

Abstract
Huntington's disease is characterized by death of striatal projection neurons. We used a Cre/Lox transgenic approach to generate an animal model in which D1 dopamine receptor (Drd1a)+ cells are progressively ablated in the postnatal brain. Striatal Drd1a, substance P, and dynorphin expression is progressively lost, whereas D2 dopamine receptor (Drd2) and enkephalin expression is up-regulated. Magnetic resonance spectroscopic analysis demonstrated early elevation of the striatal choline/creatine ratio, a finding associated with extensive reactive striatal astrogliosis. Sequential MRI demonstrated a progressive reduction in striatal volume and secondary ventricular enlargement confirmed to be due to loss of striatal cells. Mutant mice had normal gait and rotarod performance but displayed hindlimb dystonia, locomotor hyperactivity, and handling-induced electrographically verified spontaneous seizures. Ethological assessment identified an increase in rearing and impairments in the oral behaviors of sifting and chewing. In line with the limbic seizure profile, cell loss, astrogliosis, microgliosis, and down-regulated dynorphin expression were seen in the hippocampal dentate gyrus. This study specifically implicates Drd1a+ cell loss with tail suspension hindlimb dystonia, hyperactivity, and abnormal oral function. The latter may relate to the speech and swallowing disturbances and the classic sign of tongue-protrusion motor impersistence observed in Huntington's disease. In addition, the findings of this study support the notion that Drd1a and Drd2 are segregated on striatal projection neurons.
Keywords: striatum, Cre, Huntington's disease
The nigrostriatal pathway projects from the midbrain substantia nigra pars compacta to innervate the dorsal striatum. Dopamine is released from dopaminergic terminals in the striatum to regulate motor activity and eating behavior (1). Early studies suggested that dopamine D1 (Drd1a) and D2 receptors (Drd2) are segregated on striatal projection neurons; the Drd1a is expressed on substance P and dynorphin-positive striatal neurons, which project directly to the substantia nigra pars reticulata/entopeduncular complex (known as the direct pathway), whereas Drd2 is preferentially expressed on enkephalin-positive striatopallidal projecting neurons (2). Neurons within the globus pallidus then project to the subthalamic nucleus, which in turn relays to the substantia nigra pars reticulata/entopeduncular complex (known as the indirect pathway). Dopamine also modulates the activity of glutamatergic corticostriatal input on striatal projection neurons.
Idiopathic Parkinson's disease is characterized by the death of dopaminergic neurons; however, rare Parkinsonian syndromes have been identified in which the defect is associated with cell death in the dopamine-responsive neurons in the striatum (3–6). Huntington's disease (HD) (7–10), a neurodegenerative condition with motor, cognitive, and psychiatric disturbances, also involves death of dopamine receptor-expressing striatal projection neurons. Several positron emission tomography studies in HD have demonstrated a decrease of both D1- and D2-binding sites in association with volume loss in the striatum (11–13). Basic questions remain unanswered. Is the HD phenotype due to loss of the Drd1a+ or Drd2+ subpopulation of striatal neurons? What are secondary consequences, particularly on corticostriatal projection neurons, of the death of a specific population of cells within the striatum? HD animal models generated to date either use neurotoxic striatal lesioning (14) or express a CAG repeat expanded Huntingtin gene (15, 16). These approaches ultimately result in the death of cells in both Drd1a+ and Drd2+ striatal compartments.
In a previous model, we used Cre-mediated recombination to ablate neurons expressing the Drd1a gene during development (17–19). In this paradigm, most mutant (MUT) pups died in the first postnatal week, but some survived to 3 wk. MUT pups displayed periodic breathing, motor slowing, and twisting limb movements suggestive of dystonia and exhibited falls due to myoclonic jerks. Older pups (2–3 wk) showed a hyperkinetic syndrome with gait abnormality, postural instability, and limb and body jerks typical of HD (20). Although those mice model basal ganglia disease, the short lifespan of MUTac animals and potential for developmental compensation limited its value. Here, we have used another Cre-mediated strategy to ablate Drd1a-expressing neurons after birth. We expected that these mice would also manifest dystonia and motor abnormalities but were surprised to learn that they survived and had a relatively mild phenotype.
Results
Mice.
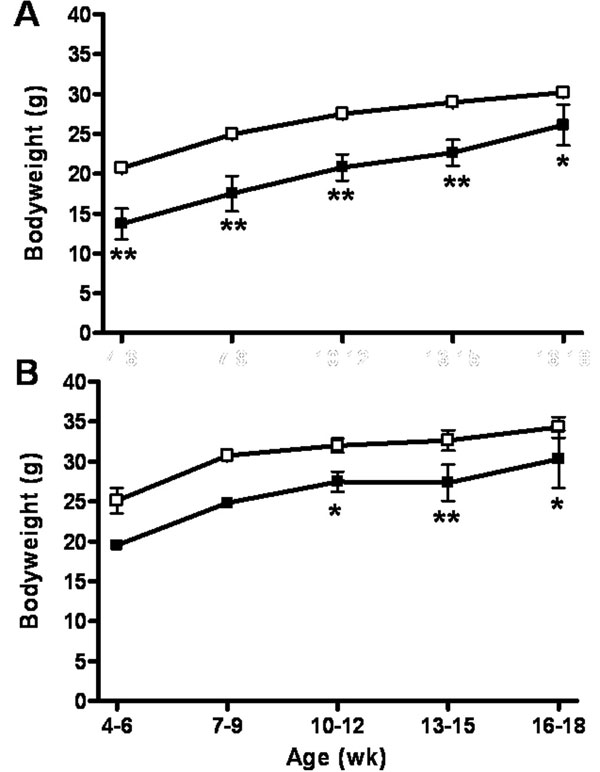
MUT mice expressing the attenuated diphtheria toxin (tox-176) gene in Drd1a+ cells were generated by crossing heterozygous calcium/calmodulin-dependent protein kinase II α promoter-driven (21) Cre-expressing transgenic mice with heterozygous tox-176 Drd1a locus knockin mice (17). Cre expression in the hippocampus, striatum, cortex, amygdala, olfactory tubercle, thalamus, and hypothalamus starts at 1–2 wk after birth, and tox-176 was expressed in Drd1a-expressing cells only in MUT mice after Cre recombination. Three other genotypes were produced as a result of mating of the parental heterozygous lines: wild-type (WT) mice, mice heterozygous for transcriptionally silenced tox-176 (HZtox), and mice heterozygous for Cre (HZcre). Overall male and female MUT mice weighed 17% and 25% less than WT littermates, respectively [supporting information (SI) Fig. 6]. MUT brains at 34 wk (n = 6) weighed 15% less than WT (n = 6). Unexpected animal death was not observed in any genotype.
Behavioral Phenotype.
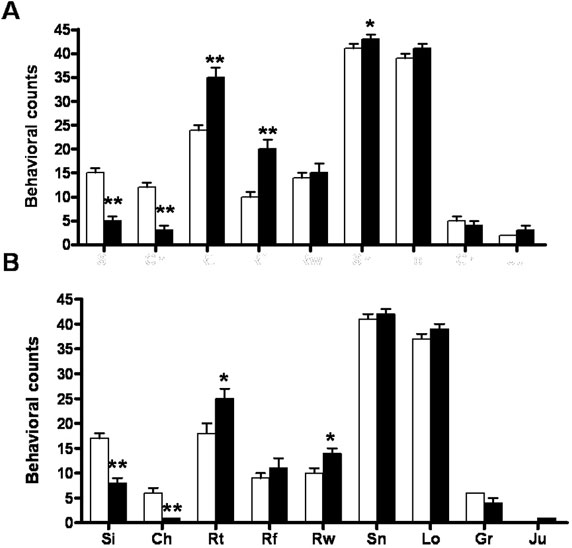
Handling-induced seizures occurred from 5 wk in MUT animals. No differences in locomotor activity were found between the three control groups (WT, HZtox, and HZcre). MUT mice of both sexes showed evidence of a hyperactive phenotype when exposed to a novel environment. They covered a greater distance (Fig. 1D), spent more time moving, and moved with increased speed compared with controls (data not shown). This hyperactivity in MUT mice was still observed in 16- to 17-wk-old animals (SI Fig. 7). No statistically significant differences were found in gait analysis or rotarod testing in MUT mice examined at 10–11 or 17–21 wk (SI Text). Separate cohorts of WT and MUT animals were assessed at two different age ranges by using a standardized ethological assay (22–24). At 6–9 wk, there was an increase in total rearing and a decrease in sifting and chewing (Fig. 1E). Sifting behavior involves the use of the snout and forepaws to displace cage bedding, and chewing was defined as being present when material was in the mouth. These changes occurred in both sexes and in older mice (16–21 wk) (SI Fig. 8). Progressive clasping of the hindlimbs was observed in a tail-suspension assay (Fig. 1 A and B). At 4 wk, MUT mice showed a subtle clasping dystonia compared with WT animals. MUT mice showed frequent paroxysmal bursts of dystonic hindlimb retraction by 12 wk, and by 14 wk, most MUT animals showed sustained hindlimb clasping associated with trunk flexion (Fig. 1C) and see SI Fig. 9.
Fig. 1.

Behavioral phenotype of MUT mice. Hindlimb clasping dystonic phenotype in MUT (A) compared with WT (B). Clasping score is determined over 14 sec (WT, □; MUT, ■). (C) Predominantly sustained hindlimb clasping was observed by 12 wk. (D) Locomotor activity data (for distance) for MUT (female, ●; male, ■) and WT (female, ○; male, □). Hyperactive phenotype of MUT (age 10–11 wk) males and females is observed over a 4-day test. (E) MUT mice (black bars) showed a decrease in sifting and chewing and an increase in total rearing. ∗∗, P < 0.01; ∗, P < 0.05 vs. WT control.
MRI and Magnetic Resonance Spectroscopy (MRS) Analysis.
Serial MRI and MRS analysis was undertaken on seven MUT and six WT mice. Striatal atrophy and secondary enlargement of the lateral ventricles were apparent in one MUT mouse at 4 wk and for the other five MUT mice from 6 wk (Fig. 2). Significant whole-brain atrophy in MUT animals was observed after 4 wk. The width of the entire brain measured at the level of the nucleus accumbens (NAc) (d4 in Fig. 2A) in MUT mice was ≈12% smaller than control littermates. There was no further reduction in total brain or cortical width (SI Fig. 10) from 4–30 wk. In contrast, there was a dramatic reduction in striatal size in the second month.
Fig. 2.

Serial MRI and MRS analysis of MUT mice. (A) Schematic diagram delineating the MRI parameters used in the study. (B) Serial changes in the size of the striatum relative to the whole brain in WT (□) and MUT (■). There is an age-dependent reduction in R value [where R = (d3−d2)/d4] in MUT compared with WT littermates. (C) MRI coronal sections of a WT mouse (C) and a MUT mouse (D) at 8 wk showing enlargement of the lateral ventricles and striatal atrophy. (E) C/C changes in striatum with time (WT, white bars; MUT, black bars). (F) NAA/creatine ratio over time. Note significant difference in C/C to 8 wk. A decrease in NAA/creatine is seen at 30 wk in MUT. ∗∗, P < 0.01; ∗, P < 0.05.
1H spectra of the striatum in MUT and WT mice was obtained at 4, 6, 8, 18, and 30 wk (Fig. 2 E and F). The choline/creatine (C/C) ratio was significantly increased in MUT compared with WT mice at 4, 6, and 8 wk. In contrast, no difference was observed in the C/C ratio at 18 and 30 wk. No significant difference in the NAA/creatine ratio was observed between MUT and WT mice at the first four time points, but a significant decrease was observed in MUT mice at 30 wk.
Seizures: Behavioral and Electroencephalographic (EEG) Phenotype.
Handling-induced seizures were observed commencing at 5 wk in MUT mice. Overnight EEG recording was performed on female mice as described (25) with infrared video monitoring (MUT n = 5, HZtox n = 1, HZcre n = 1, and WT n = 4). Multiple spontaneous seizures were recorded in two of five MUT mice. Typical seizures were associated with rhythmic electrographic spike/wave discharges followed by a period of postictal suppression of the normal background EEG activity (Fig. 3). During the seizures, MUT animals rubbed forepaws repeatedly, then fell and showed repetitive forelimb and hindlimb clonic movements, finally undergoing a second round of repetitive forepaw rubbing activity after becoming upright (see SI Movie 1 for a captured spontaneous seizure). The postictal recovery period was short (≈20 sec). The reappearance of a normal baseline EEG correlated with return of exploratory behavior. The background interictal EEG activity of all MUT mice was abnormal, with intermittent spike/wave discharges in the three mice that did not have overt seizures recorded throughout the monitoring period. WT controls had no evidence of epilepsy.
Fig. 3.

EEG phenotype of MUT mice. EEG recording of a spontaneous nocturnal seizure in a female MUT 32 wk of age. Note the seizure is demarcated between A and B.
Neuropathology in MUT Mice.
Volumetric analysis showed a 60% decrease in striatal volume of MUT mice (see SI Fig. 11). Stereological estimates of neutral red-stained sections demonstrated a 65% loss of cells in MUT mice [WT (12.87 ± 0.49) × 105 cells; MUT (4.46 ± 0.15) × 105 cells (P < 0.001)]. Cell density was found to be reduced by 17% in the striatum of MUT mice [WT (1.95 ± 0.10) × 105 cells/mm3; MUT (1.62 ± 0.07) × 105 cells/mm3 (P = 0.024)].
We also obtained estimates of volume, density, and cell number in the cortex and hippocampus of MUT animals and control littermates. Although there was no difference in cortical cell density [WT (1.94 ± 0.08) × 105 cells/mm3; MUT (2.21 ± 0.13) × 105 cells/mm3 (P = 0.162)] or total cell number [WT (12.43 ± 0.51) × 105 cells; MUT (11.51 ± 0.55) × 105 cells (P = 0.284)] in MUT mice, there was a significant decrease in the cortical volume of MUT mice [WT, 6.419 ± 0.039 mm3; MUT, 4.997 ± 0.247 mm3 (P = 0.002)]. In the hippocampus, MUT mice showed a decrease in both volume [WT mice, 0.101 ± 0.004 mm3; MUT mice, 0.039 ± 0.004 mm3 (P < 0.001)] and number of cells in the dentate gyrus (DG) [WT (1.47 ± 0.03) × 105 cells; MUT (0.42 ± 0.07) × 105 cells (P < 0.001)]. There were no differences seen in CA1/2 and CA3 regions of the hippocampus with respect to volume, cell number, or cell density (data not shown).
Staining for the astroglial marker, glial fibrillary acid protein (GFAP) was performed between 3 and 21 wk (Fig. 4). Astrocytes in MUT mice had the morphology of reactive astrocytes (between 3 and 12 wk) (Fig. 4 B and C) with large cell bodies and complex ramified cellular processes. In contrast, the much smaller number of astroglial cells identified in control mice had small cell bodies and lacked cellular processes (Fig. 4A). The number of reactive astrocytes in MUT mice increased over time, with a 20-fold higher number of GFAP+ cells at 9 wk compared with controls. The number of astroglia in MUT mice then reduced at later time points, although it always remained higher than in control mice (Fig. 4E). The astroglia in older MUT mice had a less-reactive morphology (Fig. 4D). A large number of reactive astrocytes were also seen in the hippocampus and a smaller number were seen throughout the cortex (SI Fig. 12).
Fig. 4.

Reactive astrogliosis in MUT mice. GFAP immunohistochemistry in WT (A) and MUT (B–D) at varying ages (B, 5 wk; C, 9 wk; D, 21 wk). (E) The number of GFAP-positive cells was quantified at different ages. There is significant difference (P < 0.05) between the number of cells in WT (white bars) and MUT (black bars) at all ages.
CD11b staining (see SI Materials and Methods for method) revealed the presence of activated microglia in the cortex, hippocampus, thalamus, and caudate putamen (CPu) of 3-wk MUT mice (n = 2). At 5 wk, comparatively less microglial reactivity was seen in the thalamus and CPu (n = 2), and no activated microglia were seen in the cortex or hippocampus. Older MUT mice (21 wk) were free of microglia. Activated microglia were not seen in WT mice (3–21 wk). Unlike the EIIa/Cre model (17), TUNEL staining (see SI Materials and Methods for method) showed very little evidence of apoptosis in MUT mice (2–9 wk). TUNEL-positive cells were not seen in the cortex. A small number of TUNEL-positive cells (approximately five/section) were seen in the hippocampus and thalamus [a nucleus known to express Drd1a (26)] in a 2-wk MUT.
Quantitative in Situ Hybridization.
Expression studies were undertaken on male mice of all four genotypes (HZCre, n = 8; WT, n = 7; HZtox, n = 4; MUT, n = 9) between 17 and 21 wk. Drd1a transcripts were identified in the CPu, NAc, islands of Calleja, and olfactory tubercles of control mice (HZcre and WT; Fig. 5) that was reduced in HZtox mice (63% of controls). The level of Drd1a mRNA in MUT mice was <5% of WT mice (Fig. 5 A and B). Drd1a was expressed in the DG of WT but not MUT mice. In line with the serial MRI data (Fig. 2B), the expression of Drd1a dropped progressively between 4–12.5 wk (SI Fig. 13). Drd2 mRNA was detected in the striatum of all mice (Fig. 5 C and D). Quantitative analysis demonstrated a 64% increase in expression in rostral striatum and a 49% increase in caudal striatum in MUT mice compared with controls (P < 0.05). Drd2 mRNA was up-regulated at 2 wk and remained elevated (SI Table 1). There was a significant increase in enkephalin expression levels in rostral CPu (46%) and caudal striatum (56%) of MUT mice compared with controls (P < 0.05). Enkephalin mRNA was up-regulated at 2 wk and remained elevated (SI Table 1). Dynorphin showed a differential pattern of expression in the dorsal and ventral striatum of controls (SI Table 1). A 59% decrease in dynorphin expression was seen in the NAc, a 67% decrease in the rostral Cpu, and a 75% decrease in the caudal CPu of MUT mice. An 83% decrease in dynorphin expression was apparent in the DG. Because of regional differences in expression patterns, we quantified substance P expression in different regions, the NAc, rostral, and caudal CPu. A significant decrease in substance P mRNA was observed in the NAc (58%) and rostral CPu (91%), but there was no significant difference in the caudal CPu of MUT mice.
Fig. 5.

Dopamine receptor expression. In situ hybridization for Drd1a (A, WT; B, MUT) and Drd2 (C, WT; D, MUT) at 21 wk.
Discussion
We have created a transgenic mouse model in which Drd1a+ cells are slowly but progressively ablated in the postnatal striatum, yet Drd2 single-positive cells are preserved. The cumulative cell loss provokes an up-regulation in the number of astrocytes that coincides temporally and spatially with progressive striatal atrophy. Individual astrocytes display a classical activated morphology during the phase of maximal striatal atrophy. Because the MUT line was constructed to specifically ablate Drd1a+ cells, fundamental questions relate to the specificity, extent, and timing of Drd1a+ cell killing. Stereological analysis provides clear evidence that cell death occurred, whereas Drd1a and Drd2 in situ hybridization data confirm that cell death is indeed restricted to the Drd1a+ cell compartment. Detailed behavioral analyses of MUT mice revealed a phenotype, elements of which are observed in other models of HD (15, 16, 23). We demonstrate that primary loss of a Drd1a+ subpopulation can produce a tail suspension-induced hindlimb dystonic phenotype, locomotor hyperactivity, and significant changes in oral behavioral topographies. Although striatonigral cell loss would predict bradykinesia rather than hyperkinesia (27), the locomotor hyperactivity in MUT mice is also observed in Drd1a knockout mice (28, 29). Additionally, MUT mice may lack a Drd1a/Drd2 double-positive population. Moreover, up-regulated Drd2 expression may reflect compensatory decreased signaling through the Drd2. We also show that loss of the Drd1a+ population results in spontaneous stereotyped seizures with features reminiscent of human complex partial seizures of temporal lobe origin where ictal hand automatisms are typically seen (30, 31). The seizures are likely to be related to hippocampal rather than striatal pathology.
Enlarged cerebral ventricles and striatal atrophy were identified by using noninvasive serial MRI. Significant striatal atrophy was first seen at 6 wk. Volumetric and stereological assessment undertaken on the same cohort of mice revealed significant cell loss in the striatal compartment. Cortical volume was also reduced both on MRI (see SI Fig. 10) and on formal volumetric analysis, but cell loss was not evident in the cortex. We found significant changes in the morphology of cortical neuropeptide Y-positive neurons in EIIa/Cre MUTs (19) and postulate that a decrease in soma size and/or pruning of neuritic processes are expected to account for the reduction in cortical volume. Death of striatal projection neurons may have secondary effects on corticostriatal neurons if these cortical neurons depended on striatal-derived trophic factors. Changes in cortical pyramidal neurons have been identified in other HD transgenic models (16), and it is not clear whether these changes are related to the primary disease process within the cortex or secondary to striatal pathology.
Sequential MRI studies over a 30-wk period identified progressive loss of striatal volume. In situ hybridization studies for a number of molecules were therefore undertaken between 2 and 21 wk, the time period during which major volume changes were noted on MRI analysis. The level of Drd1a (see SI Fig. 13), substance P, and dynorphin transcripts decreased gradually over this period (data not shown). In contrast to the EIIa/Cre paradigm, expression of these peptides persists in the ventral striatum. In MUT mice, Cre expression within the striatum is patchy and occurs in the postnatal brain rather than during development and at comparatively low levels. Drd1a+ cell loss is therefore slow, allowing for compensation. In the EIIa/Cre mice, Cre-mediated transgene recombination occurs at the fertilized oocyte stage (32), resulting in cell death upon Drd1a expression. In that model, there is extensive and synchronized death of the Drd1a+ population. Most MUT mice die in the first week, and only a small number survive beyond 2 wk. Because of the early demise of the animals and differences in the kinetics of Drd1a+ cell death, there may be insufficient time for compensation in substance P and dynorphin expression. The delayed expression of tox-176 in the striatum, thereby avoiding injury to Drd1a+ neuronal progenitors in the lateral ganglionic eminence (33), may also be important in explaining the phenotype differences between the two models. Furthermore, the nature of the tox-176 gene product compared with WT diphtheria toxin may be especially relevant in understanding the relative mild phenotype and potential for compensation. There are two reports that used the tox-176 gene in transgenic paradigms (34, 35). In both cases, transgenic lines were generated in which it was likely that tox-176 gene expression impaired cellular metabolism without apparent cell death. In one study (34), tox-176 expression targeted to lens cells resulted in cataract formation rather than the complete loss of lens tissue, and in the study of Ross et al. (35), tox-176 expression targeted to adipose cells resulted in lines in which fat cells survived, but mice were resistant to obesity. Finally, compensatory mechanisms made possible by the slow postnatal demise of Drd1a+ cells may include adaptive anatomical changes within the basal ganglia that ultimately may functionally reconstitute the direct pathway.
Astrocytes perform several functions that are essential for normal neuronal activity and also influence neurite outgrowth and other processes that contribute to brain recovery in the postinjury period (36). A number of studies in mice and humans have found a correlation between astrocytosis and elevated C/C ratios (37–40). Genetic disorders of astrocytes associated pathologically with astrocytosis displayed increased C/C ratios on MRS (38). The grade of malignant brain tumors is reflected in the content of astroglial elements relative to neurons and correlated with the C/C ratio in vivo (39) and in vitro (40). EIIa/Cre MUTs displayed extensive reactive gliosis (17), and so we sought to document brain astroglial activity in our adult model by using serial noninvasive MRS measurement of C/C levels. Elevated C/C levels in the brain were seen in MUT using serial MRS at 4, 6, and 8 wk (with a peak at 8 wk) but not at 18 and 30 wk. GFAP immunohistochemistry essentially mirrored the MRS results whereby the number of GFAP+ cells gradually increased over the first few months and peaked at 9 wk. In contrast, the number of astrocytes remained elevated from 12 to 21 wk, but the C/C on MRS was normal. Astrocytes at later time points had prominent GFAP-positive cell bodies and some cellular processes but did not have complex branched cellular processes apparent in the earlier time points. It appears, therefore, that the MRS finding of up-regulated C/C levels reflected the presence of activated astrocytes as C/C levels returned to normal despite persistent elevation in striatal astroglial cell counts. This serial MRS study in mice relates elevated C/C with the morphological astroglial activation state.
The functional implications of these data need to be based on what is known about the relative distribution of Drd1a and Drd2 on striatal projection neurons and other brain regions. A number of early studies using in situ hybridization immunohistochemistry (2, 41, 42) suggested that Drd1a and Drd2 were expressed on distinct populations of neostriatal projection neurons, and that there was only a small degree of overlap. Conversely, subsequent studies using highly sensitive PCR methodology concluded that the degree of overlap was substantial (43, 44). A recent study on cultured rat embryo-derived striatal neurons and adult striatal slices using complementary approaches of confocal microscopy and functional studies with D1- and D2-like selective agonists concluded that essentially all striatal projection neurons expressed both receptor subtypes but that striatopallidal neurons expressed predominantly Drd2 and striatonigral neurons expressed mainly the Drd1a subtype (45). Our results are at odds with the idea that all striatal neurons express significant levels of both receptor subtypes. The degree of striatal atrophy and associated striatal cell loss apparent in 34-wk MUT mice suggests there may be a subpopulation of neurons expressing both Drd1a and Drd2. Furthermore, given the potency of tox-176 in inhibiting cellular protein synthesis and the fact that Drd2 and enkephalin are up-regulated in the striatum, we conclude that a substantial proportion of Drd2+ cells do not have significant levels of Drd1a expression. Given the chronicity of the insult and the potential for striatal neurogenesis, as seen in rodent HD and brain ischemia models (46, 47), it is possible that the paradigm selects for replacement by, or survival of, Drd1a−/Drd2+ cells. The experimental design does not allow us to exclude the possibility that the behavioral phenotype may be due to loss of a Drd1a/Drd2 co-expressing subpopulation. A Drd2 promoter-driven Cre mouse could be used to address this question.
Drd1a expression in the cortex is at low levels in the mouse brain (see Fig. 5A and ref. 48). Despite the low Drd1a expression level, intense calcium/calmodulin-dependent protein kinase II α expression in the cortex would predict that any cells coexpressing Drd1a, even at low levels, would be killed. The up-regulated astrogliosis and microgliosis suggest a degree of cortical remodeling, and we hypothesize there may be compensatory neurogenesis as seen in other injury models (47) and selection of cells that do not express Drd1a. It is also possible that the glial response seen in the cortex is a function of remodeling of neuronal processes (49, 50). Because cortical volume does not change (on serial MRI analysis), whereas striatal volume decreases in concert with the emergence of pivotal aspects of the HD phenotype, such as hindlimb clasping, we suggest that striatal rather than primary cortical pathology explains the motor abnormalities in the model.
Neurodegenerative diseases involving progressive postnatal loss of striatal neurons can be modeled by using this double transgenic paradigm. We show that loss of the Drd1a+ compartment is sufficient to generate multiple aspects of the HD phenotype, hindlimb dystonia and hyperactivity (locomotion and rearing) and major abnormalities in oral functions of sifting and chewing. Abnormalities in oral behavioral topographies and hyperactivity may explain the persistent weight differences seen in MUT mice. The ethogram was assessed in the R6/1 line of HD transgenic mice (23), and delayed onset decreases were identified in sifting and chewing. Speech and swallowing disturbances occur in HD, and motor impersistence on tongue protrusion is a classic physical sign in HD that differentiates HD from other disorders, such as tardive dyskinesia (10). It is tempting to speculate that the oral abnormalities identified in the ethogram represent murine equivalents of the speech, swallowing, and tongue abnormalities seen in the human condition. The therapeutic implication of this study is that cell replacement and symptomatic drug strategies would need to target the D1 receptor subpopulation to reverse some aspects of HD disability.
Materials and Methods
All procedures involving the use of live animals conformed to the Australian National Health and Medical Research Council code of practice.
Animals.
Mice generated as described above were weighed from 4 to 18 wk of age. Males and females were analyzed separately with a two-way repeated-measures ANOVA with least standard difference post hoc analyses.
Behavior.
All behavioral analyses were carried out in age-matched mice with the observer blinded to genotype. Behavioral testing (locomotion, clasping, rotarod, and gait analysis) was undertaken on male (MUT, n = 19; WT, n = 13; HZtox, n = 8; and HZcre, n = 19) and female (MUT, n = 15; WT, n = 14; HZtox, n = 8; and HZcre, n = 9) mice.
Behavioral Testing.
The effect of ablating Drd1a-expressing cells on locomotor activity was examined in all four genotypes. Hindlimb clasping was assessed in all four genotypes at seven time points (4, 6, 8, 10, 12, 14, and 16 wk). Mice were observed in 2-sec time bins for 14 sec. Each mouse was allocated a score of 1 for abnormal dystonic hindlimb movement and a score of 0 in the absence of any abnormal movement. Rotarod motor performance and gait testing were performed as described in SI Materials and Methods. Ethological assessments were carried out by using a rapid time-sampling behavioral checklist technique, as described (22, 23). Ten MUT and 9–10 WT mice of each sex were used. Overnight infrared video epidural EEG monitoring of mice was performed as described (25). For detailed methods, see SI Materials and Methods and SI Table 2.
MRI and MRS.
In brief, mice were anesthetized with a 1–1.5% isoflurane–oxygen mixture during measurements using a 4.7-T MRI. A volume coil was used for excitation and a surface coil for receiving using a T2-weighted RARE sequence (repetition time (TR), 4,000 ms; echo train, 8; echo time (TE), 67 ms; field of view, 2.5 cm; 256 × 256; slice thickness, 0.6 mm; slices, 24 no gap; NEX, 16). The spectroscopic volume of interest (1.5 × 1.5 × 1.5 mm3) was placed in the CPu by visualization of the T2 images and excluded cerebrospinal fluid. The 1H-MR spectra were acquired by using a water-suppressed PRESS sequence (TR, 1,000 ms; TE, 136 ms; data points, 1,024; average, 2,048). Coronal slices were measured according to the atlas (51). The slice at bregma 0.8 mm was used to estimate the striatal volume. The R value (see Fig. 2) was calculated to assess striatal volume. MRS data were processed by using XWINNMR (Bruker, Ettlingen, Germany). The metabolite peaks were fitted for quantification, and the ratio of metabolic peaks was calculated from the integral intensity (area under peak). Linear values and metabolite concentration for each time point were compared across the two groups by using one-way ANOVA for genotype with repeated measures followed by least standard difference post hoc analysis.
Stereology, Immunohistochemistry, and in Situ Hybridization Histochemistry.
For stereology, we examined brains from the same mice assessed in the serial MRI study at 34 wk (WT, n = 5; MUT, n = 6). Striatal, cortex, and hippocampal volumetric analysis and cell counts were performed on 20-μm brain sections. For striatal analysis, sections 240 μm apart were selected to cover the extent of the striatum (bregma, 1.70 to −1.34 mm). Counts were made at regular predetermined intervals (x = 350 μm, y = 350 μm) within an unbiased counting frame of known area (50 × 40 μm = 2,000 μm2). For cortical analysis, six consecutive cortical sections (each 240 μm apart) were chosen for analysis, and the volume quoted was the sum of these six sections. The first section was isolated from the point of the appearance of corpus callosum (bregma, 0.86 mm). For hippocampal stereology, the predetermined intervals were reduced to (x = 50 μm, y = 50 μm) for Ammon's horn and (x = 40 μm, y = 40 μm) for the DG. Cells were counted on every 12th coronal section (240 μm apart), with the first section counted derived from the appearance of a complete DG (bregma, 1.34 mm). The counting frame was also reduced to accommodate the dense cell population, with a known size of 625 μm2 (x = 25 μm, y = 25 μm). GFAP staining using rabbit anti-GFAP antibody (1:450) (Dakocytomation, Carpinteria, CA) was undertaken on cryostat sections (14 μm) by using standard methods. GFAP-positive cell counts were determined on six sections. In situ hybridization histochemistry was undertaken by using published methods (52). All antisense oligonucleotides were verified against the relevant sequences by using NCBI BLAST (www.ncbi.nlm.nih.gov/BLAST). For probe sequences, see SI Materials and Methods and SI Table 3.
Supplementary Material
Acknowledgments
We thank Jim Massalas and Keith Buxton for technical assistance. J.D. is a National Health and Medical Research Council (NHMRC) Practitioner fellow. G.F.E. is an NHMRC Principal Research Fellow supported by Grant 400317. The work was funded by NHMRC Program Grant 236805 and Science Foundation Ireland Grant 02–1N1B227. Mouse behavioral analysis was undertaken in the Integrative Neuroscience Facility, Howard Florey Institute.
Abbreviations
- C/C
choline/creatine
- CPu
caudate putamen
- DG
dentate gyrus
- Drd1a
dopamine D1 receptor
- Drd2
dopamine D2 receptor
- HD
Huntington's disease
- MRS
magnetic resonance spectroscopy
- MUT
mutant
- NAc
nucleus accumbens
- EEG
electroencephalographic/electroencephalogram
- GFAP
glial fibrillary acidic protein.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0611625104/DC1.
References
- 1.Szczypka MS, Rainey MA, Palmiter RD. Nat Genet. 2000;25:102–104. doi: 10.1038/75484. [DOI] [PubMed] [Google Scholar]
- 2.Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 3.Rajput AH, Rozdilsky B, Rajput A. Can J Neurol Sci. 1991;18:275–278. doi: 10.1017/s0317167100031814. [DOI] [PubMed] [Google Scholar]
- 4.Hughes AJ, Daniel SE, Kilford L, Lees AJ. J Neurol Neurosurg Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quinn N. Multiple System Atrophy. Oxford: Butterworth-Heinemann; 1994. [Google Scholar]
- 6.Watts RL, Mirra SS, Richardson EP. Corticobasal Ganglionic Degeneration. Oxford: Butterworth-Heinemann; 1994. [Google Scholar]
- 7.Richfield EK, O'Brien CF, Eskin T, Shoulson I. Neurosci Lett. 1991;132:121–126. doi: 10.1016/0304-3940(91)90448-3. [DOI] [PubMed] [Google Scholar]
- 8.Andrews TC, Weeks RA, Turjanski N, Brooks DJ. Monitoring Disease Progression in Presymptomatic and Early Huntington's Disease: D1 and D2 Ligand PET and the Unified Huntington's Disease Rating Scale. Minneapolis: Abstract of the American Academy of Neurology; 1998. [Google Scholar]
- 9.Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhidayasiri R, Truong DD. Postgrad Med J. 2004;80:527–534. doi: 10.1136/pgmj.2004.019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Backman L, Robins-Wahlin TB, Lundin A, Ginovart N, Farde L. Brain. 1997;120:2207–2217. doi: 10.1093/brain/120.12.2207. [DOI] [PubMed] [Google Scholar]
- 12.Andrews TC, Weeks RA, Turjanski N, Gunn RN, Watkins LH, Sahakian B, Hodges JR, Rosser AE, Wood NW, Brooks DJ. Brain. 1999;122:2353–2363. doi: 10.1093/brain/122.12.2353. [DOI] [PubMed] [Google Scholar]
- 13.Pavese N, Andrews TC, Brooks DJ, Ho AK, Rosser AE, Barker RA, Robbins TW, Sahakian BJ, Dunnett SB, Piccini P. Brain. 2003;126:1127–1135. doi: 10.1093/brain/awg119. [DOI] [PubMed] [Google Scholar]
- 14.Dobrossy MD, Dunnett SB. Neuroscience. 2005;132:543–552. doi: 10.1016/j.neuroscience.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Hersch SM, Ferrante RJ. NeuroRx. 2004;1:298–306. doi: 10.1602/neurorx.1.3.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levine MS, Cepeda C, Hickey MA, Fleming SM, Chesselet MF. Trends Neurosci. 2004;27:691–697. doi: 10.1016/j.tins.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Drago J, Padungchaichot P, Wong JY, Lawrence AJ, McManus JF, Sumarsono SH, Natoli AL, Lakso M, Wreford N, Westphal H, et al. J Neurosci. 1998;18:9845–9857. doi: 10.1523/JNEUROSCI.18-23-09845.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong JY, Padungchaichot P, Massalas JS, Drago J. Neuroscience. 2000;95:1035–1041. doi: 10.1016/s0306-4522(99)00498-4. [DOI] [PubMed] [Google Scholar]
- 19.Padungchaichot P, Wong JY, Natoli AL, Massalas JS, Finkelstein DI, Lawrence AL, Drago J. Neuroscience. 2000;95:1025–1033. doi: 10.1016/s0306-4522(99)00497-2. [DOI] [PubMed] [Google Scholar]
- 20.Nance MA. J Geriatr Psychiatry Neurol. 1998;11:61–70. doi: 10.1177/089198879801100204. [DOI] [PubMed] [Google Scholar]
- 21.Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF, Schutz G. Genesis. 2001;31:37–42. doi: 10.1002/gene.1078. [DOI] [PubMed] [Google Scholar]
- 22.Ross SA, Wong JY, Clifford JJ, Kinsella A, Massalas JS, Horne MK, Scheffer IE, Kola I, Waddington JL, Berkovic SF, et al. J Neurosci. 2000;20:6431–6441. doi: 10.1523/JNEUROSCI.20-17-06431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clifford JJ, Drago J, Natoli AL, Wong JY, Kinsella A, Waddington JL, Vaddadi KS. Neuroscience. 2002;109:81–88. doi: 10.1016/s0306-4522(01)00409-2. [DOI] [PubMed] [Google Scholar]
- 24.Nally RE, McNamara FN, Clifford JJ, Kinsella A, Tighe O, Croke DT, Fienberg AA, Greengard P, Waddington JL. Neuropsychopharmacology. 2003;28:2055–2063. doi: 10.1038/sj.npp.1300259. [DOI] [PubMed] [Google Scholar]
- 25.McColl CD, Horne MK, Finkelstein DI, Wong JY, Berkovic SF, Drago J. Neuropharmacology. 2003;44:234–243. doi: 10.1016/s0028-3908(02)00369-6. [DOI] [PubMed] [Google Scholar]
- 26.Fremeau RT, Jr, Duncan GE, Fornaretto MG, Dearry A, Gingrich JA, Breese GR, Caron MG. Proc Natl Acad Sci USA. 1991;88:3772–3776. doi: 10.1073/pnas.88.9.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albin RL, Young AB, Penney JB. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 28.Xu M, Moratalla R, Gold LH, Hiroi N, Koob GF, Graybiel AM, Tonegawa S. Cell. 1994;79:729–742. doi: 10.1016/0092-8674(94)90557-6. [DOI] [PubMed] [Google Scholar]
- 29.Clifford JJ, Tighe O, Croke DT, Sibley DR, Drago J, Waddington JL. Neuropharmacology. 1998;37:1595–1602. doi: 10.1016/s0028-3908(98)00116-6. [DOI] [PubMed] [Google Scholar]
- 30.Kutlu G, Bilir E, Erdem A, Gomceli YB, Leventoglu A, Kurt GS, Karatas A, Serdaroglu A. Epilepsy Behav. 2005;6:353–359. doi: 10.1016/j.yebeh.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 31.Kramer U, Riviello JJ, Jr, Carmant L, Black PM, Madsen J, Holmes GL. Seizure. 1997;6:57–61. doi: 10.1016/s1059-1311(97)80054-4. [DOI] [PubMed] [Google Scholar]
- 32.Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Proc Natl Acad Sci USA. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohtani N, Goto T, Waeber C, Bhide PG. J Neurosci. 2003;23:2840–2850. doi: 10.1523/JNEUROSCI.23-07-02840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breitman ML, Rombola H, Maxwell IH, Klintworth GK, Bernstein A. Mol Cell Biol. 1990;10:474–479. doi: 10.1128/mcb.10.2.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ross SR, Graves RA, Spiegelman BM. Genes Dev. 1993;7:1318–1324. doi: 10.1101/gad.7.7b.1318. [DOI] [PubMed] [Google Scholar]
- 36.Sofroniew MV. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- 37.Tracey I, Dunn JF, Parkes HG, Radda GK. J Neurol Sci. 1996;141:13–18. doi: 10.1016/0022-510x(96)00135-9. [DOI] [PubMed] [Google Scholar]
- 38.Brockmann K, Dechent P, Meins M, Haupt M, Sperner J, Stephani U, Frahm J, Hanefeld F. J Neurol. 2003;250:300–306. doi: 10.1007/s00415-003-0995-2. [DOI] [PubMed] [Google Scholar]
- 39.Magalhaes A, Godfrey W, Shen Y, Hu J, Smith W. Acad Radiol. 2005;12:51–57. doi: 10.1016/j.acra.2004.10.057. [DOI] [PubMed] [Google Scholar]
- 40.Likavcanova K, Dobrota D, Liptaj T, Pronayova N, Mlynarik V, Belan V, Galanda M, Beres A, De Riggo J. Gen Physiol Biophys. 2005;24:327–335. [PubMed] [Google Scholar]
- 41.Gerfen CR, Keefe KA. Trends Neurosci. 1994;17:2–3. doi: 10.1016/0166-2236(94)90022-1. author reply, pp 4–5. [DOI] [PubMed] [Google Scholar]
- 42.Bloch B, Le Moine C. Trends Neurosci. 1994;17:3–4. doi: 10.1016/0166-2236(94)90023-x. author reply, pp 4–5. [DOI] [PubMed] [Google Scholar]
- 43.Surmeier DJ, Reiner A, Levine MS, Ariano MA. Trends Neurosci. 1993;16:299–305. doi: 10.1016/0166-2236(93)90103-s. [DOI] [PubMed] [Google Scholar]
- 44.Surmeier DJ, Song WJ, Yan Z. J Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aizman O, Brismar H, Uhlen P, Zettergren E, Levey AI, Forssberg H, Greengard P, Aperia A. Nat Neurosci. 2000;3:226–230. doi: 10.1038/72929. [DOI] [PubMed] [Google Scholar]
- 46.Tattersfield AS, Croon RJ, Liu YW, Kells AP, Faull RL, Connor B. Neuroscience. 2004;127:319–332. doi: 10.1016/j.neuroscience.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 47.Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S, Greenberg DA. Mol Cell Neurosci. 2003;24:171–189. doi: 10.1016/s1044-7431(03)00159-3. [DOI] [PubMed] [Google Scholar]
- 48.Drago J, Gerfen CR, Lachowicz JE, Steiner H, Hollon TR, Love PE, Ooi GT, Grinberg A, Lee EJ, Huang SP, et al. Proc Natl Acad Sci USA. 1994;91:12564–12568. doi: 10.1073/pnas.91.26.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parish CL, Finkelstein DI, Tripanichkul W, Satoskar AR, Drago J, Horne MK. J Neurosci. 2002;22:8034–8041. doi: 10.1523/JNEUROSCI.22-18-08034.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stanic D, Tripanichkul W, Drago J, Finkelstein DI, Horne MK. Brain Res. 2004;1023:83–91. doi: 10.1016/j.brainres.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 51.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic; 2001. [Google Scholar]
- 52.Cowen MS, Lawrence AJ. Alcohol Clin Exp Res. 2001;25:1126–1133. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}