

Abstract
The Ras signaling pathway controls important cellular responses to growth factors, and somatic mutations in RAS genes and other components of the Ras pathway, such as PTPN11 (encoding the protein-tyrosine phosphatase SHP-2) and BRAF, are found in human malignancies. Ras proteins are guanosine nucleotide-binding proteins that cycle between active guanosine triphosphate (GTP)-bound and inactive guanosine diphosphate (GDP)-bound conformations. Neoplasia-associated Ras mutations frequently affect amino acids G12, G13, or Q61 and decrease the intrinsic guanosine triphosphatase (GTPase) activity by ten- to twentyfold. The GTPase activity is crucial for Ras inactivation by hydrolysis and release of a phosphate group from Ras·GTP to produce Ras·GDP. We and others have recently discovered germline mutations in the KRAS gene in individuals diagnosed with Noonan and cardio–facio–cutaneous (CFC) syndrome, two clinically overlapping disorders characterized by short stature, distinct facial anomalies, heart defects, and other abnormalities. Noonan syndrome-associated mutations V14I and T58I K-Ras activate Ras but have milder biochemical effects than somatic mutations encountered in cancers, offering an explanation why these K-Ras lesions are tolerated during embryonic development. Together with recent findings of BRAF, MEK1, and MEK2 mutations in CFC syndrome and HRAS mutations in Costello syndrome, another clinically related disorder, it has now become clear that Noonan-like features (short stature, relative macrocephaly, facial anomalies, learning difficulties) that are found in these three related disorders are a result of constitutive activation of the Ras–Raf–extracellular signal-regulated and mitogen-activated protein kinase pathway.
Keywords: Noonan syndrome, Cardio–facio–cutaneous syndrome, Costello syndrome, Neurofibromatosis type 1, Ras signaling
RAS genes
The three human RAS genes, KRAS (isoforms A and B), NRAS, and HRAS (isoforms 1 and 2), encode small, highly conserved guanosine triphosphatases (GTPases) that relay growth signals to a number of effector proteins to control fundamental cellular pathways (reviewed in [1]). Ras proteins act as molecular switches by cycling between an active guanosine triphosphate (GTP)-bound and an inactive guanosine diphosphate (GDP)-bound state. Stimulated growth factor receptors recruit a number of adaptor proteins that activate guanosine nucleotide exchange factors (GEFs) to remove guanine nucleotides from Ras. Ras is then activated by binding to GTP, which is present at a tenfold higher concentration than GDP. In the GTP-bound state, the two switch regions of Ras (switch I and II) change their conformation. This conformational switch allows Ras to bind and activate Ras effector proteins such as Raf-1. The “on” position is turned “off” by an intrinsic GTPase activity, which hydrolyses and releases a phosphate group from Ras·GTP to produce Ras·GDP. The conformational transition of the switch I and II regions that is associated with this reaction disrupts the interaction between Ras and its effectors. The intrinsic GTPase activity of Ras is slow and accelerated about 105-fold by GTPase activating proteins (GAPs), such as neurofibromin or p120 GAP. These GAPs mediate Ras·GTP hydrolysis by inserting an arginine residue (arginine finger) into the phosphate-binding pocket of Ras (reviewed in [2, 3]; Fig. 1).
Fig. 1.

Ras cycles between an active GTP-bound and an inactive GDP-bound conformation. In the active state, the two switch regions, switch I and II, change their conformation allowing Ras to activate effector proteins. The intrinsic GTPase hydrolyzes a phosphate group to produce Ras·GDP. This reaction is accelerated by GTPase activating proteins (GAPs). A similar version of this figure has been previously published [61], republication with permission
For several decades, it has been well recognized that RAS genes are frequently mutated in human cancers (reviewed in [4]). These mutations predominantly lead to amino acid substitutions at residues G12, G13, or Q61 and lock Ras in the active GTP-bound state by diminishing the intrinsic Ras GTPase activity and/or by causing resistance to GAPs. These mutations were long believed to occur only as somatic events, and in a mouse model system, widespread expression of endogenous K-RasG12D leads to embryonic death [5]. The highest incidence of RAS mutations are found in adenocarcinomas of the pancreas (90%), the colon (50%), and the lung (30%), in thyroid tumors (50%), and in myeloid leukemia (30%) [4].
Noonan syndrome and related disorders
Noonan syndrome (NS; MIM 163950) is an autosomal dominant disorder characterized by short stature, distinct facial anomalies (Fig. 2), a typical spectrum of congenital heart defects, including pulmonic stenosis, hypertrophic cardiomyopathy, and septal defects, and developmental delays (reviewed [6]). The disorder occurs in approximately 1 out of 2,000 individuals and shares many features with the much less common disorders of Costello syndrome (CS), cardio–facio–cutaneous (CFC) syndrome, and lentigines, electrocardiographic conduction defects, ocular hypertelorism, pulmonary stenosis, abnormalities of the genitals; retarded growth resulting in short stature, and deafness (LEOPARD) syndrome (LS) [6]. Patients with these disorders have in common Noonan-like facial features, a similar spectrum of cardiac anomalies, delayed growth, and—to a variable degree—developmental retardation, which is stronger in patients with CS or CFC. In addition, each of these disorders is characterized by unique phenotypic patterns: (1) CS patients have nasal papillomata, loose skin, and a strong predisposition to tumors (mainly rhabdomyosarcoma) [7]; (2) CFC patients have ectodermal abnormalities with sparse curly hair, sparse, or absent eyelashes [8]; and (3) LS patients have multiple lentigines typically emerging during adolescence [6]. NS and LS may be familial with an autosomal dominant inheritance pattern (except for rare instances of NS where rare recessive inheritance has been suspected [9]), but most cases of these disorders and virtually all of the more severe conditions, CFC and CS, occur sporadically, suggesting dominant new mutations. Until recently, the genetic basis for these disorders was unknown. It was not clear whether they represented genetically distinct entities or if they were different (allelic) variants of a common disorder. By linkage analysis in families with NS, the disorder was mapped to chromosome 12q24 [10], and subsequently, it has been shown that approximately 50% of patients with NS carry germline mutations in PTPN11 [11]. The PTPN11 gene encodes for tyrosine-protein phosphatase (SHP-2), a phosphatase that relays growth signals from activated growth factor receptors to other signaling molecules, including Ras (reviewed in [12]). Most PTPN11 mutations are predicted to disrupt the auto-inhibition of the catalytic phosphatase domain (PTPase) by the N-terminal src-homology 2 (N-SH2) domain thereby promoting the active conformation of the protein [12]. PTPN11 mutations were not found in patients with CS or CFC syndromes (reviewed in [6]). However, specific mutations in the same gene were uncovered in patients with LS [13]. Surprisingly, in contrast to NS, LS mutants are catalytically defective and may act as dominant negative mutations [14, 15]. Molecular modeling and biochemical studies suggest that LS mutations disrupt the SHP-2 catalytic domain to result in open, inactive forms of SHP-2. Thus, the pathogenesis of LS and NS is distinct. It is unknown why the clinical phenotypes of LS and NS are similar although the underlying mutations have opposite biochemical effects. There are several possible explanations for this paradox. These are discussed in detail by Kontaridis et al. [14]: (1) The NS and LS phenotypes potentially result from differential effects of mutant SHP-2 on different receptor tyrosine kinase pathways at distinct developmental times. For example, recent work by others suggests that the main effect of NS mutants is to enhance epithelial–mesenchymal transformation/mesenchymal cell proliferation by increasing ErbB2/3 (and/or ErbB3/4) signaling. By contrast, LS mutants antagonize HB-EGF/ErbB1 signaling at later times [14]; (2) Other phenotypes common to NS and LS such as facial abnormalities and short stature might involve defective migration and/or differentiation, which might result from increased or decreased signaling involving the same pathway; (3) Yet, undetermined or poorly understood functions of SHP-2 might underlie pathogenesis of one or both of these disorders [14].
Fig. 2.

a Craniofacial phenotype of young children with NS, CFC, and CS aged between 10 and 18 months. Their genotypes are PTPN11 G503R, BRAF K499E, and HRAS G13C, respectively. Note the similarities of facial features, including hypertelorism, broad forehead, and low-set ears. Coarse facial features are particularly typical of CS. b The same disorders in older children and adolescents with the mutations PTPN11 N308D, MEK2 F57I, and HRAS G12S, respectively. Facial features become more distinct with age, although the similarities of the three syndromes are still evident. Courtesy of Prof. Rainer König, Frankfurt (images of CFC patients) and Prof. Kerstin Kutsche, Hamburg (images of CS patients)
The Noonan gene PTPN11 also acts as oncogene
One of the myeloid malignancies that has been found to be particularly related to perturbed Ras signaling is juvenile myelomonocytic leukemia (JMML), a myeloproliferative disorder (MPD) of early childhood [16], in which mutations occur in NRAS or KRAS (∼25%) or in NF1 (clinical diagnosis of neurofibromatosis type 1 in ∼11%) [17–19]. In a murine model, somatic activation of K-Ras in hematopoietic cells initiates a rapidly fatal MPD modeling JMML [20, 21]. Despite a low annual incidence estimated at 1–2 per million, JMML has attracted many clinical and basic researchers because of the severe and often lethal clinical course and the association with NS [22] and neurofibromatosis type 1 (NF1; MIM 162200; reviewed in [19]). Shortly after the discovery of PTPN11 mutations in individuals with NS, specific germline PTPN11 alterations were identified in young children with NS who developed a JMML-like disorder within the first few weeks of birth (NS/JMML) [23, 24], and somatic PTPN11 mutations were subsequently uncovered in JMML cells from 35% of children with non-syndromic JMML [23–25]. Somatic PTPN11 mutations also occur in B-cell precursor acute lymphoblastic leukemia [26] and rarely other malignancies [27]. Somatic mutations observed in patients with JMML differ from those mutations found in patients with NS/JMML and from those detected in patients with NS alone. In sporadic JMML, the most common mutation predicts an E76K substitution. This mutation has never been found in patients with NS (N308D most common) or NS/JMML (T73I most common) [6, 25]. In elegant functional experiments, several groups have shown that somatic PTPN11 mutations associated with sporadic JMML exhibit stronger biochemical and biological effects than germline PTPN11 mutations, leading to the concept that only milder SHP-2 activation may be tolerated during embryonic development [28–30]. The situation is too complex however to be explained by a simple model of strong activating somatic vs less activating germline mutations alone. Enzymatic, structural, and mathematical modeling analyses show that these mutants can affect basal activation, SH2 domain-phosphopeptide affinity, and/or substrate specificity to varying degrees, and there is no absolute correlation between the mutants’ extents of basal activation and the diseases they induce [31]. A murine knock-in Ptpn11D61G/+ model of NS has been constructed, revealing that endocardial cushions of these mice have increased activation of extracellular signal-regulated kinase [32]. This finding suggests that the phenotype exhibited by PTPN11 mutations is mediated through hyperactive Ras signaling.
Neurofibromatosis type 1
JMML is also associated with NF1 (MIM 162200), an autosomal dominant disorder that occurs in 1 of 4,000 births and is characterized by pigmentary anomalies (multiple café-au-lait spots) and a predisposition to benign and malignant tumors of mainly neurogenic origin. The disorder is caused by mutations in the NF1 tumor suppressor gene, which encodes the RasGAP neurofibromin. The incidence of JMML is increased approximately 200-fold in children with NF1 (reviewed in [33]). Loss of the normal NF1 allele (LOH, loss of heterozygosity) is common in JMML cells from children with NF1 [34, 35], and this results in severely deregulated Ras signaling and causes aberrant growth of hematopoietic progenitor colonies in vitro [36]. In addition, adoptive transfer of homozygous Nf1 mutant fetal liver cells or somatic inactivation of a conditional mutant Nf1 allele in hematopoietic cells induces a JMML-like MPD in mice [37, 38]. Remarkably, many NF1 patients have mild features reminiscent of NS [39], and some may even develop a mixed phenotype, which has prompted the definition of neurofibromatosis–NS (NFNS; MIM 601321) as a separate entity. Most patients who are diagnosed with this condition harbor NF1 mutations without an obvious genotype–phenotype correlation [40].
Additional genes mutated in Noonan syndrome
To date, the identification of additional NS genes was hampered by the fact that the vast majority of PTPN11 mutation-negative NS-patients represent sporadic cases [41], rendering genetic linkage studies impossible. However, recent findings by our group and others identified germline mutations in components of the Ras signaling pathway in individuals with NS or related syndromes [42–46]. We systematically screened patients with NS and CFC for RAS mutations after having discovered a novel de novo germline KRAS mutation in a patient with NS/JMML [42]. The mutation c.173C>T (p.T58I) detected in the index patient affects a highly conserved amino acid residue of K-Ras flanking the switch II region (amino acids 59–67) of the protein. De novo KRAS mutations were identified in 4 of 174 sporadic cases with NS previously excluded for PTPN11 mutations. All germline KRAS mutations were novel and not known to occur in human cancer. NS-associated KRAS alleles included recurrent mutations, V14I and D153V. The latter was confirmed in patients with more severe phenotypes [44, 46]. Analogous to the dual role of PTPN11 as both oncogene and developmental gene, this discovery led to the hypothesis that activating KRAS mutations do not only act as oncogenes; aberrant K-Ras can also cause developmental disorders when mutations—probably conferring relatively mild effects—emerge in the germline. To prove our hypothesis, we studied the functional properties of V14I and T58I K-Ras [42] and found both NS-associated mutants V14I and T58I to have intermediate biochemical (e.g., ability to hydrolyze Ras·GTP) and biological properties (e.g., growth behavior in response to growth factors after retroviral transfection) when compared with wild-type K-Ras and the oncogenic mutant K-Ras G12D [42].
The two aforementioned mutations, V14I and T58I, are located adjacent to amino acid residues that are typically altered in cancer (G12, G13, or Q61). They locate to regions of Ras that are known to be involved in GTP binding. By contrast, in a subset of NS patients, KRAS mutations lead to substitutions in protein regions not obviously involved in GTP binding (e.g., D153V K-Ras), indicating the existence of previously unappreciated mechanisms of Ras activation. Carta et al. [46] performed a structural analysis on the two K-Ras mutants, V152G and D153V, that these investigators identified in two patients with severe NS. Their computer-based analysis indicated that both mutations disturb the conformation of the guanine ring-binding pocket favoring the active GTP-bound conformation by increasing the guanine nucleotide dissociation rate. Additionally, these residues are predicted to be important for binding the RasGEF son of sevenless (SOS) [47]. In the meantime, we have detected a number of additional KRAS mutations associated with NS (Q22R, P34L, P34Q, I36M), all of which affect highly conserved amino acid residues and are assumed to confer (mild) gain of function [48]. In this study on additional 236 PTPN11-negative NS patients, we detected KRAS mutations in seven individuals [48], thus confirming that KRAS accounts for less than 5% of NS cases.
Genes mutated in cardio–facio–cutaneous syndrome
In our initial study, we also discovered a novel K-Ras mutation, K-Ras P34R, in 1 of 12 patients with CFC syndrome [42]. This mutation is now known to be located in another mutational hotspot associated with NS [48]. A P34R H-Ras lesion has been previously characterized by Stone et al. [49]. These investigators employed a mutagenesis strategy during which they found that H-Ras P34R binds to GTP in vivo. In vitro, H-Ras P34R is not stimulated by GAPs [49]. Considering the highly conserved G-domain structure of Ras proteins, the P34R K-Ras mutation presumably has very similar (if not identical) biochemical properties. The presence of K-Ras mutations in CFC syndrome was confirmed by Niihori et al. [44] who analyzed DNA specimens from 43 individuals with CFC syndrome and found two KRAS germline mutations (G60R and D153V) in three patients. Notably, the D153V K-Ras mutation also has been described in patients with NS [42, 46, 48]. In a more recent study, we identified additional K-Ras mutations in individuals with CFC or NS/CFC including Q22E and F156I. The observations that mutations, such as K-Ras F156I, may be associated with an overlap of NS and CFC suggest that there is not a very strict genotype–phenotype correlation [48]. Simultaneous to the finding of mutated K-Ras in CFC, Niihori et al. identified BRAF germline mutations in 16 of 43 CFC patients [36]. At the same time, another group found BRAF germline mutations in 18 of 23 CFC patients and MEK1 or MEK2 germline mutations in 3 of the remaining 5 CFC patients studied [45]. Like PTPN11 and RAS, BRAF is a known oncogene, and somatic mutations are frequently found in cancer [50].
The molecular basis of Costello syndrome
Shortly before germline KRAS mutations were described in patients with NS and CFC, HRAS germline mutations were reported to cause CS [43]. Later, other investigators also detected these mutations in the majority (∼90%) of CS patients studied [51–53]. Surprisingly, these germline lesions affect the same amino acid residues of H-Ras that are also mutated in cancer. The observation that activating oncogenic H-Ras mutations affecting codons G12 or G13 are tolerated in the germline, whereas oncogenic K-Ras mutations affecting these residues are not, underscores the notion that although H-Ras and K-Ras are structurally highly similar, they play different roles during embryonic development. Knockout studies have shown that only K-Ras is essential for embryonic development, whereas N-Ras and H-Ras are not [54]. Different phenotypes exhibited by mutations in various Ras isoforms may be due to heterogenous expression patterns of these proteins. Additionally, various Ras isoforms undergo different processing, e.g., de/repalmitoylation kinetics that regulate subcellular localization and activity of Ras isoforms [55, 56]. Although the vast majority of patients with CS harbor HRAS mutations, very recent studies have indicated that a CS phenotype may occasionally be associated with specific mutations of BRAF [57] or KRAS [48]. It remains to be determined if these patients also have the increased risk of neoplasia, which is typical of CS. Therefore, we doubt it is useful to classify these patients with mutations in Ras pathway genes other that HRAS as CS.
Neuro–cardio–facial–cutaneous syndrome
The molecular explanation of how different germline K-Ras or H-Ras mutations cause a wide spectrum of different phenotypes is still largely unknown. Activating germline K-Ras mutations may be associated with the broadest spectrum of clinical manifestations [42, 44, 46]. However, these syndromes are not just a less or more severe expression of essentially the same disorder. Therefore, it is conceivable that the different RAS mutations associated with divergent phenotypes do not only vary quantitatively in their degree of constitutive activation. Unique lesions may also have different qualitative effects on downstream signaling pathways. Additionally, modifier loci may play a role. Recently, the term ‘neuro–cardio–facial–cutaneous’ (NCFC) syndrome [58] was coined to illustrate that clinically overlapping disorders of the NS spectrum, including NF1, NS, CS, LS, and CFC syndrome, are caused by mutations in components of the Ras signaling pathway (Fig. 3). We speculate that the phenotypic variability between these disorder results from (1) different expression patterns of affected genes/isoforms and (2) variable mechanisms by which certain mutants interact with downstream effectors or regulatory proteins; these mutants therefore perturb Ras signaling in varying degrees. Further research is required to determine the basis of genotype–phenotype correlations in NS and related disorders.
Fig. 3.
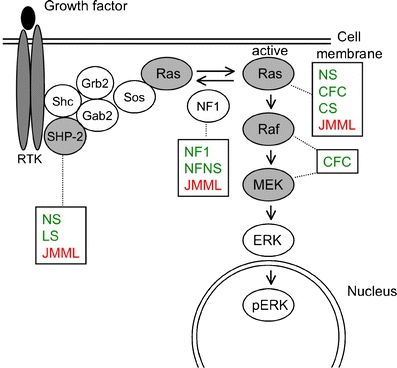
The Ras signaling pathway relays growth signals from activated growth factor receptors to the nucleus. Somatic mutations in several molecules of the pathway have been implicated in cancer. It is now recognized that germline mutations of identical molecules may cause disorders of the Noonan spectrum. A similar version of this figure has been previously published [61], republication with permission
Neurofibromatosis Noonan syndrome and NS-like features in neurofibromatosis
The rapidly increasing knowledge of the Ras signaling pathway and its relation to developmental disorders of the NS spectrum suggests that Noonan-like features are the clinical correlate of any genetic abnormality occurring in the germline and leading to a generalized mild deregulation of the Ras signaling cascade(s) during embryonic development. The new insights gained by recent research in this field may explain the phenotypic similarities between individuals with NF1 and NS. In addition to the well-known symptoms defining NF1, namely multiple café-au-lait spots and cutaneous neurofibromas, patients with NF1 are known to exhibit NS features, including relative short stature, relative macrocephaly, mild facial anomalies, thorax deformities, and learning difficulties. NFNS probably just represents the extreme end of a highly variable expression of these features [59]. Although the association of NF1 and NS was described in one family with independently segregating mutations in NF1 and PTPN11 [60], this double chance event is not the causative mechanism in many other patients with NF1 and NS-like features. The majority of patients with the NFNS phenotype have been found to harbor NF1 mutations, some of which also occur in patients with “pure” NF1 [40, 59]. We propose that the NS-like features that are often present in patients with NF1 are potentially due to NF1 haploinsufficiency leading to decreased inactivation of Ras·GTP. This might cause mild constitutional deregulation of Ras signaling, which may occasionally reach a level that leads to a NF1-NS phenotype. Frequently, patients with NF1 have mild features of NS. These may be explained by a mild activation of Ras, which is due to loss-of-function of one NF1 allele. Unknown genetic modifiers may play a role. Malignant cells arising in patients with NF1 somatically loose the wild-type NF1 allele. This second somatic hit ablates the GAP function of neurofibromin resulting in strongly enhanced Ras signaling (Fig. 4).
Fig. 4.

Model illustrating the molecular basis of how Ras signaling may be increased in patients with neurofibromatosis type 1 leading to Noonan-like feature in these patients. Although only one NF1 allele is inactive in the germline, tumors of patients with neurofibromatosis type 1 somatically lose the second, wild-type NF1 allele
Work in progress
Although the vast majority of cases with CFC and CS are explained by mutations in KRAS, BRAF, MEK1, MEK2, and HRAS, the underlying genetic defect in 50% of NS and 10–30% of CFC cases remains unknown. Based on the assumption that the NS phenotype results from hyperactive Ras signaling, we and others are currently screening DNA specimens from patients for mutations in genes encoding for other components, including negative regulators, of the Ras pathway. Analogous to PTPN11 and KRAS, currently unknown genes of the Ras pathway mutated in NS are presumably playing double roles in both development and oncogenesis. Mutations in BRAF and MEK1/2 have been excluded as major NS genes (M.Z., unpublished data).
It will be of interest to study how the different expression patterns of various RAS genes and isoforms influence clinical phenotypes when these genes/isoforms are mutated in the germline. It will be crucial to elucidate the structural mechanisms to understand how these new lesions perturb signaling on a molecular level. Characterization of the biological consequences of these mutations will be largely improved by the construction of murine knock-in models.
Note added in proof
After this paper was accepted we uncovered mutations in SOS1 encoding for the homonymous RasGEF in patients with NS. Meanwhile, however, two other groups have published elegant reports on activating SOS1 mutations in approximately 10% of cases with NS: Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, Kucherlapati RS. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2006 Dec 3; [Epub ahead of print] Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W, Ustaszewska A, Martin J, Bristow J, Carta C, Lepri F, Neri C, Vasta I, Gibson K, Curry CJ, Siguero JP, Digilio MC, Zampino G, Dallapiccola B, Bar-Sagi D, Gelb BD. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2006 Dec 3; [Epub ahead of print]”.
Abbreviations
- CFC syndrome
cardio–facio–cutaneous syndrome
- CS
Costello syndrome
- GAP
GTPase activating protein
- GDP
guanosine diphosphate
- GEF
guanosine nucleotide exchange factor
- GTP
guanosine triphosphate
- JMML
juvenile myelomonocytic leukemia
- LS
LEOPARD syndrome
- MPD
myeloproliferative disorder
- NCFC syndrome
neuro–cardio–facial–cutaneous syndrome
- NF1
neurofibromatosis type 1
- NFNS
neurofibromatosis–Noonan syndrome
- NS
Noonan syndrome
- N-SH2
n-terminal src-homology 2 domain
- NS/JMML
Noonan syndrome in association with JMML
- PTPase
phosphatase domain
Biographies
Christian P. Kratz received his medical degree from the University of Düsseldorf in Germany. He is presently an attending physician of Pediatrics of the Division of Pediatric Hematology/Oncology at the University of Freiburg. His research interests include genetic disorders that predispose to cancer or bone marrow failure.

Martin Zenker received his medical degree from the University of Erlangen-Nuremberg in Germany. He was trained as a pediatrician and human geneticist. He is currently working as an attending physician and research group leader at the Institute of Human Genetics at the University of Erlangen-Nuremberg. His research interests include the genetic basis of human malformation syndromes.

References
- 1.Repasky GA, Chenette EJ, Der CJ (2004) Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol 14:639–647 [DOI] [PubMed]
- 2.Vetter IR, Wittinghofer A (2001) The guanine nucleotide-binding switch in three dimensions. Science 294:1299–1304 [DOI] [PubMed]
- 3.Donovan S, Shannon KM, Bollag G (2002) GTPase activating poteins: critical regulators of intracellular signaling. BBA Rev Cancer 1602:23–45 [DOI] [PubMed]
- 4.Bos JL (1989) Ras oncogenes in human cancer: a review. Cancer Res 49:4682–4689 [PubMed]
- 5.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, Jacks T (2004) Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5:375–387 [DOI] [PubMed]
- 6.Tartaglia M, Gelb BD (2005) Noonan syndrome and related disorders: genetics and pathogenesis. Annu Rev Genomics Hum Genet 6:45–68 [DOI] [PubMed]
- 7.Hennekam RC (2003) Costello syndrome: an overview. Am J Med Genet C Semin Med Genet 117:42–48 [DOI] [PubMed]
- 8.Kavamura MI, Peres CA, Alchorne MM, Brunoni D (2002) CFC index for the diagnosis of cardiofaciocutaneous syndrome. Am J Med Genet 112:12–16 [DOI] [PubMed]
- 9.van der Burgt I, Brunner H (2000) Genetic heterogeneity in Noonan syndrome: evidence for an autosomal recessive form. Am J Med Genet 94:46–51 [DOI] [PubMed]
- 10.Jamieson CR, van der Burgt I, Brady AF, van Reen M, Elsawi MM, Hol F, Jeffery S, Patton MA, Mariman E (1994) Mapping a gene for Noonan syndrome to the long arm of chromosome 12. Nat Genet 8:357–359 [DOI] [PubMed]
- 11.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD (2001) Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29:465–468 [DOI] [PubMed]
- 12.Neel BG, Gu H, Pao L (2003) The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28:284–293 [DOI] [PubMed]
- 13.Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, Pizzuti A, Dallapiccola B (2002) Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet 71:389–394 [DOI] [PMC free article] [PubMed]
- 14.Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG (2006) PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem 281:6785–6792 [DOI] [PubMed]
- 15.Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V, Zampino G, Burgt I, Palleschi A, Petrucci TC, Sorcini M, Schoch C, Foa R, Emanuel PD, Gelb BD (2006) Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet 78:279–290 [DOI] [PMC free article] [PubMed]
- 16.Niemeyer CM, Arico M, Basso G, Biondi A, Cantu Rajnoldi A, Creutzig U, Haas O, Harbott J, Hasle H, Kerndrup G, Locatelli F, Mann G, Stollmann-Gibbels B, van’t Veer-Korthof ET, van Wering E, Zimmermann M (1997) Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood 89:3534–3543 [PubMed]
- 17.Flotho C, Valcamonica S, Mach-Pascual S, Schmahl G, Corral L, Ritterbach J, Hasle H, Arico M, Biondi A, Niemeyer CM (1999) RAS mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML). Leukemia 13:32–37 [DOI] [PubMed]
- 18.Kalra R, Paderanga DC, Olson K, Shannon KM (1994) Genetic analysis is consistent with the hypothesis that NF1 limits myeloid cell growth through p21ras. Blood 84:3435–3439 [PubMed]
- 19.Lauchle JO, Braun BS, Loh ML, Shannon K (2006) Inherited predispositions and hyperactive Ras in myeloid leukemogenesis. Pediatr Blood Cancer 46:579–585 [DOI] [PubMed]
- 20.Braun BS, Tuveson DA, Kong N, Le DT, Kogan SC, Rozmus J, Le Beau MM, Jacks TE, Shannon KM (2004) Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci U S A 101:597–602 [DOI] [PMC free article] [PubMed]
- 21.Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, Johnson L, Akashi K, Tuveson DA, Jacks T, Gilliland DG (2004) Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest 113:528–538 [DOI] [PMC free article] [PubMed]
- 22.Bader-Meunier B, Tchernia G, Mielot F, Fontaine JL, Thomas C, Lyonnet S, Lavergne JM, Dommergues JP (1997) Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J Pediatr 130:885–889 [DOI] [PubMed]
- 23.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD (2003) Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34:148–150 [DOI] [PubMed]
- 24.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, Curtiss NP, Gotlib J, Meshinchi S, Le Beau MM, Emanuel PD, Shannon KM (2004) Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood 103:2325–2331 [DOI] [PubMed]
- 25.Kratz CP, Niemeyer CM, Castleberry RP, Cetin M, Bergstrasser E, Emanuel PD, Hasle H, Kardos G, Klein C, Kojima S, Stary J, Trebo M, Zecca M, Gelb BD, Tartaglia M, Loh ML (2005) The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood 106:2183–2185 [DOI] [PMC free article] [PubMed]
- 26.Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, Palmi C, Carta C, Pession A, Arico M, Masera G, Basso G, Sorcini M, Gelb BD, Biondi A (2004) Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood 104:307–313 [DOI] [PubMed]
- 27.Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, Maris JM, Richardson A, Bardelli A, Sugarbaker DJ, Richards WG, Du J, Girard L, Minna JD, Loh ML, Fisher DE, Velculescu VE, Vogelstein B, Meyerson M, Sellers WR, Neel BG (2004) Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res 64:8816–8820 [DOI] [PubMed]
- 28.Schubbert S, Lieuw K, Rowe SL, Lee CM, Li X, Loh ML, Clapp DW, Shannon KM (2005) Functional analysis of leukemia-associated PTPN11 mutations in primary hematopoietic cells. Blood 106:311–317 [DOI] [PMC free article] [PubMed]
- 29.Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S, Morgan K, Boulton C, Shigematsu H, Keilhack H, Akashi K, Gilliland DG, Neel BG (2005) Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell 7:179–191 [DOI] [PubMed]
- 30.Chan RJ, Leedy MB, Munugalavadla V, Voorhorst CS, Li Y, Yu M, Kapur R (2005) Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. Blood 105(9):3737-3742 [DOI] [PMC free article] [PubMed]
- 31.Keilhack H, David FS, McGregor M, Cantley LC, Neel BG (2005) Diverse biochemical properties of Shp2 mutants: implications for disease phenotypes. J Biol Chem 280:30984–30993 [DOI] [PubMed]
- 32.Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA, Neel BG (2004) Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med 10:849–857 [DOI] [PubMed]
- 33.Side L, Emanuel P, Taylor B, Franklin J, Thompson P, Castleberry R, Shannon K (1998) Mutations of the NF1 gene in leukemias from children without evidence of neurofibromatosis, type 1. Blood 92:267–273 [PubMed]
- 34.Shannon KM, O’Connell P, Martin GA, Paderanga D, Olson K, Dinndorf P, McCormick F (1994) Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. N Engl J Med 330:597–601 [DOI] [PubMed]
- 35.Stephens K, Weaver M, Leppig KA, Maruyama K, Emanuel PD, Le Beau MM, Shannon KM (2006) Interstitial uniparental isodisomy at clustered breakpoint intervals is a frequent mechanism of NF1 inactivation in myeloid malignancies. Blood [DOI] [PMC free article] [PubMed]
- 36.Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, Lange BJ, Freedman MH, McCormick F, Jacks T, Shannon K (1996) Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 12:144–148 [DOI] [PubMed]
- 37.Largaespada DA, Brannan CI, Jenkins NA, Copeland NG (1996) Nf1 deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nat Genet 12:137–143 [DOI] [PubMed]
- 38.Le DT, Kong N, Zhu Y, Lauchle JO, Aiyigari A, Braun BS, Wang E, Kogan SC, Le Beau MM, Parada L, Shannon KM (2004) Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood 103:4243–4250 [DOI] [PubMed]
- 39.Colley A, Donnai D, Evans DG (1996) Neurofibromatosis/Noonan phenotype: a variable feature of type 1 neurofibromatosis. Clin Genet 49:59–64 [DOI] [PubMed]
- 40.De Luca A, Bottillo I, Sarkozy A, Carta C, Neri C, Bellacchio E, Schirinzi A, Conti E, Zampino G, Battaglia A, Majore S, Rinaldi MM, Carella M, Marino B, Pizzuti A, Digilio MC, Tartaglia M, Dallapiccola B (2005) NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet 77:1092–1101 [DOI] [PMC free article] [PubMed]
- 41.Zenker M, Buheitel G, Rauch R, Koenig R, Bosse K, Kress W, Tietze HU, Doerr HG, Hofbeck M, Singer H, Reis A, Rauch A (2004) Genotype–phenotype correlations in Noonan syndrome. J Pediatr 144:368–374 [DOI] [PubMed]
- 42.Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE, Nguyen H, West B, Zhang KY, Sistermans E, Rauch A, Niemeyer CM, Shannon K, Kratz CP (2006) Germline KRAS mutations cause Noonan syndrome. Nat Genet 38:331–336 [DOI] [PubMed]
- 43.Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, Matsubara Y (2005) Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet 37:1038–1040 [DOI] [PubMed]
- 44.Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G, Wieczorek D, Kavamura MI, Kurosawa K, Ohashi H, Wilson L, Heron D, Bonneau D, Corona G, Kaname T, Naritomi K, Baumann C, Matsumoto N, Kato K, Kure S, Matsubara Y (2006) Germline KRAS and BRAF mutations in cardio–facio–cutaneous syndrome. Nat Genet 38:294–296 [DOI] [PubMed]
- 45.Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA (2006) Germline mutations in genes within the MAPK pathway cause cardio–facio–cutaneous syndrome. Science 311:1287–1290 [DOI] [PubMed]
- 46.Carta C, Pantaleoni F, Bocchinfuso G, Stella L, Vasta I, Sarkozy A, Digilio C, Palleschi A, Pizzuti A, Grammatico P, Zampino G, Dallapiccola B, Gelb BD, Tartaglia M (2006) Germline missense mutations affecting KRAS Isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet 79:129–135 [DOI] [PMC free article] [PubMed]
- 47.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J (2003) Structural evidence for feedback activation by Ras. GTP of the Ras-specific nucleotide exchange factor SOS. Cell 112:685–695 [DOI] [PubMed]
- 48.Zenker M, Lehmann K, Schulz AL, Barth CF, Hansmann D, Koenig R, Korinthenberg R, Kreiss-Nachtsheim M, Meinecke P, Morlot S, Mundlos S, Quante AS, Raskin S, Schnabel D, Wehner LE, Kratz CP, Horn D, Kutsche K (2006) Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet 2006 Oct 20; [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 49.Stone JC, Colleton M, Bottorff D (1993) Effector domain mutations dissociate p21ras effector function and GTPase-activating protein interaction. Mol Cell Biol 13:7311–7320 [DOI] [PMC free article] [PubMed]
- 50.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954 [DOI] [PubMed]
- 51.Gripp KW, Lin AE, Stabley DL, Nicholson L, Scott CI, Jr., Doyle D, Aoki Y, Matsubara Y, Zackai EH, Lapunzina P, Gonzalez-Meneses A, Holbrook J, Agresta CA, Gonzalez IL, Sol-Church K (2006) HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation. Am J Med Genet A 140:1–7 [DOI] [PubMed]
- 52.Kerr B, Delrue MA, Sigaudy S, Perveen R, Marche M, Burgelin I, Stef M, Tang B, Eden OB, O’Sullivan J, De Sandre-Giovannoli A, Reardon W, Brewer C, Bennett C, Quarell O, M’Cann E, Donnai D, Stewart F, Hennekam R, Cave H, Verloes A, Philip N, Lacombe D, Levy N, Arveiler B, Black G (2006) Genotype–phenotype correlation in Costello syndrome: HRAS mutation analysis in 43 cases. J Med Genet 43:401–405 [DOI] [PMC free article] [PubMed]
- 53.Estep AL, Tidyman WE, Teitell MA, Cotter PD, Rauen KA (2006) HRAS mutations in Costello syndrome: detection of constitutional activating mutations in codon 12 and 13 and loss of wild-type allele in malignancy. Am J Med Genet A 140:8–16 [DOI] [PubMed]
- 54.Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, Bronson RT, Umanoff H, Edelmann W, Kucherlapati R, Jacks T (1997) K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev 11:2468–2481 [DOI] [PMC free article] [PubMed]
- 55.Mor A, Philips MR (2006) Compartmentalized Ras/MAPK signaling. Annu Rev Immunol 24:771–800 [DOI] [PubMed]
- 56.Rocks O, Peyker A, Kahms M, Verveer PJ, Koerner C, Lumbierres M, Kuhlmann J, Waldmann H, Wittinghofer A, Bastiaens PI (2005) An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 307:1746–1752 [DOI] [PubMed]
- 57.Rauen KA (2006) Distinguishing Costello versus cardio–facio–cutaneous syndrome: BRAF mutations in patients with a Costello phenotype. Am J Med Genet 140:1681–1683 [DOI] [PubMed]
- 58.Bentires-Alj M, Kontaridis MI, Neel BG (2006) Stops along the RAS pathway in human genetic disease. Nat Med 12:283–285 [DOI] [PubMed]
- 59.Hüffmeier U, Zenker M, Hoyer J, Fahsold R, Rauch A (2006) A variable combination of features of Noonan syndrome and neurofibromatosis type I are caused by mutations in the NF1 gene. Am J Med Genet A 140:2749–2756 [DOI] [PubMed]
- 60.Bertola DR, Pereira AC, Passetti F, de Oliveira PS, Messiaen L, Gelb BD, Kim CA, Krieger JE (2005) Neurofibromatosis–Noonan syndrome: molecular evidence of the concurrence of both disorders in a patient. Am J Med Genet A 136:242–245 [DOI] [PubMed]
- 61.Kratz CP, Schubbert S, Bollag G, Niemeyer CM, Shannon KM, Zenker M (2006) Germline mutations in components of the Ras signaling pathway in Noonan syndrome and related disorders. Cell Cycle 5:1607–1611 [DOI] [PubMed]