

Abstract
We have previously reported that group V secretory phospholipase A2 (sPLA2) amplifies the action of cytosolic phospholipase A2 (cPLA2) α in regulating eicosanoid biosynthesis by mouse peritoneal macrophages stimulated with zymosan (Satake, Y., Diaz, B. L., Balestrieri, B., Lam, B. K., Kanaoka, Y., Grusby, M. J., and Arm, J. P. (2004) J. Biol. Chem. 279, 16488–16494). To further understand the role of group V sPLA2, we studied its localization in resting mouse peritoneal macrophages before and after stimulation with zymosan and the effect of deletion of the gene encoding group V sPLA2 on phagocytosis of zymosan. We report that group V sPLA2 is present in the Golgi apparatus and recycling endosome in the juxtanuclear region of resting peritoneal macrophages. Upon ingestion of zymosan by mouse peritoneal macrophages, group V sPLA2 is recruited to the phagosome. There it co-localizes with cPLA2α, 5-lipoxygenase, 5-lipoxygenase-activating protein, and leukotriene C4 synthase. Using immunostaining for the cysteinyl leukotrienes in carbodiimide-fixed cells, we show, for the first time, that the phagosome is a site of cysteinyl leukotriene formation. Furthermore, peritoneal macrophages from group V sPLA2-null mice demonstrated a >50% attenuation in phagocytosis of zymosan particles, which was restored by adenoviral expression of group V sPLA2 but not group IIA sPLA2. These data demonstrate that group V sPLA2 contributes to the innate immune response both through regulation of eicosanoid generation in response to a phagocytic stimulus and also as a component of the phagocytic machinery.
Phospholipase A2 (PLA2)2 hydrolyzes the sn-2 position of cell membrane phospholipids to release free fatty acids and lysophospholipids. Where the fatty acid is arachidonic acid, it serves as the precursor for biosynthesis of leukotrienes and prostaglandins (1). Free fatty acids also may signal through peroxisome proliferator-activated receptors (2), and lysophospholipids activate cell surface G protein-coupled receptors (3). The family of PLA2 enzymes includes the cytosolic PLA2 (cPLA2) isoforms that utilize a catalytic serine (4, 5). cPLA2α is critical for the release of arachidonic acid for leukotriene and prostaglandin generation (6, 7). The calcium-independent isoforms of PLA2, which also use a catalytic serine, have been implicated in cell membrane remodeling (8), spermatogenesis (9), and regulation of store-operated calcium channels (10). There are 10 mammalian secretory PLA2 enzymes (11–14) that use a catalytic histidine and that have been implicated in amplifying cPLA2α-dependent eicosanoid generation (15–17) and in regulating secretory granule exocytosis (18). In certain circumstances, sPLA2 enzymes may regulate eicosanoid generation independently from cPLA2α (19). Certain sPLA2 isoforms may act as ligands for cell surface receptors (20). However, the roles of the sPLA2 enzymes have largely been inferred from their biochemical properties, the effects of their overexpression in transfected cells, or the use of pharmacologic inhibitors. To elucidate the biological functions of one of the sPLA2 enzymes, group V sPLA2, we generated mice in which the gene encoding this sPLA2 was deleted by homologous recombination (15).
Macrophages contribute to the innate immune response to invading microorganisms by engulfing them through the process of phagocytosis (21). Subsequent fusion of lysosomes with the phagosome to form phagolysosomes and generation of reactive oxygen species leads to killing of the ingested pathogen (22). The process of phagocytosis in mouse peritoneal macrophages results in the formation of leukotriene (LT) C4 and prostaglandin E2 that initiate and amplify the ensuing inflammatory response (23–26). The synthesis of eicosanoids in response to phagocytosis of zymosan requires the provision of the substrate, arachidonic acid, by cPLA2α (27). Mouse peritoneal macrophages derived from group V sPLA2-null mice have a 50% attenuation of eicosanoid generation in response to zymosan compared with wild-type macrophages, consistent with a role for group V sPLA2 in amplifying the action of cPLA2α (15). Further, the early phase of plasma extravasation in response to zymosan, which is dependent on cysteinyl leukotrienes acting at the type 1 CysLT receptor, is markedly attenuated in Group V sPLA2-null mice (15). Thus, group V sPLA2 contributes to the innate immune response through the regulation of eicosanoid generation in response to a phagocytic stimulus, thereby regulating the ensuing inflammatory response. We now report that group V sPLA2 translocates to the phagosome upon zymosan stimulation of mouse peritoneal macrophages where it co-localizes with the other enzymes of leukotriene biosynthesis. We further show that group V sPLA2 regulates phagocytosis of zymosan independent of its effects on eicosanoid generation.
EXPERIMENTAL PROCEDURES
Materials
Zymosan A, paraformaldehyde, 1-ethyl-3-(3-dimethylamino-propyl) carbodiimide (EDAC), bovine serum albumin, and saponin were from Sigma. The 5-lipoxygenase inhibitor, AA861, and the FLAP antagonist, MK886, were from Biomol (Plymouth Meeting, PA). The following antibodies were used: affinity-purified rabbit polyclonal antibody directed to group V sPLA2 (28), mouse monoclonal IgG1 antibodies against GS28 and Syntaxin 6 (Stressgen, Victoria, Canada), mouse monoclonal IgG1 against the transferrin receptor (Zymed Laboratories Inc., San Francisco, CA), rat monoclonal IgG2a against lysosome-associated membrane protein (LAMP) 1 (BD Pharmingen, San Jose, CA), rat monoclonal IgG1 to the cysteinyl leukotrienes (clone 6E7; Sigma), rabbit polyclonal antibody against cPLA2 (29), rabbit polyclonal antibodies against FLAP and 5-LO (Dr. J. Evans, Merck Research Laboratories, Rahway, NJ), rabbit polyclonal antibody against LTC4 synthase (Dr. Bing Lam, Brigham and Women’s Hospital, Boston, MA). Rabbit polyclonal antibodies were cleared of cross-reactivity to yeast by incubating with zymosan overnight at 4 °C. FITC-conjugated donkey anti-rabbit IgG, Cy3-conjugated donkey anti-mouse IgG, Cy3-conjugated donkey anti-rat IgG (each against heavy and light chains with minimal cross-reactivity to other species), and normal donkey serum were from Jackson ImmunoResearch (West Grove, PA). Group V sPLA2-null mice (15) and cPLA2α-null mice (7) have been described. LTC4 synthase-null mice were from Dr. Bing Lam (24). Adenoviral constructs carrying the cDNAs encoding group V sPLA2 and group IIA sPLA2 were previously described (16). Each viral construct carried the cDNA encoding the sPLA2 in tandem with a cassette encoding green fluorescent protein (GFP); the control adenovirus expressed the Escherichia coli LacZ gene, encoding β-galactosidase, in tandem with GFP.
Cell Culture
The peritoneal cavities of mice were flushed with 5 ml of ice-cold PCG buffer (25 mM Pipes, 110 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 g/liter glucose, pH 7.4). After washing, 3.3 × 105 cells were plated on sterile glass coverslips in 24-well tissue culture plates in 500 μl of PCG buffer containing 10% fetal bovine serum, 2 mM L -glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin and incubated overnight at 37 °C with 5% CO2. Nonadherent cells were removed by washing three times with PCG buffer.
Synchronized Phagocytosis
After removing nonadherent cells, the macrophages were incubated for another hour at 37 °C. The cells were then washed once with cold PCG buffer and cooled on ice for 5 min before the addition of zymosan (10 particles/cell) for 15 min on ice. The cells were then warmed by adding warm PCG buffer containing 10% fetal bovine serum, 2 mM L -glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin and incubated at 37 °C for the indicated period of time (30). In all of the experiments, nonopsonized zymosan was used.
Immunofluorescence Analysis
Macrophages were fixed with 2% paraformaldehyde in phosphate-buffered saline for 15 min at room temperature, washed once in Hanks’ balanced salt solution without Mg2+ or Ca2+ containing 0.1% bovine serum albumin (HBA), permeabilized with 0.025% saponin in phosphate-buffered saline for 10 min at room temperature, and washed twice with HBA. The cells were then blocked in HBA containing 5% normal donkey serum (blocking buffer) for 1 h at room temperature. The macrophages were incubated with 5 μg/ml rabbit polyclonal antibody to group V sPLA2 (28) and mouse mAb GS28 (2 μg/ml), mouse mAb against Syntaxin 6 (5 μg/ml), mouse mAb against the transferrin receptor (2.5 μg/ml), or rat mAb against LAMP-1 (2.5 μg/ml) in blocking buffer for 2 h at room temperature. Negative control cells were instead incubated for 2 h with rabbit IgG, mouse IgG, or rat IgG (Jackson Immunoresearch) in place of specific primary antibodies. After 2 h, the cells were washed extensively with HBA and incubated for 1 h at room temperature with FITC-conjugated donkey anti-rabbit IgG (1:400) and Cy3-conjugated donkey anti-mouse IgG (1:200) or Cy3-conjugated donkey anti-rat IgG (1:200). The cells were washed five times with HBA, mounted in Vectashield™ mounting medium (Vector Laboratories, Burlingame, CA), and imaged using a Nikon C1 confocal system combined with an Eclipse TE2000-U inverted microscope with 60× oil PlanApo NA 1.4 objective. Conventional Nomarski differential contrast images were taken using a SPOT-RT digital camera. The data were analyzed by Photoshop image software. To determine the localization of the enzymes of leukotriene biosynthesis after phagocytosis, macrophages were fixed and permeabilized as described above before and after incubation with zymosan and incubated with antibodies directed to group V sPLA2, cPLA2α, 5-lipoxygenase, FLAP, or LTC4 synthase. After washing, the cells were incubated with FITC- or Cy3-conjugated donkey anti-rabbit IgG (1:400) and imaged using a Nikon Eclipse TE2000-U microscope coupled with a Spot-RT Digital Camera.
Intracellular Localization of LTC4 after Zymosan Phagocytosis
To detect CysLTs at the site of their generation, we used a modification of a previous protocol (31). Briefly, macrophages were activated with zymosan as described above. At the end of the incubation, the medium was removed, and the cells were quickly fixed and permeabilized with a solution of 0.25% EDAC, 2% paraformaldehyde for 15 min at room temperature. The cells were washed three times with HBA, incubated for 2 h at room temperature with a rat mAb anti-Cys-LTs (1:100) or a rat IgG1 (BD Pharmingen, San Diego, CA) in blocking buffer, washed in HBA five times, and then incubated with Cy3-conjugated donkey anti-rat IgG (1:200) in blocking buffer for 1 h and imaged by confocal microscopy. In selected experiments, before the addition of zymosan, the cells were incubated for 5 min with AA861 (10 μM ) or MK886 (10 μM ).
Measurement of Phagocytosis
Peritoneal macrophages were obtained from group V sPLA2-null mice and from their wild-type littermates. Macrophages were used from mice on a C57BL/6×129 Sv background, from mice bred to a BALB/c background for 3–10 generations and from mice bred to a C57BL/6 background for 10 generations. Time-dependent zymosan stimulation was followed by staining with Diff-Quick (Dade Behring Inc., Newark, DE) and visualization under the light microscope. The phagocytic index was calculated by dividing the number of phagosomes by the total number of cells in a field, which was multiplied by the percentage of phagocytosing cells (30).
Introduction of PLA2 Enzymes into Peritoneal Macrophages
Cells obtained from peritoneal lavage were incubated on glass coverslips with adenoviral vectors for 48 h in PCG buffer containing 2% fetal bovine serum, 2 mM L -glutamine, and 15% L cell-conditioned medium as a source of macrophage colony-stimulating factor. The efficiency of adenovirus-mediated gene transfer was assessed by the expression of GFP using fluorescence microscopy. Wells in which 60–70% of cells were GFP-positive were used for subsequent experiments in which cells were washed and stimulated with Zymosan (10 ppc), fixed with 2% paraformaldehyde, and imaged using fluorescence microscopy. The phagocytic index was calculated for green fluorescence protein-positive cells.
Statistical Analyses
We performed two-way analysis of variance using the Prism 4 software to compare phagocytosis between populations of peritoneal macrophages derived from gene-disrupted mice and their wild-type controls. Where statistical significance was apparent, we performed post-hoc analyses of individual time points using paired Student’s t test.
RESULTS
Group V sPLA2 Co-localizes with Markers of the Golgi Apparatus and the Recycling Endosome
We have previously demonstrated, using immunofluorescence microscopy with a specific antibody, that group V sPLA2 is present in the cytoplasm and in the juxtanuclear region of mouse peritoneal macrophages (15). The specificity of the staining was evident from the lack of any staining of peritoneal macrophages from group V sPLA2-null mice (15). To ascertain the nature of the juxtanuclear region where group V sPLA2 resides, we double-labeled macrophages with a rabbit polyclonal antibody specific to group V sPLA2 (15, 28) and with monoclonal antibodies specific for particular subcellular compartments. We found that group V sPLA2 partially co-localizes with a cis-Golgi marker, GS28 (Fig. 1A), and to a lesser extent with Syntaxin 6, a trans-Golgi network marker (Fig. 1B). In addition to the Golgi apparatus, the juxtanuclear region contains recycling endosomes, subcellular organelles that localize in close proximity to the nucleus whose function is to recycle protein to the cell surface. Dual label immunofluorescence of group V sPLA2 with the transferrin receptor, an endosomal marker, showed strong co-localization in the juxtanuclear region but not elsewhere in the cell (Fig. 1C). Group V sPLA2 did not co-localize with protein-disulfide isomerase, a marker of the endoplasmic reticulum (Fig. 1D). Negative control experiments revealed no cross-reactivity of donkey anti-rabbit IgG with mouse mAbs or of donkey anti-mouse IgG with rabbit IgG (data not shown).
FIGURE 1. Subcellular localization of group V sPLA2.
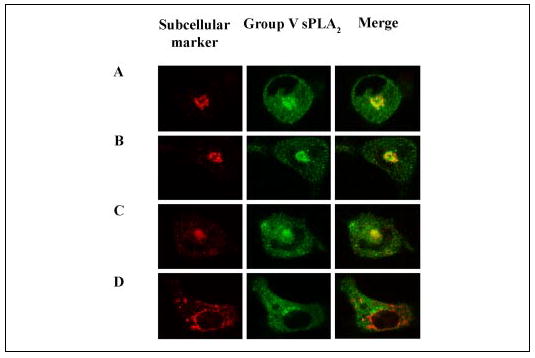
Peritoneal macrophages isolated from BALC/c mice were fixed with 2% paraformaldehyde, permeabilized with 0.025% saponin, and incubated with rabbit anti-group V sPLA2 and mAbs to GS28 (A), Syntaxin 6 (B), the transferrin receptor (C), or protein-disulfide isomerase (D). The cells were washed and incubated with FITC-conjugated donkey anti-rabbit IgG and Cy3-conjugated donkey anti-mouse IgG. Confocal images were obtained as described under “Experimental Procedures. ” Single color images are shown for the subcellular marker (Cy3, left panels) and group V sPLA2 (FITC, middle panels). The right panels are the merged images for both FITC and Cy3 staining.
Group V sPLA2 Translocates to the Phagosome during Phagocytosis
We have previously reported a role for group V sPLA2 in the generation of eicosanoids by mouse peritoneal macrophages after ingestion of zymosan (15). The correct temporal and spatial location of the enzymes of eicosanoid biosynthesis is critical for their function, yet little is known about the location of group V sPLA2 during eicosanoid biosynthesis. We therefore examined the location of endogenous group V sPLA2 in macrophages following ingestion of zymosan. Group V sPLA2 translocated from its juxtanuclear resting position to the forming phagosomes within 5 min of zymosan phagocytosis, localizing in the forming phagocytic cup (50% of cells) or completely surrounding the phagocytosed zymosan particles (Fig. 2A). By 15 min group V sPLA2 completely surrounded the phagocytosed particles, whereas juxtanuclear staining for group V sPLA2 was still visible in about 10% of the cells. At 30 min to 1 h, there was complete loss of juxtanuclear staining for group V sPLA2, which was most prominent surrounding peripheral phagosomes with loss of immunostaining around phagosomes that had moved away from the periphery. To confirm the localization of group V sPLA2 to phagosomes, we determined whether group V sPLA2 co-localized with LAMP-1, a lysosomal protein that moves to the maturing phagosome to form the phagolysosome (32) (Fig. 2B). In resting cells LAMP-1 had mainly lysosomal staining, as expected. By 30 min after zymosan stimulation, LAMP-1 co-localized with group V sPLA2 around ingested zymosan particles, consistent with the movement of group V sPLA2 to the phagosome.
FIGURE 2. Translocation of group V sPLA2 to the phagosome.
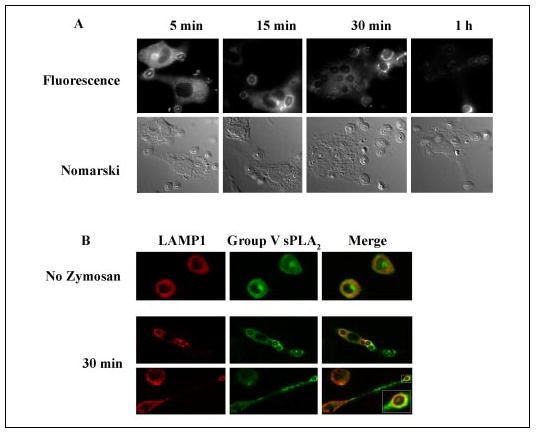
A, peritoneal macrophages isolated from BALB/c mice were stimulated with unopsonized zymosan (10 particles/cell) for 5, 15, 30, or 60 min, fixed in 2% paraformaldehyde, permeabilized with 0.025% saponin, and incubated with rabbit anti-group V sPLA2 followed by FITC-conjugated donkey anti-rabbit IgG. Immunofluorescence images are paired with the corresponding Nomarski image. B, peritoneal macrophages isolated from BALB/c mice were left unstimulated or were stimulated with unopsonized zymosan (10 particles/cell) for 30 min, fixed in 2% paraformaldehyde, permeabilized with 0.025% saponin, and incubated with rabbit anti-group V sPLA2 and mAb to LAMP-1. The cells were washed and incubated with FITC-conjugated donkey anti-rabbit IgG and Cy3-conjugated donkey anti-rat IgG. Confocal images were obtained as described under “Experimental Procedures. ” Autofluorescence of zymosan particles is visible within each phagosome.
Because group V sPLA2 amplifies cPLA2α-dependent eicosanoid biosynthesis in response to zymosan and because cPLA2α has been reported to translocate to the phagosome upon ingestion of zymosan, we determined whether other enzymes of eicosanoid biosynthesis also translocate to the phagosome (Fig. 3). Preliminary kinetic analyses of leukotriene biosynthesis revealed detectable leukotriene generation within 15 min, which peaked at 3 h as previously reported (15) (Fig. 3A). Extrapolation from these data indicated that the maximal rate of LTC4 biosynthesis occurs within the first hour of zymosan stimulation. Therefore, immunostaining for eicosanoid biosynthetic enzymes was performed at these early time points. Representative data are shown for 15 min and 1 h. 5-LO was present in both the cytoplasm and the nucleus of resting macrophages and was visible at the forming phagosome within 5 min. By 15 min the protein was also visible in a perinuclear location (Fig. 3B). At 60 min 5-LO was visualized mainly surrounding the most peripheral zymosan particles. FLAP localizes to the nuclear envelope of resting cells (33). 5 min after zymosan stimulation FLAP was prominently visible at the phagosome and was still evident in the perinuclear location where it remained visible at 15 and 30 min. Staining at the phagosome was most prominent in newly formed, peripheral phagosomes, whereas phagosomes further from the plasma membrane showed only faint and diffuse staining. Perinuclear staining for FLAP was indistinct by 60 m. LTC4S, an integral perinuclear membrane protein with homology to FLAP (34), translocated to the phagosome with kinetics that appeared slower compared with FLAP. Immunostaining for LTC4 was weakly visible around ~50% of the ingested particles at 5 min (data not shown), was clearly visible at 15 min, and increased to 100% of particles by 30–60 m. In contrast to FLAP, strong perinuclear immunostaining for LTC4 was visible at all time points studied (up to 60 min after zymosan stimulation).
FIGURE 3. The phagosome is a site of LTC4 biosynthesis.

A, kinetics of LTC4 generation by mouse peritoneal macrophages stimulated with zymosan (10 particles/cell). B, peritoneal macrophages isolated from BALB/c mice were stimulated with unopsonized zymosan (10 particles/cell) for 15 and 60 min, fixed in 2% paraformaldehyde, permeabilized with 0.025% saponin, and incubated with specific antibodies for each of the enzymes of LTC4 biosynthesis followed by FITC-conjugated secondary antibodies. C, to demonstrate LTC4 formation at the phagosome, the cells were fixed with EDAC and immunostained with rat mAb anti-Cys-LTs followed by Cy3-conjugated donkey anti-rat IgG. Immunofluorescence images are paired with the corresponding Nomarski or bright field image.
Immunodetection of LTC4 at the Phagosome
To definitively establish whether the phagosome is a site of LTC4 biosynthesis, we modified a previously published protocol (31) to visualize intracellular LTC4 at its site of formation (Fig. 3C). LTC4 is not detectable in any location in unstimulated cells, consistent with its formation de novo in response to an inflammatory stimulus (data not shown). Five min after zymosan phagocytosis, 20% of the cells showed perinuclear and/or periphagosome staining, indicating the presence of LTC4 in the perinuclear region and at the phagosome. Interestingly, at 15 min almost 100% of the phagocytosed particles demonstrated bright periphagosome staining, whereas perinuclear LTC4 was visible in some cells (Fig. 3C). By 60 min LTC4 was most prominent in the most peripheral, most recently formed phagosomes, and perinuclear staining was no longer present. The specificity of the staining for LTC4 was demonstrated by the lack of staining in macrophages isolated from LTC4 synthase-null mice and in wild-type macrophages pretreated for 5 min with AA861, a 5-LO inhibitor, or MK886, a FLAP inhibitor (data not shown).
Group V sPLA2-null Peritoneal Macrophages Are Defective in Phagocytosis
Because group V sPLA2 translocated to the forming phagosome, we quantified phagocytosis in group V sPLA2-null macrophages. Interestingly, before the addition of zymosan, adherent peritoneal macrophages from group V sPLA2-null cells had a narrower and more elongated appearance compared with wild-type control cells (Fig. 4A). After stimulation with zymosan, group V sPLA2-null cells tended to retain this narrower configuration (Fig. 4A) and appeared to ingest fewer particles of zymosan than wild-type cells. Formal quantification of the phagocytosis of zymosan over 3 h confirmed that macrophages lacking group V sPLA2 were markedly impaired in their ability to ingest zymosan (Fig. 4B; n = 3; p = 0.016 by analysis of variance). The impairment of phagocytosis by group V sPLA2-null macrophages was not strain-specific, being observed when cells were derived from mice bred to a BALB/c background (for 3–10 generations), from mice bred to a C57BL/6 background (for 10 generations), or from the first generation of C57BL/6×129 group V sPLA2-null mice (data not shown). By comparison, peritoneal macrophages derived from cPLA2α-null mice bred to a BALB/c background (Fig. 5A) and from LTC4 synthase-null mice bred to a C57BL/6 background (Fig. 5B) had no significant defect in phagocytosis compared with cells from wild-type strain-matched littermates (n = 3; p = 0.86 and p = 0.53, respectively, by analysis of variance).
FIGURE 4. Phagocytosis of zymosan by group V sPLA2-null macrophages.
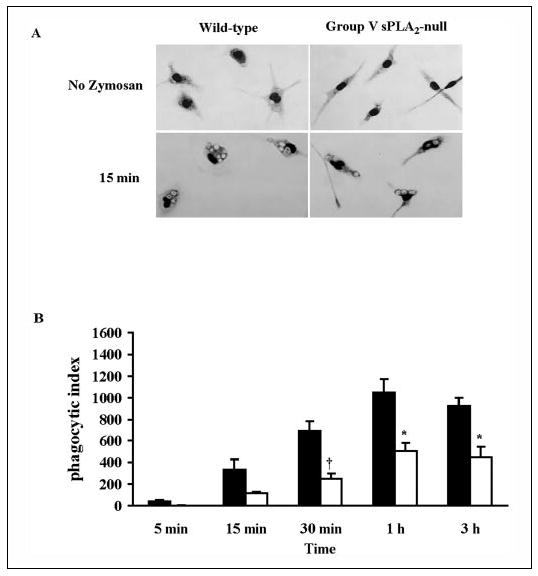
A, light microscopy image of peritoneal macrophages derived from group V sPLA2-null mice (BALB/c N3) and wild-type littermates stained with Diff-Quick before and 15 min after the addition of zymosan (10 particles/cell). B, peritoneal macrophages from group V sPLA2- null mice (open bars) and from their wild-type littermate controls (filled bars) bred to a BALB/c background for three generations were incubated with 10 particles of zymosan/cell for 5 min to 3 h and stained with Diffquick. The phagocytic index was calculated as described under “Experimental Procedures. ” †, p < 0.02; *, p < 0.05 using Student’s paired t test.
FIGURE 5. Phagocytosis of zymosan by macrophages lacking cPLA2α (A) or LTC4 synthase (B).
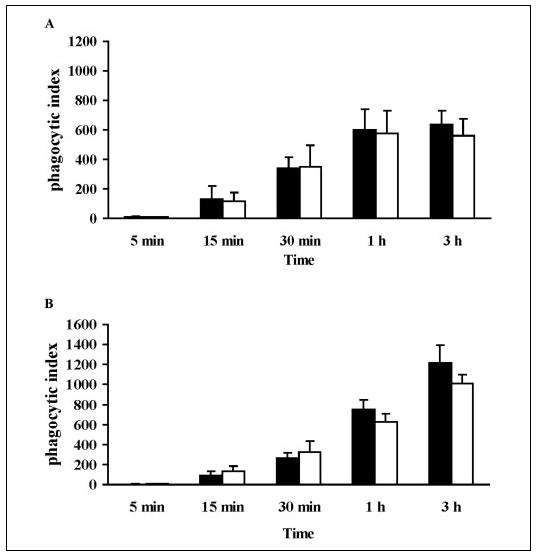
Peritoneal macrophages from cPLA2α-null mice (A, open bars) and LTC4 synthase-null mice (B, open bars) and from their wild-type littermate controls (filled bars) were incubated with 10 particles of zymosan/cell for 5 min to 3 h and stained with Diffquick. The phagocytic index was calculated as described under “Experimental Procedures.”
Expression of Group V sPLA2, but Not Group IIA sPLA2, in Group V sPLA2-null Macrophages Restores Phagocytosis
The embryonic stem cells used to derive group V sPLA2-null mice were derived from 129 mice. Certain strains of mice, including 129 and C57BL/6, but not BALB/c, have a natural disruption of the gene encoding group IIA sPLA2 because of insertion of a thymidine residue in the open reading frame (35). The genes for group IIA sPLA2 and group V sPLA2 are tightly linked on the distal portion of mouse chromosome 4 (36). A search of the map of mouse chromosome 4 at the National Center for Biotechnology and Information website indicates that these two genes are separated by less than 20 kilobases. Therefore the disrupted group IIA sPLA2 gene is always carried with the disrupted group V sPLA2 gene during backcrosses with minimal chance of their separation by a recombination event (Table 1). Indeed, we confirmed by PCR that group V sPLA2-null mice crossed to a BALB/c background for 10 generations carried the disrupted group IIA sPLA2 gene (data not shown). To confirm that the reduced capacity for phagocytosis in group V sPLA2-null mice was due to a lack of group V sPLA2 rather than group IIA sPLA2 or the combined deletion of both proteins, we used adenoviral constructs to reconstitute expression of each sPLA2 in peritoneal macrophages of group V sPLA2-null mice bred to a BALB/c background for 10 generations. As illustrated in Fig. 6, reconstitution of group V sPLA2 by adenoviral transfer augmented the phagocytic index of group V sPLA2-null macrophages (N10 BALB/c) by 47.21 ± 10.72% (mean ± S.D. for five time points in two separate experiments; p = 0.03 by analysis of variance) compared with cells infected with adenovirus expressing LacZ in tandem with GFP. Reconstitution of group IIA sPLA2 failed to augment the capacity of these cells to phagocytose zymosan.
TABLE 1. Strain-dependent expression of group IIA sPLA2 and group V sPLA2.
The expression (+) or disruption (−) of the genes encoding group IIA sPLA2 and group V sPLA2 is shown for group V sPLA2-null mice and their wild-type (WT) littermates bred to a C57BL/6 or BALB/c background. Expression is also indicated for group V sPLA2-null cells, derived from mice bred to a BALB/c background, reconstituted with adenovirally expressed group IIA sPLA2 (Group IIA sPLA2-Ad) or group V sPLA2 (Group V sPLA2-Ad).
| Group IIA sPLA2 | Group V sPLA2 | |
|---|---|---|
| C57BL/6 | ||
| Group V sPLA2-null | − | − |
| Littermate WT | − | + |
| BALB/c | ||
|
| ||
| Group V sPLA2-null | − | − |
| Littermate WT | + | + |
| Group IIA sPLA2-Ad | + | − |
| Group V sPLA2-Ad | − | + |
FIGURE 6. Effects on phagocytosis of expressing group V sPLA2 and group IIA sPLA2 in group V sPLA2-null peritoneal macrophages.
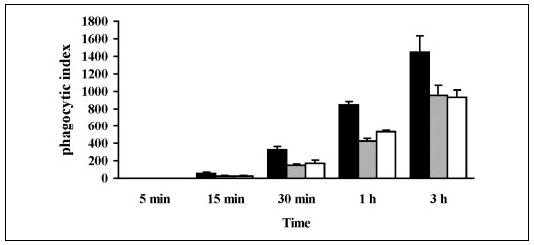
Peritoneal macrophages obtained from group V sPLA2-null mice bred to a BALB/c background (N10) were infected with adenovirus carrying group V sPLA2 (black bars), group IIA sPLA2 (gray bars), or LacZ (open bars) and GFP. After 48 h of infection cells were washed, stimulated with 10 particles/cell of zymosan for 5 min to 3 h and fixed with 2% paraformaldehyde. The cells were imaged as described under “Experimental Procedures, ” and the phagocytic index was calculated for GFP-positive cells
DISCUSSION
Macrophages are an integral part of the host innate immune response and one of the first lines of defense against certain pathogens. They rapidly ingest pathogens, internalizing them in phagosomes that later recruit lysosomal proteins to form phagolysosomes in which the pathogens are killed by an array of proteases and the generation of reactive oxygen species (21). The process of phagocytosis also elicits the rapid generation of proinflammatory mediators, including prostaglandins and leukotrienes derived from arachidonic acid, that elicit an acute inflammatory response (23–26).
Previous work has firmly established that leukotrienes and prostaglandins are formed in a perinuclear location in response to intracellular calcium flux elicited by diverse cell membrane receptors and calcium ionophores (31, 33, 37–39). We have previously reported that group V sPLA2 is prominently expressed in a perinuclear location in mouse mast cells (28) and peritoneal macrophages (15), and we speculated that this would facilitate its interaction with translocated cPLA2α. Our present data confirm that group V sPLA2 is expressed in the Golgi of adherent mouse peritoneal macrophages, co-localizing with GS28, a cis-Golgi marker, and Syntaxin 6, a marker of the trans Golgi network. This is consistent with our prior results in which immunoelectron microscopy revealed group V sPLA2 in Golgi cicsternae of the mast cell (28). Interestingly, group V sPLA2 also co-localizes in the perinuclear region of peritoneal macrophages with the transferrin receptor, which is a marker of the recycling endosomal pathway. It has been suggested that the location of group V sPLA2 in the Golgi may simply be a reflection of its synthesis, especially when overexpressed by transfection. Similarly, its co-expression with the transferrin receptor may reflect its secretion from the cell and subsequent internalization. However, its location in the Golgi would facilitate its interaction with translocated cPLA2α, and as we show in this paper, its location in the endosomal compartment allows it to co-localize with the other enzymes of leukotriene biosynthesis in the forming phagosome.
Using a gene disruption approach, we previously reported a role for group V sPLA2 in amplifying eicosanoid biosynthesis by mouse peritoneal macrophages stimulated with zymosan (15). We were therefore interested to investigate its subcellular trafficking during phagocytosis and found that it rapidly moved to the forming phagosomes (Fig. 2). Leslie and co-workers (30) recently reported that cPLA2α translocates to the phagocytic cup of mouse peritoneal macrophages in response to ingestion of zymosan. cPLA2α is essential for the release of arachidonic acid from mouse peritoneal macrophages in response to zymosan (27). The demonstration that group V sPLA2 translocates to the forming phagosome (Fig. 2) along with cPLA2α (30) (Fig. 3) prompted us to test the hypothesis that the other enzymes of leukotriene biosynthesis translocate to the phagosome and that this is a site of leukotriene biosynthesis. We found that 5-LO, FLAP, and LTC4 synthase were each detected at the forming phagosome as early as 5 min after the ingestion of zymosan particles and were prominent in this location during the first hour after zymosan stimulation (Fig. 3B), the time of maximal LTC4 biosynthesis (Fig. 3A). The kinetics of translocation and predominant location varied between enzymes. Thus, LTC4 synthase retained a prominent perinuclear location in addition to its presence in the forming phagosome. Weaker perinuclear staining was retained for FLAP and was also transiently appreciated for 5-LO at 5 and 15 min, similar to the weak and transient perinuclear staining for cPLA2α reported in mouse macrophages after ingestion of zymosan (30) and consistent with the expected movement of these enzymes to the perinuclear region in response to intracellular calcium flux. To definitively establish that LTC4 was synthesized at the phagosome, we examined the location of LTC4 itself. We adapted a protocol published by Weller and co-workers (31) in which EDAC is used to fix LTC4 to cellular proteins at its site of formation within the cell before its export by specific carrier systems (40). Consistent with the results of immunostaining for the enzymes of its synthesis, we demonstrated the presence of LTC4 at the phagosome within 5 min of the onset of zymosan ingestion. Interestingly, transient perinuclear LTC4 could also be detected, consistent with the transient perinuclear detection of 5-LO (Fig. 3B) and cPLA2α (30), although this was less prominent than that observed at the phagosome.
Previous work has demonstrated that the plasma membrane does not have sufficient surface area to provide membrane for efficient phagocytosis, which requires the recruitment of membrane from other subcellular compartments, mainly early endosomes (41). The contribution of the ER is controversial (41–43). It is intriguing to note that this not only provides the membrane that is physically required for phagocytosis but also results in the assembly of the complete machinery necessary for leukotriene biosynthesis and indeed results in substantial leukotriene biosynthesis that is most prominent at the phagosome. The result is that leukotriene biosynthesis proceeds not merely as the inevitable consequence of calcium flux causing synthesis in a perinuclear location, but rather the biosynthesis of leukotrienes is physically linked to the process of phagocytosis. We can only speculate on the teleology of these events. Leukotrienes are prominent proinflammatory mediators that are essential for efficient host defense against certain pathogens. Mice that lack 5-LO are more susceptible to death from pulmonary infection with Klebsiella pneumoniae (44) at least in part because of impaired phagocytosis in neutrophils unable to synthesize LTB4. Neutrophils from mice that lack cPLA2α are less able to kill E. coli (45), an effect that was attributed to impaired platelet activating factor biosynthesis. Thus, leukotriene generation in response to phagocytosis may not only amplify the ensuing inflammatory response but also contribute to the killing of the ingested pathogens. Cysteinyl leukotrienes have also been implicated in the migration of dendritic cells (46) providing a link between leukotriene biosynthesis by professional phagocytes and the adaptive immune response. It remains to be determined whether our findings extend to dendritic cells.
Because group V sPLA2 localizes in the wall of the phagosome within minutes of zymosan stimulation (Fig. 2), we assessed its contribution to phagocytosis. Indeed, macrophages from mice deficient in group V sPLA2 had an attenuated capacity for phagocytosis of zymosan, suggesting that the enzyme is required for efficient phagocytosis (Fig. 5). It has been reported that leukotrienes stimulate actin polymerization (47–50). We therefore speculated that group V sPLA2 might be acting through provision of leukotrienes at the forming phagosomal membrane. However, macrophages deficient in leukotriene C4 synthase or cPLA2α, which is required for zymosan-induced leukotriene generation (27), demonstrated no deficiency in phagocytosis (Fig. 5). Girotti et al. (30) also reported that inhibition of cPLA2α did not impair the phagocytosis of zymosan by mouse peritoneal macrophages. Collectively, these data indicate that group V sPLA2 does not contribute to phagocytosis through the provision of leukotrienes or other eicosanoids. Our data contrast with those from Peters-Golden and co-workers (44), who reported that alveolar macrophages from 5-LO-null mice had impaired phagocytosis and killing of bacteria leading to increased lethality from K. pneumonia. This could be largely corrected by exogenous LTB4 (44), LTC4 (51), or 5-hydroxyeicosatetraenoic acid (51). The role of LTB4 appeared to be restricted to phagocytosis of IgG-opsonized bacteria and was not observed for complement-coated or unopsonized bacteria (52). Neutrophils from 5-LO-null mice were also defective in phagocytosis of opsonized bacteria, but in contrast to alveolar macrophages this could be corrected only by exogenous LTB4 and not other 5-lipoxygenase products (60). That we did not show a role for endogenous leukotrienes in ingestion of unopsonized zymosan by mouse peritoneal macrophages may relate to the nature of the cell (peritoneal versus alveolar macrophages) or the target (unopsonized zymosan versus opsonized bacteria). Ongoing work is focused on determining the extent of the defect in phagocytosis in group V sPLA2-null mice.
The allele carrying the disrupted group V sPLA2 gene was derived from a 129 embryonic stem cell and therefore carried the closely linked gene encoding group IIA sPLA2, which is disrupted in both 129 and C57BL/6 mice (35). Thus, group V sPLA2-null mice bred to a BALB/c background are effectively double knockouts of both group V sPLA2 and group IIA sPLA2 compared with their wild-type littermates (Table 1). The observation that phagocytosis is defective in peritoneal macrophages derived from group V sPLA2-null mice bred to a C57BL/6 background, in which both wild-type and knockout mice lack group IIA sPLA2, suggested that group V sPLA2 was indeed required for efficient phagocytosis. To definitively prove that the defect in phagocytosis in group V sPLA2-null macrophages is due to the loss of group V sPLA2, we used adenoviral constructs to reconstitute group V sPLA2 expression. The demonstration that expression of group V sPLA2, but not group IIA sPLA2, restored the phagocytic capacity of group V sPLA2-null macrophages (Fig. 6) confirms its role in phagocytosis. That group IIA sPLA2 did not substitute for group V sPLA2 indicates that these enzymes do not serve merely redundant functions. This observation also suggests that certain structural features of group V sPLA2, such as its capacity for interfacial binding to phosphatidylcholine (which it does not share with group IIA sPLA2) as opposed to its capacity to bind to proteoglycans (which is shared with group IIA sPLA2) are important in determining the translocation and/or function of the enzyme at the phagosome. Nevertheless, this speculation requires experimental confirmation.
Dennis and co-workers (53–55) have extensively characterized the contribution of group V sPLA2 and cPLA2α to the release of arachidonic acid and prostaglandin E2 generation in the P388D1 macrophage. When these cells are stimulated with lipopolysaccharide, there is increased steady state RNA for group V sPLA2 that requires the action of cPLA2α and that in turn induces COX-2 expression (56). Using immunofluoresence staining, these authors revealed that 18 h after stimulation with lipopolysaccharide, group V sPLA2 was present in granules that co-immunostained for COX-2 and caveolin-2 (57). Studies by Cho and co-workers (58) indicated that exogenous group V sPLA2 triggered LTB4 biosynthesis by human neutrophils through activating cPLA2α. Subsequent internalization of group V sPLA2 occurred after release of arachidonic acid and was proposed as a degradation pathway for the enzyme. In contrast, exogenous group V sPLA2 triggered LTC4 biosynthesis in human eosinophils independent of cPLA2α (59). Using a fluorescent substrate, the authors demonstrated the action of group V sPLA2 in hydrolyzing cell membrane phospholipids first at the surface of the eosinophil and then at the nuclear envelope to which 5-LO had translocated. Taken together these studies suggest that group V sPLA2 may act in an autocrine or paracrine fashion at different subcellular locations in the cell, depending on the cell type and the nature of the activating stimulus. Where one PLA2 induces the expression of the other, their co-localization is not required. Nevertheless, in circumstances of rapid eicosanoid generation that is regulated by the coordinate action of sPLA2 and cPLA2α, as in our present studies, their co-localization seems logical. Our studies reveal for the first time the association of group V sPLA2 at the phagosome where it couples to cPLA2α to amplify leukotriene biosynthesis and where it facilitates phagocytosis through mechanisms that are independent of cPLA2α.
In summary, we provide novel data to indicate that group V sPLA2 is present not only in Golgi compartments but also in the recycling endosome of mouse peritoneal macrophages. We provide the first demonstration that the phagosome is a site of leukotriene biosynthesis. We further show for the first time that group V sPLA2 is required for efficient phagocytosis of zymosan particles and provide evidence that this requirement is likely independent of its role in eicosanoid generation.
The abbreviations used are
- PLA2
phospholipase A2
- cPLA2
cytosolic phospholipase A2
- sPLA2
secretory phospholipase A2
- LAMP
lysosome-associated membrane protein
- LT
leukotriene
- EDAC
1-ethyl-3-(3-dimethylamino-propyl) carbodiimide
- 5-LO
5-lipoxygenase
- FITC
fluorescein isothiocyanate
- GFP
green fluorescent protein
- Pipes
1,4-piperazinediethanesulfonic acid
- mAb
monoclonal antibody
References
- 1.Soberman RJ, Christmas P. J Clin Investig. 2003;111:1107–1113. doi: 10.1172/JCI18338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keller H, Dreyer C, Medin J, Mahfoudi A, Ozato K, Wahli W. Proc Natl Acad Sci U S A. 1993;90:2160–2164. doi: 10.1073/pnas.90.6.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu K, Baudhuin LM, Hong G, Williams FS, Cristina KL, Kabarowski JHS, Witte ON, Xu Y. J Biol Chem. 2001;276:41325–41335. doi: 10.1074/jbc.M008057200. [DOI] [PubMed] [Google Scholar]
- 4.Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. Cell. 1991;65:1043–1051. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- 5.Pickard RT, Strifler BA, Kramer RM, Sharp JD. J Biol Chem. 1999;274:8823–8831. doi: 10.1074/jbc.274.13.8823. [DOI] [PubMed] [Google Scholar]
- 6.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Nature. 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 7.Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA, Sapirstein A. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 8.Balsinde J, Balboa MA, Dennis EA. J Biol Chem. 1997;272:29317–29321. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- 9.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. J Biol Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 11.Gelb MH, Valentin E, Ghomashchi F, Lazdunski M, Lambeau G. J Biol Chem. 2000;275:39823–39826. doi: 10.1074/jbc.C000671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valentin E, Koduri RS, Scimeca JC, Carle G, Gelb MH, Lazdunski M, Lambeau G. J Biol Chem. 1999;274:19152–19160. doi: 10.1074/jbc.274.27.19152. [DOI] [PubMed] [Google Scholar]
- 13.Valentin E, Ghomashchi F, Gelb MH, Lazdunski M, Lambeau G. J Biol Chem. 1999;274:31195–31202. doi: 10.1074/jbc.274.44.31195. [DOI] [PubMed] [Google Scholar]
- 14.Ishizaki J, Suzuki Y, Higashino K, Yokota Y, Ono T, Kawamoto K, Fujii N, Arita H, Hanasaki K. J Biol Chem. 1999;274:24973–24979. doi: 10.1074/jbc.274.35.24973. [DOI] [PubMed] [Google Scholar]
- 15.Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, Arm JP. J Biol Chem. 2004;279:16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han WK, Sapirstein A, Hung CC, Alessandrini A, Bonventre JV. J Biol Chem. 2003;278:24153–24163. doi: 10.1074/jbc.M300424200. [DOI] [PubMed] [Google Scholar]
- 17.Mounier CM, Gelb F, Lindsay MR, James S, Singer AG, Parton RG, Gelb MH. J Biol Chem. 2004:M313019200. doi: 10.1074/jbc.M313019200. [DOI] [PubMed] [Google Scholar]
- 18.Enomoto A, Murakami M, Valentin E, Lambeau G, Gelb MH, Kudo I. J Immunol. 2000;165:4007–4014. doi: 10.4049/jimmunol.165.7.4007. [DOI] [PubMed] [Google Scholar]
- 19.Akiba S, Hatazawa R, Ono K, Kitatani K, Hayama M, Sato T. J Biol Chem. 2001;276:21854–21862. doi: 10.1074/jbc.M010201200. [DOI] [PubMed] [Google Scholar]
- 20.Lambeau G, Ancian P, Barhanin J, Lazdunski M. J Biol Chem. 1994;269:1575–1578. [PubMed] [Google Scholar]
- 21.Underhill DM, Ozinsky A. Annu Rev Immunol. 2002;20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- 22.Deleo FR, Allen LA, Apicella M, Nauseef WM. J Immunol. 1999;163:6732–6740. [PubMed] [Google Scholar]
- 23.Maekawa A, Austen KF, Kanaoka Y. J Biol Chem. 2002;277:20820–20824. doi: 10.1074/jbc.M203163200. [DOI] [PubMed] [Google Scholar]
- 24.Kanaoka Y, Maekawa A, Penrose JF, Austen KF, Lam BK. J Biol Chem. 2001;276:22608–22613. doi: 10.1074/jbc.M103562200. [DOI] [PubMed] [Google Scholar]
- 25.Rouzer CA, Scott WA, Cohn ZA, Blackburn P, Manning JM. Proc Natl Acad Sci U S A. 1980;77:4928–4932. doi: 10.1073/pnas.77.8.4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rouzer CA, Scott WA, Kempe J, Cohn ZA. Proc Natl Acad Sci U S A. 1980;77:4279–4282. doi: 10.1073/pnas.77.7.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gijon MA, Spencer DM, Siddiq AR, Bonventre JV, Leslie CC. J Biol Chem. 2000;275:20146–20156. doi: 10.1074/jbc.M908941199. [DOI] [PubMed] [Google Scholar]
- 28.Bingham CO, III, Fijneman RJA, Friend DS, Goddeau RP, Rogers RA, Austen KF, Arm JP. J Biol Chem. 1999;274:31476–31484. doi: 10.1074/jbc.274.44.31476. [DOI] [PubMed] [Google Scholar]
- 29.de Carvalho MS, McCormack FX, Leslie CC. Arch Biochem Biophys. 1993;306:534–540. doi: 10.1006/abbi.1993.1549. [DOI] [PubMed] [Google Scholar]
- 30.Girotti M, Evans JH, Burke D, Leslie CC. J Biol Chem. 2004;279:19113–19121. doi: 10.1074/jbc.M313867200. [DOI] [PubMed] [Google Scholar]
- 31.Bandeira-Melo C, Phoofolo M, Weller PF. J Biol Chem. 2001;276:22779–22787. doi: 10.1074/jbc.M101436200. [DOI] [PubMed] [Google Scholar]
- 32.Rabinowitz S, Horstmann H, Gordon S, Griffiths G. J Cell Biol. 1992;116:95–112. doi: 10.1083/jcb.116.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woods JW, Evans JF, Ethier D, Scott S, Vickers PJ, Hearn L, Heibein JA, Charleson S, Singer II. J Exp Med. 1993;178:1935–1946. doi: 10.1084/jem.178.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lam BK, Penrose JF, Rokach J, Xu K, Baldasaro MH, Austen KF. Eur J Biochem. 1996;238:606–612. doi: 10.1111/j.1432-1033.1996.0606w.x. [DOI] [PubMed] [Google Scholar]
- 35.Kennedy BP, Payette P, Mudgett J, Vadas P, Pruzanski W, Kwan M, Tang C, Rancourt DE, Cromlish WA. J Biol Chem. 1995;270:22378–22385. doi: 10.1074/jbc.270.38.22378. [DOI] [PubMed] [Google Scholar]
- 36.Tischfield JA, Xia YR, Shih DM, Klisak I, Chen J, Engle SJ, Siakotos AN, Winstead MV, Seilhamer JJ, Allamand V. Genomics. 1996;32:328–333. doi: 10.1006/geno.1996.0126. [DOI] [PubMed] [Google Scholar]
- 37.Woods JW, Coffey MJ, Brock TG, Singer II, Peters-Golden M. J Clin Investig. 1995;95:2035–2046. doi: 10.1172/JCI117889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spencer AG, Woods JW, Arakawa T, Singer II, Smith WL. J Biol Chem. 1998;273:9886–9893. doi: 10.1074/jbc.273.16.9886. [DOI] [PubMed] [Google Scholar]
- 39.Evans JH, Spencer DM, Zweifach A, Leslie CC. J Biol Chem. 2001;276:30150–30160. doi: 10.1074/jbc.M100943200. [DOI] [PubMed] [Google Scholar]
- 40.Lam BK, Xu K, Atkins MB, Austen KF. Proc Natl Acad Sci U S A. 1992;89:11598–11602. doi: 10.1073/pnas.89.23.11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Touret N, Paroutis P, Terebiznik M, Harrison RE, Trombetta S, Pypaert M, Chow A, Jiang A, Shaw J, Yip C, Moore HP, van der WN, Houben D, Peters PJ, de Chastellier C, Mellman I, Grinstein S. Cell. 2005;123:157–170. doi: 10.1016/j.cell.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 42.Greenberg S, Grinstein S. Curr Opin Immunol. 2002;14:136–145. doi: 10.1016/s0952-7915(01)00309-0. [DOI] [PubMed] [Google Scholar]
- 43.Aderem A. Cell. 2002;110:5–8. doi: 10.1016/s0092-8674(02)00819-x. [DOI] [PubMed] [Google Scholar]
- 44.Bailie MB, Standiford TJ, Laichalk LL, Coffey MJ, Strieter R, Peters-Golden M. J Immunol. 1996;157:5221–5224. [PubMed] [Google Scholar]
- 45.Rubin BB, Downey GP, Koh A, Degousee N, Ghomashchi F, Nallan L, Stefanski E, Smart BP, Lindsay TF, Cherepanov V, Vachon E, Kelvin D, Sadilek M, Brown GE, Yaffe MB, Plumb J, Grinstein S, Glogauer M, Gelb MH. J Biol Chem. 2005;280:7519–7529. doi: 10.1074/jbc.M407438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robbiani DF, Finch RA, Jager D, Muller WA, Sartorelli AC, Randolph GJ. Cell. 2000;103:757–768. doi: 10.1016/s0092-8674(00)00179-3. [DOI] [PubMed] [Google Scholar]
- 47.Peppelenbosch MP, Tertoolen LGJ, Hage WJ, de Laat SW. Cell. 1993;74:565–575. doi: 10.1016/0092-8674(93)80057-l. [DOI] [PubMed] [Google Scholar]
- 48.Massoumi R, Larsson C, Sjolander A. J Cell Science. 2002;115:3509–3515. doi: 10.1242/jcs.115.17.3509. [DOI] [PubMed] [Google Scholar]
- 49.Saegusa S, Tsubone H, Kuwahara M. Eur J Pharmacol. 2001;413:163–171. doi: 10.1016/s0014-2999(01)00773-7. [DOI] [PubMed] [Google Scholar]
- 50.Omann GM, Rengan R, Hoffman JF, Linderman JJ. J Immunol. 1995;155:5375–5381. [PubMed] [Google Scholar]
- 51.Mancuso P, Standiford TJ, Marshall T, Peters-Golden M. Infect Immun. 1998;66:5140–5146. doi: 10.1128/iai.66.11.5140-5146.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mancuso P, Peters-Golden M. Am J Respir Cell Mol Biol. 2000;23:727–733. doi: 10.1165/ajrcmb.23.6.4246. [DOI] [PubMed] [Google Scholar]
- 53.Balsinde J, Balboa MA, Dennis EA. J Biol Chem. 2000;275:22544–22549. doi: 10.1074/jbc.M910163199. [DOI] [PubMed] [Google Scholar]
- 54.Balsinde J, Shinoharo H, Lefkowitz LJ, Johnson CA, Balboa MA, Dennis EA. J Biol Chem. 1999;274:25967–25970. doi: 10.1074/jbc.274.37.25967. [DOI] [PubMed] [Google Scholar]
- 55.Balsinde J, Balboa MA, Dennis EA. Proc Natl Acad Sci U S A. 1998;95:7951–7956. doi: 10.1073/pnas.95.14.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shinohara H, Johnson CA, Balsinde J, Dennis EA. J Biol Chem. 2000;274:12263–12268. doi: 10.1074/jbc.274.18.12263. [DOI] [PubMed] [Google Scholar]
- 57.Balboa MA, Shirai Y, Gaietta G, Ellisman MH, Balsinde J, Dennis EA. J Biol Chem. 2003;278:48059–48065. doi: 10.1074/jbc.M305904200. [DOI] [PubMed] [Google Scholar]
- 58.Kim KP, Rafter JD, Bittova L, Han SK, Snitko Y, Munoz NM, Leff AR, Cho W. J Biol Chem. 2001;276:11126–11134. doi: 10.1074/jbc.M004604200. [DOI] [PubMed] [Google Scholar]
- 59.Munoz NM, Kim YJ, Meliton AY, Kim KP, Han SK, Boetticher E, O’Leary E, Myou S, Zhu X, Bonventre JV, Leff AR, Cho W. J Biol Chem. 2003;278:38813–38820. doi: 10.1074/jbc.M302476200. [DOI] [PubMed] [Google Scholar]
- 60.Mancuso P, Nana-Sinkam P, Peters-Golden M. Infect Immun. 2001;69:2011–2016. [Google Scholar]