

Abstract
For over three decades, the design of linear peptide ligands often has incorporated cyclic constraints to improve potency, receptor selectivity, proteolytic stability and biodistribution. Its importance has been so well established that modern day schemes for ligand-based drug design often start with cyclization of linear peptides to rigidify peptide structure, to limit its conformational possibilities, and to find key pharmacophore elements in three-dimensional space. In the past several years, cyclic constraints have been used to develop ligands with improved efficacy, binding affinity, biostability and receptor selectivity for α-melanocyte-stimulating hormone (α-MSH). Furthermore, potent cyclic α-MSH analogues, such as MT-II and SHU-9119, have made structure–activity relationship studies and molecular modeling more useful for creating new three-dimensional, topographical pharmacophore templates.
Introduction: constrained cyclic peptides
The design and study of constrained cyclic peptides has had a revolutionary impact in the field of peptide chemistry, receptor–ligand interactions, and drug design in the past three decades [1–6]. Cyclic analogues of linear, biologically active peptides of, for example, somatostatins, melanotropins and enkephalins, have demonstrated that cyclic peptides possess potential to: (1) increase agonist and antagonist potency; (2) withstand proteolytic degradation; (3) increase receptor selectivity; (4) enhance bioavailability; and (5) provide conformational insight for receptor binding (see, for example [1,7,8]). Constrained cyclic peptides have been used to stabilize secondary structures and have been modelled to mimic β-turns [1–3,6–8] and other peptide structures. Results from these studies have been exploited to expand the possibilities of developing peptidomimetics with increased efficacy and binding affinity to protein receptors [1,4,7,9].
If the structure of a receptor’s protein-binding site is known, one can design potential ligands by theoretically synthesizing a ligand that can complement the binding pocket by an induced fit [4,9]. In principle, this makes ligand design easier because the shape of the ligand simply has to fit the shape of the binding site. The ideal ligand in return, should either increase bioactivity or inhibit receptor response, making it an efficacious agonist and antagonist respectively. The known structures of many bioactive peptide hormones and neurotransmitters, the first of which was oxytoxin, and their proposed receptor-binding sites can now be found in searchable databases such as the RCBS Protein Data Bank (PDB) [10]. Although recent advances in crystallography and NMR have greatly increased the number of three-dimensional (3-D) structures found in these databases, the percentage of solved structures compared with the number of known proteins is less than ideal [11,12]. Hence, drug design derived from 3-D structural conformation is not feasible for proteins such as G-protein-coupled receptors (GPCRs) whose structures have not been solved. Here, constrained cyclic peptides have proven useful in two main ways. First, cyclic peptides possess less structural freedom than linear peptides and, hence, the ligand can be ‘locked’ into a limited number of possible conformations allowing one to derive testable hypothesis in the design of better receptor pharmacophores [3,4,6,11]. Secondly, the restraint placed on a cyclic peptide can create a ‘fit’ for ligand–receptor recognition that increases binding affinity, efficacy and receptor selectivity.
Factors affecting 3-D conformation
The spatial conformation of a peptide begins with its backbone. The atoms that map the backbone are the α carbons (Cα), the carbonyl carbons (C′), and the amide nitrogens (N). The pharmacophore elements within a potent ligand are then dictated by the topographical structure of the side chains and where they exist in 3-D space [13].
The backbone conformation in a peptide is governed by a series of torsional angles, denoted as φ, ψ, ω, and the side chain conformations by chi (χ) torsional angles. Bond rotation around C′–N–Cα–C′ is defined by the φ angle. The torsional angle for rotation around N–Cα–C′–N is known as the ψ angle. The torsional angle between C′i and C′i+1 is referred to as the ω angle. Torsional angles coding the relationship between side chains and the backbone are designated as χ1 for N–Cα–Cβ–Cγ, χ2 for Cα–Cβ–Cγ–Cδ, etc. As with all other molecules, peptide ligands must obey certain rules for steric and electrostatic interactions. Thus, a peptide chain is restricted to certain allowable or energetically favourable conformations. Ramachandran and co-workers were the first to qualitatively evaluate the contribution of peptide sequences for backbone secondary structures [14,15]. They showed that a plot of the φ angle versus the ψ angle revealed tendencies for most amino acids to fall within specific regions of φ, ψ space. In addition to spatially labeling allowable torsional angles for amino residues, the Ramachandran plot defines regions for secondary structure. For example, the right-handed α helix tends to have a φ angle around − 63° and a ψ angle around −42°.
It should now be noted that although the number of possible conformations for a small, linear peptide sequence can be limited by certain steric hindrances and allowable torsional angles, the overall number often is still large because of the flexibility of the peptide chain. To further restrict the number of possible conformations into a handful of allowed backbone structures, which future ligands can be modelled after, other restrictions such as cyclic constraints must be incorporated [2,3]. Figure 1 outlines a systematic approach to peptidomimetic design [2–4,7]. It must be mentioned that conformationally constrained peptides, whether involving a global constraint using cyclization, or a local constraint with incorporation of specialized amino acids, all play a crucial role in designing functional ligands [4,7,8,13].
Figure 1.
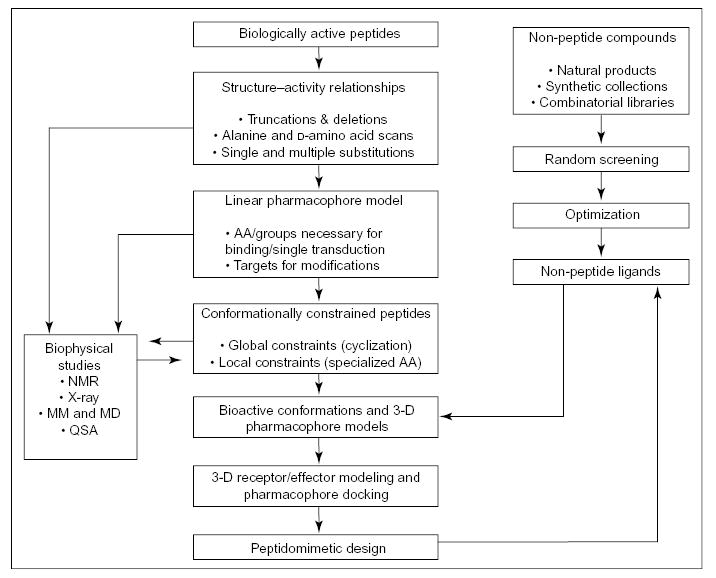
Systematic approach to peptidomimetic design. Adapted from [7] with permission. © 2002 Nature: Reviews Drug Discovery. www.nature.com/reviews.
Global constraint cyclization
There are four main ways to form constrained cyclic peptides. These cyclizations, as illustrated in Figure 2, involve bridging between (1) two side chains; (2) a side chain to N-terminus; (3) a side chain to C-terminus; or (4) two backbone residues either C-terminal to N-terminal, or backbone internal residue to internal residue (the N and Cα can be used as sites for cyclization). The simplest method used to introduce global constraint into a peptide chain is formation of a covalent bond between two distant segments in the sequence. Such cyclic introduction can be made with lactam bridges, disulfide bridges and other chemical moieties including use of spacer regions in constrained portions of a molecule [2,3,7,8,16]. Furthermore, ring size can be controlled depending on the region for cyclization.
Figure 2.
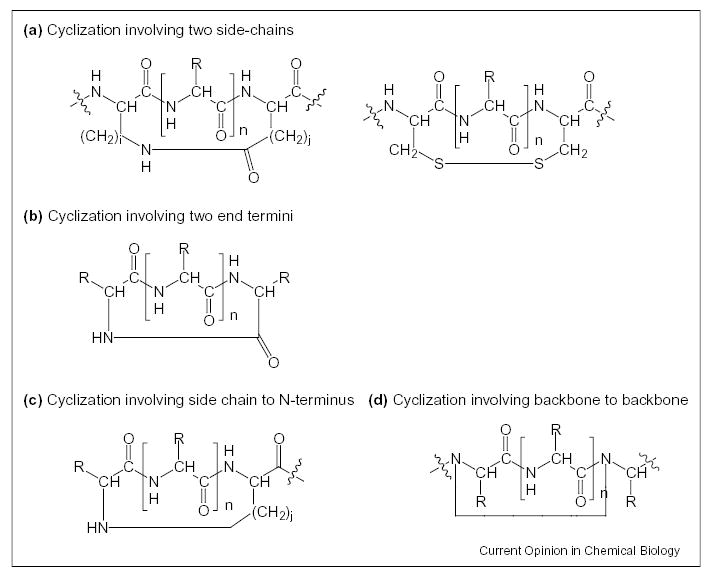
Types of covalent cyclization. (a) Cyclization involving two side chains. (b) Cyclization involving two end termini. (c) Cyclization involving side chain to N-terminus (or C-terminus). (d) Cyclization involving side chain to backbone to backbone (or side chain to backbone).
Cyclization of a peptide chain can dramatically change the overall conformation of a ligand favouring certain secondary structures. Table 1 lists some types of structural constraint that lead to specific secondary elements. Greatly reducing the degrees of freedom in a newly formed cyclic structure can stabilize the bioactive conformation of the ligand.
Table 1.
Incorporation of certain structural constraints can introduce or stabilize specific secondary conformations.
| Constraints | Secondary structures |
|---|---|
| Disulfide bridging | β-turns, γ-turns, α-helices, β-sheets |
| Lactam bridging (side-chain to side-chain or side-chain to N- or C-terminus) | α-helices, β-turns |
| Backbone to backbone cyclization | β-turns, γ-turns, β-sheets |
α-Melanocyte-stimulating hormone (historical background)
An excellent example of the increased potency, efficacy and other desirable pharmacological and biological properties due to substitution and cyclization of a linear parent peptide ligand can be seen in the studies of α-melanocyte-stimulating hormone (α-MSH; see Box 1 for a glossary showing peptide sequences). This tridecapeptide was first isolated in the pituitary gland, with primary physiological function in skin pigmentation [17,18]. However, more recently it has been shown that α-MSH also affects feeding behaviour, male and female sexual behaviour, erectile function, immune response, memory and learning, and other biological effects [19–21,22●,23,24●, 25]. This mini-review focuses on work from our laboratory in which cyclic constraints were used to explore a variety of templates and the effects of these constraints on bioactivity.
Box 1. Peptide structures covered in this article
- α-MSH
Ac-Ser1-Tyr2-Ser3-Met4-Glu5-His6-Phe7-Arg8-Trp9-Gly10-Lys11-Pro12-Val13-NH2
- CTAP/CTOP
H-d-Phe-c[Cys-Tyr-d-Trp-XXX-Thr-Pen]-Thr-NH2, where XXX = Arg, Orn, respectively
- MT-I
Ac-Ser1-Tyr2-Ser3-Nle4-Glu5-His6-D-Phe7-Arg8-Trp9-Gly10-Lys11-Pro12-Val13-NH2
- MT-II
Ac-Nle-c[Asp-His-d-Phe-Arg-Trp-Lys]-NH2
- penta-c[Asp-5-ClAtc-DPhe-Arg-Trp-Lys]-NH2
CH3CH2CH2CH2C(=O)-c[Asp-5-ClAtc-DPhe-Arg-Trp-Lys]-NH2
- PG-931
Ac-Nle-c[Asp-Pro-d-Phe-Arg-Trp-Lys]-Pro-Val-NH2
- SHU-9119
Ac-Nle-c[Asp-His-d-Nal(2′)-Arg-Trp-Lys]-NH2
One of the first α-MSH analogues reported to have increased potency by up to 60 times the activity of native α-MSH tested in frog skin was MT-I. Replacement of Met with Nle at the 4 position and substituting Phe with d-Phe at the 7 position increased the hydrophobicity and created a hypothesized reverse β-turn conformation that improved potency and efficacy [26]. Although MT-I was super potent, it lacked selectivity for melanocortin receptor [27]. This initial study led to a series of cyclic analogues which were designed to obtain a constrained cyclic peptide that would have the necessary conformation held rigidly in space, and which would be a selective agonist (or antagonist) for directed receptor(s) [28].
It was found very early on that the tetrapeptide, His-Phe-Arg-Trp formed the essential active core for α-MSH [29]. Hence, new cyclic analogues were fashioned to incorporate this tetrapeptide into various rings. The first cyclic analogues to be designed involved the replacement of Met4 and Gly10 with cysteine residues to form a disulfide bond. The resulting c[Cys4, Cys10]-α-MSH analogue was also found to be superpotent but lacked prolonged biological potency [28]. It wasn’t until the late 1980s when the first successful shortened sequence and cyclic constrained analogue of α-MSH was developed. The new compound, MT-II, was found to be a superpotent agonist in the lizard, frog and tyrosinase assays, with prolonged biological potency in frog skin assays as well [30,31]. Recent studies of MT-II in the sciatic nerve crush lesion and cisplatin-induced neuropathy in rats found MT-II also to be an effective agent in nerve regeneration and neuroprotection [32●].
All the first-generation cyclic analogues possessed great potency, but lacked selectivity to specific melanocortin receptor sites. Replacement of d-Phe in MT-II with d-Nal-(2′) led to SHU-9119, the first selective antagonist for MC3R and MC4R (the human melanocortin 3 and 4 receptors), although potency at the MC1R remained [33,34]. Recently it has been reported that through steady-state and time-resolved fluorescence spectroscopy studies, both the d-Nal and Trp residues show signs of deep lipid bilayer penetration, an important role for selective hMC4R binding [35]. Furthermore, appropriately labeled derivatives of MT-II and SHU-9119 could be used to evaluate the biological transduction mechanism of these ligands by two-photon fluorescence spectroscopy [36●].
α-MSH cyclic analogues
The discovery of MT-II and SHU-9119 began an era of ligand design directed toward obtaining peptide and peptidomimetic ligands (agonists and antagonists) for MC1R, MC3R, MC4R and MC5R (also, non-peptide ligands were sought with considerable success but are not reviewed). Several analogues based on the MT-II template have been reported that are more selective and highly potent (e.g. [37,38●,39–43]. For example, penta-c[Asp-5-ClAtc-d-Phe-Arg-Trp-Lys]-NH2 and penta-c[Asp-5-ClAtc-d-Phe-Cit-Trp-Lys]-NH2, closely related analogues of MT-II were generated by replacing His6 with 5-ClAtc and substitution of Arg8 with Cit to the parent MT-II. Both these analogues were found to be potent hMC4R agonists with weak partial agonist activities in hMC1R, hMC3R and hMC5R in vitro agonist assays [38●].
Hruby et al. found that although PG-931 was considerably less potent than MT-II, its selectivity for hMC4R versus hMC3R was at least 100-fold higher, making the cyclic peptide a potentially useful ligand for distinguishing binding site activities between the hMC4 and hMC3 receptors [42].
In a separate study, a series of β-substituted prolines replaced His6 and d-Phe7 in the MT-II scaffold to test the relationship between conformationally and topographically constrained ligands and selective receptor binding [44]. Overall, binding affinities for the new analogues were weak compared with the parent ligand, suggesting that the newly introduced constraint resulted in an unfavourable 3-D arrangement in chi space. Interestingly, increasing the bulky substituents on proline caused selectivity for hMC5R to increase. These studies demonstrate that hMC3 and hMC4 receptors are more easily affected by conformational changes to the ligands with reduced binding affinity, whereas hMC5R seems to be capable of accommodating bulkier residues [44].
Recent SAR studies
Extensive NMR studies in conjunction with structure–activity relationships (SAR) of α-MSH analogues and molecular modeling have been used to obtain novel peptidomimetic ligand designs (e.g. [44]). For example, Sun et al. [45] reported the synthesis of one of the most constrained and hMC4R-selective peptide analogues to date, [penta-cyclo(D-K)-Asp-Apc-(D)Phe-Arg-(2S,3S)-β-methylTrp-Lys-NH2]. The backbone is reported to feature a β-turn that incorporates the pharmacophore region needed for activity. The rigidity of the conformation causes the Asp residue to form a protrusion that apparently is neither compatible with nor adaptable to the hMC1R binding site [45]. Further examination of conformational and topographical space should prove fruitful in the design of more selective ligands for the melanocortin receptors since it appears that each receptor has its own special requirements.
Development of chimeric analogues
Another approach we have taken is to place the key structural moieties of α-MSH on novel templates. The idea is to use the key pharmacophores of well-established receptor recognition moieties, and link them together on alternate templates to examine whether selectivity or potency can be increased. We have previously called these ligands chimeric analogues. Recent examples in this context are the novel chimeric melanotropin–deltor-phin analogues by Han et al. [46]. The chimeric ligand was designed based on the main pharmacophore elements of melanotropin and deltorphin, and had the initial peptide sequence His-Phe-Arg-Trp-Glu-Val-Val-Gly-NH2. This structure was then cyclized to reduce the number of possible conformation by incorporating a lactam bridge between the acidic functional group of a glutamic acid side-chain and the free amide group at the N-terminus. Figure 3 depicts the design for this chimeric analogue. Two of the 19 chimeric analogues made [46] are listed in Table 2. GXH-32B and GXH-38B were found to have the most potent agonist and antagonist activities, respectively, at each of the MC receptors. GXH-32B was found to be a potent agonist with 30-fold higher binding affinity than GXH-15, which was a weak antagonist for MC1R with an IC50 of 8.2 μM. Subsequent substitution of the dipeptide, β-Ala-His, at the N-terminus with Gly-Cpg (cyclopentylglycine) gave GXH-38B, which was found to be the most potent antagonist. Although GXH-38B carried antagonist activity for all the melanocortin receptors, it was particularly selective for MC1R. Table 3 illustrates the potency of GXH-32B and GXH-38B at three human MC receptor sites.
Figure 3.
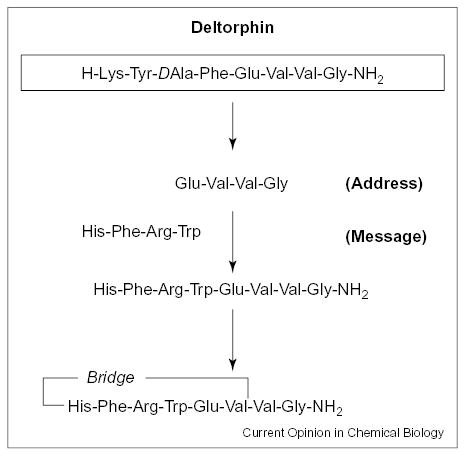
Design of novel chimeric melanotropin analogues.
Table 2.
High-throughput screening of chimeric α-MSH analogues at Xenopus frog MC1R.
| Peptide | Sequence | EC50 (nM) | IC50 nMa (0.5 nm α-MSH) |
|---|---|---|---|
| GXH-32B | c[β-Ala-His-d-Nal(2′)-Arg-Trp-Glu]-Val-Val-Gly-NH2 | 2 (SE 0.3) | None |
| GXH-38B | c[Gly-Cpg-D-Nal(2 μ)-Arg-Trp-Glu]-Val-Val-Gly-NH2 | > 1000 | 43 (SE 5) |
IC50 is the competitive binding in the presence of 500 pM of α-MSH. From [46].
Table 3.
Bioassay results of selected analogues at human MC receptors.
| EC50 or IC50a (nM) |
||||
|---|---|---|---|---|
| Peptide | Sequence | MC1 | MC3 | MC4 |
| GXH-32B | c[β-Ala-His-d-Nal(2′)-Arg-Trp-Glu]-Val-Val-Gly-NH2 | 75 | 3.8 | 77 |
| GXH-38B | c[Gly-Cpg-d-Nal(2′)-Arg-Trp-Glu]-Val-Val-Gly-NH2 | 12 | 44 | 1300 |
EC50 for GXH-32B; IC50 for GXH-38B. From [46].
Other chimeric analogues involving α-MSH and somatostatin derivatives have been examined [47]. Using CTAP and CTOP, which are μ opioid receptor antagonists derived from somatostatin, a series of 25 chimeric ligand were synthesized using the design template H-D-Phe-c[XXX-YYY-ZZZ-Arg-Trp-AAA]-Thr-NH2 [47], and several were shown to be potent agonists.
Conclusion
Incorporation of cyclic constraints into peptidic scaffolds, when properly designed, can provide novel ligands with novel biological profiles, especially with previous insight into key pharmacophore structural moieties. Constrained cyclic peptides with appropriate backbone or template rigidity and relatively fixed side-chains in chi space often have enhanced efficacy, selectivity, binding affinity and bioavailability, as outlined and demonstrated in many other studies (e.g. [1,7]). In addition, highly potent and selective ligands have helped elucidate proposed bioactive conformations at the receptor-binding sites. This, in return, further facilitates identification of the 3-D relationships of key pharmacophores, and contributes greatly to a better understanding for design and synthesis of peptides and peptide mimetics [7]. A major issue with these peptides is the appropriate methods for drug delivery of these ligands. Because most of the biological actions of these compounds appear to be hormonal and/or neurotransmitter-like in their biological actions, a bolus administration, whether oral or otherwise, may be counter indicated, and slow and/or pulsatile administration may be needed for a potent response.
Acknowledgments
The support of some of the work from our laboratory by grants from the US Public Health Service is gratefully acknowledged.
References
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Rizo J, Gierasch LM. Constrained peptides: models of bioactive peptides and protein substructures. Annu Rev Biochem. 1992;61:387–418. doi: 10.1146/annurev.bi.61.070192.002131. [DOI] [PubMed] [Google Scholar]
- 2.Hruby VJ. Conformational restrictions of biologically active peptides via amino acid side chain groups. Life Sci. 1982;31:189–199. doi: 10.1016/0024-3205(82)90578-1. [DOI] [PubMed] [Google Scholar]
- 3.Kessler H. Peptide conformations. Part 19. Conformation and biological effects of cyclic peptides. Angew Chem. 1982;94:509–520. [Google Scholar]
- 4.Marshall GR. A hierarchical approach to peptidomimetic design. Tetrahedron. 1993;49:3547–3558. [Google Scholar]
- 5.Jackson S, DeGrado W, Dwivedi A, Parthasarathy A, Higley A, Krywko J, Rockwell A, Markwalder J, Wells G, Wexler R, et al. Template-constrained cyclic peptides: design of high-affinity ligands for GPIIb/IIIa. J Am Chem Soc. 1994;116:3220–3230. [Google Scholar]
- 6.Hruby VJ, Al-Obeidi F, Kazmierski WM. Emerging approaches in the molecular design of receptor selective peptide ligands: conformational, topographical and dynamic considerations. Biochem J. 1990;268:249–262. doi: 10.1042/bj2680249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hruby VJ. Designing peptide receptor agonists and antagonists. Nat Rev Drug Discov. 2002;1:847–858. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- 8.Rose GD, Gierasch LM, Smith JA. Turns in peptides and proteins. Adv Prot Chem. 1985;37:1–109. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 9.Sawyer TK: Peptidomimetic and non-peptide drug discovery: impact of structure based design. In Structure Based Drug Design: Diseases, Targets, Techniques and Developments. Edited by Veerapandian P. Marcel Dekker; 1997:599–634.
- 10.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang H, Guranovic V, Dutta S, Feng Z, Berman HM, Westbrook JD. Automated and accurate deposition of structures solved by X-ray diffraction to the protein data bank. Acta Crystallogr D Biol Crystallogr. 2004;60:1833–1839. doi: 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]
- 12.Bourne PE, Westbrook J, Berman HM. The protein data bank and lessons in data management. Briefings Bioinform. 2004;5:23–30. doi: 10.1093/bib/5.1.23. [DOI] [PubMed] [Google Scholar]
- 13.Hruby VJ, Li G, Haskell-Luevano C, Shenderovich MD. Design of peptides, proteins, and peptidomimetics in chi space. Biopolymers. 1997;43:219–266. doi: 10.1002/(SICI)1097-0282(1997)43:3<219::AID-BIP3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 14.Ramachandran GN, Sasisekharan V. Conformation of polypeptides and proteins. Adv Prot Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 15.Ho BK, Thomas A, Brasseur R. Revisiting the Ramachandran plot: hard-sphere repulsion, electrostatics, and H-bonding in the α-helix. Protein Sci. 2003;12:2508–2522. doi: 10.1110/ps.03235203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilon C, Halle D, Chorev M, Selincer Z, Byk G. Backbone cyclization: a new method for conferring conformational constraint on peptides. Biopolymers. 1991;31:745–750. doi: 10.1002/bip.360310619. [DOI] [PubMed] [Google Scholar]
- 17.Harris JI, Lerner AB. Amino acid sequence of α-melanocyte stimulating hormone. Nature. 1957;179:1346–1347. doi: 10.1038/1791346a0. [DOI] [PubMed] [Google Scholar]
- 18.Steelman SL, Kelly TL, Norgello H, Weber GF. Occurrence of melanocyte stimulating hormone (MSH) in a transplantable pituitary tumor. Proc Soc Exp Biol Med. 1956;92:392–394. doi: 10.3181/00379727-92-22488. [DOI] [PubMed] [Google Scholar]
- 19.Cone RD (Ed): The Melanocortin Receptors. Humana Press; 2000: 1–551.
- 20.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 21.Wessells H, Fuciarelli K, Hansen J, Hadley ME, Hruby VJ, Dorr R, Levine N. Synthetic melanotropic peptide initiates erections in men with psychogenic erectile dysfunction: double-blind, placebo controlled crossover study. J Urology. 1998;160:389–393. [PubMed] [Google Scholar]
- 22●.Wessells H, Hruby VJ, Hackett J, Han G, Balse-Srinivasan P, Vanderah TW. Ac-Nle-c[Asp-His-DPhe-Arg-Trp-Lys]-NH2 induces penile erection via brain and spinal melanocortin receptors. Neuroscience. 2003;118:755–762. doi: 10.1016/s0306-4522(02)00866-7. Provides direct experimental evidence that constrained melanotropin peptides induced penile erection by both brain and spinal melanocortin receptors. Use of both agonists and antagonists aid in dissecting the bioactivities. [DOI] [PubMed] [Google Scholar]
- 23.Martin WJ, MacIntyre DE. Melanocortin receptors and erectile function. Eur Urology. 2004;45:706–713. doi: 10.1016/j.eururo.2003.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Olszewski PK, Wirth MM, Grace MK, Levine AS, Giraudo SQ. Evidence of interactions between melanocortin and opioid systems in regulation of feeding. Neuroreport. 2001;12:1727–1730. doi: 10.1097/00001756-200106130-00042. [DOI] [PubMed] [Google Scholar]
- 25●.MacNeil DJ, Howard AD, Guan X, Fong TM, Nargund RP, Bednarek MA, Goulet MT, Weinberg DH, Strack AM, Marsh DJ, et al. The role of melanocortins in body weight regulation: opportunities for the treatment of obesity. Eur J Pharmacol. 2002;440:141–157. doi: 10.1016/s0014-2999(02)01425-5. Reviews the evidence that melanocortin receptor ligands for the MC3 and MC4 receptors have great potential for energy homeostasis and body weight regulation. Other potential uses of melanotropin ligands are discussed. [DOI] [PubMed] [Google Scholar]
- 26.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. [Nle4,D-Phe7]-α-Melanocyte stimulating hormone: a highly potent α -melanotropin with ultralong biological activity. Proc Natl Acad Sci. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haskell-Luevano C, Miwa H, Dickinson C, Hruby VJ, Yamada T, Gantz I. Binding and cAMP studies of melanotropin peptides with the cloned human peripheral melanocortin receptor, hMC1R. Biochem Biophys Res Commun. 1994;204:1137–1142. doi: 10.1006/bbrc.1994.2581. [DOI] [PubMed] [Google Scholar]
- 28.Sawyer TK, Hruby VJ, Darman PS, Hadley ME. [4-Half-cystine, 10-half-cystine]-α-melanocyte stimulating hormone: a cyclic α-melanotropin exhibiting superagonist biological activity. Proc Natl Acad Sci. 1982;79:1751–1755. doi: 10.1073/pnas.79.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, de Lauro Castrucci A-M, Hintz MF, Riehm JR, Rao RR. α-Melanotropin: the minimum active sequence in the frog skin bioassay. J Med Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- 30.Al-Obeidi F, Hadley ME, Pettitt BM, Hruby VJ. Design of a new class of superpotent cyclic α-melanotropins based on quenched dynamic simulations. J Am Chem Soc. 1989;111:3413–3416. [Google Scholar]
- 31.Al-Obeidi F, de L Castrucci AM, Hadley ME, Hruby VJ. Potent and prolonged-acting cyclic lactam analogs of α-melanotropin: design based on molecular dynamics. J Med Chem. 1989;32:2555–2561. doi: 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]
- 32●.Vogelaar CF, Vrinten DH, Hoekman MFM, Brakkee JH, Burbach JPH, Hamers FPT. Sciatic nerve regeneration in mice and rats: recovery of sensory innervation is followed by a slowly retreating neuropathic pain-like syndrome. Brain Res. 2004;1027:67–72. doi: 10.1016/j.brainres.2004.08.036. Suggests that melanotropic peptides can aid in nerve regeneration, which in turn can reduce pain. [DOI] [PubMed] [Google Scholar]
- 33.Hruby VJ, Lu D, Sharma SD, de L Castrucci A, Kesterson RA, Al-Obeidi FA, Hadley ME, Cone RD. Cyclic lactam α-melanotropin analogs of Ac-Nle4-cyclo[Asp5,D-Phe7,Lys10]-α-melanocyte-stimulating hormone-(4-10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 34.Schioth HB, Muceniece R, Mutulis F, Prusis P, Lindeberg G, Sharma SD, Hruby VJ, Wikberg JES. Selectivity of cyclic [D-Nal7] and [D-Phe7] substituted MSH analogs for the melanocortin receptor subtypes. Peptides. 1997;18:1009–1013. doi: 10.1016/s0196-9781(97)00079-x. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez RM, Ito AS, Schioth HB, Lamy MT. Structural study of melanocortin peptides by fluorescence spectroscopy: identification of α-(2-naphthyl)-d-alanine as a fluorescent probe. Biochim Biophys Acta. 2003;1623:13–20. doi: 10.1016/s0304-4165(03)00152-1. [DOI] [PubMed] [Google Scholar]
- 36●.Cai M, Stankova M, Pond SJK, Mayorov AV, Perry JW, Yamamura HI, Trivedi D, Hruby VJ. Real time differentiation of G-protein coupled receptor (GPCR) agonist and antagonist by two photon fluorescence laser microscopy. J Am Chem Soc. 2004;126:7160–7161. doi: 10.1021/ja049473m. Two-photon fluorescence laser microscopy and other spectroscopic methods can be used to examine G-protein-coupled receptor transduction mechanisms directly in living cells. Using this method, it was possible to distinguish directly agonist from antagonist bioactivity, and it can be developed for high-throughput screening. [DOI] [PubMed] [Google Scholar]
- 37.Balse-Srinivasan P, Grieco P, Cai M, Trivedi D, Hruby VJ. Structure-activity relationships of α-MSH analogues at the human melanocortin MC3, MC4, and MC5 receptors. Discovery of highly selective hMC3R, hMC4R, and hMC5R analogues. J Med Chem. 2003;46:4965–4973. doi: 10.1021/jm030119t. [DOI] [PubMed] [Google Scholar]
- 38●.Cheung AW, Danho W, Swistok J, Qi L, Kurylko G, Rowan K, Yeon M, Franco L, Chu X, Chen L, Yagaloff K. Structure-activity relationship of cyclic peptide penta-c[Asp-His6-DPhe7-Arg8-Trp9-Lys]-NH2 at the human melanocortin-1 and -4 receptors: His6 substitution. Bioorg Med Chem Letts. 2003;13:1307–1311. doi: 10.1016/s0960-894x(03)00114-8. The constrained, cyclic MT-II pharmacophore is modified to produce highly selective agonist ligands for the hMC1R and hMC4R. Use of novel amino acids. [DOI] [PubMed] [Google Scholar]
- 39.Haskell-Luevano C, Lim S, Yuan W, Cone RD, Hruby VJ. Structure-activity studies of the melanocortin antagonist SHU 9119 modified at the 6, 7, 8, and 9 positions. Peptides. 2000;21:49–57. doi: 10.1016/s0196-9781(99)00167-9. [DOI] [PubMed] [Google Scholar]
- 40.Grieco P, Han G, Hruby VJ: New dimensions in the design of potent and receptor selective melanotropin analogues. In Peptides for the New Millenium. Edited by Fields GB, Tam JP, Barany G. Kluwer Acad. Publ; 2000:541–542.
- 41.Schioth HB, Kask A, Mutulis F, Muceniece R, Mutule I, Mutule I, Mandrika I, Wikberg JES. Novel selective melanocortin 4 receptor antagonist induces food intake after peripheral administration. Biochem Biophy Res Commun. 2003;301:399–405. doi: 10.1016/s0006-291x(02)03065-6. [DOI] [PubMed] [Google Scholar]
- 42.Grieco P, Balse-Srinivasan P, Han G, Weinberg D, MacNeil T, Van der Ploeg LHT, Hruby VJ. Extensive structure-activity studies of lactam derivatives of MT-II and SHU-9119: their activity and selectivity at human melanocortin receptors 3, 4, and 5. J Pept Res. 2003;62:199–206. doi: 10.1034/j.1399-3011.2003.00087.x. [DOI] [PubMed] [Google Scholar]
- 43.Cai M, Cai C, Mayorov AV, Xiong C, Cabello CM, Soloshonok VA, Swift JR, Trivedi D, Hruby VJ. Biological and conformational study of α-substituted prolines in MT-II template: steric effects leading to human MC5 receptor selectivity. J Pept Res. 2004;63:116–131. doi: 10.1111/j.1399-3011.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- 44.Ying J, Kover KE, Gu X, Han G, Trivedi DB, Kavarana MJ, Hruby VJ. Solution structures of cyclic melanocortin agonists and antagonists by NMR. Biopolymers. 2003;71:696–716. doi: 10.1002/bip.10596. [DOI] [PubMed] [Google Scholar]
- 45.Sun H, Greeley DN, Chu XJ, Cheung A, Danho W, Swistok J, Wang Y, Zhao C, Chen L, Fry DC. A predictive pharmacophore model of human melanocortin-4 receptor as derived from the solution structures of cyclic peptides. Bioorg Med Chem. 2004;12:2671–2677. doi: 10.1016/j.bmc.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 46.Han G, Quillan JM, Carlson K, Sadee W, Hruby VJ. Design of novel chimeric melanotropin-deltorphin analogues. discovery of the first potent human melanocortin 1 receptor antagonist. J Med Chem. 2003;46:810–819. doi: 10.1021/jm020355o. [DOI] [PubMed] [Google Scholar]
- 47.Han G, Haskell-Luevano C, Kendall L, Bonner G, Hadley ME, Cone RD, Hruby VJ. De novo design, synthesis, and pharmacology of alpha-melanocyte stimulating hormone analogues derived from somatostatin by a hybrid approach. J Med Chem. 2004;47:1514–1526. doi: 10.1021/jm030452x. [DOI] [PubMed] [Google Scholar]