

Abstract
Holoprosencephaly (HPE) is the most common structural malformation of the forebrain and face in humans. Our current understanding of the pathogenesis of HPE attempts to integrate genetic susceptibility, evidenced by mutations in the known HPE genes, with the epigenetic influence of environmental factors. Mutations or deletions of the human TGIF gene have been associated with HPE in multiple population cohorts. Here we examine the functional effects of all previously reported mutations, and describe four additional variants. Of the eleven sequence variations in TGIF, all but four can be demonstrated to be functionally abnormal. In contrast, no potentially pathogenic sequence alterations were detected in the related gene TGIF2. These results provide further evidence of a role for TGIF in HPE and demonstrate the importance of functional analysis of putative disease-associated alleles.
Keywords: TGIF, TGIF2, corepressor, HPE, complex inheritance
Introduction
Holoprosencephaly (HPE) is best understood as a failure in the generation of, or response to, midline signals that normally instruct the developing prosencephalon to divide into paired left and right hemispheres and subcortical structures [1,2]. This incompletely understood process is etiologically heterogeneous and can be perturbed by both genetic and environmental causes, either individually or more likely in combination. Clinically, there is a nearly continuous spectrum of malformations consistent with HPE, and this variable expressivity and/or penetrance is clearly demonstrable in all well documented familial cases segregating a particular HPE mutation. Families frequently manifest a wide range of phenotypes, such as, typically severe HPE with perinatal lethality, or microforms (such as microcephaly, closely spaced eyes, single central incisor), or even clinically unaffected individuals. Most investigators consider HPE to be consistent with autosomal dominant inheritance of a major susceptibility locus, although X-linked, autosomal recessive, and digenic inheritance have been suggested in isolated cases [3,4]. With few exceptions, such as ZIC2 and GLI2 [5–7], prediction of phenotype based on the type or nature of mutation has been elusive for HPE, as well as for an increasingly large number of unrelated genetic disorders [8,9]. We hypothesized that for some conditions, including HPE, alterations in modifier genes or interactions with environmental factors contribute to the variable phenotype in HPE and other disorders (multiple–hit hypothesis) [10]. TGIF [11] is one of several genes associated with HPE, including Sonic Hedgehog (SHH), SIX3, ZIC2, GLI2 and potentially TDGF1 and PATCHED [5–7,12–16]. Although several of these genes are within the Sonic Hedgehog pathway, having been studied because SHH was the first HPE gene to be identified, there is currently limited understanding of the potential interactions between these HPE susceptibility genes. In all cases to date, only heterozygous sequence changes or hemizygosity have been detected in these genes.
TGIF (5′TG3′) interacting factor (or TGFβ-induced factor, OMIM #602630) is a transcriptional repressor and member of the TALE (Three aminoacid loop extension) class of atypical homeodomain proteins [17]. The TGIF gene is located on 18p11.3 within the HPE4 minimal critical region (OMIM #142946) defined by the comparison of several cytogenetic rearrangements leading to the loss of 18p in association with the presence of HPE or its microforms [18,19]. Mutations in the human TGIF gene have been identified exclusively among HPE patients in several studies and include deletions of the entire gene due to cytogenetically visible [20] or microscopic rearrangements (21,22), missense and nonsense sequence changes [11,23,24]. However, attempts to model HPE in mice utilizing targeted inactivation of the Tgif gene have failed to recapitulate the clinical findings of HPE seen in humans [25,26, 52, 53, and this study]. Consequently, it became important for us to determine the potential mutational spectrum of the TGIF and the related TGIF2 genes [27,28,29] among our HPE patients. Furthermore, it is important to evaluate the functional effects of these putative disease-associated alleles, since these findings have a direct bearing on genetic counseling, as well as evaluations aimed at the study of gene-gene and gene-environment interactions.
TGIF was first identified as a 272 aminoacid (NP_775300) homeodomain transcription factor that competed with the binding of RXR to the DR-1 RXRE in the rat cellular retinol binding protein II (CrbpII) promotor [30,31]; however, the physiological significance of this competition is poorly understood, since most retinoid response elements lack direct binding sites for TGIF. Subsequent investigations have described multiple modes of repression for TGIF, affecting the magnitude of TGFβ-induced responses as a corepressor of Smad2 or Smad3 [32–35], as well as a distinct role as a corepressor of retinoic acid mediated changes in gene expression through interactions with RXR nuclear receptors [26]. Recent studies demonstrate that TGIF also has a more general role as a corepressor of RXR nuclear receptors through a protein-protein interaction, and the recruitment of additional corepressors into a multiprotein complex. Similar multiprotein complexes are seen with Smad2 and TGIF, and can include the recruitment of histone deacetylase (HDAC), CtBP and mSin3. These studies suggest that the repression effects are likely to be mediated by gene-targeted changes in chromatin remodeling. In general, TGIF acts to attenuate, or limit the extent of TGFβ or retinoid responses; however, limited information is available on a potential direct role as a transcription factor, beyond its ability to participate as a molecular switch from gene activation to gene repression. There may well be other functions of TGIF and TGIF2 yet to be described.
Members of the TGFβ family of secreted signaling molecules play diverse roles in intercellular signaling and developmental programs (reviewed in [36]). These TGFβ ligands bind to heterodimeric Type I/Type II cell surface receptors leading to the phosphorylation, and activation of Smad 2 and/or 3. These activated Smads bind to the common Smad 4 and translocate to the nucleus to influence gene expression. Activated Smad complexes are directed to transcriptional targets through interactions with additional transcription factors, such as FoxH1, and via direct DNA binding by the Smads. Target genes can be either activated in part by the recruitment of coactivators, such as p300/CBP, and the extent of this activation can be limited by interactions with corepressors like TGIF. It is the balance between competing activator and repressor activities that ultimately determines the magnitude of the TGFβ response within the cell.
It was initially attractive to speculate that mutations in TGIF affected the functioning of Nodal [11], or related TGFβ factors, since genes in these pathways are intimately involved in the development of the vertebrate organizer and its midline derivatives (such as the notochord and prechordal plate) that are considered essential organizing centers for specification of all three vertebrate axes. Defective Nodal signaling can result in cyclopic phenotypes that can resemble some of the more extreme forms of HPE seen in humans. Given the role of TGIF as a corepressor, loss of TGIF function would lead to an increase, rather than a decrease in signaling. However, it may be that too much or too little Nodal signaling could result in defects in axis formation and potentially generate HPE-like phenotypes.
The second described role for TGIF, as a modulator of retinoid responses, is perhaps a more attractive model since retinoic acid is a well-described teratogen resulting in HPE both in humans and animal models. Furthermore, targeted disruption of murine Tgif leads to aberrant activation of retinoid-responsive gene expression and an increase in sensitivity to excess retinoids [26]. However, even complete elimination of Tgif function in homozygous null mice does not fully reflect the morphological abnormalities attributed to the loss of a single TGIF allele in humans [25, 26, 52, 53]. The potential basis of these species differences (mouse vs. human) is poorly understood. Interestingly, there are also minor phenotypic differences between mouse lines established in different laboratories, which are attributed to genetic modifiers between strains.
We set out to examine the mutational spectrum of the human TGIF gene and putative promoter region using a more highly sensitive method of dHPLC screening than had been used in our original studies; we applied the same technology to the evaluation of TGIF2 as a potential candidate gene, since this related gene shares many of the functional attributes of TGIF and might compensate for decreased activity of TGIF in certain tissues or developmental contexts.
Results
A total of 435 patients with HPE were available in our collection for mutational analysis of the TGIF gene. These samples are representative of the full clinical spectrum of HPE phenotypes seen in non-syndromic, cytogenetically normal individuals, and include sporadic or familial forms analyzed irrespective of the status of mutations detectable in unrelated HPE candidate genes. A minimum of 95 normal control individuals were studied in parallel to the patient sample set. New sample accrual has more than compensated for sample depletion making this the most comprehensive analysis of TGIF to date. Previously detected mutations were confirmed through this dHPLC study, and four additional sequence variants were identified. Table 1 describes the molecular and clinical findings of all eleven TGIF variations identified by our lab, or by others.
Table 1.
Summary of the clinical findings and functional consequences of mutations in the TGIF gene
| Transcriptional repression | ||||||
|---|---|---|---|---|---|---|
| Mutation | Inheritance | Defects | TGFβ dependent | RXR-dependent | Clinical findings | Reference |
| c.83C>T (p.S28C) | Paternal | No CtBP interaction | decreased | decreased | H, CNPAS, SCI, ACC, DD | [11, 39] |
| c.133G>T (p.E45X) | De novo | Truncation | No | No | lobar HPE, median CL/P, MC | This report |
| [c.140_141delTG] + | Paternal | p.S46fs causes truncation | No | No | Semilobar HPE, CL/P | This report |
| [c.228C>A; p.H76Q] | Maternal | Likely normal | Yes | Yes | ||
| c.177C>G (p.Y59X) | Paternal | Truncation | No | No | Semilobar HPE, MC, H; father with H, lateral CL | [23] |
| c.187C>G (p.P63A) | De novo | Likely mis-folded | No | No | Lobar HPE, CL/P, MR | [11] |
| c.268C>T (p.R90C) | De novo | Likely mis-folded | No | No | Alobar HPE, H, median CL, PMA | [24] |
| c.778delC (K259fs) | Paternal | Likely mis-folded | No | decreased | Proband referred for HPE; father anosmia, H | This report |
| c.320A>T (p.Q107L) | Maternal? | Likely normal | Yes | Yes | Proband unavailable; mother MC, CL/P, MR | [23] |
| c.451A>G (p.T151A) | Unknown | p.T151A likely normal | Yes | Yes | Semilobar HPE, MC, H, midline CL; father not available | [11] |
| c.485C>T (p.S162F) | Paternal | p.S162F likely normal | Yes | Yes | Proband with encephalocele, ACC midline CL; normal father also positive | [10] |
Abbreviations: H, hypotelorism; CNPAS, congenital nasal pyriform aperture stenosis; SCI, single central incisor; ACC, agenesis corpus callosum; DD, developmental delay; CL/P, cleft lip and palate; MC, microcephaly; HPE, holoprosencephly; PMA, premaxillary agenesis; MR. mental retardation.
Case reports
The first mutation, p.E45X, was seen in a male proband presenting with lobar HPE, atypical ventricles with small frontal horns, hypothalamic and caudate fusion, diabetes insipidus, seizures, premaxillary agenesis, microcephaly, absent nasal root and septum with a depressed nasal tip (Fig. 1A and B). This heterozygous sequence change (Fig. 1C) predicts a premature termination of the protein within the first α helix of the homeodomain. Both parents were tested with normal results, indicating that this case represents a de novo mutation.
Fig. 1.
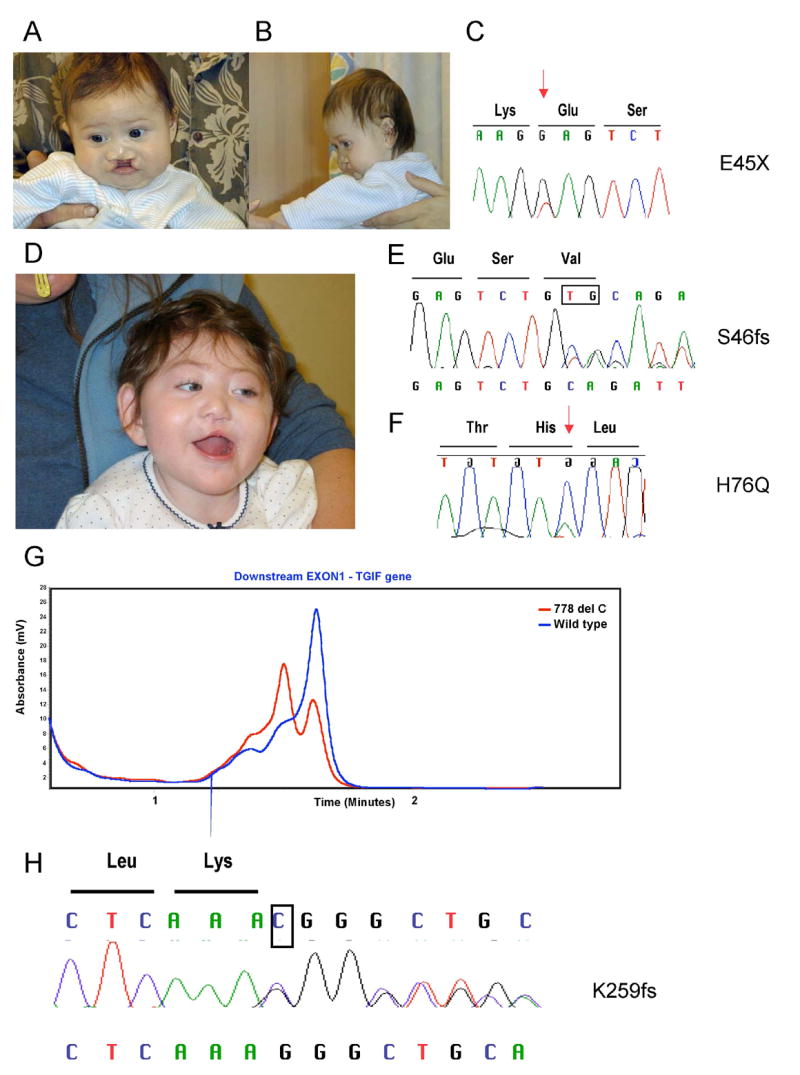
Facial photographs of the first proband (A and B) with the pE45X mutatiom (C). Patient two (D) carries two different variations the S46fs (E) and H76Q (F). Red arrows indicate the missense changes and the deleted bases are boxed. The expected chromatogram from the normal allele is above and the frameshifted allele is below. A representative dHPLC chromatogram from patient three (G) shows heterozygosity for the normal allele (blue) and the variant allele (red). In panel H, the deleted base is boxed and the expected sequence from the normal allele is above and the frameshifted allele is below the chromatogram.
Interestingly, the second and third mutations were detected in the same female patient (Fig. 1D). To our knowledge, this represents the first case of HPE with two alterations in the same gene. She presented with semilobar HPE, absence of the olfactory tracts, diabetes insipidus, dystonia, unilateral cleft lip, flat nasal bridge, premaxilla dysgenesis, and microcephaly. One variation, p.S46fs, was inherited from her father, while the second variation, p.H76Q, was inherited from her mother (Fig. 1E and F). Both parents were reported to be non-consanguinous, and normal by routine physical exam.
An unrelated HPE patient was also identified with the same p.H76Q sequence change. In order to confirm that these cases were independent examples of the identical sequence change (c.228C>A), we examined the promoter region, 5′ UTR and exon 3 for polymorphic variants; we could demonstrate that these cases were indeed distinct and differed at four common polymorphic loci.
The fourth novel sequence variation detected in our study by dHPLC (Fig. 1G) was identified in a male infant who died at 5 days of age and diagnosed with HPE. This variation predicts a frameshift at position 259 of the COOH-terminus of the TGIF protein (Fig. 1H) with an extension of 57 aminoacids not present in the native protein and a protein 44 aminoacids longer. This change was also detected in the father who displayed hypotelorism and anosmia, features consistent with HPE microforms.
HPE-associated variations in TGIF are family-specific alterations
We detected a substantial number of single nucleotide polymorphisms in the human TGIF gene during the course of our investigation (Table 2). Many of these polymorphisms have been described by the investigation of TGIF as a candidate gene for myopia in a distinct population [37,38]. In contrast to the disease-associated alleles, the majority of these polymorphisms were detectable with comparable frequencies among patients, as well as controls. Nevertheless, all detected sequence variations were assessed for their potential role in affecting protein expression or function; none were selected by us for further analysis.
Table 2.
Common sequence variations in the human TGIF gene
| Frequency in patients with HPE | Frequency in controls | |||||
|---|---|---|---|---|---|---|
| Polymorphism | Region | Hetero | Homo | Hetero | Homo | Two sided P value |
| c.-961T>C | Promoter | 8/348 | 8/118 | 0.02 | ||
| c.-945dupC | Promoter | 3/348 | 2/118 | 0.44 | ||
| c.-920T>A | Promoter | 2/348 | 0/118 | 0.39 | ||
| c.-783A>T | Promoter | 0/348 | 1/118 | 0.09 | ||
| c.-777G>A | Promoter | 1/348 | 0/118 | 0.56 | ||
| c.-767T>G | Promoter | 2/348 | 0/118 | 0.41 | ||
| c.-713G>A | Promoter | 72/348 | 24/118 | 1/118 | 1.00 | |
| c.-711G>C | Promoter | 3/348 | 6/118 | 0.0037# | ||
| c.-696G>T | Promoter | 46/348 | 1/348 | 9/118 | 0.11 | |
| c.-693T>C | Promoter | 1/348 | 3/118 | 0.023 | ||
| c.-678G>T | Promoter | 1/348 | 0/118 | 0.56 | ||
| c.-613A>G | Promoter | 1/331 | 0/118 | 0.55 | ||
| c.-611A>G | Promoter | 2/311 | 2/111 | 0.28 | ||
| c.-478G>C | Promoter | 2/331 | 0/118 | 0.40 | ||
| c.-427G>C | Promoter | 1/343 | 0/118 | 0.55 | ||
| c.-284A>T | 5′UTR | 7/343 | 11/93 | 0.0001# | ||
| c.-214dupG | 5′UTR | 3/343 | 0/93 | 0.37 | ||
| c.-33C>A | 5′UTR | 101/424 | 2/424 | 32/94 | 2/94 | 0.045 |
| c.16+9G>A | Intronic | 1/424 | 0/94 | 0.66 | ||
| c.16+86_88 delTGG | Intronic | 2/424 | 0/94 | 0.50 | ||
| c.16+173C>T | Intronic | 1/424 | 0/94 | 0.63 | ||
| c.244-106T>G | Intronic | 1/181 | not tested | N/A | ||
| c.244-119C>G | Intronic | 31/181 | 4/181 | not tested | N/A | |
| c.244-128delT | Intronic | 1/181 | not tested | N/A | ||
| c.244-49T>C | Intronic | 0/181 | 1/8 | 0.00001# | ||
| c.244-18G>A | Intronic | 1/181 | not tested | N/A | ||
| c.60C>T | Coding (p.S20S) | 1/419 | 2/94 | 0.03 | ||
| c.174T>A | Coding (p.R58R) | 0/419 | 1/94 | 0.04 | ||
| c.420A>G | Coding (p.P140P)* | 61/404 | 4/404 | 14/95 | 1/95 | 1.00 |
| c.487C>T | Coding (p.P163S) | 26/404 | 2/404 | 11/95 | 0.06 | |
| c.488C>T | Coding (p.P163L)* | 41/404 | 1/404 | 3/95 | 0.03 | |
| c.489G>A | Coding (p.P163P) | 7/404 | 3/95 | 1/95 | 0.40 | |
| c.887T>G | Coding (p.V192V) | 16/404 | 10/95 | 0.02 | ||
| c.657T>G | Coding (p.T219T)* | 21/404 | 1/404 | 14/95 | 0.0006# | |
| c.711C>A | Coding (p.P237P) | 1/404 | 0/94 | 0.88 | ||
Sequence variations also seen in TGIF as a candidate for myopia (38).
Variations found significantly more frequently in normal controls.
Lack of evidence of a role for TGIF2 in HPE
We also performed mutational screening of the two principal coding exons of the TGIF2 gene in a slightly larger cohort of 496 patients with HPE. In contrast to the potentially pathogenic variations seen with TGIF, the only sequence variants that we identified involved a wobble (p.R96R) in an HPE proband (c.228C>T). In the normal control samples, we detected a single individual with a −7_−10inv in the immediate vicinity of the 5′UTR upstream of the initiator methionine. Both changes were considered likely rare variants and not studied further. Like many genes evaluated as potential HPE candidate genes, TGIF2 is not associated with pathogenic mutations in humans.
Strategy for functional analysis
Our original study linking TGIF mutations with HPE described significantly impaired repressor function for p.S28C, decreased DNA binding for p.P63R, and suggested hypomorphic activities for the p.T151A and p.S162F variants [11]. The p.S28C mutation was further shown to impair the interaction with CtBP due to the disruption of a conserved PLDLS motif in the NH2-terminal repression domain [39]. In addition, three more potential mutations have been described: one nonsense (p.Y59X) and two missense changes, p.Q107L and p.R90C ([23, 24], see Table 1). In order to better understand the significance of these sequence variations, we performed functional studies of all eleven described TGIF alterations. Over-expression of high levels of human TGIF and its potential human binding partners (DNA target sequences, CtBP, mSin3, Smad3, and RXRα) in COS1 cells was chosen as the most sensitive test of the ability of the mutant TGIF forms to interact with these factors; furthermore, this allowed us to detect quantitative differences among the TGIF variants in relative expression levels and stability (see below). These over-expression studies were then supplemented with reporter assays of TGFβ and RXR-dependent responses according to published procedures.
HPE mutations within the homeodomain of TGIF affect DNA binding
TGIF can bind to DNA via interaction with other DNA-binding proteins or direct binding to a TGIF consensus site. To determine the effect of the HPE mutations on the ability of TGIF to bind DNA, we performed DNA pulldown assays. Lysates from transfected COS1 cells were incubated with biotinylated double-stranded DNA oligonucleotide comprised of a portion of the p15 promoter sequence, which contains a TGIF binding site (Fig. 2). TGIF protein bound to biotinylated oligo was isolated on streptavidin agarose, run on SDS-PAGE gels, and western blotted for TGIF. Wildtype TGIF was pulled down by the wildtype oligo, but not by an oligo containing a mutated TGIF binding site that was used as a negative control. p.S28C, p.T151A, and p.S162F bound DNA as well as wildtype. p.K259 frame shift mutant showed reduced binding, but this is likely due to its low expression level in general. The above mutations do not affect the homeodomain and therefore do not affect DNA binding. p.H76Q and p.Q107L are expressed as well as wildtype TGIF and do not exhibit significantly reduced DNA binding ability. p.P63A and p.R90C do not bind DNA detectably in this assay. In addition we noticed that the expression of these two mutants in the lysate fraction is reduced compared to wildtype. Lysate and pellet expression controls show that more of p.P63A and p.R90C are present in the pellet fraction than in the lysate, indicating that these mutations render TGIF relatively insoluble, possibly due to misfolding. However, the expression level of these proteins in the soluble fraction is similar to that of p.K259fs, which binds DNA, suggesting that the P63A and R90C mutations disrupt the ability of TGIF to bind to its consensus site.
Fig. 2.
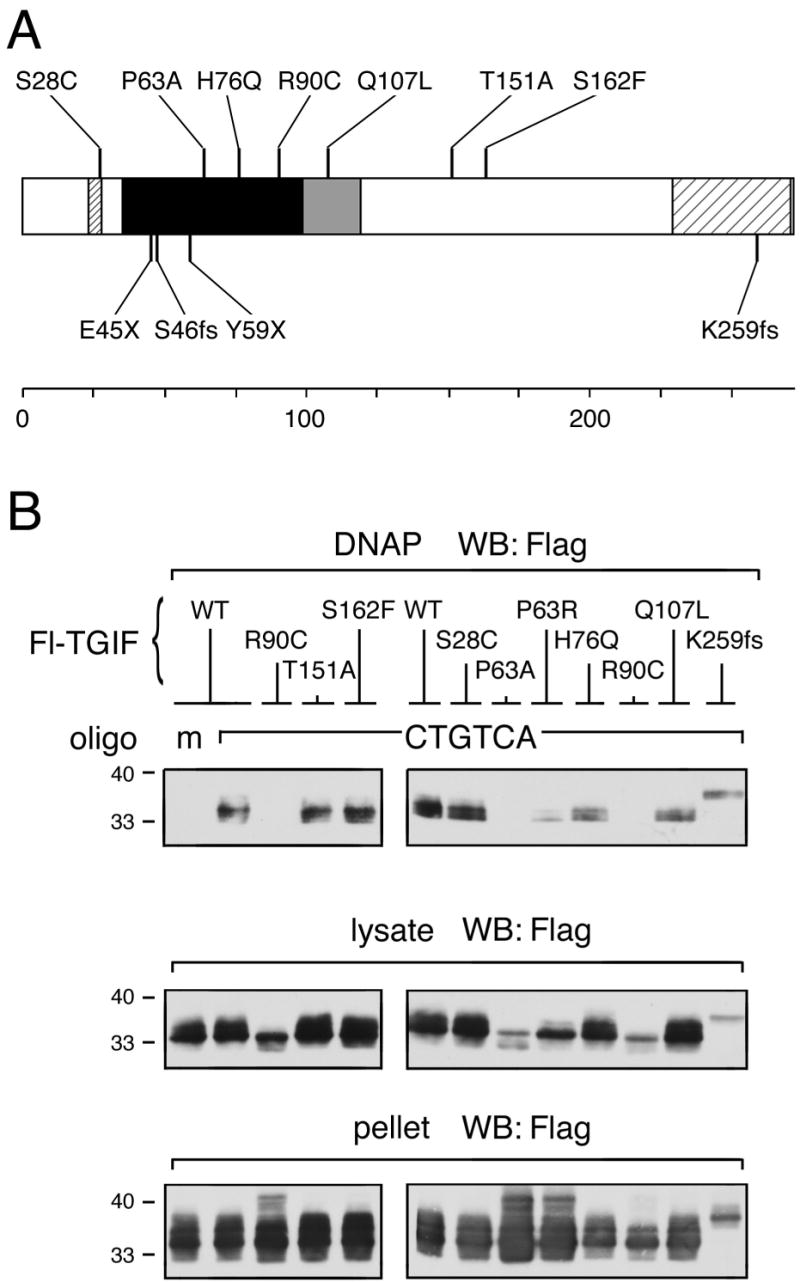
(A) Summary of the positions of the studied mutations within the TGIF protein. Note that the P63R mutation [11] was incorrect; instead, the observed sequence change predicts P63A. (B) HPE mutations in the homeodomain of TGIF affect DNA binding ability. COS1 cells were transfected with a Flag-tagged TGIF wildtype or HPE mutant construct as indicated. 36 hours after transfection, cell lysates were incubated with biotinylated double-stranded oligonucleotide containing either mutated (m) or consensus TGIF binding site (CTGTCA). TGIF protein bound to DNA was isolated on Streptavidin agarose and analyzed by western blot for the presence of TGIF. These experiments were performed repeatedly to assure consistency of the results. Portions of each lysate and pellet sample were subjected to direct western blot analysis to monitor protein expression and solubility (below).
TGIF HPE mutants interact with corepressors
TGIF contains a PLDLS motif near its N-terminus, through which TGIF interacts with the corepressor CtBP to regulate transcription. It has previously been shown that the HPE mutation p.S28C, located within the CtBP binding motif, abolishes the interaction between TGIF and CtBP. The remaining HPE mutants were assayed for CtBP interaction by immunoprecipitation of Flag-TGIF proteins followed by western blot for coprecipitating His6-CtBP. The HPE point mutants interact with CtBP as well as wildtype TGIF (Fig. 3A). Frame shift and truncation HPE mutations appear to weaken interaction with CtBP, in part due to decreased expression levels of these constructs (Fig. 3B). Although there is clearly a detectable interaction between the p.Y59X truncation mutant and CtBP, perhaps suggesting that this mutant form of TGIF might be able to titrate CtBP away from its normal functions.
Fig. 3.
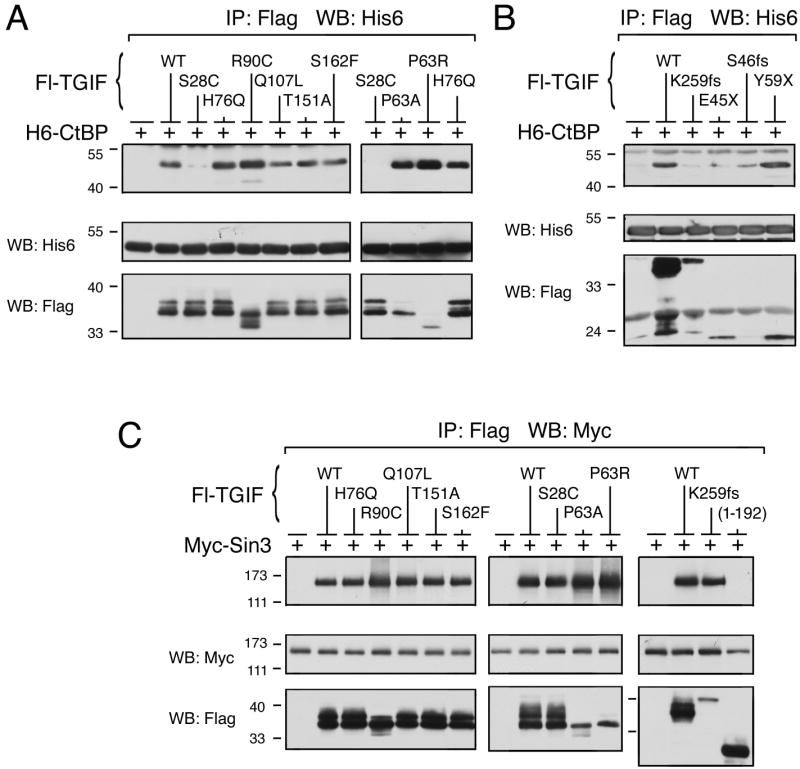
TGIF HPE mutants interact with corepressors. (A and B) COS1 cells were transfected with His6-CtBP and Flag-TGIF wildtype or HPE mutant, as indicated. Protein complexes were isolated on anti-Flag agarose and analyzed by western blot for the presence of co-precipitating CtBP. A portion of the lysates was subjected to direct western blot analysis to monitor protein expression (below). (C) COS1 cells were transfected with Myc-epitope tagged Sin3 expression construct, together with the indicated Flag-tagged TGIF constructs or control vector. Protein complexes were isolated on anti-Flag agarose and analyzed by western blot for the presence of co-precipitating Sin3. A portion of the lysates was subjected to direct western blot analysis to monitor protein expression (below).
It is known that TGIF interacts with the Sin3 corepressor through an interaction domain in its C-terminus. To determine whether HPE mutations in TGIF affect Sin3 interaction, COS1 cells were cotransfected with Myc-Sin3 and Flag-TGIF constructs, as indicated. Lysates were immunoprecipitated for Flag-TGIF then western blotted for coprecipitating Myc-Sin3 (Fig. 3C). A C-terminal deletion of TGIF that eliminates the Sin3 binding domain of TGIF, and therefore abolishes interaction, was used as a negative control in this assay. All of the TGIF HPE mutants assayed interact with Sin3. HPE truncation mutants were not assayed, as they do not contain the Sin3 interaction domain. Therefore, other than the loss of CtBP interaction with the p.S28C mutation, TGIF HPE point mutants are able to interact with transcriptional corepressors.
TGIF HPE mutants interact with Smad3 but exhibit a decreased ability to repress TGFβ-activated transcription
TGIF has previously been shown to regulate TGFβ-activated transcription via interaction with Smad2 and Smad3. An immunoprecipitation assay was used to determine whether HPE mutations affect the ability of TGIF to interact with Smads. All of the HPE mutants tested retain the ability to interact with Smad3. However, p.R90C and p.K259 frame shift expression levels are low, decreasing the amount of co-precipitating Smad3 (Fig. 4A).
Fig. 4.
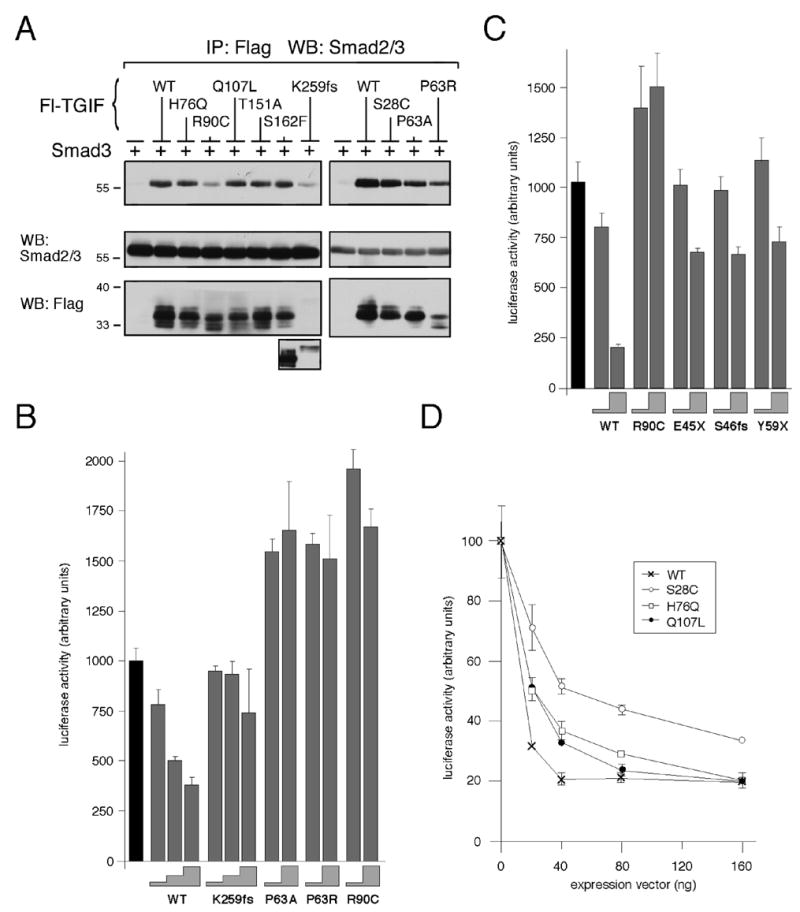
TGIF HPE mutants interact with Smad3 and affect repression of TGFβ-activated transcription. (A) COS1 cells were transfected with untagged Smad3 and Flag-TGIF wildtype or HPE mutants or control vector, as indicated. Protein complexes were isolated on anti-Flag agarose and analyzed by western blot for the presence of coprecipitating Smad3. A portion of the lysates was subjected to direct western blot analysis to monitor protein expression (below). Due to the low expression level of the K259fs construct, a longer exposure of the lysate western blot is shown below. (B, C and D) HepG2 cells were transfected with a TGFβ-responsive luciferase reporter, 3TP-lux, and increasing amounts of TGIF wildtype or HPE mutant expression constructs. All cells were treated with 100 pM TGFβ prior to analysis. Firefly luciferase activity was normalized to Renilla luciferase and is represented in arbitrary units, as the mean +/− s.d. of duplicate transfections. (B) HepG2 cells were transfected with 10 ng and 50, or 10, 25 and 50 ng of TGIF expression construct (the height of the grey steps indicate the amount of input TGIF DNA: 10, 25 or 50 ng). The black bar indicate the activity of the reporter alone, without TGIF (C) 10 or 100 ng of TGIF constructs were transfected (grey steps). (D) Dose-dependent repression of luciferase activity by TGIF is shown as a graph of luciferase activity in arbitrary units as the mean +/− s.d. of duplicate transfections as a function of the amount (in ng) of TGIF expression vector transfected.
To this point, we have shown that TGIF HPE mutants retain the ability to interact with transcriptional co-regulators. To test whether TGIF HPE mutants retain the ability to repress transcription, luciferase assays in HepG2 cells using a TGFβ-responsive transcriptional reporter in the presence of TGFβ were performed (Fig. 4B, C and D). With increasing amounts of transfected TGIF wildtype, increasing repression of TGFβ-activated transcription of the reporter was observed. p.K259fs repressed TGFβ-activated transcription, but not as well as wildtype. p.P63A, p.P63R, and p.R90C do not repress TGFβ-activated transcription at all in this assay (Figure 3B). In the case of p.R90C, this may be due to a lack of recruitment to the reporter due to decreased interaction with the Smads (Fig. 4A). The three HPE truncation mutations of TGIF did not significantly repress TGFβ-activated transcription (Fig. 4C). Loss of CtBP interaction due to the p.S28C mutation decreases the ability of TGIF to repress TGFβ-activated transcription, as previously shown (Fig. 3A). p.H76Q and p.Q107L do not affect transcriptional repression by TGIF in this assay (Fig. 4D).
TGIF HPE mutants interact with RXRα but affect repression of retinoic acid regulated transcription
TGIF interacts with nuclear receptor complexes through interaction of the TGIF homeodomain with the ligand-binding domain of RXRα. It has recently been shown that as a result of this interaction, TGIF represses retinoic acid-regulated transcription, predominantly in the absence of retinoic acid [26]. Exposure of cell cultures to 9-cis retinoic acid (9C-RA) for 24 hours prior to analysis favors the disassociation of TGIF-RXRα repressor complexes and permits nearly full reporter activity. TGIF HPE mutants were first assayed for interaction with RXRα by immunoprecipitation followed by western blot. The three HPE truncation mutations of TGIF do not contain much of the homeodomain and were therefore presumed negative for RXRα interaction and not assayed. The remaining HPE mutants interact with RXRα, although p.R90C interacts only weakly (Fig. 5A).
Fig. 5.
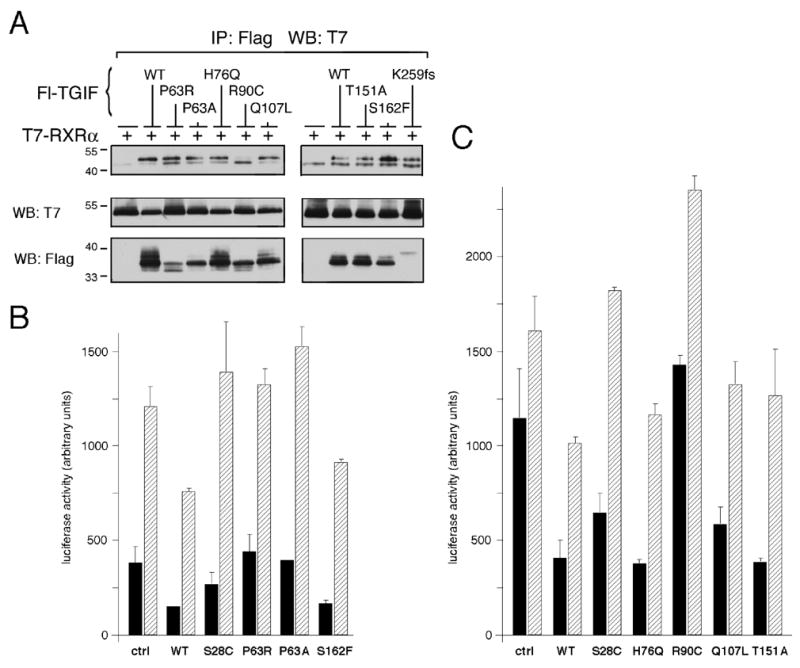
TGIF HPE mutants interact with RXRα and affect repression of retinoic acid regulated transcription. (A) COS1 cells were transfected with T7-RXRα and Flag-TGIF wildtype or HPE mutant construct, as indicated. Protein complexes in lysates were isolated on anti-Flag agarose and analyzed by western blot for the presence of co-precipitating RXRα. Coprecipitating RXR is indicated with an arrow, the Ig heavy chain with a bar. A portion of the lysates was subjected to direct western blot analysis to monitor protein expression (below). (B and C) HepG2 cells were transfected with a DR1-TATA-luc luciferase reporter and RXRα (control and experimental), together with the indicated TGIF wildtype or HPE mutant expression constructs in the experimental lanes. Cells were either left untreated (black bars) or treated with 9-cis-retinoic acid (9C-RA) for 24 hours prior to analysis (striped bars). Reporter luciferase activity (normalized to Renilla luciferase activity) was assayed 40 hours after transfection and is presented, in arbitrary units, as the mean +/− s.d., of duplicate transfections.
Since TGIF HPE mutations do not affect interaction with nuclear receptors via RXRα, we next performed luciferase assays to determine whether HPE mutants repress retinoic acid regulated transcription. TGIF wildtype represses transcriptional activity from a DR1-TATA-luc reporter in the absence of ligand and to a lesser extent in the presence of retinoic acid for 24 hours prior to assay (Fig. 5B and C). p.S28C does not repress well in the absence and not at all in the presence of ligand (Fig. 5B and C). p.P63R and p.P63A do not repress transcription from this reporter, despite the fact that these mutants interact with RXRα (Fig. 5B). This interaction may represent the small amount of these proteins which folds correctly, perhaps stabilized by the presence of overexpressed RXRα, however, it appears that this is not enough to significantly repress gene expression when expressed at lower levels. p.H76Q, p.Q107L, p.T151A, and p.S162F all repress to an extent comparable to that of TGIF wildtype both in the absence and presence of ligand (Fig. 5B and C). p.R90C does not repress transcription from a retinoic acid regulated reporter, possibly due to the weak interaction of this mutant with RXRα (Fig. 5C).
Shh/Tgif double knockout mice do not exhibit HPE
Previous studies had indicated that mice homozygous null for Tgif were viable, fertile and did not display HPE [25, 26, 52, 53]. However, studies of human HPE subjects had suggested a potential genetic interaction between TGIF and SHH [10, 11, 43]. To test whether these HPE genes interact genetically to cause HPE in mice, Shh heterozygotes were crossed with Tgif homozygous mutants to obtain Shh/Tgif double heterozygotes in a mixed C57BL6/J × 129Sv/J strain background. Shh/Tgif double heterozygotes were intercrossed and genotypes of the offspring at weaning, at postnatal day 21 (P21) were analyzed. Due to the fact that Shh homozygous mutants die in utero or perinatally, mice with these genotypes were not expected at P21. All other genotypes resulting from these crosses were observed at the expected frequencies, indicating that in a mixed strain background Shh/Tgif double knockouts are viable (Fig. 6A). Shh and Tgif heterozygotes were bred for six generations onto a pure C57BL6/J strain background, which is more sensitive to a variety of phenotypes. Knowing that Shh homozygous mutants have an HPE phenotype by 10.5 days postcoitum (dpc), Tgif heterozygotes were crossed to Shh/Tgif double heterozygotes and embryos were analyzed at 10.5 dpc for developmental defects. All genotypes were observed at the expected frequencies in these embryos (Fig. 6B), indicating that Shh/Tgif double knockouts are viable at this stage in a relatively pure background strain. Furthermore, very few embryos exhibited a phenotype at this stage and no embryos observed had an HPE phenotype (Fig. 6C). Some defects in growth or anterior development were observed in these embryos at a low frequency, although these defects appeared to correlate better with the presence of the Tgif mutation, than Shh (Fig. 6C–G). From this breeding data, it appears that Shh and Tgif null mutations do not cause a synthetic enhancement of HPE in mice (see also [25]).
Fig. 6.
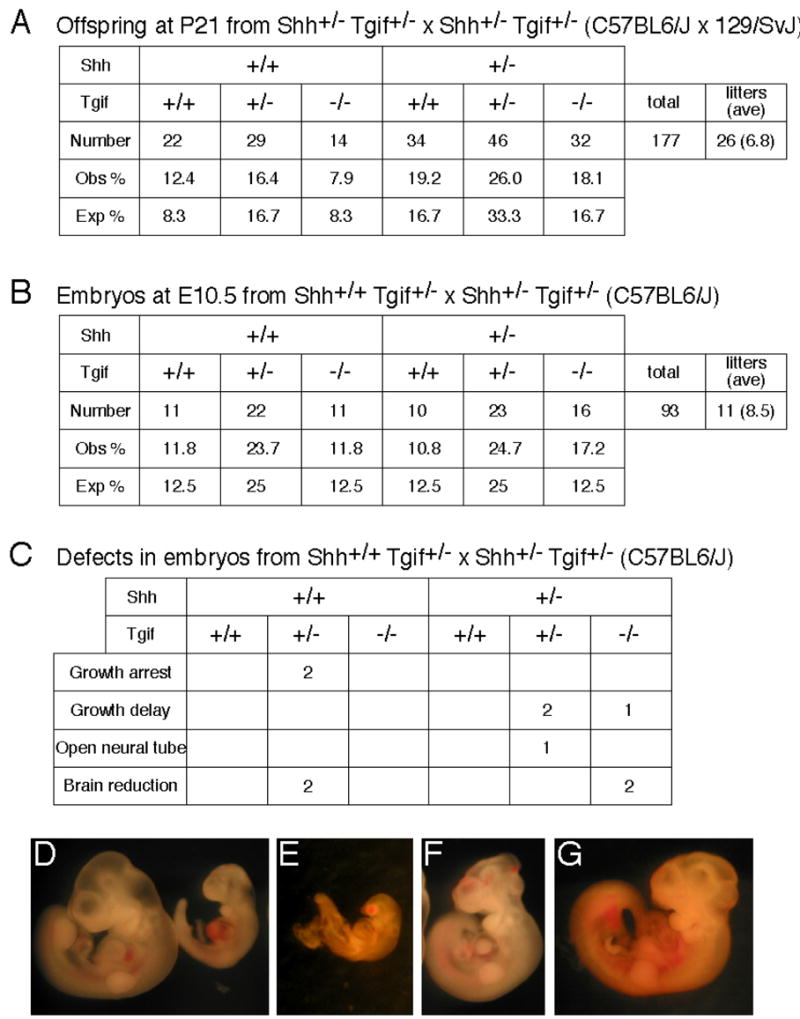
Analysis of Shh/Tgif double mutant mice. (A) Genotyping data at postnatal day 21 (P21) of offspring obtained from Shh/Tgif double heterozygote intercrosses in a mixed strain background. Numbers of mice with each genotype, the percentage of the total number of mice with each genotype observed, and the expected percentage of mice with each genotype, based on Mendelian ratios, are shown. (B) Genotyping data of embryos at 10.5 dpc obtained from Tgif heterozygotes crossed with Shh/Tgif double heterozygotes in a C57BL6/J strain background. The number of embryos with each genotype observed and the expected frequency of each genotype are shown, as in A. (C) The number of embryos of each genotype from the breeding in B with each type of observed defect is shown. (D–G) Examples of defective embryos. (D) Wildtype (left) 10.5 dpc embryo and a littermate with growth delay (right). (E) Growth arrested embryo. (F). Embryo with open neural tube defect. (G) Embryo with brain reduction.
Discussion
HPE is an extremely common developmental anomaly occurring in 1:250 pregnancies and 1:16,000 live-born infants [2,3,4]. Most cases are apparently sporadic and associated with de novo cytogenetic rearrangements in up to 50% of described cases [2,40]. Furthermore, recent studies demonstrate that a substantial fraction of apparently cytogenetically normal cases harbor occult microdeletions of HPE genes detected in live-born infants [21], and even more frequently in fetal samples [22]. After a decade of mutational analysis, only a quarter of HPE cases can be demonstrated to have mutations or deletions of an HPE-associated gene. When functional analysis has been performed [6, 7, 11, 15, 44, 46, 48, this study] the majority of these individual changes have been demonstrated to be pathogenic.
We now conclude that the majority of human TGIF mutations are also functionally abnormal. Four of these mutations are early trunctions or frame-shifts that might be predicted to be unstable, or non-functional, even if we did not know the physiological role associated with HPE. These results are also in substantial agreement with the results obtained using wild-type and a subset of mutant forms of TGIF to correct a newly described proliferative defect in Tgif null murine embryonic fibroblasts [53]. However, it should be emphasized that none of the described functions of Tgif have been definitively linked to a known pathogenetic mechanism leading to HPE. Therefore, further studies will be necessary to clarify the role of Tgif in mice, as well as TGIF in humans.
In three of our cases of TGIF mutations associated with HPE the genetic alteration also occurs de novo. Furthermore, the four additional mutations with a loss-of-function phenotype are family-specific changes seen exclusively in these particular HPE families, and not in controls or unrelated populations including other HPE families. This latter observation deserves particular emphasis, since it is typical of all pathogenic HPE mutations described to date [5–7, 11–16, 19–24, 41–46]. Given the decreased viability of patients with HPE, it is unlikely that a pathogenic HPE mutation would achieve genetic equilibrium in a population. Reproductive fitness is also compromised in HPE given the neurological burden of the extreme forms, suggesting that the clinical manifestations of parents with mutated alleles will be biased towards those with few HPE manifestations. Unlike recessive disorders (two deleterious mutations in the same gene and with common mutations accounting for a substantial fraction of disease alleles), HPE behaves as an apparent autosomal dominant disorder with reduced penetrance and variable expression, associated with novel, family-specific genetic alterations. Whether or not this reflects the “first hit” or the “second hit” is subject to debate. However, these pathological mutations do uncover a susceptibility for developing HPE, upon which other genetic or environmental factors can modify the spectrum of clinical manifestations.
The HPE4 locus, which includes the human TGIF gene, was originally defined by cytogenetic aberrations affecting 18p. However, hemizygosity for 18p- has a low penetrance of 10% [19,40]. This is in contrast to the nearly complete penetrance of hemizygosity for HPE2 (SIX3) and the ~50% penetrance of HPE3 (SHH) (see [2] for review; [14,19,40]). This low concordance HPE with del(18p) was originally interpreted as either the deletion of an autosomal dominant gene or the unmasking of a common recessive allele through hemizygosity. An alternative interpretation would be that the dysfunction of HPE4 is necessary, but not entirely sufficient to cause HPE.
Careful examination of mice with targeted disruption of the Tgif gene reveals that Tgif +/− mice are predisposed to malformations, even though the typical HPE phenotype is not reproduced by alterations of Tgif function alone. Rather, these mice are more susceptible to exposure to excess retinoic acid and through the loss of Tgif display dysregulation of their retinoid responses [26]. This difference between mice and humans with respect to susceptibility to HPE has never been adequately explained. Mice heterozygous for mutations in Shh, for example, are phenotypically normal; whereas heterozygous SHH mutations are the most common genetic changes associated with HPE in humans. Furthermore, mice homozygous null for Shh are clearly cyclopic; however, they also display limb and other severe malformations rarely encountered in humans (47). Mice with two pathogenic lesions in the same HPE gene, or within the same developmental program, are uniformly severely affected and lack the variability in the phenotype that the digenic model of HPE in humans is intended to explain. One possible explanation for these discrepancies is the tendency to deliberately control the environment of inbred strains of mice experimentally. It may be useful to use Tgif null mice (or other mouse models of HPE) to systematically evaluate the potential contributions of teratogens, drug ingestion, maternal cholesterol levels, alcohol exposure, maternal diabetes, or other epigenetic factors that are implicated in the causation of HPE in humans. Attempting to explain HPE entirely on the basis of genetics may be misguided.
Our analysis of the spectrum of mutations in the human TGIF gene indicates that pathologically significant variations are detected in the coding region that severely affect the stability and activity of the mutated alleles as assessed by a variety of functional assays. The frameshift and truncation variants, in particular, indicate that the likely mechanism is loss-of-function; there is currently no evidence for gain-of-function properties of these variants in co-transfection studies with mutant and wild-type TGIF constructs (D. Wotton, unpublished observations).
Essentially the same three categories of mutation: loss-of-function (e.g. p.P63R;) hypomorphic (e.g. p.S28C) and seemingly normal variants (e.g. p.T151A and p.S162F) were described in our original report (see Table 1). Additional apparently functionally normal variants are also detected in this expanded study. The p.Q107L variant was described in a mother of an HPE case who demonstrated microsigns consistent with the HPE spectrum; however, it could not be demonstrated that the proband also carried the mutation. We now can say that the p.Q107L variant is most likely an extremely rare polymorphism of uncertain significance. This highlights one of the challenges for genetic counseling in HPE. The detection of a rare, apparently family-specific, sequence change in an HPE-associated gene is consistent with, but not proof of, the pathogenic nature of the mutation. Despite the fact that this change occurs in a highly conserved domain of the TGIF-like proteins, predictions of functional significance based on these findings alone can lead to spurious conclusions. Similarly, the p.H76Q variant is within the conserved homeodomain between helix 2 and 3. This histidine residue is conserved among the TGIF-like family members, but not generally among all homeodomain proteins. Furthermore, it is seen in more than one HPE family, but with different flanking single nucleotide polymorphisms. Therefore, a structure-function interpolation is difficult to make in this case. Only a functional analysis reveals that the p.H76Q variation is largely normal in its activity. The apparent compound heterozygosity (p.S46fs and p.H76Q) is best interpreted as a rare coincidence, with the p.S46fs being the only allele that can be demonstrated to be clearly functionally abnormal. Interestingly, these same parents had a second at-risk pregnancy monitored by high-resolution ultrasound based on the family history of HPE. The structurally normal fetus was later shown to have inherited the p.H76Q allele. Furthermore, in the second patient with HPE, also with a p.H76Q variant, the cause of the disorder may be the diabetes mellitus present in the mother during the pregnancy. The importance of detailed functional analysis of detectable mutations from these examples is clear. It impacts directly on scientific interpretations of pathogenicity, inheritance modeling (monogenic vs. digenic), and genetic counseling.
We have suggested that HPE is an example of a condition in which multiple genetic and environmental influences can affect the severity of the phenotype and that multiple hits are required for severe manifestations of this disorder [10]. A strict digenic model predicts that HPE is observed only when there are two or more defects in the same, or intersecting, developmental program. However, there is little known about the potential genetic interactions between the set of HPE genes presently recognized. The “multiple hit” hypothesis, as proposed originally, is less strict and allows for environmental factors as well. Both models have the appeal that it could help to explain the wide variability in clinical manifestations in HPE families. However, pathologically significant HPE mutations are empirically relatively uncommon, despite the high incidence of HPE as a human malformation suggesting that a strictly digenic model for HPE is unlikely. Either there are a tremendous number of HPE-influencing genes, each with their own modest effect, or more likely there are relatively common genetic or environmental modifiers in the general population, such that the abrupt alteration of an HPE gene within a given family is sufficient to cause susceptibility to disease. It is interesting to note that one of the proposed genetic compound cases involves the p.T151A variant of TGIF ([11], and this study) and a second mutation in SHH. Given that the p.T151A variant cannot be convincingly demonstrated to be abnormal (and furthermore, any defect in SHH activity is yet to be proven) these apparent proof of principal cases need to be interpreted with caution.
The evidence that silent modifiers can have a dramatic effect on HPE manifestations has recently been reported in mice with targeted disruption of the Cdo (aka Cdon) gene [54]. In the 129/Sv background, these mice manifest microform HPE with a single central incisor; whereas, in the C57BL/6 strain they exhibit cebocephaly and semilobar HPE. Although the modifier responsible has yet to be identified, these authors argue that similar modifiers are likely to exist in human populations. Perhaps part of the discrepancy observed between humans and mice in sensitivity to reductions in TGIF may be explained by modifier loci that are presently unrecognized.
Our investigations of the role of TGIF in HPE are entirely consistent with reduced penetrance and/or variable expression in an autosomal dominant model of inheritance of relatively recent mutations in HPE susceptibility genes within affected families. Nevertheless, our studies do not preclude the action of additional genetic or environmental influences. Developmental anomalies, such as HPE, might be expected to display complex inheritance. The premise that there is no such thing as “simple” Mendelian inheritance is gaining increasing empirical support [8,9]. We would maintain that the potential complexity of developmental abnormalities could be expected to be even more intricate than a “simple” metabolic pathway. The developmental programs that establish the basic body plan depend on complex interactions between different tissues, and unfold in a coordinated sequence over time. Elegant studies in the developing chick demonstrate that virtually the entire spectrum of HPE phenotypes can be generated based on the timing of inhibition of the hedgehog signaling pathway with teratogens, such as cyclopamine [49]. Different aspects of the HPE brain and craniofacial malformations are sensitive to inhibition in different tissues and at different times. Therefore, it is not just the integrity of the signaling pathways themselves (influenced primarily by genetic variation) but the way the entire sequence of events play out during embryogenesis that is essential. Perhaps some of the species differences in phenotypes and observed gene dosage sensitivities may ultimately be attributed to differences in when and where the HPE genes are active and/or environmental and genetic modifiers. HPE is a default state of brain development; a large number of events need to occur, in a proper sequence, to modify this program leading to normal brain and craniofacial formation.
Materials and methods
Patient samples and mutational analysis
Genomic DNA was extracted from peripheral blood or transformed lymphoblastoid cell lines by standard methods. All patients with HPE and parents were recruited into an NHGRI IRB approved research protocol in accordance with their ethical guidelines and supervision.
PCR amplification, dHPLC analysis WAVE™ and WAVEMAKER™ (Transgenomic, Omaha, NE), amplicon purification Quiagen PCR purification kit (Quiagen, Valencia, CA), and DNA sequencing Big Dye™ version 3.1 terminator cycle sequencing on an ABI 3100 (Applied Biosystems, Foster City, CA) instrument were performed according to the manufactures instructions, essentially as previously described [50]. The reference sequence for the TGIF cDNA was NM_003244 (http://ncbi.nih.gov) corresponding to the most prevalent transcript (variant 4) and mutation nomenclature adopts the conventions used by this journal. The 272 aminoacid TGIF protein (NP_775300 isoform c) is the ortholog of the rat and murine proteins previously studied. Alternatively spliced variants are described in the most recent build of the UCSC genome browser (http://genome.ucsc.edu/); however, their functional significance has not been demonstrated.
Amplification of genomic DNA was performed in 35 μl reaction volumes, using 60–100 ng of genomic DNA, 200 μM dNTP, 20 pmol of each primer, 1× PCR buffer (Invitrogen, Carlsbad, CA), 0.5× enhancer (Invitrogen, Carlsbad, CA), 1.5 mM MgSO4 (Invitrogen, Carlsbad, CA), and 2.5 U of AmpliTaq (Applied Biosystems). All reactions were performed in a PTC-225 thermocycler (MJ Research, Waltham, MA). PCR cycling parameters were 95°C for 4 minutes, followed by 95°C for 30 sec, 56°C for 30 sec, 72°C for 1 min, for 50 cycles, with a final step of 72°C for 7 minutes. One half of the PCR reaction was used for dHPLC analysis and the remainder was stored for direct DNA sequencing.
Primers were designed for the putative promoter region of TGIF as previously defined [51]. The genomic sequence of TGIF is NT_010859. Amplification of TGIF was accomplished with six primer pairs: TGIF-A1 (5′GGCAGAGACGTTTAAAGAGC3′ and 5′CAACAGATGGAAAAAAGGACACC3′; 443 bp; promoter part 1); TGIF-A2 (5′AGAGCAGGGCCAGTAGAGTTC3′ and 5′AGGAGGGAAGGTACAGGAGG3′; 432 bp; promoter part 2); TGIF-A3 (5′CCGAGGGACGAGTGACAGCG3′ and 5′ACACAGGGGATAAGCGAACG3′; 415 bp; 5′UTR); TGIF-EX1 (5′ATCAGAGCGTCCTGTTTAGC3′ and 5′TTTCTTGACAACGTGCCAGC3′; 410 bp; TGIF Exon 1); TGIF-EX2 (5′CAATAGTTGCTGTGCTTATAAAGC3′ and 5′GAGTGGCAAGGAGCTTAATGA3′; 378 bp; TGIF Exon 2); In order to avoid the amplification of the TGIF pseudogene (LOC126052, chromosome 19q13.32) the entire exon 3 was amplified, and because its many polymorphisms, it was sequenced directly. TGIF-EX3 (5′ATTCTCAGAACCCGTTGGCTG3′ and 5′AATTCATCTCTTGCCTTCACC3′; 705 bp; TGIF Exon 3); TGIF-A7F 5′TGGCTCGTCCATCAGTGATC3′ and TGIF-A6R 5′ACTGGCAGAGAGAGAAAGGGAC3′ were used as TGIF Exon 3 internal sequencing primers.
Primers were also designed for the coding region of the human TGIF2 gene based on the genomic sequence NT_011362 on chromosome 20q. Both coding exons were examined using the primers TGIF2-A1 (5′GTACGTGCTAATGATGTTCCC3′ and 5′AGCTGGAATAGGACTAGAACC3′; 332 bp; TGIF2-Exon 2); TGIF2-A2 (5′GCCCATAGCTGTTTTAGATTAAGC3′ and 5′AAAACGGAAACCCAGGACAGCTG3′; 632 bp; TGIF2-Exon 3. The reference sequence for TGIF2 is NM_021809 that predicts two coding exons similar to TGIF: ie. “exon 2-like” and “exon 3-like” without a comparable exon 1. The strategy was similar to that of TGIF to avoid false amplification of the pseudogene on chromosome 1.
Plasmids
Expression constructs were prepared in pCMV5, the same wild-type TGIF construct previously reported, by site-directed mutagenesis (Transponics, York, PA). Successful introduction of sequence variations was confirmed by bi-directional sequencing. All experiments reported here use a TGIF construct tagged at the NH2-terminus with FLAG. His6-CtBP, Myc-Sin3, Smad3, and T7-RXRα expression constructs have been described previously. Creation of the retinoid-responsive pGL2-DR1-TATA-luc and the TGFβ-responsive 3TP-lux transcriptional reporters was described previously.
DNA pulldown assays
COS1 cells were maintained in DMEM with 10% BGS (Hyclone), and transfected using LipofectAmine (Invitrogen). 36 hours after transfection, cells were lysed by sonication in 50 mM HEPES, pH 7.4, 10% glycerol, 100 mM NaCl, 0.1% Tween-20 with protease and phosphatase inhibitors. Lysates were incubated at 4°C with double-stranded biotinylated oligonucleotide containing a TGIF consensus or mutated consensus binding site and poly(dI-dC).poly(dI-dC) competitor. Protein complexes bound to the labeled DNA were precipitated with Streptavidin-agarose (Novagen). Following SDS-polyacrylamide gel electrophoresis, proteins were electroblotted to Immobilon-P (Millipore) and incubated with antiserum specific for Flag (Sigma). Proteins were visualized with horseradish peroxidase-conjugated goat anti-mouse Ig (Pierce) and ECL (Amersham Pharmacia Biotech).
Immunoprecipitation and western blotting
COS1 cells were maintained and transfected as above. 36 hours after transfection, cells were lysed by sonication in 50 mM HEPES, pH 7.4, 10% glycerol, 100 mM NaCl, 0.1% Tween-20 with protease and phosphatase inhibitors. Immunocomplexes were precipitated with Flag M2-agarose (Sigma). Following SDS-polyacrylamide gel electrophoresis, proteins were electroblotted to Immobilon-P (Millipore) and incubated with antisera specific for Flag (Sigma), His6 (Covance), Myc (9E10), Smad2/3 (Upstate), or T7 (Novagen). Proteins were visualized with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit Ig (Pierce) and ECL (Amersham Pharmacia Biotech).
Luciferase assays
HepG2 cells were maintained in DMEM with 10% FBS, and transfected using Exgen 500 (MBI Fermentas) according to the manufacturer’s instructions. Cells were transfected with 3TP-lux or DR1-TATA-luc reporter, phCMVRLuc, and the indicated TGIF expression constructs. Cells were treated either with 100 pM TGFβ or 10−8 M 9cis-retinoic acid, as appropriate. 48 hours after transfection, firefly luciferase activity was assayed using a luciferase assay kit (Promega) and Renilla luciferase was assayed with 0.09 μM coelenterazine (Biosynth), using a Berthold LB953 luminometer.
Shh/Tgif double knockout mice
Mice with both alleles of exon 2 of the Sonic hedgehog (Shh) locus flanked by LoxP sites, obtained from Jackson Labs (129-Shhtm2Amc/J; (ref Andrew McMahon), were bred to an Adenovirus EIIa-Cre transgenic mouse (Tg(EIIa-cre)C5379Lmgd; Jackson Labs) so that the Shh coding sequence was recombined out at the 2 cell zygote stage, resulting in offspring heterozygous for a Shh null allele in all cell types. Shh heterozygotes were crossed with mice homozygous for a Tgif null allele on a mixed C57BL6/J × 129/SvJ strain background. Mice doubly heterozygous for Shh/Tgif null alleles were intercrossed and the genotypes of offspring were analyzed upon weaning at P21. Separately, Shh heterozygotes were backcrossed with pure C57BL6/J mice for 6 generations then crossed with mice homozygous null for Tgif on a C57BL6/J N6 background. Shh/Tgif C57BL6/J double heterozygotes were crossed with Tgif heterozygotes and embryos were analyzed at 10.5 dpc for developmental defects. Genomic tail or yolk sac DNA was purified by HotShot, or using a Promega Wizard purification kit, and was analyzed by PCR using primers specific for wildtype and mutant alleles of Shh and Tgif. Procedures were approved by the Animal Care and Use Committee of the University of Virginia.
Acknowledgments
We are indebted to the families who participated in this research, and the support by the DIR, NHGRI (K. B. E.-J., F.L., E.R. and M.M.), the Fogarty Training Grant DE43TW05503 (supporting, in part, K. B. E.-J.), and to the March of Dimes (grant number 6-FY04-77, to D.W.). We thank Mauricio Arcos-Burgos and Sabina Domene for the statistical analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Muenke M, Beachy PA. Genetics of ventral forebrain development and holoprosencephaly. Cur Opin Genet Dev. 2000;10:262–269. doi: 10.1016/s0959-437x(00)00084-8. [DOI] [PubMed] [Google Scholar]
- 2.Muenke M, Beachy PA. Holoprosencephaly. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Disease. 8. Vol. 4. McGraw-Hill; New York, NY: 2001. pp. 6203–6230. [Google Scholar]
- 3.Ming JE, Muenke M. From Homer to hedgehog. Clin Genet. 1998;53:155–163. doi: 10.1111/j.1399-0004.1998.tb02666.x. [DOI] [PubMed] [Google Scholar]
- 4.Cohen M. Perspectives on holoprosencephaly. Part 1. Epidemiology, genetics, and syndromology. Am J Medical Genet. 1989;40:211–235. doi: 10.1002/tera.1420400304. [DOI] [PubMed] [Google Scholar]
- 5.Brown SA, Warburton D, Brown LY, Yu C-y, Roeder ER, Stengel-Rutkowski S, Hennekam RCM, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–183. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- 6.Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gilessen-Kaesbach G, Roeder E, Ruiz i Altaba A, Muenke M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephly-like features. Proc Natl Acad Sci, USA. 2003;100:13424–13429. doi: 10.1073/pnas.2235734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roessler E, Ermilov AN, Grange DK, Wang A, Grachtchouk M, Dluglosz AA, Muenke M. A previously unidentified amino terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum Mol Genet. 2005;14:2181–2188. doi: 10.1093/hmg/ddi222. [DOI] [PubMed] [Google Scholar]
- 8.Dipple KM, McCabe ERB. Phenotypes of patients with “simple” Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet. 2000;66:1729–1735. doi: 10.1086/302938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dipple KM, McCabe ERB. Modifier genes convert “simple” mendelian disorder to complex traits. Mol Genet Metabol. 2000;71:43–50. doi: 10.1006/mgme.2000.3052. [DOI] [PubMed] [Google Scholar]
- 10.Ming JE, Muenke M. Multiple hits during early embryonic development; digenic diseases and holoprosencephly. Am J Hum Genet. 2002;71:1017–1032. doi: 10.1086/344412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richeri-Costa A, Zackai EH, Massague J, Muenke M, Elledge SJ. Mutations in TGIF cause holoprosencephaly and link NODAL signaling to human neural axis determination. Nat Genet. 2000;25:205–208. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- 12.Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, Mitchell HF, Donis-Keller H, Helms C, Hing AV, Heng HHQ, Koop B, Martindale D, Rommens JM, Tsui LC, Scherer SW. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet. 1996;14:353–356. doi: 10.1038/ng1196-353. [DOI] [PubMed] [Google Scholar]
- 13.Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- 14.Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire T, Richieri-Costa A, Gillessen-Kaesbach G, Zackai EH, Rommens J, Muenke M. Missense mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22:196–198. doi: 10.1038/9718. [DOI] [PubMed] [Google Scholar]
- 15.de la Cruz J, Bamford RN, Burdine RD, Roessler E, Barkovich AJ, Donnai D, Schier AF, Muenke M. A loss of function mutation in the CFC domain of TDGF-1 is associated with human forebrain defects. Hum Genet. 2002;110:422–428. doi: 10.1007/s00439-002-0709-3. [DOI] [PubMed] [Google Scholar]
- 16.Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, Stratton E, Bale SJ, Muenke M. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet. 2002;110:297–301. doi: 10.1007/s00439-002-0695-5. [DOI] [PubMed] [Google Scholar]
- 17.Burglin TR. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucl Acids Res. 1997;25:4173–4180. doi: 10.1093/nar/25.21.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frezal J, Schinzel A. Report of the committee on clinical disorders, chromosome aberrations, and uniparental disomy. Cyto Cell Genet. 1991;58:986–1052. [Google Scholar]
- 19.Overhauser J, Mitchell HF, Zackai EH, Tick DB, Rojas K, Muenke M. Physical mapping of the holoprosencephaly critical region in 18p11.3. Am J Hum Genet. 1995;57:1080–1085. [PMC free article] [PubMed] [Google Scholar]
- 20.Chen CP, Chern SR, Wang W, Lee CC, Chen WL, Chen LF, Chang TY, Tzen TYCY. Prenatal diagnosis of partial monosomy 18p (18p11.2 to pter) and trisomy 21q (21q22.3 to qter) with alobar holoprosencephaly and premaxillary agenesis. Prenat Diagn. 2001;21:346–350. doi: 10.1002/pd.63. [DOI] [PubMed] [Google Scholar]
- 21.Bendavid C, Haddad BR, Griffin A, Huizing M, Dubourg C, Gicquel I, Cavalli LR, Pasquier L, Long R, Ouspenskaia M, Odent S, Lacbawan F, David V, Muenke M. Multicolor FISH and quantitative PCR can detect submicroscopic deletions in holoprosencephaly patients with a normal karyotype. J Med Genet. 2005;43:496–500. doi: 10.1136/jmg.2005.037176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bendavid C, Dubourg C, Gicquel I, Pasquier L, Saugler-Veber P, Durou MR, Jaillard S, Frebourg T, Haddad BR, Henry C, Odent S, David V. Molecular evaluation of foetuses with holoprosencephaly shows high incidence of microdeletions in the HPE genes. Hum Genet. 2005;119:1–8. doi: 10.1007/s00439-005-0097-6. [DOI] [PubMed] [Google Scholar]
- 23.Aguilella C, Dubourg C, Attia-Sobol J, Vigneron J, Blayau M, Pasquier L, Lazaro L, Odent S, David V. Molecular screening of the TGIF gene in holoprosencephly: identification of two novel mutations. Hum Genet. 2003;112:131–134. doi: 10.1007/s00439-002-0862-8. [DOI] [PubMed] [Google Scholar]
- 24.Chen CP, Chern SR, Du SH, Wang W. Molecular diagnosis of a novel heterozygous 268C>T (R90C) mutation in the TGIF gene in a fetus with holoprosencephaly and premaxillary agenesis. Prenat Diagn. 2002;22:5–7. doi: 10.1002/pd.202. [DOI] [PubMed] [Google Scholar]
- 25.Shen J, Walsh JCA. Targeted disruption of Tgif, the mouse ortholog of a human holoprosencephaly gene, does not result in holoprosencephaly in mice. Mol Cell Biol. 2005;25:3639–3647. doi: 10.1128/MCB.25.9.3639-3647.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartholin L, Powers SE, Melhuish TA, Lasse S, Weinstein M, Wotton D. TGIF inhibits retinoid signaling. Mol Cell Biol. 2005;26:990–1001. doi: 10.1128/MCB.26.3.990-1001.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melhuish TA, Gallo CM, Wotton D. TGIF2 interacts with histone deacetylase 1 and represses transcription. J Biol Chem. 2001;276:32109–32114. doi: 10.1074/jbc.M103377200. [DOI] [PubMed] [Google Scholar]
- 28.Melhuish TA, Wotton D. A retained intron in the Tgif2 gene. BMC Mol Biol. 2006;7:2. doi: 10.1186/1471-2199-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin L, Zhou Y, Kuang C, Lin L, Chen Y. Expression pattern of TG-interacting factor 2 during mouse development. Gene Exp Patterns. 2005;5:457–462. doi: 10.1016/j.modgep.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 30.Bertolino E, Reimund B, Wildt-Perinic D, Clerc RG. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif. J Biol Chem. 1995;270:31178–31188. doi: 10.1074/jbc.270.52.31178. [DOI] [PubMed] [Google Scholar]
- 31.Bertolino E, Wildt S, Richards G, Clerc RG. Expression of a novel murine homeobox gene in the developing cerebellar external granular layer during its proliferation. Dev Dyn. 1996;205:410–420. doi: 10.1002/(SICI)1097-0177(199604)205:4<410::AID-AJA5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 32.Wotton D, Lo RS, Lee S, Massague J. A Smad transcriptional corepressor. Cell. 1999;97:29–39. doi: 10.1016/s0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- 33.Wotton D, Lo RS, Swaby LA, Massague J. Multiple modes of repression by the Smad transcriptional corepressor TGIF. J Biol Chem. 1999;274:37105–37110. doi: 10.1074/jbc.274.52.37105. [DOI] [PubMed] [Google Scholar]
- 34.Lo RS, Wottton D, Massague J. Epidermal growth factor signaling via Ras controls the Smad transcriptional co-repressor TGIF. EMBO J. 2001;20:128–136. doi: 10.1093/emboj/20.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wotton D, Knoepfler PS, Laherty CD, Eisenman RN, Massague J. The Smad transcriptional corepressor TGIF recruits mSin3. Cell Growth Diff. 2001;12:457–463. [PubMed] [Google Scholar]
- 36.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 37.Scavello GS, Paluru PC, Ganter WR, Young TL. Sequence variants in the transforming growth β-induced factor (TGIF) gene are not associated with high myopia. Invest Ophthal Vis Sci. 2004;45:2091–2097. doi: 10.1167/iovs.03-0933. [DOI] [PubMed] [Google Scholar]
- 38.Lam DSC, Lee WS, Leung YF, Tam POS, Fan DSP, Fan BJ, Pang CP. TGFβ-induced factor; a candidate gene for high myopia. Invest Ophthal Vis Sci. 2003;44:1012–1015. doi: 10.1167/iovs.02-0058. [DOI] [PubMed] [Google Scholar]
- 39.Melhuish TA, Wotton D. The interaction of the carboxyl terminus-binding protein with the Smad corepressor TGIF is disrupted by a holoprosencephaly mutation in TGIF. J Biol Chem. 2000;275:39762–39766. doi: 10.1074/jbc.C000416200. [DOI] [PubMed] [Google Scholar]
- 40.Schinzel A. In: Catalog of unbalanced chromosome aberrations in man. De Gruyter, editor. Berlin; New York: 1984. pp. 604–609. [Google Scholar]
- 41.Roessler E, Ward DE, Gaudenz K, Belloni E, Scherer SW, Donnai D, Siegel-Bartelt J, Tsui LC, Muenke M. Cytogenetic rearrangements involving the loss of the Sonic Hedgehog gene at 7q36 cause holoprosencephaly. Hum Genet. 1997;100:172–181. doi: 10.1007/s004390050486. [DOI] [PubMed] [Google Scholar]
- 42.Roessler E, Belloni E, Gaudenz K, Vargas F, Scherer SW, Tsui LC, Muenke M. Mutations in the carboxy-terminal domain of Sonic Hedgehog cause holoprosencephaly. Hum Mol Genet. 1997;6:1847–1853. doi: 10.1093/hmg/6.11.1847. [DOI] [PubMed] [Google Scholar]
- 43.Nanni L, Ming JE, Bocian M, Steinhaus K, Bianchi DW, de Die-Smulders C, Giannotti A, Imaizumi K, Jones KL, Del Campo M, Martin RA, Meinecke P, Pierpont MEM, Robin NH, Young ID, Roessler E, Muenke M. The mutational spectrum of the Sonic Hedgehog gene in holoprosencephly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet. 1999;8:2479–2488. doi: 10.1093/hmg/8.13.2479. [DOI] [PubMed] [Google Scholar]
- 44.Traiffort E, Dubourg C, Faure H, Rognan D, Odent S, Durou MR, David V, Ruat M. Functional characterization of Sonic Hedgehog mutations associated with holoprosencephaly. J Biol Chem. 2004;279:42889–42897. doi: 10.1074/jbc.M405161200. [DOI] [PubMed] [Google Scholar]
- 45.Marini M, Cusano R, De Biasio P, Caroli F, Lerone M, Silengo M, Ravazzolo R, Seri M, Camera G. Previously undescribed nonsense mutation in SHH caused autosomal dominant holorosencephaly with wide intrafamilial variability. Am J Med Genet. 2003;117A:112–115. doi: 10.1002/ajmg.a.10163. [DOI] [PubMed] [Google Scholar]
- 46.Brown L, Paraso M, Arkell R, Brown SA. In vitro analysis of partial loss-of-function ZIC2 mutations in holoprosencephaly: alanine tract expansion modulates DNA binding and transactivation. Hum Mol Genet. 2005;14:411–420. doi: 10.1093/hmg/ddi037. [DOI] [PubMed] [Google Scholar]
- 47.Chiang C, Litingtung Y, Lee E, Young K, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 48.Maity T, Fuse N, Beachy PA. Molecular mechanisms of Sonic hedgehog mutant effects in holoprosencephaly. Proc Natl Acad Sci, USA. 2005;102:17026–17031. doi: 10.1073/pnas.0507848102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cordero D, Marcucio R, Hu D, Gaffield W, Tapadia M, Helms JA. Temporal perturbations in sonic hedgehog signaling elicit the spectrum of holoprosencephaly phenotypes. J Clin Invest. 2004;114:485–494. doi: 10.1172/JCI19596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schimmenti LA, de la Cruz J, Lewis RA, Karkera JD, Manligas GS, Roessler E, Muenke MM. Novel mutation in Sonic Hedgehog in non-syndromic colobomatous micropthalmia. Am J Med Genet. 2003;116A:215–221. doi: 10.1002/ajmg.a.10884. [DOI] [PubMed] [Google Scholar]
- 51.Chen F, Ogawa K, Nagarajan RP, Zhang M, Kuang C, Chen CY. Regulation of TG-interacting factor by transforming growth factor-β. Biochem J. 2003;371:257–263. doi: 10.1042/BJ20030095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin JZ, McKinney P, Ding J. Expression and functional analysis of Tgif during mouse midline development. Dev Dyn. 2006;235:547–553. doi: 10.1002/dvdy.20642. [DOI] [PubMed] [Google Scholar]
- 53.Mar L, Hoodless PA. Embryonic fibroblasts from mice lacking Tgif were defective in cell cycling. Mol Cell Biol. 2006;26:4302–4310. doi: 10.1128/MCB.02156-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zang W, Kang J-S, Cole F, Jeong M, Krauss RS. Cdo functions at multiple points in the Sonic hedgehog pathway, and Cdo-deficient mice accurately model human holoprosencephly. Dev Cell. 2006;10:657–665. doi: 10.1016/j.devcel.2006.04.005. [DOI] [PubMed] [Google Scholar]