

Abstract
The translation initiation GTPase eukaryotic translation initiation factor 5B (eIF5B) binds to the factor eIF1A and catalyzes ribosomal subunit joining in vitro. We show that rapid depletion of eIF5B in Saccharomyces cerevisiae results in the accumulation of eIF1A and mRNA on 40S subunits in vivo, consistent with a defect in subunit joining. Substituting Ala for the last five residues in eIF1A (eIF1A-5A) impairs eIF5B binding to eIF1A in cell extracts and to 40S complexes in vivo. Consistently, overexpression of eIF5B suppresses the growth and translation initiation defects in yeast expressing eIF1A-5A, indicating that eIF1A helps recruit eIF5B to the 40S subunit prior to subunit joining. The GTPase-deficient eIF5B-T439A mutant accumulated on 80S complexes in vivo and was retained along with eIF1A on 80S complexes formed in vitro. Likewise, eIF5B and eIF1A remained associated with 80S complexes formed in the presence of nonhydrolyzable GDPNP, whereas these factors were released from the 80S complexes in assays containing GTP. We propose that eIF1A facilitates the binding of eIF5B to the 40S subunit to promote subunit joining. Following 80S complex formation, GTP hydrolysis by eIF5B enables the release of both eIF5B and eIF1A, and the ribosome enters the elongation phase of protein synthesis.
The initiation of protein synthesis in eukaryotes is a complex series of events orchestrated by initiation factors (eIFs). In vitro studies using purified translation initiation factors have led to the elucidation of the translation initiation pathway in which the different factors associate with the 40S ribosomal subunit and guide the binding of Met-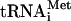 and mRNA. While the essential in vitro functions of the different factors are well established, the precise timing of factor binding to and release from the ribosome is less well understood. Moreover, not all of the in vitro-defined functions of the factors have been confirmed in vivo. The first step in translation initiation involves the binding of factor eIF2 to GTP and Met-
and mRNA. While the essential in vitro functions of the different factors are well established, the precise timing of factor binding to and release from the ribosome is less well understood. Moreover, not all of the in vitro-defined functions of the factors have been confirmed in vivo. The first step in translation initiation involves the binding of factor eIF2 to GTP and Met-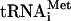 forming a ternary complex. The ternary complex along with factors eIF1, eIF1A, eIF3, and eIF5 binds to the 40S ribosome, forming the 43S complex (reviewed in reference 16). The 43S complex then binds to an mRNA near the 5′ cap, forming a 48S complex that scans along the mRNA in a 3′ direction in search of an AUG start codon. In the 48S complex, some of the eIF2-GTP is hydrolyzed to eIF2-GDP + Pi (2). Upon AUG codon recognition, GTP hydrolysis is completed, and Pi is released, converting the 48S complex from an open to a closed complex with Met-
forming a ternary complex. The ternary complex along with factors eIF1, eIF1A, eIF3, and eIF5 binds to the 40S ribosome, forming the 43S complex (reviewed in reference 16). The 43S complex then binds to an mRNA near the 5′ cap, forming a 48S complex that scans along the mRNA in a 3′ direction in search of an AUG start codon. In the 48S complex, some of the eIF2-GTP is hydrolyzed to eIF2-GDP + Pi (2). Upon AUG codon recognition, GTP hydrolysis is completed, and Pi is released, converting the 48S complex from an open to a closed complex with Met-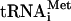 in the P site of the 40S subunit (29). The release of Pi from the 48S complex appears to be coupled with displacement of eIF1 from its binding site near the P site (2, 22). In addition, the majority of eIF2 either dissociates from or repositions on the 48S complex following AUG codon recognition (32). Intriguingly, factors eIF3, eIF1, and eIF1A appear to be retained on the 48S complex following eIF2 release (38).
in the P site of the 40S subunit (29). The release of Pi from the 48S complex appears to be coupled with displacement of eIF1 from its binding site near the P site (2, 22). In addition, the majority of eIF2 either dissociates from or repositions on the 48S complex following AUG codon recognition (32). Intriguingly, factors eIF3, eIF1, and eIF1A appear to be retained on the 48S complex following eIF2 release (38).
Conversion of the 48S complex to a functional 80S ribosome requires joining of the large 60S ribosomal subunit in a reaction catalyzed by factor eIF5B (30). In the yeast Saccharomyces cerevisiae, eIF5B is encoded by the nonessential FUN12 gene. Cells lacking eIF5B display an extremely slow-growth phenotype, an altered polysome profile consistent with a defect in translation initiation, and are unable to grow under amino acid starvation conditions due to the failure to derepress translation of the GCN4 mRNA (8). While in vitro reconstitution experiments have demonstrated the role for eIF5B in promoting subunit joining (3, 30, 36), in vivo data supporting this precise function have not been reported. As eIF3 and eIF1 are not present on the 80S complex following subunit joining (38), it is thought that these factors dissociate during subunit joining.
eIF1A is a eukaryotic ortholog of the bacterial translation initiation factor IF1, which binds in the ribosomal A site, and eIF5B is an ortholog of the factor IF2. Like bacterial IF1, eIF1A possesses a conserved β-barrel fold (5, 34); however, the eukaryotic factor has in addition a helical element that packs against the core domain and the N- and C-terminal nonstructured tails. eIF1A functionally coordinates with eIF1 and promotes 43S complex formation, as well as ribosomal scanning and AUG start codon recognition (12, 23, 29). In addition to these early roles in the translation pathway, eIF1A physically and functionally interacts with eIF5B (9, 24, 28). The C terminus of eIF1A binds to eIF5B, and structural analysis revealed that the eIF1A C-terminal residues pack into a groove formed by helices H13 and H14 in C-terminal domain IV of eIF5B (24). This eIF5B-eIF1A interaction appears to be important for protein synthesis. First, overexpression of eIF1A exacerbates the slow-growth phenotype in strains lacking eIF5B (9). Second, deletion of or substitution of Ala for the five C-terminal residues (DIDDI) in yeast eIF1A impairs 80S complex formation and eIF5B ribosome-dependent GTPase activity in vitro (1). Third, deletion of helix H14 impairs eIF5B GTPase and ribosome-joining activities (1). As eIF1A binds to the 40S subunit early in the translation initiation pathway, it has been proposed that the eIF5B-eIF1A interaction helps recruit eIF5B to the 48S complex. Following subunit joining, GTP hydrolysis by eIF5B triggers a switch enabling release of the factor from the 80S ribosome (36). The timing of eIF1A release from the ribosome has not been examined, and it is unknown whether the eIF5B-eIF1A interaction enables the release of eIF1A in concert with eIF5B.
In this report we utilize a degron approach in which eIF5B is tagged with an unstable ubiquitin fusion protein to rapidly deplete eIF5B in yeast cells (11, 18). Analysis of translation initiation complexes from these cells revealed the accumulation of 48S complexes, providing in vivo evidence that eIF5B promotes ribosomal subunit joining. Consistent with a direct interaction between eIF5B and eIF1A, C-terminal mutations in these factors impaired the ability of eIF1A to recruit eIF5B to the 48S complex. Finally, blocking GTP hydrolysis by eIF5B led to the accumulation of both eIF1A and eIF5B on the 80S products of subunit joining both in vivo and in vitro. Based on these findings, we propose a model in which eIF1A helps recruit eIF5B to the 48S complex to promote subunit joining and that the subsequent release of eIF1A is dependent on GTP hydrolysis and the release of eIF5B from the 80S ribosome.
MATERIALS AND METHODS
Plasmids and yeast strains.
The plasmids and yeast strains employed in these studies are listed in Table 1 and 2, respectively. The fun12-td integration plasmid (pC2845) was constructed by inserting an XhoI-flanked PCR (primers JF1, 5′CCGCTCGAGATGGGGAAAAAGAGTAAAAAG3′, and JF2, 5′CCGCTCGAGCTTTCTGGTCAATAGCTTACC3′) product containing the first 720 nucleotides of the eIF5B open reading frame (ORF) into the XhoI site of pPW66R (17). Plasmid pC2846 was generated by subcloning the Eco47III-BamHI fragment encoding ΔN-eIF5B-ΔH14 from pC2283 between the SmaI and BamHI sites, a modified version of the vector pEGKT (25) in which the SmaI site precedes the BamHI site. The kanMX4 gene was amplified by PCR using SmaI primers JF64 (5′TCCCCCGGGGGACTTGCCTCGTCCCCGCCGGGTCACGCGGCC3′) and JF65 (5′TCCCCCGGGGGAGAATCGACAGCAGTATAGCGACCAGCATTC3′) and then inserted into the SmaI site in p180, creating the GCN4-lacZ, KanMX plasmid pC2847.
TABLE 1.
Plasmids used in this study
| Plasmid | Descriptiona | Source or reference |
|---|---|---|
| pC2845 | Integrating URA3 plasmid containing PCUP1-UBI-R-DHFRts-HA-fun12-td | This study |
| YCplac33 | sc URA3 backbone | 13 |
| pC1107 | sc URA3 FUN12-FL in YCplac33 backbone | This study |
| pC1217 | sc URA3 ΔN-FUN12-FL in YCplac33 backbone | 36 |
| pC2283 | sc URA3 ΔN-fun12-ΔH14 in YCplac33 backbone | This study |
| pC2298 | sc URA3 fun12-ΔH14 in YCplac33 backbone | This study |
| pC1293 | sc URA3 ΔN-fun12-T439A in YCplac33 backbone | 36 |
| pC1285 | hc URA3 FUN12-FL in YEplac195 backbone | This study |
| p3499 | sc LEU2 TIF11-FL in YCplac111 backbone | 12 |
| p4409 | sc LEU2 tif11-149AAAAA153-FL (5A) in YCplac111 backbone | 12 |
| pC1834 | GST galactose-inducible vector backbone | 25 |
| pC1842 | ΔN-FUN12-FL in pC1834 backbone | 36 |
| pC2846 | ΔN-fun12-ΔH14-FL in pC1834 backbone | This study |
| pC1843 | ΔN-fun12-T439A in pC1834 backbone | 36 |
| p180 | sc URA3 GCN4-lacZ with WT leader | 15 |
| pC2847 | sc URA3-KANMX4 GCN4-lacZ with WT leader | This study |
| p3498 | sc LEU2 TIF11 FUN12 in pRS315 backbone | 28 |
Single-copy (sc) or high-copy-number (hc) plasmids.
TABLE 2.
Yeast strains used in this study
| Strain | Genotypea | Source or reference |
|---|---|---|
| YAJ42 | MATα leu2-3,-112 ura3-52 trp1Δ gcn2Δ ubr1::PGAL1-UBR1-TRP1 | 17 |
| J259 | MATα leu2-3,-112 ura3-52 trp1Δ gcn2Δ ubr1::PGAL1-UBR1-TRP1PCUP1-UBI-R-DHFRts-HA-fun12-td::URA3::fun12 | This study |
| J260 | MATα leu2-3,-112 ura3-52 trp1Δ gcn2Δ ubr1::PGAL1-UBR1-TRP1fun12::KANMX4 | This study |
| J261 | MATα leu2-3,-112 ura3-52 trp1Δ gcn2Δ ubr1::PGAL1-UBR1-TRP1rpl16b::KANMX4 | This study |
| J262 | MATα leu2-3,-112 ura3-52 trp1Δ gcn2Δ ubr1::PGAL1-UBR1-TRP1PCUP1-UBI-R-DHFRts-HA-fun12-td::URA3::fun12 rpl16b::KANMX4 | This study |
| H2971 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [p3498: sc TIF11 FUN12 LEU2] | 28 |
| J263 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1107: sc FUN12 URA3] [p3499: sc TIF11 LEU2] | This study |
| J264 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1107: sc FUN12 URA3] [p4409: sc tif11-5A LEU2] | This study |
| J265 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1285: hc FUN12 URA3] [p3499: sc TIF11 LEU2] | This study |
| J266 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1285: hc FUN12 URA3] [p4409: tif11-5A LEU2] | This study |
| J267 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1834: GST URA3] [p3499: sc TIF11 LEU2] | This study |
| J268 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1834: GST URA3] [p4409: sc tif11-5A LEU2] | This study |
| J269 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1842: GST-ΔN-FUN12 URA3] [p3499: sc TIF11 LEU2] | This study |
| J270 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1842: GST-ΔN-FUN12 URA3] [p4409: sc tif11-5A LEU2] | This study |
| J271 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC2846: GST-ΔN-fun12-ΔH14 URA3] [p3499: sc TIF11 LEU2] | This study |
| J272 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1107: sc FUN12URA3] [p3499: sc TIF11 LEU2] | This study |
| J273 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC2298: sc fun12-ΔH14 URA3] [p3499: sc TIF11 LEU2] | This study |
| J274 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1217: sc ΔN-FUN12 URA3] [p3499: sc TIF11 LEU2] | This study |
| J275 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC2283: sc ΔN-fun12-ΔH14 URA3] [p3499: sc TIF11 LEU2] | This study |
| J276 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1217: sc ΔN-FUN12 URA3] [p4409: sc tif11-5A LEU2] | This study |
| J277 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [YCplac33: sc URA3][p3499: sc TIF11 LEU2] | This study |
| J278 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [YCplac33: sc URA3][p4409: sc tif11-5A LEU2] | This study |
| J279 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC2283: sc ΔN-fun12-ΔH14 URA3] [p4409: sc tif11-5A LEU2] | This study |
| J280 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1293: sc ΔN-fun12-Τ439Α URA3] [p3499: sc TIF11 LEU2] | This study |
| J281 | MATα ura3-52 leu2-3,112 fun12::hisG tif11::hisG [pC1293: sc ΔN-fun12-Τ439Α URA3] [p4409: sc tif11-5A LEU2] | This study |
sc, single copy.
The SacI-SalI fragment from pC1005 (9) encoding Flag-eIF5B was subcloned between the same sites of the low-copy-number URA3 vector YCplac33, creating pC1107. Next, an Eco47III site was introduced at residues 28 to 29 of Flag-eIF5B in pC1107. Deletion of the internal Eco47III fragment from the resulting plasmid yielded plasmid pC1286 encoding ΔN-eIF5B. Derivatives of pC1107 and pC1286 encoding eIF5B-ΔH14 (pC2298) and ΔN-eIF5B-ΔH14 (pC2283), respectively, were constructed by site-directed mutagenesis, deleting the residues encoding Leu-975 to the C terminus (Glu-1002) of eIF5B.
Yeast strain J259 was generated by the integration of pC2845 (linearized with MscI) into the FUN12 locus of strain YAJ42. The fun12::KanMX4 module from the fun12Δ strain in the yeast genome deletion collection (Research Genetics) was amplified by PCR using primers JF47 (5′TTTGGATCTTGGCACCTTTATAACGAATGA3′) and JF50 (5′GCTAACTCCACGTCGTTCACTTTCTCTCGA3′) that hybridize ∼1 kb upstream and downstream, respectively, of the FUN12 ORF. The PCR product was used to delete FUN12 from strain J259, generating strain J260 by selecting transformants on YEPD (yeast extract-peptone-dextrose) medium containing 0.2 g/liter kanamycin. The RPL16B gene (YNL069c) encoding rpL16b was deleted from strains J261, YAJ42, and J262 by transforming with a PCR product obtained upon amplifying the rpl16b::KANMX4 cassette from the yeast genome deletion collection by using primers JF38 (5′CGCTGCTGTCTTTGCATCCTTGACA3′) and JF39 (5′ATATGGTGAAAATTCGAAGTCTCTA3′) that hybridize at ∼1 kb 5′ and 3′, respectively, of the RPL16B ORF. Standard techniques were employed for yeast transformations (14), gene replacement (33), plasmid shuffling (7), and yeast medium preparation (35).
Sucrose density gradient analysis of ribosomal complexes.
Formaldehyde cross-linking of cells, whole-cell extract (WCE) preparation, and sucrose density gradient analysis were performed as described previously (27). Briefly, cells were grown to log phase, fixed with 1% formaldehyde, and then lysed with glass beads. WCEs were separated on 4 to 45% sucrose gradients subjected to centrifugation at 39,000 rpm for 2.5 h or on 7 to 30% sucrose gradients that were spun at 41,000 rpm for 5 h (for 40S complex resolution). For Western and Northern analyses, gradients were fractionated, and 600-μl samples were collected. Proteins in individual fractions were precipitated with ethanol, and washed pellets were resuspended in 1× sodium dodecyl sulfate (SDS) loading buffer (Invitrogen), followed by separation on 4 to 20% Tris-glycine polyacrylamide gels (Criterion, Bio-Rad). Immunoblot analysis was performed using antibodies that recognize the following yeast proteins: eIF5B (9), eIF1A (28), eIF3b, eIF5, eIF2γ (31), eIF2α (10), and RPS22 (kindly provided by Jan van't Riet). RNA in individual fractions was isolated as described elsewhere (17), and the mRNA probe was directed against RPL14B (27).
43S-mRNA and 80S complex formation assays.
43S-mRNA complexes were formed by incubating 3× TC (2.4 μM GDPNP-Mg2+ or GTP-Mg2+, 2.4 μM eIF2, 2.4 μM Met-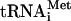 , 1× reaction buffer [30 mM HEPES-KOH, [pH 7.4, 100 mM potassium acetate, 3 mM magnesium diacetate, 2 mM dithiothreitol]) with 3× factor mixture (1.2 μM 40S, 2.4 μM eIF1, 1.2 μM eIF1A, 2.4 μM mRNA) for 2 min at 26°C, followed by the addition of 3× initiation mixture (2.4 μM eIF5, 6 mM GDPNP-Mg2+, and 3 μM eIF5B when appropriate), and incubation for 20 min at 26°C. When 80S complexes were formed, 43S-mRNA complexes were prepared with GTP-Mg2+, and 1.2 μM 60S ribosomes were included with 3× initiation mixture. After the 20-min incubation, 10 μl of the reaction mixture was mixed with 2 μl of loading dye and run in duplicate on native polyacrylamide gels. After running for 1 h at 25 W, the gels were analyzed in the following way: one duplicate was electrotransferred onto nitrocellulose paper at 300 mA for 2 h. Western blotting was performed as described above. The other duplicate was stained with Coomassie blue for 5 min, and then the 48S and 80S bands were located and excised with a razor blade. These gel slices were mechanically sheared by hand in 2× SDS sample buffer, boiled, and then subjected to SDS-polyacrylamide gel electrophoresis (PAGE), followed by Western analysis.
, 1× reaction buffer [30 mM HEPES-KOH, [pH 7.4, 100 mM potassium acetate, 3 mM magnesium diacetate, 2 mM dithiothreitol]) with 3× factor mixture (1.2 μM 40S, 2.4 μM eIF1, 1.2 μM eIF1A, 2.4 μM mRNA) for 2 min at 26°C, followed by the addition of 3× initiation mixture (2.4 μM eIF5, 6 mM GDPNP-Mg2+, and 3 μM eIF5B when appropriate), and incubation for 20 min at 26°C. When 80S complexes were formed, 43S-mRNA complexes were prepared with GTP-Mg2+, and 1.2 μM 60S ribosomes were included with 3× initiation mixture. After the 20-min incubation, 10 μl of the reaction mixture was mixed with 2 μl of loading dye and run in duplicate on native polyacrylamide gels. After running for 1 h at 25 W, the gels were analyzed in the following way: one duplicate was electrotransferred onto nitrocellulose paper at 300 mA for 2 h. Western blotting was performed as described above. The other duplicate was stained with Coomassie blue for 5 min, and then the 48S and 80S bands were located and excised with a razor blade. These gel slices were mechanically sheared by hand in 2× SDS sample buffer, boiled, and then subjected to SDS-polyacrylamide gel electrophoresis (PAGE), followed by Western analysis.
Other biochemical methods.
For glutathione S-transferase (GST) pulldown assays, cells were grown in synthetic minimal (SD) medium, collected, resuspended in breaking buffer (20 mM Tris-HCl [pH 7.4], 1 mM magnesium acetate, 0.1% Triton X-100, 100 mM KCl), and lysed with glass beads as described previously (28). Glutathione-Sepharose (Amersham Biosciences) was prepared according to the manufacturer's instructions and incubated with WCEs at 1:10 final volume for 3 h at 4°C. The glutathione-Sepharose beads were collected, washed three times for 5 min each in breaking buffer, and prepared for SDS-PAGE and Western blotting as described above. GST antibodies were obtained from Sigma. β-Galactosidase assays of GCN4-lacZ expression were performed as described previously (15).
RESULTS
Depletion of eIF5B results in accumulation of 48S complexes.
As fun12Δ yeast strains lacking eIF5B are viable, with a severely slow-growth phenotype, the depletion of eIF5B in a degron-tagged strain is expected to impair but not block cell growth. An FUN12 degron (fun12-td) allele was constructed by inserting upstream of and in-frame with the eIF5B open reading frame a DNA cassette carrying the copper-regulated CUP1 promoter driving the expression of ubiquitin linked to a temperature-sensitive allele of DHFR, followed by a hemagglutinin (HA) tag (PCUP1-UBI-R-DHFRts-HA tag). In addition, a galactose-inducible version of the ubiquitin E3-ligase UBR1 was introduced into the yeast strain. When grown in raffinose medium supplemented with copper, where UBR1 expression is low and the CUP1 promoter is induced, the fun12-td strain exhibited a growth rate similar to that of the isogenic wild-type (WT) strain in which eIF5B is expressed under the control of its native promoter (Fig. 1A). Between 4 and 8 hours after a shift to nonpermissive conditions, a galactose medium lacking copper, where UBR1 expression is induced and the CUP1 promoter is repressed, growth of the degron strain was significantly impaired (Fig. 1A). Importantly, a plasmid carrying the WT FUN12 gene encoding eIF5B complemented the impaired growth of the fun12-td strain under restrictive conditions (data not shown). Western analyses of whole-cell extracts confirmed that the impaired growth of the fun12-td strain correlated with depletion of the protein (data not shown). Thus, the impaired growth of the fun12-td strain under nonpermissive conditions appears to be due to the loss of eIF5B. To ensure near-complete depletion of eIF5B in our experiments, cells were cultured for 8 hours under restrictive conditions.
FIG. 1.

Depletion of degron-tagged eIF5B leads to polysome runoff and accumulation of halfmer ribosomes with bound mRNA and eIF1A. (A) Cultures of the fun12-td strain J259 and the isogenic WT strain YAJ42 were grown at 25°C under permissive conditions (SC medium containing 2% raffinose and 100 μM CuSO4) to an OD600 of ∼0.2, split in halves, and grown under permissive or nonpermissive conditions (SC medium containing 2% galactose, lacking CuSO4), as indicated. (B) Polysome analysis of WT (YAJ42), fun12-td (J259), and eIF5BΔ (J260) strains. Cultures of cells were grown under permissive conditions as described for panel A to an OD600 of ∼0.2, split in halves, and transferred to either permissive or nonpermissive conditions and grown for 6 h at 25°C. Cells were cross-linked with 1% formaldehyde, and WCEs were separated on 4.5% to 45% sucrose gradients by centrifugation at 39,000 rpm for 2.5 h. Gradients were fractionated while scanning at 254 nm to visualize the indicated ribosomal species. The halfmer shoulder on the 80S peak in the fun12-td strain was magnified for clarity (inset) and is represented in the schematic; the same portion of the 80S peak in the eIF5BΔ strain was also magnified (inset). (C) WCEs prepared from strains J261 (rpl16bΔ) and J262 (rpl16bΔ, fun12-td) were cultured under nonpermissive conditions as described for panel B and then separated on 7 to 30% sucrose gradients by centrifugation at 41,000 rpm for 5 h. One hundred-microliter aliquots (17% of the total aliquot) of the gradient fractions and a portion of the starting WCE (In, input) were subjected to Western analysis using antibodies against the indicated proteins. For Northern analyses, total RNA was isolated from 72% of each fraction and from the WCE and subjected to Northern analysis using probes to detect RPL41A mRNA (mRNA) and Met-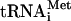 , as indicated. The amounts of the various factors and RNAs in the 40S fractions from multiple experiments were quantified, normalized to the amount of RPS22 (40S levels), and then expressed as a ratio of values obtained in the fun12-td versus the WT strain.
, as indicated. The amounts of the various factors and RNAs in the 40S fractions from multiple experiments were quantified, normalized to the amount of RPS22 (40S levels), and then expressed as a ratio of values obtained in the fun12-td versus the WT strain.
To examine translational activity, WCEs from WT, eIF5BΔ, and fun12-td strains grown under permissive or restrictive conditions were subjected to velocity sedimentation in sucrose density gradients. The polysome content was analyzed by continuously monitoring the optical density at 254 nm (OD254) while fractionating the gradients. Peaks corresponding to free 40S subunits, free 60S subunits, and 80S monosomes and polysomes were observed in the extracts from WT cells grown under permissive and nonpermissive conditions (Fig. 1B, left column). In contrast, extracts from eIF5BΔ cells exhibited a nearly complete loss of polysomes and a large accumulation of the 80S peak corresponding to translating monosomes and inactive 80S couples not bound to mRNA (Fig. 1B, right column). This result is consistent with previous observations and indicates a strong defect in translation initiation. Under conditions of limiting translation initiation, the ribosomes in polysomes terminate translation, disengage from mRNAs, and accumulate as vacant 80S couples awaiting recruitment into new rounds of translation. Likewise, when grown under restrictive conditions, the fun12-td strain exhibited a loss of polysomes and an accumulation of the 80S peak as observed in the eIF5BΔ cells (Fig. 1B, middle column). However, careful examination of the profile revealed the presence of a small shoulder peak sedimenting slightly faster than the 80S peak. This so-called “halfmer” peak was observed in every trial of this experiment (>4 times, and never in the control strains) and is indicative of a defect in ribosomal subunit joining, representing mRNAs with one translating 80S monosome and one 40S subunit stalled at the AUG start codon, waiting for 60S subunit joining (Fig. 1B, inset). The appearance of the halfmer peak was restricted to the fun12-td strain and was not observed in the WT strain or in the eIF5BΔ strain from the same genetic background (Fig. 1B).
To confirm the accumulation of 48S complexes (40S subunits bound to mRNA) in the fun12-td strain, cells were first fixed with formaldehyde to cross-link factors, mRNAs and tRNAs to ribosomes. Following fractionation of WCEs on sucrose gradients, individual fractions were subjected to Western and Northern analyses to monitor initiation factor, 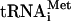 , and mRNA association with the 40S subunit. In order to enhance visualization of the 40S peak, the RPL16B gene (YNL069c), one of two genes encoding the 60S subunit protein L16, was deleted from the fun12-td strain. The decrease in the levels of L16b results in a reduction of the abundance of 60S subunits, increasing the amount of free 40S subunits and 48S complexes. In cells expressing WT eIF5B, factors eIF5B, eIF1A, eIF3, eIF2, and eIF5 as well as Met-
, and mRNA association with the 40S subunit. In order to enhance visualization of the 40S peak, the RPL16B gene (YNL069c), one of two genes encoding the 60S subunit protein L16, was deleted from the fun12-td strain. The decrease in the levels of L16b results in a reduction of the abundance of 60S subunits, increasing the amount of free 40S subunits and 48S complexes. In cells expressing WT eIF5B, factors eIF5B, eIF1A, eIF3, eIF2, and eIF5 as well as Met-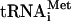 were associated with 40S subunits (denoted by the presence of ribosomal protein RPS22), consistent with an accumulation of 43S and/or 48S complexes (Fig. 1C, left panel). To monitor the presence of mRNA on the 40S subunits, we probed for the RPL41A mRNA by Northern analysis. As described previously, mRNP complexes containing the short RPL41A mRNA are readily resolved from 40S complexes, thus enabling this mRNA to serve as a good marker for general mRNA binding to 40S subunits (27). As expected, the RPL41A mRNA peaked in the 40S fraction as well as in the 80S fraction (Fig. 1C, left panel), indicating the presence of 48S complexes and monosomes, respectively.
were associated with 40S subunits (denoted by the presence of ribosomal protein RPS22), consistent with an accumulation of 43S and/or 48S complexes (Fig. 1C, left panel). To monitor the presence of mRNA on the 40S subunits, we probed for the RPL41A mRNA by Northern analysis. As described previously, mRNP complexes containing the short RPL41A mRNA are readily resolved from 40S complexes, thus enabling this mRNA to serve as a good marker for general mRNA binding to 40S subunits (27). As expected, the RPL41A mRNA peaked in the 40S fraction as well as in the 80S fraction (Fig. 1C, left panel), indicating the presence of 48S complexes and monosomes, respectively.
Depletion of eIF5B from the fun12-td strain had little effect on the ratio of eIF3, eIF2, and eIF5 relative to that of 40S subunits to the WT strain (Fig. 1C). Likewise, the ratio of Met-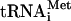 to 40S subunits was nearly equivalent in the WT and the fun12-td strains (Fig. 1C). In contrast, the ratio of mRNA to 40S subunits was ∼fivefold higher in the fun12-td strain than in the WT strain. Similarly, the ratio of eIF1A to 40S subunits was increased fivefold in the fun12-td strain (Fig. 1C). The increased amount of mRNA associated with 40S subunits suggests an accumulation of 48S complexes in the fun12-td strain; however, the fact that eIF2 and eIF3 do not accumulate to the same extent as mRNA indicates a novel form of 48S complex lacking eIF2 and eIF3 but containing eIF1A and mRNA. This putative 48S complex is predicted to form following AUG codon recognition and release of eIF2. The presence of eIF1A in the 48S complexes is consistent with the increased affinity of eIF1A for 40S subunits containing mRNA versus free 40S subunits (21); alternatively, or in addition, eIF1A release from the 48S complexes may require eIF5B function. This accumulation of 48S complexes and the presence of halfmer polysomes in the fun12-td strain provide the first in vivo evidence supporting the biochemically defined role for eIF5B in ribosomal subunit joining.
to 40S subunits was nearly equivalent in the WT and the fun12-td strains (Fig. 1C). In contrast, the ratio of mRNA to 40S subunits was ∼fivefold higher in the fun12-td strain than in the WT strain. Similarly, the ratio of eIF1A to 40S subunits was increased fivefold in the fun12-td strain (Fig. 1C). The increased amount of mRNA associated with 40S subunits suggests an accumulation of 48S complexes in the fun12-td strain; however, the fact that eIF2 and eIF3 do not accumulate to the same extent as mRNA indicates a novel form of 48S complex lacking eIF2 and eIF3 but containing eIF1A and mRNA. This putative 48S complex is predicted to form following AUG codon recognition and release of eIF2. The presence of eIF1A in the 48S complexes is consistent with the increased affinity of eIF1A for 40S subunits containing mRNA versus free 40S subunits (21); alternatively, or in addition, eIF1A release from the 48S complexes may require eIF5B function. This accumulation of 48S complexes and the presence of halfmer polysomes in the fun12-td strain provide the first in vivo evidence supporting the biochemically defined role for eIF5B in ribosomal subunit joining.
Suppression of the translation defect and the Slg− and Gcn− phenotypes in the eIF1A-5A C-terminal mutant by overexpression of eIF5B.
The accumulation of eIF1A on 48S complexes in the fun12-td strain (Fig. 1C) combined with the results of prior studies revealing a direct interaction between the C termini of eIF5B and eIF1A (9, 24, 28) led us to test the functional importance of the eIF5B-eIF1A interaction. Replacing the last five residues of eIF1A (residues 149 to 153, encoding DIDDI) with alanine (the mutant named eIF1A-5A) resulted in a slow-growth (Slg−) phenotype on synthetic complete (SC) medium containing all amino acids (Fig. 2A, left panel, rows 1 and 3). This Slg− phenotype was attributed to a defect in translation initiation, as polysome profile analysis revealed a loss of polysomes and an accumulation of 80S monosomes in the eIF1A-5A mutant (Fig. 2B). In WT cells with actively translating ribosomes, the ratio of polysomes to monosomes (P/M) was 1.14. In contrast, the P/M ratio in the eIF1A-5A mutant cells was reduced to 0.39 (Fig. 2B). The reduction in P/M ratio in the eIF1A mutant is consistent with a defect in translation initiation, which leads to a slower formation of 80S complexes. Accordingly, elongating ribosomes terminate translation, detach from the mRNA, and accumulate as inactive 80S couples awaiting recruitment into another round of translation.
FIG. 2.

Overexpression of eIF5B suppresses the translation defects associated with the eIF1A-5A mutant. (A) Analysis of cell growth and GCN4 expression. Isogenic WT (J263) and eIF1A-5A mutant (J264) strains expressing eIF5B from single-copy (sc) or high-copy-number (hc) plasmids were grown to saturation, and 4 μl of serial dilutions (at OD600 of 1.0, 0.1, 0.01, 0.001, and 0.0001) were spotted on SC medium or SC medium containing 10 mM 3-AT (SC + 3-AT). Plates were incubated for 2 or 3 days at 30°C, as indicated. The GCN4-lacZ plasmid pC2847 was introduced into the strains, and cells were grown and β-galactosidase activities were determined as described previously (15). R, cells were grown under nonstarvation conditions in SD medium, where GCN4 expression is repressed; DR, cells were grown under starvation conditions (SD + 10 mM 3-AT), where GCN4 expression is derepressed. The β-galactosidase activities represent mean values (with standard errors/deviations in parentheses) of nmol ONPG (o-nitrophenyl-β-d-galactopyranoside) cleaved min−1 mg−1 from three cultures. (B) Polysome analysis. WCEs of the strains described for panel A were separated and analyzed as described for Fig. 1B. The polysome/monosome ratios (P/M) were calculated by measuring the area in the combined polysome fractions and the 80S peak.
In addition to the general growth defect, the eIF1A-5A mutation blocked growth on medium containing 3-aminotriazole (3-AT), an inhibitor of histidine biosynthesis that causes amino acid starvation (Fig. 2A, middle panel, rows 1 and 3). Growth on medium containing 3-AT requires elevated expression of GCN4, encoding a transcriptional activator of amino acid biosynthetic gene expression. Expression of GCN4 is under translational control, and cells lacking eIF5B are unable to derepress GCN4 expression (Gcn− phenotype) and fail to grow on 3-AT medium (8). Consistent with the 3-AT-sensitive (3-ATs) phenotype in the eIF1A-5A mutant, the fivefold derepression of expression from a GCN4-lacZ reporter in WT cells treated with 3-AT was blocked in cells expressing the eIF1A-5A mutant (Fig. 2A, right panel, rows 1 and 3). Thus, the eIF1A-5A mutation confers a Gcn− phenotype.
As the C terminus of eIF1A directly interacts with eIF5B (9, 24, 28), we hypothesized that the phenotypes associated with the eIF1A-5A mutation were due to impaired eIF5B binding. To test this idea, we overexpressed full-length eIF5B in the eIF1A-5A mutant cells. Overexpression of eIF5B strongly suppressed the slow-growth phenotype and nearly completely suppressed the 3-ATs phenotype in cells expressing the eIF1A-5A mutant (Fig. 2A). Consistent with these growth phenotypes, overexpression of eIF5B resulted in a significant restoration of both the GCN4 expression (Fig. 2A) and the P/M ratio (Fig. 2B) in eIF1A-5A cells. The eIF1A-5A mutation did not impair eIF2α phosphorylation, a critical element in GCN4 translational control, and likewise, overexpression of eIF5B did not affect eIF2α phosphorylation in strains expressing eIF1A-5A, nor did it affect the levels of eIF1A-5A (data not shown). As overexpression of eIF5B had no effect on cells expressing WT eIF1A, we propose that overexpression of eIF5B restores both general translation and gene-specific translational regulation in the eIF1A-5A mutant either by promoting the eIF1A-eIF5B interaction by mass action or by enabling eIF5B to function in the absence of an interaction with eIF1A.
Mutations in the eIF5B and eIF1A C termini impair direct binding of eIF5B to eIF1A and the recruitment of eIF5B to 40S complexes in vivo.
Previous studies have revealed that the binding of eIF5B to eIF1A was mediated by the C termini of the two proteins. To characterize the function of this interaction in vivo, we deleted the eIF5B C-terminal helix H14 (mutant ΔH14 lacking residues 974 to 1002), which helps form a binding pocket for the C terminus of eIF1A (24). As predicted, deletion of the H14 helix blocked the ability of eIF5B to interact with eIF1A in a yeast two-hybrid assay (data not shown). To directly assess eIF1A binding to eIF5B, we expressed GST-eIF5B397-1002, GST-eIF5B397-974 (ΔH14), or GST in yeast cells also expressing Flag-tagged eIF1A or the eIF1A-5A mutant. Whereas neither eIF1A nor eIF1A-5A coprecipitated with GST, precipitation of GST-eIF5B397-1002 from crude cell extracts resulted in the coprecipitation of eIF1A (Fig. 3A, lane 10) but not the eIF1A-5A mutant (Fig. 3A, lane 12). Likewise, GST-eIF5B-ΔH14 failed to pull down eIF1A (Fig. 3A, lane 14) or eIF1A-5A (data not shown). Thus, these pulldown assays confirm the important roles of eIF1A and eIF5B C termini in mediating the interaction between these factors.
FIG. 3.

eIF1A-5A and eIF5B-ΔH14 mutations block the eIF1A-eIF5B interaction and impair binding of eIF5B to the 40S subunit in vivo. (A) GST pulldown assay. Yeast strain derivatives of H2971 expressing Flag-tagged eIF1A (eIF1A-FL) or eIF1A-5A (5A-FL) were transformed with plasmids designed to express GST, GST-eIF5B397−1002 (GST-eIF5B), or GST-eIF5B397-974 (GST-5B-ΔH14) under the control of the galactose-regulated GAL1 promoter. Transformants were grown in SC plus galactose medium to induce GST or GST-eIF5B expression, and WCEs were incubated with GST-Sepharose beads. After washing the bound proteins, the pellet (P), supernatant fractions (S; 2%), and 2% of the WCE inputs (lysate) were analyzed by immunoblotting using anti-Flag (eIF1A) and anti-GST (eIF5B) antisera, as indicated. The positions of the various GST fusion proteins are indicated by arrowheads: black, GST-ΔN-eIF5B; white, GST-ΔN-eIF5B-ΔH14; gray, GST. (B) Derivatives of yeast strain H2971 expressing full-length eIF5B1-1002 (row 1), eIF5B1-794 (eIF5B-ΔH14, row 2), eIF5B397-1002 (ΔN-eIF5B, row 3) or eIF5B397-974 (ΔN-eIF5B-ΔH14, row 4), as indicated, were grown to saturation, and 4 μl of serial dilutions (at OD600 of 1.0, 0.1, 0.01, 0.001, and 0.0001) were spotted on SC medium and incubated at 30°C for 2 days. (C) Derivatives of yeast strain H2971 expressing eIF1A or the eIF1A-5A mutant (5A) and either eIF5B397-1002 (ΔN-eIF5B), no eIF5B (eIF5BΔ), or eIF5B397-974 (ΔH14), as indicated, were grown to saturation, spotted on SC medium as described for panel B, and incubated at 30°C for 2 or 4 days. The GCN4-lacZ plasmid pC2847 was introduced into the strains, and cells were grown and β-galactosidase activities were determined as described in the legend to Fig. 2A. (D) Polysome and Western blot analysis. Yeast strains J274 (ΔN-eIF5B + eIF1A), J276 (ΔN-eIF5B + eIF1A-5A), and J275 (ΔN-eIF5B-ΔH14 + eIF1A) were grown in SC medium to an OD600 of ∼1.0 and then fixed with 1% formaldehyde. WCEs were prepared and analyzed for polysome content (upper panels) and factor binding to the 40S subunit (lower panels) as described in the legend to Fig. 1B and C.
Interestingly, the deletion of helix H14 had only a modest effect on the growth rate of yeast (Fig. 3B, rows 1 and 2). Previous work demonstrated that the N-terminal ∼396 residues of yeast eIF5B are not essential for growth (9, 20); however, the eIF5B N terminus is believed to promote ribosomal binding perhaps through a direct interaction with the 40S subunit or possibly through eIF1A (28). As shown in Fig. 3B (rows 1 and 3), yeasts expressing full-length eIF5B1-1002 and eIF5B397-1002 (ΔN) grow at similar rates (Fig. 3B, rows 1 and 3). To restrict our analysis to the C-terminal interaction between eIF5B and eIF1A and to avoid potentially redundant functions of the eIF5B N terminus, we examined the impact of the eIF5B-ΔH14 and the eIF1A-5A mutations in the context of ΔN-eIF5B (lacking residues 1 to 396). Deleting helix H14 from ΔN-eIF5B resulted in a significant slow-growth phenotype (Fig. 3B, row 4, and Fig. 3C, row 3); likewise, the eIF1A-5A mutation caused a slow-growth phenotype in strains expressing ΔN-eIF5B (Fig. 3C, compare rows 1 and 2 or 7). However, coexpression of the eIF1A-5A and the ΔN-eIF5B-ΔH14 mutants in the same strain resulted in growth rates comparable to that in cells expressing WT eIF1A and either ΔN-eIF5B-ΔH14 or no eIF5B (Fig. 3C, rows 3 to 7). Thus, the eIF1A-5A mutation did not exacerbate the phenotype associated with the ΔH14 mutation in eIF5B, indicating that the 5A and ΔH14 mutations affect the same step in protein synthesis.
Further evidence for the functional redundancy of the eIF1A-5A and eIF5B-ΔH14 mutations came from the analysis of GCN4 expression. In cells expressing WT eIF1A and ΔN-eIF5B, GCN4-lacZ expression increased ∼eightfold under derepressing conditions (Fig. 3C, row 1). As observed previously, this high level of expression of GCN4 under derepressing conditions was blocked in cells expressing eIF1A-5A (Fig. 3C, row 2) and in cells lacking eIF5B (Fig. 3C, row 5). The ΔN-eIF5B-ΔH14 mutation likewise blocked the high-level expression of the GCN4-lacZ reporter under derepressing conditions (Fig. 3C, row 3). Importantly, introduction of the eIF1A-5A mutant into strains lacking eIF5B (Fig. 3C, row 6) or expressing ΔN-eIF5B-ΔH14 (Fig. 3C, row 4) did not further impair GCN4 expression. These results are consistent with the notion that the primary function of the DIDDI residues in eIF1A is to bind the C terminus of eIF5B.
As eIF1A is part of the 48S complex that recognizes the AUG start codon on an mRNA, we hypothesized that the binding of eIF5B to eIF1A may recruit eIF5B to the preinitiation complex. Accordingly, we predicted that the disruption of the eIF1A-eIF5B interaction by the 5A and ΔH14 mutations would impair the binding of eIF5B to the 40S subunit in vivo. Following formaldehyde cross-linking of whole cells, WCEs were separated on 4.5 to 45% sucrose gradients to examine the polysome profiles (Fig. 3D, upper panels) and on 7 to 30% sucrose gradients, followed by Western analysis of individual fractions to monitor translation factor binding to the 40S ribosomal subunit (Fig. 3D, lower panels). Consistent with the growth defects observed in cells expressing eIF1A-5A or ΔN-eIF5B-ΔH14, the polysome profiles from 4.5 to 45% sucrose revealed polysome runoff and accumulation of 80S ribosomes, indicative of a translation initiation defect in these strains (Fig. 3D, upper panels). In extracts from cells expressing ΔN-eIF5B and WT eIF1A, factors eIF5B, eIF1A, and eIF2α cosedimented with the 40S marker RPS22p (Fig. 3D, bottom left panel). Analysis of the extracts from cells expressing the eIF1A-5A or the ΔN-eIF5B-ΔH14 mutant showed that eIF1A and eIF2α continued to sediment with the 40S subunit. This result is consistent with the notion that eIF1A and eIF2 bind to the 40S subunit early in the translation initiation pathway and with the results of previous studies showing that the eIF1A-5A mutation did not affect the formation of 43S preinitiation complexes (1). However, less eIF5B was bound to the 40S subunit and more of the factor was found at the top of the gradient in the extracts from the eIF1A-5A (61% reduction of eIF5B on the 40S compared to that of WT eIF1A) and ΔN-eIF5B-ΔH14 (68% reduction in factor bound to the 40S) mutants (Fig. 3D, lower middle and right panels). Thus, the eIF1A-5A and ΔN-eIF5B-ΔH14 mutations impair the eIF1A-dependent recruitment of eIF5B to the 48S preinitiation complex.
The eIF1A-5A mutation suppresses the slow-growth phenotype and 80S accumulation of the GTPase-defective eIF5B-T439A mutant.
The eIF5B-T439A mutant lacks GTPase activity and is thought to form dead-end initiation complexes due to its impaired dissociation from the 80S ribosome following subunit joining. In vitro, eIF5B-T439A promoted ribosomal subunit joining and stabilized Met-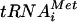 binding to 80S ribosomes, supporting the idea that eIF5B-T439A, due to its failure to hydrolyze GTP, is stuck on the ribosome following subunit joining. Consistent with these in vitro findings, yeast cells expressing ΔN-eIF5B-T439A grew very slowly (36), even more slowly than cells lacking eIF5B (Fig. 4B, rows 1 and 3). To examine the impact of the T439A mutation on eIF5B binding to ribosomes in vivo, we used formaldehyde cross-linking followed by sucrose gradient and Western analyses. In WT cells, ΔN-eIF5B was distributed across the gradient, with a small percentage of the factor cofractionating with 40S and 80S ribosomes (Fig. 4A, left panel). Factors eIF1A and eIF2α were mainly localized at the top of the gradient and the 40S fractions, though a small amount of eIF1A cofractionated with the 80S monosome. In contrast, ΔN-eIF5B-T439A was found at the top of the gradient and was then present in a clear peak comigrating with the 80S ribosomes (Fig. 4A, right panel). In relation to the input amount of eIF5B, there was an ∼1.5-fold increase in the amount of ΔN-eIF5B-T439A versus WT ΔN-eIF5B bound to the 80S fractions. Interestingly, eIF1A was found at the top of the gradient and cofractionating with eIF5B in the 80S peak in cells expressing ΔN-eIF5B-T439A (Fig. 4A, right panel). This accumulation of ΔN-eIF5B-T439A on 80S ribosomes in vivo is consistent with the idea that GTP hydrolysis by eIF5B is required for the factor's release from the ribosome following subunit joining. Moreover, the cofractionation of eIF1A with eIF5B and 80S ribosomes suggests that eIF1A release from the 80S ribosome is coupled to eIF5B GTP hydrolysis and the release of eIF5B.
binding to 80S ribosomes, supporting the idea that eIF5B-T439A, due to its failure to hydrolyze GTP, is stuck on the ribosome following subunit joining. Consistent with these in vitro findings, yeast cells expressing ΔN-eIF5B-T439A grew very slowly (36), even more slowly than cells lacking eIF5B (Fig. 4B, rows 1 and 3). To examine the impact of the T439A mutation on eIF5B binding to ribosomes in vivo, we used formaldehyde cross-linking followed by sucrose gradient and Western analyses. In WT cells, ΔN-eIF5B was distributed across the gradient, with a small percentage of the factor cofractionating with 40S and 80S ribosomes (Fig. 4A, left panel). Factors eIF1A and eIF2α were mainly localized at the top of the gradient and the 40S fractions, though a small amount of eIF1A cofractionated with the 80S monosome. In contrast, ΔN-eIF5B-T439A was found at the top of the gradient and was then present in a clear peak comigrating with the 80S ribosomes (Fig. 4A, right panel). In relation to the input amount of eIF5B, there was an ∼1.5-fold increase in the amount of ΔN-eIF5B-T439A versus WT ΔN-eIF5B bound to the 80S fractions. Interestingly, eIF1A was found at the top of the gradient and cofractionating with eIF5B in the 80S peak in cells expressing ΔN-eIF5B-T439A (Fig. 4A, right panel). This accumulation of ΔN-eIF5B-T439A on 80S ribosomes in vivo is consistent with the idea that GTP hydrolysis by eIF5B is required for the factor's release from the ribosome following subunit joining. Moreover, the cofractionation of eIF1A with eIF5B and 80S ribosomes suggests that eIF1A release from the 80S ribosome is coupled to eIF5B GTP hydrolysis and the release of eIF5B.
FIG. 4.

The eIF1A-5A mutation blocks the growth defect and 80S accumulation of the eIF5B-T439A GTPase-defective mutant. (A) eIF5B-T439A accumulates on 80S ribosomes in vivo. Yeast strains J274 (ΔN-eIF5B) and J280 (ΔN-eIF5-T439A) were grown in SC medium to an OD600 of ∼1.0 and fixed with 1% formaldehyde, and then WCEs were separated on 4.5% to 45% sucrose gradients by centrifugation at 39,000 rpm for 2.5 h. Gradients were fractionated while being scanned at 254 nm to visualize the indicated ribosomal species (upper panels). One hundred-microliter aliquots (17% of total) of the gradient fractions and a portion of the starting WCE (In, input) were subjected to Western analysis using antibodies against the indicated proteins. (B) Serial dilutions of derivatives of yeast strain H2971 expressing eIF1A (rows 1 and 3) or the eIF1A-5A mutant (rows 2 and 4) and either no eIF5B (eIF5BΔ, rows 1 and 2) or eIF5B397-1002-T439A (ΔN-eIF5B-T439A, rows 3 and 4) were spotted on SC medium and grown at 30°C for 4 days. (C) WCEs from formaldehyde-fixed yeast strains J280 (ΔN-eIF5B-T439A + eIF1A) and J281 (ΔN-eIF5B-T439A + eIF1A-5A) were prepared, and initiation factor binding to ribosomal species was determined by sucrose gradient and Western analysis, as described for panel A.
Expression of ΔN-eIF5B-T439A in yeast resulted in a severe inhibition of translation initiation, as revealed by the loss of polysomes and the accumulation of inactive 80S couples (associated 40S and 60S subunits, lacking mRNA and Met-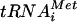 [36]) (Fig. 4A). As eIF5B binds to vacant 80S couples in vitro (36), the cosedimentation of ΔN-eIF5B-T439A with the 80S peak on sucrose gradients (Fig. 4A) could represent spurious binding of ΔN-eIF5B-T439A to vacant 80S couples rather than the specific recruitment of ΔN-eIF5B-T439A to ribosomes during the translation initiation pathway. Results from two experiments indicated that at least a fraction of ΔN-eIF5B-T439A was recruited to the ribosome via the normal translation initiation pathway. First, the slow-growth phenotype associated with the ΔN-eIF5B-T439A mutation was partially suppressed by the eIF1A-5A mutation (Fig. 4B). Expression of ΔN-eIF5B-T439A exacerbated the slow-growth phenotype observed in cells expressing WT eIF1A and lacking WT ΔN-eIF5B (Fig. 4B, compare rows 1 and 3). Significantly, in the presence of the eIF1A-5A mutant, ΔN-eIF5B-T439A was no longer toxic and the double mutant grew at nearly the same rate as cells lacking eIF5B (Fig. 4B, rows 2 to 4). The fact that the eIF1A-5A mutation suppresses the toxicity associated with the ΔN-eIF5B-T439A mutation is consistent with the notion that ΔN-eIF5B-T439A is recruited into the translation initiation pathway via interaction with the eIF1A C terminus. Second, whereas the majority of the input ΔN-eIF5B-T439A was associated with 80S ribosomes in sucrose gradient analyses of extracts from cells expressing WT eIF1A (Fig. 4C, left panel), less ΔN-eIF5B-T439A was bound to the 80S complexes (∼20% decrease), and the factor redistributed to the 40S peak and top of the gradient in extracts from the eIF1A-5A mutant cells (Fig. 4C, right panel). This loss of binding of ΔN-eIF5B-T439A to 80S ribosomes in strains expressing eIF1A-5A supports the idea that the eIF5B-eIF1A interaction is critical for the recruitment and perhaps the stable binding of eIF5B to the ribosome.
[36]) (Fig. 4A). As eIF5B binds to vacant 80S couples in vitro (36), the cosedimentation of ΔN-eIF5B-T439A with the 80S peak on sucrose gradients (Fig. 4A) could represent spurious binding of ΔN-eIF5B-T439A to vacant 80S couples rather than the specific recruitment of ΔN-eIF5B-T439A to ribosomes during the translation initiation pathway. Results from two experiments indicated that at least a fraction of ΔN-eIF5B-T439A was recruited to the ribosome via the normal translation initiation pathway. First, the slow-growth phenotype associated with the ΔN-eIF5B-T439A mutation was partially suppressed by the eIF1A-5A mutation (Fig. 4B). Expression of ΔN-eIF5B-T439A exacerbated the slow-growth phenotype observed in cells expressing WT eIF1A and lacking WT ΔN-eIF5B (Fig. 4B, compare rows 1 and 3). Significantly, in the presence of the eIF1A-5A mutant, ΔN-eIF5B-T439A was no longer toxic and the double mutant grew at nearly the same rate as cells lacking eIF5B (Fig. 4B, rows 2 to 4). The fact that the eIF1A-5A mutation suppresses the toxicity associated with the ΔN-eIF5B-T439A mutation is consistent with the notion that ΔN-eIF5B-T439A is recruited into the translation initiation pathway via interaction with the eIF1A C terminus. Second, whereas the majority of the input ΔN-eIF5B-T439A was associated with 80S ribosomes in sucrose gradient analyses of extracts from cells expressing WT eIF1A (Fig. 4C, left panel), less ΔN-eIF5B-T439A was bound to the 80S complexes (∼20% decrease), and the factor redistributed to the 40S peak and top of the gradient in extracts from the eIF1A-5A mutant cells (Fig. 4C, right panel). This loss of binding of ΔN-eIF5B-T439A to 80S ribosomes in strains expressing eIF1A-5A supports the idea that the eIF5B-eIF1A interaction is critical for the recruitment and perhaps the stable binding of eIF5B to the ribosome.
eIF1A dissociation from the 80S ribosome following subunit joining is coupled to eIF5B GTP hydrolysis and release.
The in vivo data presented thus far support a model in which eIF1A bound to the 40S subunit recruits eIF5B, which in turn promotes 60S subunit joining. Consistent with these results, Acker et al. demonstrated that the interaction between the eIF5B and eIF1A C termini is required for efficient subunit joining in vitro and enhances the rate of GTP hydrolysis by eIF5B by ∼20-fold (1). To further explore the role of the eIF5B-eIF1A interaction, we modified the 80S complex formation assay in order to monitor the binding of ΔN-eIF5B and eIF1A to 48S and 80S complexes. As typically performed, the 80S formation assay monitors the conversion of 48S complexes containing [35S]Met-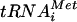 to 80S complexes by separating the reaction products on nondenaturing gels and using autoradiography to visualize the [35S]Met-
to 80S complexes by separating the reaction products on nondenaturing gels and using autoradiography to visualize the [35S]Met-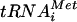 (3). To monitor the binding of eIF1A and ΔN-eIF5B to the 48S and 80S complexes, the proteins in the nondenaturing gels were transferred to nitrocellulose membranes and subjected to immunoblot analysis using anti-eIF1A and anti-eIF5B antisera. Consistent with the known binding of eIF1A to 43S and 48S complexes, eIF1A was readily detected on the 40S complexes formed in the presence of eIF1, eIF1A, eIF5, and eIF2-Met-
(3). To monitor the binding of eIF1A and ΔN-eIF5B to the 48S and 80S complexes, the proteins in the nondenaturing gels were transferred to nitrocellulose membranes and subjected to immunoblot analysis using anti-eIF1A and anti-eIF5B antisera. Consistent with the known binding of eIF1A to 43S and 48S complexes, eIF1A was readily detected on the 40S complexes formed in the presence of eIF1, eIF1A, eIF5, and eIF2-Met-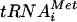 ternary complexes (Fig. 5A, lane 4). Interestingly, the addition of ΔN-eIF5B in the absence of 60S subunits did not affect the binding of eIF1A to the 40S complexes (Fig. 5A, lane 5), and ΔN-eIF5B was not observed on the 40S fraction (Fig. 5A, lane 5). (The presence of a small amount of ΔN-eIF5B on 80S complexes in Fig. 5A, lane 5 [see bottom panel where a smaller fraction of each reaction was loaded on the gel], suggests that the 40S fractions used in these assays contained a small amount of 60S subunits that were converted to 80S complexes in the presence of ΔN-eIF5B.) Importantly, the detection of eIF1A and ΔN-eIF5B on the 40S and 80S complexes was specific, as neither the anti-eIF1A nor the anti-eIF5B antisera cross-reacted with purified 40S or 60S subunits or 80S ribosomes (Fig. 5A, lanes 1 to 3).
ternary complexes (Fig. 5A, lane 4). Interestingly, the addition of ΔN-eIF5B in the absence of 60S subunits did not affect the binding of eIF1A to the 40S complexes (Fig. 5A, lane 5), and ΔN-eIF5B was not observed on the 40S fraction (Fig. 5A, lane 5). (The presence of a small amount of ΔN-eIF5B on 80S complexes in Fig. 5A, lane 5 [see bottom panel where a smaller fraction of each reaction was loaded on the gel], suggests that the 40S fractions used in these assays contained a small amount of 60S subunits that were converted to 80S complexes in the presence of ΔN-eIF5B.) Importantly, the detection of eIF1A and ΔN-eIF5B on the 40S and 80S complexes was specific, as neither the anti-eIF1A nor the anti-eIF5B antisera cross-reacted with purified 40S or 60S subunits or 80S ribosomes (Fig. 5A, lanes 1 to 3).
FIG. 5.

Release of eIF5B and eIF1A following 80S complex formation in vitro is dependent on GTP hydrolysis by eIF5B. (A) Native polyacrylamide gel monitoring incorporation of eIF1A and eIF5B into 43S-mRNA (48S) and 80S complexes. 48S complexes were assembled using purified 40S subunits, Met-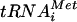 , eIF1, eIF1A, eIF2, eIF5, and mRNA in the presence of GTP and the presence (lane 5) or absence (lane 4) of ΔN-eIF5B (see Materials and Methods). 80S complexes were formed from 48S complexes by adding 60S subunits and either WT ΔN-eIF5B (lane 6) or ΔN-eIF5B-T439A (lane 7) in the presence of GDPNP. Purified 40S, 60S, and 80S ribosomes were loaded in lanes 1, 2, and 3, respectively. The native gel was transferred to a nitrocellulose membrane, and Western analysis was carried out using antibodies recognizing yeast eIF1A (upper panel) or eIF5B (middle panel). The positions of 80S and 48S (43S-mRNA) complexes are indicated. (Lower panel) To better resolve the amount of eIF5B on the 80S complexes, 10-fold less of the reaction mixtures from a duplicate experiment was separated on a native gel and subjected to Western analysis to detect eIF5B. (B) SDS-PAGE analysis of 80S and 48S complexes isolated from a native gel. 80S complexes were formed and analyzed as described for panel A, using ΔN-eIF5B or ΔN-eIF5B-T439A in the presence of GTP or GDPNP as indicated. The gel was stained with Coomassie blue to reveal the locations of the 48S and 80S complexes. The complexes were excised, denatured in loading buffer containing SDS, and subjected to SDS-PAGE and immunoblot analysis using the antibodies that recognize eIF5B, eIF1A, 40S subunit protein RPS22, and 60S subunit protein PUB2, as indicated. Lanes 1 to 4 contain the 80S complexes; lanes 5 to 8 contain the 48S complexes. The lower RPS22 panel is a longer exposure of the blot in the upper panel. (Lower panel) The relative amounts of eIF5B and eIF1A in 80S fractions (lanes 1 and 2) of three experiments were quantified and normalized to the amounts obtained in the presence of GDPNP (lane 1).
, eIF1, eIF1A, eIF2, eIF5, and mRNA in the presence of GTP and the presence (lane 5) or absence (lane 4) of ΔN-eIF5B (see Materials and Methods). 80S complexes were formed from 48S complexes by adding 60S subunits and either WT ΔN-eIF5B (lane 6) or ΔN-eIF5B-T439A (lane 7) in the presence of GDPNP. Purified 40S, 60S, and 80S ribosomes were loaded in lanes 1, 2, and 3, respectively. The native gel was transferred to a nitrocellulose membrane, and Western analysis was carried out using antibodies recognizing yeast eIF1A (upper panel) or eIF5B (middle panel). The positions of 80S and 48S (43S-mRNA) complexes are indicated. (Lower panel) To better resolve the amount of eIF5B on the 80S complexes, 10-fold less of the reaction mixtures from a duplicate experiment was separated on a native gel and subjected to Western analysis to detect eIF5B. (B) SDS-PAGE analysis of 80S and 48S complexes isolated from a native gel. 80S complexes were formed and analyzed as described for panel A, using ΔN-eIF5B or ΔN-eIF5B-T439A in the presence of GTP or GDPNP as indicated. The gel was stained with Coomassie blue to reveal the locations of the 48S and 80S complexes. The complexes were excised, denatured in loading buffer containing SDS, and subjected to SDS-PAGE and immunoblot analysis using the antibodies that recognize eIF5B, eIF1A, 40S subunit protein RPS22, and 60S subunit protein PUB2, as indicated. Lanes 1 to 4 contain the 80S complexes; lanes 5 to 8 contain the 48S complexes. The lower RPS22 panel is a longer exposure of the blot in the upper panel. (Lower panel) The relative amounts of eIF5B and eIF1A in 80S fractions (lanes 1 and 2) of three experiments were quantified and normalized to the amounts obtained in the presence of GDPNP (lane 1).
Incubating the components of the 48S complex (40S, eIF1, eIF1A, eIF2, Met-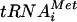 , mRNA, and GTP) with 60S subunits and either ΔN-eIF5B or GTPase-defective ΔN-eIF5B-T439A, both in the presence of nonhydrolyzable GDPNP, resulted in the accumulation of ΔN-eIF5B on 80S complexes (Fig. 5A, lanes 6 to 7; the smaller loadings in the bottom panel provide a better quantitative analysis). These results are consistent with previous findings using [35S]Met-
, mRNA, and GTP) with 60S subunits and either ΔN-eIF5B or GTPase-defective ΔN-eIF5B-T439A, both in the presence of nonhydrolyzable GDPNP, resulted in the accumulation of ΔN-eIF5B on 80S complexes (Fig. 5A, lanes 6 to 7; the smaller loadings in the bottom panel provide a better quantitative analysis). These results are consistent with previous findings using [35S]Met-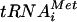 to monitor 80S complex formation, and they support the in vivo data demonstrating that eIF5B associates with 80S complexes. Interestingly, eIF1A, which was present on the 48S complexes (Fig. 5A, lanes 4 and 5), was largely retained in the 80S complexes (Fig. 5A, lanes 6 and 7). Because both ΔN-eIF5B and eIF1A were present on the 80S complexes, we proposed that eIF1A release following subunit joining was dependent on eIF5B release. To test this hypothesis, 48S complexes formed in the presence of GTP were mixed with 60S subunits and WT ΔN-eIF5B in the presence of GTP or GDPNP. The reaction mixture products were separated on nondenaturing gels, and the 80S and 48S complexes were excised from the gels, separated by SDS-PAGE, and analyzed by immunoblot analysis using anti-eIF5B and anti-eIF1A antisera. In these experiments we also monitored the abundance of 40S and 60S subunits by using antisera directed against the ribosomal proteins RPS22 (40S) and PUB2 (60S). As shown in Fig. 5B, lane 1, both ΔN-eIF5B and eIF1A accumulated on 80S complexes formed in the presence of GDPNP. Relative to the amount of ribosomal proteins, the amount of ΔN-eIF5B and eIF1A recovered in 80S complexes formed in the presence of GTP was reduced by 40 to 70% compared to the amount of factors bound in the presence of GDPNP (Fig. 5B, lane 2, and bottom panel). The absence of detectable RPS22 and eIF1A in the 48S fractions (Fig. 5B, lanes 5 and 6) indicates nearly quantitative conversion of 48S complexes to 80S complexes in these in vitro assays. Our results are consistent with the idea that eIF5B GTPase activity is required for release of the factor from 80S complexes following subunit joining. Moreover, the coupled loss of both eIF5B and eIF1A from the 80S complexes supports the idea that GTP hydrolysis releases eIF5B from the 80S complexes, enabling eIF1A to dissociate as well.
to monitor 80S complex formation, and they support the in vivo data demonstrating that eIF5B associates with 80S complexes. Interestingly, eIF1A, which was present on the 48S complexes (Fig. 5A, lanes 4 and 5), was largely retained in the 80S complexes (Fig. 5A, lanes 6 and 7). Because both ΔN-eIF5B and eIF1A were present on the 80S complexes, we proposed that eIF1A release following subunit joining was dependent on eIF5B release. To test this hypothesis, 48S complexes formed in the presence of GTP were mixed with 60S subunits and WT ΔN-eIF5B in the presence of GTP or GDPNP. The reaction mixture products were separated on nondenaturing gels, and the 80S and 48S complexes were excised from the gels, separated by SDS-PAGE, and analyzed by immunoblot analysis using anti-eIF5B and anti-eIF1A antisera. In these experiments we also monitored the abundance of 40S and 60S subunits by using antisera directed against the ribosomal proteins RPS22 (40S) and PUB2 (60S). As shown in Fig. 5B, lane 1, both ΔN-eIF5B and eIF1A accumulated on 80S complexes formed in the presence of GDPNP. Relative to the amount of ribosomal proteins, the amount of ΔN-eIF5B and eIF1A recovered in 80S complexes formed in the presence of GTP was reduced by 40 to 70% compared to the amount of factors bound in the presence of GDPNP (Fig. 5B, lane 2, and bottom panel). The absence of detectable RPS22 and eIF1A in the 48S fractions (Fig. 5B, lanes 5 and 6) indicates nearly quantitative conversion of 48S complexes to 80S complexes in these in vitro assays. Our results are consistent with the idea that eIF5B GTPase activity is required for release of the factor from 80S complexes following subunit joining. Moreover, the coupled loss of both eIF5B and eIF1A from the 80S complexes supports the idea that GTP hydrolysis releases eIF5B from the 80S complexes, enabling eIF1A to dissociate as well.
Analysis of 80S complexes formed in the presence of the ΔN-eIF5B-T439A mutant provided further support for this idea. Consistent with the inability of ΔN-eIF5B-T439A to hydrolyze GTP, both ΔN-eIF5B-T439A and eIF1A were retained on 80S complexes formed in the presence of GDPNP or GTP (Fig. 5B, lanes 3 and 4). The small amount of eIF1A found on 48S complexes formed in the presence of ΔN-eIF5B-T439A and GDPNP (Fig. 5B, lane 7) likely reflects turnover products in which the GDPNP associates with eIF2, forming dead-end 48S complexes that cannot release eIF2 and participate in subunit joining. Taken together, these in vitro studies reveal the presence of eIF1A in 80S complexes following subunit joining and indicate that eIF1A release from the 80S complex is dependent on the dissociation of eIF5B.
DISCUSSION
eIF5B catalyzes ribosomal subunit joining in vivo.
The function of eIF5B to promote ribosomal subunit joining was first revealed in reconstituted mammalian in vitro translation assays (30). The mammalian eIF5B functionally substituted for the yeast factor, restoring the growth and in vitro translation activity of strains lacking eIF5B (20, 30). Consistently, yeast eIF5B was also shown to catalyze ribosomal subunit joining in vitro (3, 36). Given this strong in vitro evidence defining the function of eIF5B, it was surprising that yeast cells lacking eIF5B did not show a subunit-joining defect in vivo. Here we show that rapid depletion of eIF5B in a fun12-td strain results in the accumulation of 48S translation initiation complexes consisting of 40S subunits, Met-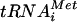 , and eIF1A bound to an mRNA (Fig. 1). These in vivo results indicate that, in cells rapidly depleted of eIF5B, subunit joining becomes a rate-limiting step for translation initiation. Interestingly, we did not observe an accumulation of mRNA-bound 40S subunits or halfmer species in strains lacking eIF5B (Fig. 1B and data not shown). A modest reduction in 40S subunit levels in the eIF5BΔ strain (8) (Fig. 1B, note the small size of the 40S peak in the eIF5B strain compared to that in the WT and fun12-td strains) may offset the subunit-joining defect and block the appearance of halfmer ribosomes, indicating that, in contrast to the fun12-td strain, subunit joining is not the sole limiting step in the eIF5BΔ strain.
, and eIF1A bound to an mRNA (Fig. 1). These in vivo results indicate that, in cells rapidly depleted of eIF5B, subunit joining becomes a rate-limiting step for translation initiation. Interestingly, we did not observe an accumulation of mRNA-bound 40S subunits or halfmer species in strains lacking eIF5B (Fig. 1B and data not shown). A modest reduction in 40S subunit levels in the eIF5BΔ strain (8) (Fig. 1B, note the small size of the 40S peak in the eIF5B strain compared to that in the WT and fun12-td strains) may offset the subunit-joining defect and block the appearance of halfmer ribosomes, indicating that, in contrast to the fun12-td strain, subunit joining is not the sole limiting step in the eIF5BΔ strain.
In addition, it is noteworthy that eIF5B is not essential in yeast. Thus, both an eIF5B-catalyzed and an uncatalyzed mechanism of subunit joining must operate in yeast. Perhaps there is a lag in the switch from the eIF5B-catalyzed to the uncatalyzed mechanism of subunit joining following depletion of eIF5B, and this lag accounts for the accumulation of halfmers. Accordingly, the impaired function of earlier steps in the translation pathway in eIF5BΔ strains, perhaps due to the reduction in 40S subunit levels, may prevent the accumulation of 48S complexes. As the only documented in vitro function of eIF5B is catalysis of ribosomal subunit joining, additional studies will be necessary to determine whether eIF5B influences the rates or efficiencies of earlier steps in the translation initiation pathway or whether eIF5BΔ strains adapt to the translation initiation defect by decreasing the abundance of 40S subunits via a quality control mechanism similar to that recently described for rRNAs in yeast (19).
Interaction of the eIF5B and eIF1A C termini helps recruit eIF5B to the 40S subunit prior to subunit joining.
Release of Pi and eIF1 upon AUG start codon recognition triggers the release of eIF2 and fixes the reading frame for translation (2). Concurrent with these events, eIF1A binding to the 40S subunit is enhanced, indicating a conformational change in the complex (21, 22). As eIF1A is bound tightly to the scanning 40S subunit (21), this enhanced binding likely reflects a change in the eIF1A-binding site on the 40S subunit, perhaps through the acquisition of new factor-ribosome interactions. Previous studies revealed an interaction between eIF2 and the N terminus of eIF1A that must be broken upon eIF2 release from the 48S complex (28). We propose that following release of eIF2, the eIF1A C terminus serves as a docking site to recruit eIF5B to the 48S complex.
Previous studies revealed that eIF1A and eIF5B bind directly to each other, and the binding sites were mapped to the C termini of the two factors (9). Structural studies confirmed these in vitro and yeast two-hybrid assay results, demonstrating that the last five residues of eIF1A (DIDDI) bind in a groove formed by the C-terminal helices H13 and H14 of eIF5B (24). It has been proposed that this interaction may both promote eIF5B recruitment to the 40S subunit and coordinate the coupled release of the factors from the 80S ribosome following subunit joining. Our studies provide additional support for these two roles of the eIF5B-eIF1A interaction. The eIF1A-5A mutation impaired cell growth, GCN4 expression, and general translation initiation, as revealed by the impaired polysome formation in vivo (Fig. 2). Consistent with the idea that the 5A mutation impaired eIF5B binding, each of these phenotypes was suppressed by overexpression of eIF5B. Furthermore, direct examination of eIF5B binding to 40S subunits in formaldehyde-fixed cells demonstrated that the eIF1A-5A mutation, as well as the eIF5B-ΔH14 mutation, impaired eIF5B binding to 40S complexes (Fig. 3C). It is noteworthy that eIF5B binding to 40S complexes was detected only in extracts from cells fixed with formaldehyde. We suspect that in the absence of covalent cross-linking, the binding of eIF5B to 40S complexes is disrupted during sedimentation through a sucrose gradient.
Further support for the role of eIF1A in recruiting eIF5B to the 48S complex came from studies of the eIF5B-T439A GTPase mutant. While cells lacking eIF5B grew slowly, expression of eIF5B-T439A caused a more severe slow-growth phenotype likely due to the failure of eIF5B-T439A to dissociate from 80S complexes following subunit joining. Consistent with the notion that eIF1A-5A fails to recruit eIF5B to the 40S subunit, this eIF1A mutation suppressed the toxic effects associated with the expression of eIF5B-T439A and rescued growth to the level observed in the absence of eIF5B (Fig. 4B). Thus, in the eIF1A-5A mutant strain, subunit joining proceeds through the eIF5B-independent pathway as neither WT eIF5B nor eIF5-T439A is recruited efficiently to the 40S subunit.
In addition to the binding of the eIF5B C terminus to eIF1A, the eIF5B N terminus contributes to interactions with eIF1A. Deletion of eIF1A C-terminal residues 130 to 153 or 108 to 153 abolished the interaction with N-terminally truncated eIF5B (lacking residues 1 to 377) but not with full-length eIF5B in WCEs (28). However, it is important to note that the interaction between eIF1A-Δ108 to -153 and ΔN-eIF5B was not observed in postribosomal supernatant fractions. Thus, the interaction of the eIF5B N terminus with eIF1A may be weak and detectable only when both factors are bound to the ribosome, or the interaction may not be direct and instead is bridged by the ribosome. The latter idea is consistent with the reported ribosome-binding activity of the bacterial IF2 N-terminal region (26). In light of this potential ribosome-binding activity of the eIF5B N-terminal region, it is interesting to note that the ΔH14 mutation conferred neither a Gcn− nor a slow-growth phenotype in full-length eIF5B (Fig. 3B and data not shown). We propose that the eIF5B N-terminal region and C-terminal helix H14 perform redundant roles, likely in recruiting eIF5B to the 40S ribosome. Thus, both deletion of the eIF5B N terminus and disruption of the eIF1A-eIF5B interaction by either the eIF1A-5A or the eIF5B-ΔH14 mutation are required to impair eIF5B function. However, the more severe slow-growth phenotype of the strain expressing ΔN-eIF5B-ΔH14 plus WT eIF1A versus the strain coexpressing the eIF1A-5A and ΔN-eIF5B mutants (Fig. 3C) indicates that the eIF5B C terminus may have functions in addition to binding to eIF1A.
Given the apparently redundant functions of the eIF5B N and C termini in mediating recruitment to the 40S subunit, it is paradoxical that the eIF1A-5A mutation confers a slow-growth and a Gcn− phenotype in the presence of full-length eIF5B (Fig. 2A). We propose that although the N terminus of eIF5B contributes to 40S binding, the interaction with eIF1A is critical for accurate and efficient translation initiation. Indeed, Acker et al. showed that the interaction between eIF1A and ΔN-eIF5B was important for efficient subunit joining in vitro (1). We propose that the 5A mutation in eIF1A physically occludes eIF5B binding to eIF1A and thus the 40S subunit. In addition, the eIF1A C terminus may have additional functions following eIF5B binding (1). Consistent with the former interpretation, eIF1A-5A failed to bind to GST-eIF5B even when the proteins were overexpressed (Fig. 3A). Taken together, we propose that the C terminus of eIF1A serves as a docking site for eIF5B, which also binds directly to the ribosome via other low-affinity interactions. Accordingly, the eIF1A-5A mutation interferes with eIF5B binding and impairs translation initiation and yeast cell growth. Overexpression of eIF5B suppresses the growth defect (Fig. 2A) not by restoring eIF5B binding to eIF1A-5A per se but rather by enabling eIF5B to productively engage the ribosome in the absence of the eIF1A interaction.
eIF1A dissociation from the 80S ribosome following subunit joining is dependent on release of eIF5B following GTP hydrolysis.
The in vitro experiments presented in Fig. 5 show for the first time that both eIF1A and eIF5B are present on the 80S ribosome following subunit joining. Moreover, the release of eIF1A from the 80S ribosome is coupled to GTP hydrolysis and dissociation of eIF5B. In reactions containing WT eIF5B and nonhydrolyzable GDPNP, both eIF5B and eIF1A accumulated on 80S complexes. In contrast, in reactions containing GTP, where eIF5B can hydrolyze the nucleotide and release from the ribosome, the amounts of both eIF5B and eIF1A on the 80S ribosome were smaller (Fig. 5B, lane 1 versus lane 2 and lower panel). Consistent with these findings, levels of eIF1A and eIF5B accumulation on 80S ribosomes were equivalent in the presence of GDPNP and GTP in reactions containing the GTPase-defective eIF5B-T439A mutant (Fig. 5B, lanes 3 to 4). These in vitro findings are supported by in vivo data obtained from the fun12-td strain. The 48S complexes that accumulate upon destruction of eIF5B contain elevated amounts of eIF1A and mRNA but not other factors like eIF2, eIF5, and eIF3 (Fig. 1D). We propose that this in vivo snapshot reveals the translation initiation intermediate following GTP hydrolysis and Pi release by eIF2 in which eIF2, eIF5, eIF3, and eIF1 dissociate from the 48S complex, leaving eIF1A in the A site to recruit eIF5B. Though our experiments involved formaldehyde cross-linking of factors to ribosomal complexes in vivo, we cannot rule out the possibility that GTP hydrolysis by eIF2, rather than dissociating the factors from the 80S ribosome, simply lowered their ribosomal binding affinity such that they were removed during the high-speed velocity sedimentation in the sucrose gradients due to incomplete cross-linking of these factors to the 40S subunit. Finally, in further support of the notion that eIF5B release is required for eIF1A release from the 80S product of subunit joining, both eIF5B and eIF1A were observed to cofractionate with 80S monosomes in extracts from formaldehyde cross-linked cells expressing eIF5B-T439A (Fig. 4A).
Genetic data provide additional support for the proposed coupled release of eIF5B and eIF1A from the 80S ribosome. Overexpression of eIF1A exacerbated the growth defect of strains lacking eIF5B or expressing a C-terminally truncated form of eIF5B that fails to interact with eIF1A (9). Thus, in the eIF5B-independent translation initiation pathway operating in cells lacking eIF5B, subunit joining and eIF1A release may be rate limiting. Accordingly, overexpression of eIF1A results in the accumulation of eIF1A on the 80S ribosome, blocking the subsequent steps in the pathway. It is noteworthy that in bacteria, release of IF2 requires IF1 (6), suggesting that the coupled release of eIF5B and eIF1A is an evolutionarily conserved process.
Combining the results of our study with those obtained by Unbehaun et al. (38) and Algire et al. (2) provides new details on the release of initiation factors from the ribosome during the translation initiation pathway. Recognition of the start codon during scanning triggers the release or transfer of eIF1 to a new site on the 48S complex, and this is coupled with the release of Pi and eIF2-GDP, which may be associated with eIF5 (37). The eIF1A remains bound in the A site of the ribosome and serves as a docking site for eIF5B. It is interesting to note that we were unable to detect eIF5B binding to the 48S complexes assembled in vitro (Fig. 5); however, the in vivo cross-linking experiments apparently stabilized this interaction (Fig. 3D). As eIF3 and eIF1 are not present on the 80S product of subunit joining, it can be proposed that eIF5B binding to the 48S complex and subsequent 60S subunit joining displaces eIF1 and eIF3, resulting in the accumulation of 80S complexes containing eIF1A and eIF5B. Cryo-electron microscopy images of the bacterial 70S ribosome with IF1 in the A site, IF2-GDPNP occupying the factor-binding site and contacting both the GTPase center on the large subunit and fMet-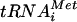 in the P site, and IF3 near the E site (4) support our findings that eIF1A and eIF5B are simultaneously bound to the 80S ribosome. We propose that eIF5B acts like a plug to prevent the release of eIF1A from the 80S complex, such that GTP hydrolysis and release of eIF5B are necessary steps for the release of eIF1A. It is also possible that GTP hydrolysis by eIF5B plays an active role in releasing eIF1A from the 80S ribosome. However, our previous description of eIF5B mutations that lower ribosome-binding affinity and bypass the requirement for GTP hydrolysis (36) indicate that this putative GTP hydrolysis-dependent function of eIF5B is not essential. Finally, the C-terminally labeled form of eIF1A that works as well as unlabeled eIF1A in promoting the early steps of the translation pathway (1, 22) does not support subunit joining, likely because the label interferes with eIF5B interactions. Thus, the precise timing of eIF1A and eIF5B release in relation to GTP hydrolysis by eIF5B will require the development of novel labeled forms of eIF1A and eIF5B that can be used for kinetic analyses of these last steps in the translation initiation pathway.
in the P site, and IF3 near the E site (4) support our findings that eIF1A and eIF5B are simultaneously bound to the 80S ribosome. We propose that eIF5B acts like a plug to prevent the release of eIF1A from the 80S complex, such that GTP hydrolysis and release of eIF5B are necessary steps for the release of eIF1A. It is also possible that GTP hydrolysis by eIF5B plays an active role in releasing eIF1A from the 80S ribosome. However, our previous description of eIF5B mutations that lower ribosome-binding affinity and bypass the requirement for GTP hydrolysis (36) indicate that this putative GTP hydrolysis-dependent function of eIF5B is not essential. Finally, the C-terminally labeled form of eIF1A that works as well as unlabeled eIF1A in promoting the early steps of the translation pathway (1, 22) does not support subunit joining, likely because the label interferes with eIF5B interactions. Thus, the precise timing of eIF1A and eIF5B release in relation to GTP hydrolysis by eIF5B will require the development of novel labeled forms of eIF1A and eIF5B that can be used for kinetic analyses of these last steps in the translation initiation pathway.
Acknowledgments
We thank Alan Hinnebusch for many helpful discussions and comments on the manuscript, Byung-Sik Shin and Joo-Ran Kim for the eIF5B-ΔH14 constructs and for helpful suggestions, and members of the Dever and Hinnebusch laboratories for helpful discussions.
This work was supported in part by the Intramural Research Program of the NIH, NICHD (to T.E.D.), and by American Cancer Society grant RSG-03-156-01-GMC (to J.R.L.).
Footnotes
Published ahead of print on 22 January 2007.
REFERENCES
- 1.Acker, M. G., B. S. Shin, T. E. Dever, and J. R. Lorsch. 2006. Interaction between eukaryotic initiation factors 1A and 5B is required for efficient ribosomal subunit joining. J. Biol. Chem. 281:8469-8475. [DOI] [PubMed] [Google Scholar]
- 2.Algire, M. A., D. Maag, and J. R. Lorsch. 2005. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell 20:251-262. [DOI] [PubMed] [Google Scholar]
- 3.Algire, M. A., D. Maag, P. Savio, M. G. Acker, S. Z. Tarun, Jr., A. B. Sachs, K. Asano, K. H. Nielsen, D. S. Olsen, L. Phan, A. G. Hinnebusch, and J. R. Lorsch. 2002. Development and characterization of a reconstituted yeast translation initiation system. RNA 8:382-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen, G. S., A. Zavialov, R. Gursky, M. Ehrenberg, and J. Frank. 2005. The cryo-EM structure of a translation initiation complex from Escherichia coli. Cell 121:703-712. [DOI] [PubMed] [Google Scholar]
- 5.Battiste, J. B., T. V. Pestova, C. U. T. Hellen, and G. Wagner. 2000. The eIF1A solution structure reveals a large RNA-binding surface important for scanning function. Mol. Cell 5:109-119. [DOI] [PubMed] [Google Scholar]
- 6.Benne, R., N. Naaktgeboren, J. Gubbens, and H. O. Voorma. 1973. Recycling of initiation factors IF-1, IF-2 and IF-3. Eur. J. Biochem. 32:372-380. [DOI] [PubMed] [Google Scholar]
- 7.Boeke, J. D., J. Trueheart, G. Natsoulis, and G. R. Fink. 1987. 5-Fluoroorotic acid as a selective agent in yeast molecular genes. Methods Enzymol. 154:164-175. [DOI] [PubMed] [Google Scholar]
- 8.Choi, S. K., J. H. Lee, W. L. Zoll, W. C. Merrick, and T. E. Dever. 1998. Promotion of Met-tRNAiMet binding to ribosomes by yIF2, a bacterial IF2 homolog in yeast. Science 280:1757-1760. [DOI] [PubMed] [Google Scholar]
- 9.Choi, S. K., D. S. Olsen, A. Roll-Mecak, A. Martung, K. L. Remo, S. K. Burley, A. G. Hinnebusch, and T. E. Dever. 2000. Physical and functional interaction between the eukaryotic orthologs of prokaryotic translation initiation factors IF1 and IF2. Mol. Cell. Biol. 20:7183-7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dever, T. E., L. Feng, R. C. Wek, A. M. Cigan, T. D. Donahue, and A. G. Hinnebusch. 1992. Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68:585-596. [DOI] [PubMed] [Google Scholar]
- 11.Dohmen, R. J., P. Wu, and A. Varshavsky. 1994. Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science 263:1273-1276. [DOI] [PubMed] [Google Scholar]
- 12.Fekete, C. A., D. J. Applefield, S. A. Blakely, N. Shirokikh, T. Pestova, J. R. Lorsch, and A. G. Hinnebusch. 2005. The eIF1A C-terminal domain promotes initiation complex assembly, scanning and AUG selection in vivo. EMBO J. 24:3588-3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gietz, R. D., and A. Sugino. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527-534. [DOI] [PubMed] [Google Scholar]
- 14.Gietz, R. D., A. R. Willems, and R. A. Woods. 1995. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11:355-560. [DOI] [PubMed] [Google Scholar]
- 15.Hinnebusch, A. G. 1985. A hierarchy of trans-acting factors modulate translation of an activator of amino acid biosynthetic genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 5:2349-2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinnebusch, A. G. 2000. Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes, p. 185-243. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 17.Jivotovskaya, A. V., L. Valasek, A. G. Hinnebusch, and K. H. Nielsen. 2006. Eukaryotic translation initiation factor 3 (eIF3) and eIF2 can promote mRNA binding to 40S subunits independently of eIF4G in yeast. Mol. Cell. Biol. 26:1355-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Labib, K., J. A. Tercero, and J. F. Diffley. 2000. Uninterrupted MCM2-7 function required for DNA replication fork progression. Science 288:1643-1647. [DOI] [PubMed] [Google Scholar]
- 19.LaRiviere, F. J., S. E. Cole, D. J. Ferullo, and M. J. Moore. 2006. A late-acting quality control process for mature eukaryotic rRNAs. Mol. Cell 24:619-626. [DOI] [PubMed] [Google Scholar]
- 20.Lee, J. H., S. K. Choi, A. Roll-Mecak, S. K. Burley, and T. E. Dever. 1999. Universal conservation in translation initiation revealed by human and archaeal homologs of bacterial translation initiation factor IF2. Proc. Natl. Acad. Sci. USA 96:4342-4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maag, D., M. A. Algire, and J. R. Lorsch. 2006. Communication between eukaryotic translation initiation factors 5 and 1A within the ribosomal pre-initiation complex plays a role in start site selection. J. Mol. Biol. 356:724-737. [DOI] [PubMed] [Google Scholar]
- 22.Maag, D., C. A. Fekete, Z. Gryczynski, and J. R. Lorsch. 2005. A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol. Cell 17:265-275. [DOI] [PubMed] [Google Scholar]
- 23.Maag, D., and J. R. Lorsch. 2003. Communication between eukaryotic translation initiation factors 1 and 1A on the yeast small ribosomal subunit. J. Mol. Biol. 330:917-924. [DOI] [PubMed] [Google Scholar]
- 24.Marintchev, A., V. G. Kolupaeva, T. V. Pestova, and G. Wagner. 2003. Mapping the binding interface between human eukaryotic initiation factors 1A and 5B: a new interaction between old partners. Proc. Natl. Acad. Sci. USA 100:1535-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell, D. A., T. K. Marshall, and R. J. Deschenes. 1993. Vectors for the inducible overexpression of glutathione S-transferase fusion proteins in yeast. Yeast 9:715-722. [DOI] [PubMed] [Google Scholar]
- 26.Moreno, J. M. P., J. Kildsgaard, I. Siwanowicz, K. K. Mortensen, and H. U. Sperling-Petersen. 1998. Binding of Escherichia coli initiation factor IF2 to 30S ribosomal subunits: a functional role for the N-terminus of the factor. Biochem. Biophys. Res. Commun. 252:465-471. [DOI] [PubMed] [Google Scholar]
- 27.Nielsen, K. H., B. Szamecz, L. Valasek, A. V. Jivotovskaya, B. S. Shin, and A. G. Hinnebusch. 2004. Functions of eIF3 downstream of 48S assembly impact AUG recognition and GCN4 translational control. EMBO J. 23:1166-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olsen, D. S., E. M. Savner, A. Mathew, F. Zhang, T. Krishnamoorthy, L. Phan, and A. G. Hinnebusch. 2003. Domains of eIF1A that mediate binding to eIF2, eIF3 and eIF5B and promote ternary complex recruitment in vivo. EMBO J. 22:193-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pestova, T. V., and V. G. Kolupaeva. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 16:2906-2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pestova, T. V., I. B. Lomakin, J. H. Lee, S. K. Choi, T. E. Dever, and C. U. T. Hellen. 2000. The joining of ribosomal subunits in eukaryotes requires eIF5B. Nature 403:332-335. [DOI] [PubMed] [Google Scholar]
- 31.Phan, L., X. Zhang, K. Asano, J. Anderson, H. P. Vornlocher, J. R. Greenberg, J. Qin, and A. G. Hinnebusch. 1998. Identification of a translation initiation factor 3 (eIF3) core complex, conserved in yeast and mammals, that interacts with eIF5. Mol. Cell. Biol. 18:4935-4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pisarev, A. V., V. G. Kolupaeva, V. P. Pisareva, W. C. Merrick, C. U. Hellen, and T. V. Pestova. 2006. Specific functional interactions of nucleotides at key −3 and +4 positions flanking the initiation codon with components of the mammalian 48S translation initiation complex. Genes Dev. 20:624-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothstein, R. J. 1983. One-step gene disruption in yeast. Methods Enzymol. 101:202-211. [DOI] [PubMed] [Google Scholar]
- 34.Sette, M., P. van Tilborg, R. Spurio, R. Kaptein, M. Paci, C. O. Gualerzi, and R. Boelens. 1997. The structure of the translational initiation factor IF1 from E. coli contains an oligomer-binding motif. EMBO J. 16:1436-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherman, F., G. R. Fink, and C. W. Lawrence. 1974. Methods of yeast genetics, p. 61-64. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 36.Shin, B. S., D. Maag, A. Roll-Mecak, M. S. Arefin, S. K. Burley, J. R. Lorsch, and T. E. Dever. 2002. Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell 111:1015-1025. [DOI] [PubMed] [Google Scholar]
- 37.Singh, C. R., B. Lee, T. Udagawa, S. S. Mohammad-Qureshi, Y. Yamamoto, G. D. Pavitt, and K. Asano. 2006. An eIF5/eIF2 complex antagonizes guanine nucleotide exchange by eIF2B during translation initiation. EMBO J. 25:4537-4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Unbehaun, A., S. I. Borukhov, C. U. Hellen, and T. V. Pestova. 2004. Release of initiation factors from 48S complexes during ribosomal subunit joining and the link between establishment of codon-anticodon base-pairing and hydrolysis of eIF2-bound GTP. Genes Dev. 18:3078-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]