

Abstract
Myokymia is characterized by spontaneous, involuntary muscle fiber group contraction visible as vermiform movement of the overlying skin. Myokymia with episodic ataxia is a rare, autosomal dominant trait caused by mutations in KCNA1, encoding a voltage-gated potassium channel. In the present study, we report a family with four members affected with myokymia. Additional clinical features included motor delay initially diagnosed as cerebral palsy, worsening with febrile illness, persistent extensor plantar reflex, and absence of epilepsy or episodic ataxia. Mutation analysis revealed a novel c.676C>A substitution in the potassium channel gene KCNA1, resulting in a T226K nonconservative missense mutation in the Kv1.1 subunit in all affected individuals. Electrophysiological studies of the mutant channel expressed in Xenopus oocytes indicated a loss of function. Co-expression of WT and mutant cRNAs significantly reduced whole-oocyte current compared to expression of WT Kv1.1 alone.
Keywords: Myokymia, Potassium channel, Mutation
Introduction
Myokymia is defined as spontaneous involuntary muscle fiber group contraction, is visible as vermiform movement of the overlying skin, and may be hereditary or acquired. Electromyography (EMG) shows continuous motor unit activity with spontaneous discharges of high intraburst frequency. Myokymia occurs in autoimmune disease with antibodies to voltage-gated potassium channel subunits in Isaacs–Merton syndrome and Morvan fibrillary chorea [1]. Hereditary myokymia with episodic ataxia type 1 (OMIM #160120) arises from mutations in the voltage-gated potassium channel KCNA1 [2]. The ataxia is characterized by profound attacks of cerebellar ataxia lasting seconds to minutes. Partial epilepsy has been reported in some families with KCNA1-associated disease [3, 4]. Another rare cause of myokymia with neonatal epilepsy but lacking episodic ataxia results from a KCNQ2 potassium channel mutation [5]. Reduced frequency and severity of myokymia and episodic ataxia have been reported after treatment with acetazolamide or carbamazepine [3, 6].
Approximately a dozen missense mutations and one truncating mutation in KCNA1 have been reported [3, 4, 7–10]. The functional consequences of human KCNA1 mutations causing typical myokymia and EA1 have been reported [11], much less is known about the functional consequences of mutations causing unusual KCNA1-associated phenotypes.
In the present study, we describe the clinical and genetic features of a new family with hereditary myokymia lacking epilepsy or episodic ataxia. Molecular studies of KCNA1 in the proband revealed a c.676C>A transversion resulting in the substitution of Lys for Thr at codon 226 (T226K) and a c.1355A>C transversion resulting in Y452S. Further genetic studies showed that all affected family members carried the T226K mutation, which was absent in unaffected family members and ethnically matched normal controls, whereas the Y452S substitution was present in both affected and unaffected family members and normal controls. In transfected mammalian cells, electrophysiologic studies demonstrated functional effects of the c.676C>A transversion with no currents above background levels recorded from oocytes injected with the mutant subunit. Co-injection of the wild-type and mutant cRNAs produced significantly smaller whole-oocyte currents than injection with the WT cRNA alone.
Materials and methods
Clinical data
The family pedigree is shown in Fig. 1. Six family members (two unaffected and four affected) were available for clinical and genetic studies approved by the Yale Human Investigations Committee.
Fig. 1.
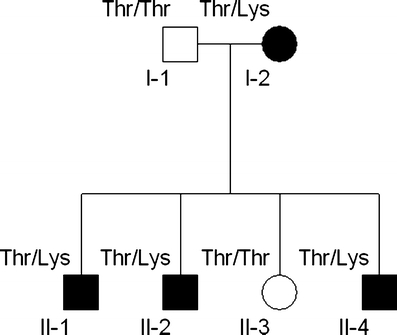
Family pedigree. Squares indicate males, circles indicate females. Blackened symbols denote individuals with myokymia. Alleles at codon 226 are indicated (Thr = wild-type, Lys = mutation)
Subject II-1
The proband, a 13-year-old boy (II-1), was the product of an unremarkable pregnancy and delivery. He was diagnosed with cerebral palsy because of leg stiffness and delayed walking at 18 months old. Cognitive development was normal; his medical history was significant for esotropia. At 4 years old, he was hospitalized with increasing leg pain, stiffness, and inability to walk during the course of a flu-like illness. Creatine kinase (CK) was elevated at 520 U/l acutely (normal <195 U/l), but was normal when repeated interictally. Examination showed periorbital myokymia, mild abdominal and leg muscle hypertrophy, leg stiffness, spastic gait, hyperreflexia, and bilateral Babinski sign. Magnetic resonance imaging (MRI) of the brain and spine were normal. Metabolic studies including electrolytes, amino and organic acids, carnitine, ammonia, lactate, and thyroid functions were normal. Routine studies of cerebral spinal fluid and an electroencephalogram were normal. Needle EMG of deltoid and iliopsoas muscles showed irregular, polyphasic continuous motor unit discharges with normal interference pattern.
Additional family members
Six family members were evaluated. Four affected members (mother and three sons) had myokymia on clinical examination, EMG examination, or both. Nerve conduction velocities were normal. There was no history of seizures or episodic ataxia in any of them. The mother (I-2, Fig. 1) is a 40-year-old woman who had had muscle twitching since childhood, hyperreflexia, bilateral Babinski sign, and tendency to run on her toes. Her husband is unaffected and had a normal neurological exam. A 12-year-old son (II-2) also had delayed walking, leg stiffness, and was diagnosed with cerebral palsy. At age 3 years, he had an episode of prolonged generalized paralysis with no muscle response to tetanic stimulation after general anesthesia for esotropia surgery. He recovered within 1 day. MRI of the brain and spine were normal. An 8-year-old daughter (II-3) is unaffected. A 5-year-old son (II-4) had symptoms identical to his brothers and was hospitalized twice for episodes of increased muscle stiffness with presumed viral gastroenteritis, elevated CK during both episodes (606 U/l and 997 U/l) but had normal CKs measured while well. Carbamazepine treatment resulted in marked improvement of muscle symptoms in three of the brothers who remain asymptomatic. The mother, with milder symptoms, declined treatment. Three family members with myokymia had esotropia; one patient required three eye surgeries.
Molecular studies
Genomic DNA was extracted from blood samples or buccal swabs. The coding region of the single-exon 1,448-bp gene KCNA1 (GenBank sequence NM_000217) was amplified. Polymerase chain reactions (PCRs) were performed in a 25-μl reaction volume containing 50 ng genomic DNA, 50 ng of each primer, 1.5 mM MgCl2, 200 μM dNTPs, 1× PCR buffer (Finnzyme), and 2.5 U of Finnzyme using a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA). Cycling parameters consisted of a modified touchdown (stepdown) protocol as follows: an initial hot start denaturation at 94°C for 5 min (hot start); 94°C for 45 s, annealing temperature (three cycles each at 70, 67, 64, 61,and 58°C) for 45 s, and extension at 72°C for 2 min for a total of 15 cycles followed by 25 cycles of 94°C for 45 s, 55°C for 45 s, 72°C for 1 min; a final extension step of 72°C for 10 min followed by a 4°C hold. PCR products were separated by electrophoresis on a 1% agarose gel and visualized by ethidium bromide staining. PCR products were purified by spin column (Qiagen, Valencia, CA) or by enzymatic method (ExoSAP-IT, USB, Cleveland, OH) and analyzed for mutations by automated sequencing (Applied Biosystems, Foster City, CA). The c.1355A>C transversion resulted in the creation of a DdeI restriction site, so the presence of the sequence variant was tested by sequencing and confirmed with restriction digest in all subjects. Restriction digests were performed with DdeI (New England Biolabs, Beverly, MA) at 37°C for 2 h.
Oocyte expression and electrophysiological studies
Human Kv1.1 cDNA was subcloned into Kpn1 and HindIII sites of psGEM. The Kv1.1-T226K mutation was introduced by site-directed mutagenesis and confirmed by automated sequencing. cRNAs were synthesized using an mMessage mMachine kit (Ambion, Austin, TX) and quantified by spectroscopy.
Oocytes were isolated from Xenopus laevis and defolliculated by collagenase treatment. Each oocyte was injected with either 5 ng of human Kv1.1-WT, 5 ng Kv1.1-T226K, or 2.5 ng WT plus 2.5 ng T226K cRNA. Whole-oocyte currents were measured with two electrode voltage clamp techniques (Oocyte Clamp, Warner Instruments, Hamden, CT) with constant perfusion (1 ml/min, solution exchange\3 s) after 16 h. Data were sampled at 1 kHz and filtered at 0.25 kHz. Standard bath solution was ND-96 (in mM): 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES/NaOH, pH 7.5.
Results
Mutation analysis of KCNA1
Mutation analysis of KCNA1 identified two sequence variants: a c.676C>A transversion resulting in the substitution of Lys for Thr at codon 226 (T226K) and a c.1355A>C transversion resulting in Y452S and creating a DdeI restriction site. Either a Tyr (NM_000217) or a Ser (AAI01734) had been reported at codon 452, but the population frequency and association with disease were unknown. Therefore, we tested all six family members (four affected, two unaffected) and 10 normal controls for the SNP and found that all 16 subjects were homozygous for the Y452S substitution, compatible with a common polymorphism. In contrast, all (4/4) affected subjects were heterozygous for the previously unreported T226K missense mutation, which was absent in both unaffected family members and absent in 190 chromosomes from ethnically matched normal controls (Fig. 2).
Fig. 2.
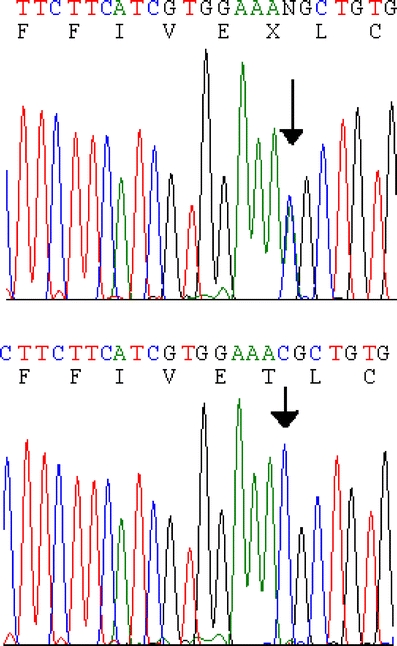
Sequence chromatograms demonstrating c.676C>A resulting in T226K substitution in KCNA1. A DNA sequence from an affected patient (I-2) heterozygous for the mutation (top) and from a normal control (bottom) are presented
Electrophysiological studies of the human Kv1.1 mutant The genetic studies of the segregation of the T226K mutation favored it as a pathological mutation, but given that two missense mutations were identified, we sought additional supporting evidence. We tested whether the missense mutation alters human Kv1.1 channel function by expressing Kv1.1-T226K in Xenopus oocytes. No currents above background levels were recorded from Kv1.1-T226K cRNA-injected oocytes, whereas Kv1.1 wild-type channels produced typical delayed rectifier currents (Fig. 3). We then mimicked the heterozygous situation by co-injecting equal amounts of Kv1.1 WT and T226K mutant cRNAs. Co-injection produced significantly smaller whole-oocyte currents than injection with Kv1.1 WT cRNA alone (Fig. 3c and d). These results indicate that the T226K mutation significantly reduced efflux of potassium ions during depolarization and is therefore likely to result in increased muscle cell excitability.
Fig. 3.
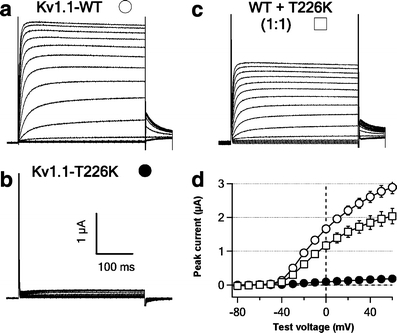
Missense mutation T226K induces loss of function of human Kv1.1 channels expressed in Xenopus oocytes. a–c Representative family of whole-oocyte currents recorded from oocytes expressing human Kv1.1-WT (a), Kv1.1-T226K (b), and both Kv1.1 WT and T226K (c). Currents were evoked by a series of 350-ms test pulses from −80 to +60 mV in 10-mV increments from a hold potential of −90 mV. Tail currents were recoded at −40 mV. Scale bar, 100 ms and 1 μA. d I–V curves for three experimental conditions in a–c. Data were collected for 10–15 oocytes in each group. The peak currents in a and c are significantly different at test voltages between 0 and +60 mV (P < 0.05)
Discussion
Mutations in KCNA1 are typically associated with episodic ataxia type 1 (EA1), characterized by profound cerebellar ataxia lasting seconds to minutes and interictal myokymia [6]. The expanded phenotypic spectrum associated with proven KCNA1 mutation includes a family with prolonged episodes of episodic ataxia and epilepsy, but without myokymia [12]. Other families have been reported with myokymia and adducted thumbs, congenital contractures, kyphoscoliosis, or epilepsy with myokymia [4, 7–10]. We report a family with autosomal dominant myokymia lacking either ataxia or epilepsy. The mother had self-described lifelong “muscle twitchiness” but never sought medical attention for her condition until the hospitalizations of her sons. Myokymia in the periorbital region and on EMG were the features that led to genetic studies of KCNA1.
Although the stiffness, toe walking, and esotropia could be caused by continuous muscle contraction, the extensor plantar reflex [observed on multiple exams by experienced two neurologists (LRM and FMH)] is a new finding for KCNA1-associated disease and suggests involvement of the corticospinal tract. The KCNA1 knockout mouse has been reported to exhibit altered inhibition of layer V pyramidal neocortical neurons [13].
KCNA1 on chromosome 12p13 encodes Kv1.1, a potassium channel subunit. Delayed-rectifier voltage-gated-type potassium channels are important in the rapid neuronal repolarization after an action potential and are multimeric proteins containing four subunits around a central pore. The Kv1.1 potassium channel subunit is expressed during development in neuronal and glial cells in the central and peripheral nervous systems [14]. Kv1.1 plays an important role in regulating neuronal excitability in the central nervous system, as shown by studies of the KCNA1 knockout mouse with an increased susceptibility to seizures [15].
The present family has generalized myokymia due to a T226K mutation in the voltage-gated potassium channel KCNA1. Unusual clinical features include pyramidal dysfunction and worsening with febrile illness or anaesthesia. One previously reported 3-year-old boy presented in a similar fashion to our patients with a 12-h episode of inability to walk, increased tone, and elevated CK during a febrile illness [3]. No anesthesia exposure was reported. That child had a P244H mutation in the intracellular loop between transmembrane segments 2 and 3. The whole-cell current amplitudes elicited by injection of P244H did not differ from wild-type amplitudes. Co-injection of P244H plus an equal amount of wild-type cRNA yielded a peak current amplitude that was 200% of wild-type alone.
We identified a novel, nonconservative T226K missense mutation that is located in the second transmembrane domain of Kv1.1 and is invariant from Drosophila melanogaster to mice and humans [6]. In contrast to the functional effects reported for the P244H mutation, heterologous expression of the T226K mutation identified in the present family showed that the mutant potassium channel expressed in oocytes demonstrated no current above background levels, and its co-expression with the wild-type channel significantly reduced whole-oocyte current, compatible with a severe effect of the mutation. The T226K mutation is likely to reduce efflux of potassium ions during depolarization, thereby increasing cellular excitability. The phenotypic similarity between the P244H and the T226K mutations contrasts with their biophysical effects. Therefore, additional factors must contribute to the functional effects of these mutations. Kv1.1 forms heteromeric channels with other subunits and interacts with other accessory subunits [16]. In addition to their well-known role as ion channels that regulate neuronal firing, potassium channels have also been shown to activate enzymes linked to intracellular signaling pathways [16, 17]. One of these novel interactions may contribute to differences in the manifestation of disease-causing mutations.
Acknowledgment
The authors thank the family members for their participation and Dr. Dimitri M. Kullmann (Institute of Neurology, University College of London) for the Kv1.1 clone. FMH was supported by a Paul Beeson Physician Faculty Scholar Award from the American Federation for Aging Research. HC, CvH, and LKK were supported by NIH grant DC01919 to LKK.
Contributor Information
Haijun Chen, Email: Haijun.chen@yale.edu.
Christian von Hehn, Email: Christian.vonhehn@yale.edu.
Leonard K. Kaczmarek, Email: Leonard.kaczmarek@yale.edu
Laura R. Ment, Email: laura.ment@yale.edu
Barbara R. Pober, Email: Barbara.pober@childrens.harvard.edu
Fuki M. Hisama, Phone: +1-617-3554697, Email: fuki.hisama@childrens.harvard.edu
References
- 1.Vernino S, Lennon VA (2002) Ion channel and striational antibodies define a continuum of autoimmune neuromuscular hyperexcitability. Muscle Nerve 26:702–707 [DOI] [PubMed]
- 2.Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M (1994) Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 8:136–140 [DOI] [PubMed]
- 3.Eunson LH, Rea R, Zuberi SM, Youroukos S, Panayiotopoulos CP, Liguori R, Avoni P, McWilliam RC, Stephenson JB, Hanna MG, Kullmann DM, Spauschus A (2000) Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol 48:647–656 [DOI] [PubMed]
- 4.Zuberi SM, Eunson LH, Spauschus A, De Silva R, Tolmie J, Wood NW, McWilliam RC, Stephenson JP, Kullmann DM, Hanna MG (1999) A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain 122(Pt 5):817–825 [DOI] [PubMed]
- 5.Dedek K, Kunath B, Kananura C, Reuner U, Jentsch TJ, Steinlein OK (2001) Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci U S A 98:12272–12277 [DOI] [PMC free article] [PubMed]
- 6.van Dyke DH, Griggs RC, Murphy MJ, Goldstein MN (1975) Hereditary myokymia and periodic ataxia. J Neurol Sci 25:109–118 [DOI] [PubMed]
- 7.Comu S, Giuliani M, Narayanan V (1996) Episodic ataxia and myokymia syndrome: a new mutation of potassium channel gene Kv1.1. Ann Neurol 40:684–687 [DOI] [PubMed]
- 8.Kinali M, Jungbluth H, Eunson LH, Sewry CA, Manzur AY, Mercuri E, Hanna MG, Muntoni F (2004) Expanding the phenotype of potassium channelopathy: severe neuromyotonia and skeletal deformities without prominent episodic ataxia. Neuromuscul Disord 14:689–693 [DOI] [PubMed]
- 9.Klein A, Boltshauser E, Jen J, Baloh RW (2004) Episodic ataxia type 1 with distal weakness: a novel manifestation of a potassium channelopathy. Neuropediatrics 35:147–149 [DOI] [PubMed]
- 10.Scheffer H, Brunt ER, Mol GJ, van der Vlies P, Stulp RP, Verlind E, Mantel G, Averyanov YN, Hofstra RM, Buys CH (1998) Three novel KCNA1 mutations in episodic ataxia type I families. Hum Genet 102:464–466 [DOI] [PubMed]
- 11.Zerr P, Adelman JP, Maylie J (1998) Characterization of three episodic ataxia mutations in the human Kv1.1 potassium channel. FEBS Lett 431:461–464 [DOI] [PubMed]
- 12.Lee H, Wang H, Jen JC, Sabatti C, Baloh RW, Nelson SF (2004) A novel mutation in KCNA1 causes episodic ataxia without myokymia. Human Mutat 24:536 [DOI] [PubMed]
- 13.van Brederode JF, Rho JM, Cerne R, Tempel BL, Spain WJ (2001) Evidence of altered inhibition in layer V pyramidal neurons from neocortex of Kcna1-null mice. Neuroscience 103:921–929 [DOI] [PubMed]
- 14.Hallows JL, Tempel BL (1998) Expression of Kv1.1, a Shaker-like potassium channel, is temporally regulated in embryonic neurons and glia. J Neurosci 18:5682–5691 [DOI] [PMC free article] [PubMed]
- 15.Rho JM, Szot P, Tempel BL, Schwartzkroin PA (1999) Developmental seizure susceptibility of kv1.1 potassium channel knockout mice. Dev Neurosci 21:320–327 [DOI] [PubMed]
- 16.Kaczmarek LK (2006) Policing the ball: a new potassium channel subunit determines inactivation rate. Neuron 49:642–644 [DOI] [PubMed]
- 17.Kaczmarek LK (2006) Non-conducting functions of voltage-gated ion channels. Nat Rev Neurosci 7:761–771 [DOI] [PubMed]