in which we compared a proteomic analysis to gene expression analysis, we found additional changes at the protein level without a corresponding change in RNA levels (6). Thus, future studies using a proteomic approach may uncover additional factors leading to altered mitochondrial electron transport and fatty acid beta-oxidation.
Skeletal muscle is a major site of glucose disposal, accounting for up to 80% of glucose uptake in humans after a glucose load (13, 31). Insulin stimulates glucose uptake in most tissues, and peripheral insulin resistance is an early stage in humans predisposed to type 2 diabetes (27, 31). Mice lacking the insulin-sensitive glucose transporter GLUT4 specifically in muscle develop insulin resistance and glucose intolerance, showing that muscle glucose uptake is indeed important for glucose homeostasis in the mouse (55). Surprisingly, MIRKO mice have normal whole-body glucose homeostasis (8). This can in part can be explained by a shunting of glucose from the muscle to adipose tissue (22) and also suggests that other, IR-independent signals can regulate glucose uptake in muscle.
IGF-1, presumably acting through IGF-1R, has been demonstrated to have a hypoglycemic effect in mice lacking IR (14). Furthermore, IGF-1R activity and insulin- and IGF-1-stimulated glucose uptake were found to be increased in myotubes lacking IR (46). Although IGF-1R protein level is unchanged in MIRKO muscle (8), it is possible that normal IGF-1R signaling could compensate for the absence of IR in stimulating glucose uptake. However, we show here that MI2RKO mice maintain normal glucose homeostasis, despite a combined deficiency of IR and IGF-1R in muscle. This observation contrasts with the results of a study of a transgenic mouse model expressing a dominant-negative form of IGF-1R in muscle, leading to functional interference of both the IR and the IGF-1R due to the formation of hybrid receptors (17). These mice develop insulin resistance associated with elevated serum insulin as early as 2 weeks of age and become hyperglycemic at 5 weeks of age. The reason for the phenotypic discrepancy between MI2RKO mice and the IGF-1R dominant-negative transgenic mice is not clear. One possibility is that the discrepancy was due to differences in the genetic background of the two mouse models. The IGF-1R dominant-negative transgenic mice have a FVB background, whereas the MI2RKO mice are on a mixed (C57BL/6J, 129 Sv, and FVB) background. Another possibility is that when highly overexpressed, the dominant-negative IGF-1R interferes with signaling through other tyrosine kinases to alter muscle glucose metabolism. Finally, it is possible that there is some minimal residual IR/IGF-1R signaling in MI2RKO mice, which is sufficient to prevent the development of systemic insulin resistance and type 2 diabetes.
In conclusion, we have shown that combined deficiency in IR and IGF-1R in cardiac and skeletal muscle leads to early death from heart failure in the mouse and that cardiac muscle-specific deletion of IR signaling is most important in this process. At these young ages, combined deletion of IR and IGF-1R produces no abnormalities in glucose homeostasis but do produce a coordinated down-regulation of electron transport and mitochondrial fatty acid beta-oxidation. Therefore, a defect in ATP production may be an important contributor to the cardiomyopathy observed in MI2RKO mice. There is also a myocardial growth defect leading to reduced heart mass relative to body size. These factors, possibly combined with reduced inotropic effect due to absent IGF-1 signaling, may account for the development of overt heart failure. Defining the precise roles of each of these mechanisms will require further study.
Acknowledgments
We thank Christian Rask-Madsen (Joslin Diabetes Center) for valuable advice and discussions, Rohit N. Kulkarni (Joslin Diabetes Center) for providing the DLox mice, Roderick Bronson (Harvard Medical School) and Christopher Cahill (Joslin Diabetes Center's EM core) for expert help with histological analyses, and Stephane Gesta and Jeremie Boucher for help with quantitative RT-PCR. We also thank the Foster Animal Facility at Brandeis University for excellent animal care.
This work was supported by grant DK 31036 and the Joslin DERC grant.
Footnotes
Published ahead of print on 22 December 2006.
REFERENCES
- 1.Accili, D., J. Drago, E. J. Lee, M. D. Johnson, M. H. Cool, P. Salvatore, L. D. Asico, P. A. Jose, S. I. Taylor, and H. Westphal. 1996. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat. Genet. 12:106-109. [DOI] [PubMed] [Google Scholar]
- 2.Aihara, Y., M. Kurabayashi, Y. Saito, Y. Ohyama, T. Tanaka, S. Takeda, K. Tomaru, K. Sekiguchi, M. Arai, T. Nakamura, and R. Nagai. 2000. Cardiac ankyrin repeat protein is a novel marker of cardiac hypertrophy: role of M-CAT element within the promoter. Hypertension 36:48-53. [DOI] [PubMed] [Google Scholar]
- 3.Bang, M. L., R. E. Mudry, A. S. McElhinny, K. Trombitas, A. J. Geach, R. Yamasaki, H. Sorimachi, H. Granzier, C. C. Gregorio, and S. Labeit. 2001. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J. Cell Biol. 153:413-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belke, D. D., S. Betuing, M. J. Tuttle, C. Graveleau, M. E. Young, M. Pham, D. Zhang, R. C. Cooksey, D. A. McClain, S. E. Litwin, H. Taegtmeyer, D. Severson, C. R. Kahn, and E. D. Abel. 2002. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J. Clin. Investig. 109:629-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blakesley, V. A., A. Scrimgeour, D. Esposito, and D. Le Roith. 1996. Signaling via the insulin-like growth factor-I receptor: does it differ from insulin receptor signaling? Cytokine Growth Factor Rev. 7:153-159. [DOI] [PubMed] [Google Scholar]
- 6.Bluher, M., L. Wilson-Fritch, J. Leszyk, P. G. Laustsen, S. Corvera, and C. R. Kahn. 2004. Role of insulin action and cell size on protein expression patterns in adipocytes. J. Biol. Chem. 279:31902-31909. [DOI] [PubMed] [Google Scholar]
- 7.Brown, G. C. 1992. Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem. J. 284:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brüning, J. C., M. D. Michael, J. N. Winnay, T. Hayashi, D. Horsch, D. Accili, L. J. Goodyear, and C. R. Kahn. 1998. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2:559-569. [DOI] [PubMed] [Google Scholar]
- 9.Condorelli, G., A. Drusco, G. Stassi, A. Bellacosa, R. Roncarati, G. Iaccarino, M. A. Russo, Y. Gu, N. Dalton, C. Chung, M. V. Latronico, C. Napoli, J. Sadoshima, C. M. Croce, and J. Ross, Jr. 2002. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc. Natl. Acad. Sci. USA 99:12333-12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costa, J., M. Borges, C. David, and C. A. Vaz. 2006. Efficacy of lipid lowering drug treatment for diabetic and non-diabetic patients: meta-analysis of randomised controlled trials. BMJ 332:1115-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crackower, M., G. Oudit, I. Kozieradzki, R. Sarao, H. Sun, T. Sasaki, E. Hirsch, A. Suzuki, T. Shioi, J. Irie-Sasaki, R. Sah, H. Cheng, V. Rybin, G. Lembo, L. Fratta, A. Oliveira-dos-Santos, J. Benovic, C. R. Kahn, S. Izumo, S. Steinberg, M. Wymann, P. Backx, and J. Penninger. 2002. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 110:737-749. [DOI] [PubMed] [Google Scholar]
- 12.Dahlquist, K. D., N. Salomonis, K. Vranizan, S. C. Lawlor, and B. R. Conklin. 2002. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat. Genet. 31:19-20. [DOI] [PubMed] [Google Scholar]
- 13.DeFronzo, R. A. 1997. Pathogenesis of type 2 diabetes: metabolic and molecular implications for identifying diabetes genes. Diabetes Rev. 5:177-269. [Google Scholar]
- 14.Di Cola, G., M. H. Cool, and D. Accili. 1997. Hypoglycemic effect of insulin-like growth factor-1 in mice lacking insulin receptors. J. Clin. Investig. 99:2538-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang, Z. Y., J. B. Prins, and T. H. Marwick. 2004. Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocrine Rev. 25:543-567. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez, A. M., J. Dupont, R. P. Farrar, S. Lee, B. Stannard, and D. Le Roith. 2002. Muscle-specific inactivation of the IGF-I receptor induces compensatory hyperplasia in skeletal muscle. J. Clin. Investig. 109:347-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez, A. M., J. K. Kim, S. Yakar, J. Dupont, C. Hernandez-Sanchez, A. L. Castle, J. Filmore, G. I. Shulman, and D. Le Roith. 2001. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev. 15:1926-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giles, T. D. 2003. The patient with diabetes mellitus and heart failure: at-risk issues. Am. J. Med. 115(Suppl. 8A):107S-110S. [DOI] [PubMed] [Google Scholar]
- 19.Holzenberger, M., P. Leneuve, G. Hamard, B. Ducos, L. Perin, M. Binoux, and Y. Le Bouc. 2000. A targeted partial invalidation of the insulin-like growth factor I receptor gene in mice causes a postnatal growth deficit. Endocrinology 141:2557-2566. [DOI] [PubMed] [Google Scholar]
- 20.Jeyaseelan, R., C. Poizat, R. K. Baker, S. Abdishoo, L. B. Isterabadi, G. E. Lyons, and L. Kedes. 1997. A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J. Biol. Chem. 272:22800-22808. [DOI] [PubMed] [Google Scholar]
- 21.Joshi, R. L., B. Lamothe, N. Cordonnier, K. Mesbah, E. Monthioux, J. Jami, and C. Bucchini. 1996. Targeted disruption of the insulin receptor gene in the mouse results in neonatal lethality. EMBO J. 15:1542-1547. [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, J. K., M. D. Michael, S. F. Previs, O. D. Peroni, F. Mauvais-Jarvis, S. Neschen, B. B. Kahn, C. R. Kahn, and G. I. Shulman. 2000. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J. Clin. Investig. 105:1791-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krenz, M., and J. Robbins. 2004. Impact of beta-myosin heavy chain expression on cardiac function during stress. J. Am. Coll. Cardiol. 44:2390-2397. [DOI] [PubMed] [Google Scholar]
- 24.Laakso, M. 2001. Cardiovascular disease in type 2 diabetes: challenge for treatment and prevention. J. Intern. Med. 249:225-235. [DOI] [PubMed] [Google Scholar]
- 25.Laustsen, P. G., W. S. Lane, V. Bennett, and G. E. Lienhard. 2001. Association of protein kinase C(lambda) with adducin in 3T3-L1 adipocytes. Biochim. Biophys. Acta 1539:163-172. [DOI] [PubMed] [Google Scholar]
- 26.Liao, R., M. Jain, L. Cui, J. D'Agostino, F. Aiello, I. Luptak, S. Ngoy, R. M. Mortensen, and R. Tian. 2002. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 106:2125-2131. [DOI] [PubMed] [Google Scholar]
- 27.Lillioja, S., D. M. Mott, M. Spraul, R. Ferraro, J. E. Foley, E. Ravussin, W. C. Knowler, P. H. Bennett, and C. Bogardus. 1993. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus: prospective studies of Pima Indians. N. Engl. J. Med. 329:1988-1992. [DOI] [PubMed] [Google Scholar]
- 28.Liu, J. P., J. Baker, J. A. Perkins, E. J. Robertson, and A. Efstratiadis. 1993. Mice carrying null mutations of the genes encoding insulin-like growth factor I (lgf-1) and type 1 IGF receptor (lgf1r). Cell 75:59-72. [PubMed] [Google Scholar]
- 29.Louvi, A., D. Accili, and A. Efstratiadis. 1997. Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev. Biol. 189:33-48. [DOI] [PubMed] [Google Scholar]
- 30.Lowell, B. B., and G. I. Shulman. 2005. Mitochondrial dysfunction and type 2 diabetes. Science 307:384-387. [DOI] [PubMed] [Google Scholar]
- 31.Martin, B. C., J. Warram, A. S. Krolewski, R. N. Bergman, J. S. Soeldner, and C. R. Kahn. 1992. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet 340:925-929. [DOI] [PubMed] [Google Scholar]
- 32.McMullen, J. R., T. Shioi, W. Y. Huang, L. Zhang, O. Tarnavski, E. Bisping, M. Schinke, S. Kong, M. C. Sherwood, J. Brown, L. Riggi, P. M. Kang, and S. Izumo. 2004. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J. Biol. Chem. 279:4782-4793. [DOI] [PubMed] [Google Scholar]
- 33.Mootha, V. K., C. M. Lindgren, K. F. Eriksson, A. Subramanian, S. Sihag, J. Lehar, P. Puigserver, E. Carlsson, M. Ridderstrale, E. Laurila, N. Houstis, M. J. Daly, N. Patterson, J. P. Mesirov, T. R. Golub, P. Tamayo, B. Spiegelman, E. S. Lander, J. N. Hirschhorn, D. Altshuler, and L. C. Groop. 2003. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34:267-273. [DOI] [PubMed] [Google Scholar]
- 34.Mora, A., A. M. Davies, L. Bertrand, I. Sharif, G. R. Budas, S. Jovanovic, V. Mouton, C. R. Kahn, J. M. Lucocq, G. A. Gray, A. Jovanovic, and D. R. Alessi. 2003. Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J. 22:4666-4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nadal-Ginard, B., and V. Mahdavi. 1989. Molecular basis of cardiac performance. Plasticity of the myocardium generated through protein isoform switches. J. Clin. Investig. 84:1693-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nathan, D. M., P. A. Cleary, J. Y. Backlund, S. M. Genuth, J. M. Lachin, T. J. Orchard, P. Raskin, and B. Zinman. 2005. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 353:2643-2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patti, M. E., A. J. Butte, S. Crunkhorn, K. Cusi, R. Berria, S. Kashyap, Y. Miyazaki, I. Kohane, M. Costello, R. Saccone, E. J. Landaker, A. B. Goldfine, E. Mun, R. DeFronzo, J. Finlayson, C. R. Kahn, and L. J. Mandarino. 2003. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 100:8466-8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petersen, K. F., S. Dufour, D. Befroy, R. Garcia, and G. I. Shulman. 2004. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 350:664-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petersen, K. F., S. Dufour, and G. I. Shulman. 2005. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2:e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poornima, I. G., P. Parikh, and R. P. Shannon. 2006. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ. Res. 98:596-605. [DOI] [PubMed] [Google Scholar]
- 41.Pyle, W. G., and R. J. Solaro. 2004. At the crossroads of myocardial signaling: the role of Z-discs in intracellular signaling and cardiac function. Circ. Res. 94:296-305. [DOI] [PubMed] [Google Scholar]
- 42.Ren, J., W. K. Samson, and J. R. Sowers. 1999. Insulin-like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J. Mol. Cell. Cardiol. 31:2049-2061. [DOI] [PubMed] [Google Scholar]
- 43.Saetrum Opgaard, O., and P. H. Wang. 2005. IGF-I is a matter of heart. Growth Horm. IGF Res. 15:89-94. [DOI] [PubMed] [Google Scholar]
- 44.Saltiel, A. R., and C. R. Kahn. 2001. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799-806. [DOI] [PubMed] [Google Scholar]
- 45.Scheinowitz, M., G. Kessler-Icekson, S. Freimann, R. Zimmermann, W. Schaper, E. Golomb, N. Savion, and M. Eldar. 2003. Short- and long-term swimming exercise training increases myocardial insulin-like growth factor-I gene expression. Growth Horm. IGF Res. 13:19-25. [DOI] [PubMed] [Google Scholar]
- 46.Shefi-Friedman, L., E. Wertheimer, S. Shen, A. Bak, D. Accili, and S. R. Sampson. 2001. Increased IGFR activity and glucose transport in cultured skeletal muscle from insulin receptor null mice. Am. J. Physiol. Endocrinol. Metab. 281:E16-E24. [DOI] [PubMed] [Google Scholar]
- 47.Shioi, T., P. M. Kang, P. S. Douglas, J. Hampe, C. M. Yballe, J. Lawitts, L. C. Cantley, and S. Izumo. 2000. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 19:2537-2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shioi, T., J. R. McMullen, P. M. Kang, P. S. Douglas, T. Obata, T. F. Franke, L. C. Cantley, and S. Izumo. 2002. Akt/protein kinase B promotes organ growth in transgenic mice. Mol. Cell. Biol. 22:2799-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shiojima, I., M. Yefremashvili, Z. Luo, Y. Kureishi, A. Takahashi, J. Tao, A. Rosenzweig, C. R. Kahn, E. D. Abel, and K. Walsh. 2002. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J. Biol. Chem. 277:37670-37677. [DOI] [PubMed] [Google Scholar]
- 50.Tardiff, J. C., T. E. Hewett, S. M. Factor, K. L. Vikstrom, J. Robbins, and L. A. Leinwand. 2000. Expression of the beta (slow)-isoform of MHC in the adult mouse heart causes dominant-negative functional effects. Am. J. Physiol. Heart Circ. Physiol. 278:H412-H419. [DOI] [PubMed] [Google Scholar]
- 51.Uwaifo, G. I., and R. E. Ratner. 2003. The roles of insulin resistance, hyperinsulinemia, and thiazolidinediones in cardiovascular disease. Am. J. Med. 115(Suppl. 8A):12S-19S. [DOI] [PubMed] [Google Scholar]
- 52.von Lewinski, D., K. Voss, S. Hulsmann, H. Kogler, and B. Pieske. 2003. Insulin-like growth factor-1 exerts Ca2+-dependent positive inotropic effects in failing human myocardium. Circ. Res. 92:169-176. [DOI] [PubMed] [Google Scholar]
- 53.Wang, X., and B. Seed. 2003. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 31:e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yechoor, V. K., M. E. Patti, K. Ueki, P. G. Laustsen, R. Saccone, R. Rauniyar, and C. R. Kahn. 2004. Distinct pathways of insulin-regulated versus diabetes-regulated gene expression: an in vivo analysis in MIRKO mice. Proc. Natl. Acad. Sci. USA 101:16525-16530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zisman, A., O. D. Peroni, D. Abel, M. D. Michael, F. Mauvais-Jarvis, B. B. Lowell, J. F. P. Wojtaszewski, M. F. Hirshman, A. Virkamaki, L. J. Goodyear, C. R. Kahn, and B. B. Kahn. 2000. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 6:924-928. [DOI] [PubMed] [Google Scholar]
- 56.Zolk, O., M. Frohme, A. Maurer, F. W. Kluxen, B. Hentsch, D. Zubakov, J. D. Hoheisel, I. H. Zucker, S. Pepe, and T. Eschenhagen. 2002. Cardiac ankyrin repeat protein, a negative regulator of cardiac gene expression, is augmented in human heart failure. Biochem. Biophys. Res. Commun. 293:1377-1382. [DOI] [PubMed] [Google Scholar]
- 57.Zou, Y., S. Evans, J. Chen, H. C. Kuo, R. P. Harvey, and K. R. Chien. 1997. CARP, a cardiac ankyrin repeat protein, is downstream in the Nkx2-5 homeobox gene pathway. Development 124:793-804. [DOI] [PubMed] [Google Scholar]