

Abstract
Biomarkers are likely to be important in the study of Alzheimer disease (AD) for a variety of reasons. A clinical diagnosis of Alzheimer disease is inaccurate even among experienced investigators in about 10% to 15% of cases, and biomarkers might improve the accuracy of diagnosis. Importantly for the development of putative disease-modifying drugs for Alzheimer disease, biomarkers might also serve as indirect measures of disease severity. When used in this way, sample sizes of clinical trials might be reduced, and a change in biomarker could be considered supporting evidence of disease modification. This review summarizes a meeting of the Alzheimer’s Association’s Research Roundtable, during which existing and emerging biomarkers for AD were evaluated. Imaging biomarkers including volumetric magnetic resonance imaging and positron emission tomography assessing either glucose utilization or ligands binding to amyloid plaque are discussed. Additionally, biochemical biomarkers in blood or cerebrospinal fluid are assessed. Currently appropriate uses of biomarkers in the study of Alzheimer disease, and areas where additional work is needed, are discussed.
Keywords: Alzheimer disease, amyloid beta, cerebrospinal fluid, clinical trials, cytokines, isoprostanes, positron emission tomography, tau, volumetric magnetic resonance imaging
INTRODUCTION
The development of new therapies for Alzheimer disease (AD) and other neurodegenerative conditions has become of increasing societal importance given our aging population and increasing longevity, combined with the fact that this disease typically begins late in life. Various biomarkers1,2 can be used in a variety of ways to allow new therapies to be developed more quickly and to increase the probability of success in the pivotal trials ultimately needed to gain new drug approval by regulatory agencies. On November 11–12, 2004, a meeting of the Alzheimer’s Association Research Roundtable was held with experts on biomarkers associated with AD. This report summarizes the information presented and discussed at that meeting. A discussion of the use of biomarkers in the diagnosis of pre-symptomatic AD was also held and will be reported separately.
Biomarkers may be applied to drug development for AD in a number of distinct ways. First, they may be applied as additional diagnostic measures in a population clinically identified as having AD. Sensitivity, specificity, and positive predictive value all must be considered for such an application.
One of the most important uses of biomarkers in drug development is as an indirect measure of disease severity. A number of points should be established for such use: the marker must have a scientific rationale (eg, tau in cerebrospinal fluid [CSF]), the biomarker should change with disease progression in longitudinal observational studies,3 and the marker must be measurable and reproducible. Unlike typical diagnostic measures, when biomarkers are used for this purpose, high specificity is not required. These biomarkers can be of both scientific and regulatory value. Particularly in mid-phase trials, biomarkers can be used to identify appropriate dosage, improve safety assessments, demonstrate pharmacological activity, and identify preliminary evidence of efficacy.
ALZHEIMER’S DISEASE NEUROIMAGING INITIATIVE
The National Institute of Aging has initiated the Alzheimer’s Disease Neuroimaging Initiative (ADNI), a large observational study of patients with AD, patients with mild cognitive impairment (MCI), and cognitively normal volunteers to assess longitudinal changes in AD biomarkers. Current trials of investigational treatments require large sample sizes and long treatment durations because cognitive measures do not easily reflect disease-modifying effects of treatment.
Many groups, including pharmaceutical companies, have great interest in using imaging and other biomarkers for treatment trials; however, current data are from many institutions using different methods and different subjects.
Outcome measures included in ADNI are based on previous studies assessing AD biomarkers. Volumetric magnetic resonance imaging (vMRI) will be a major focus of the study, based largely on data from patients with AD and MCI. These data suggest that MCI is a transitional state between normal and early diagnosable disease, and progression from MCI to AD is reflected by changes in brain volumes. Longitudinal studies of patients with diagnosed AD show greater rates of change in hippocampal and temporal horn volumes than are seen with normal aging, based on currently available data.4 [18]fluoro-deoxy-glucose (FDG) positron emission tomography (PET) will also be used in ADNI. For patients with AD, decline in glucose utilization as determined by FDG PET imaging is progressive, correlates with dementia severity, and predicts a histopathological diagnosis of AD.1,5–7
Based on these and other data, ADNI was established as a longitudinal, prospective naturalistic study of early AD, mild cognitive impairment, and normal aging. MRI, PET, biochemical biomarkers, and clinical data will be included in the final database, which will be placed in the public domain. ADNI’s goals include the following: (1) identify the best biomarkers for early diagnosis; (2) identify the best biomarkers for following disease progression and monitoring treatment response; (3) develop surrogate endpoints for clinical trials; and (4) establish methods for the multisite acquisition, quality determinations, and processing of biomarker and clinical data. About two-thirds of the funding for the study is from the NIA, and the remaining one-third, via the NIH Foundation, is from the pharmaceutical industry, imaging equipment manufacturers, and nonprofit organizations. Additional information regarding the study can be found at the ADNI web site (http://www.loni.ucla.edu/ADNI/).
APPLICATIONS OF BIOMARKERS TO CLINICAL TRIAL DESIGNS
A key issue in the design of trials of investigational drugs for AD is the ability to distinguish between symptomatic effects of drugs and effects that are due to modification of the underlying disease process. A trial design that measures delay of an end point, such as the onset of disease, cannot distinguish between these two possibilities. Designs that may allow conclusions regarding disease modification include those incorporating a randomized start, randomized withdrawal, or a persistent difference in slope. Randomized start and randomized withdrawal designs are illustrated in Figure 1. The underlying assumption for both of these trial designs is that the group initially randomized to treatment has a slowing of underlying disease progression. Thus, using the randomized withdrawal design, even if a purely symptomatic effect of the drug were present, the treated group would not return to the same cognitive scores as the placebo group after drug withdrawal. Similarly, when active treatment is started in the placebo group using the randomized start design, the cognitive scores of the group originally assigned to placebo would not reach those of the group originally assigned to active treatment.
FIGURE 1.
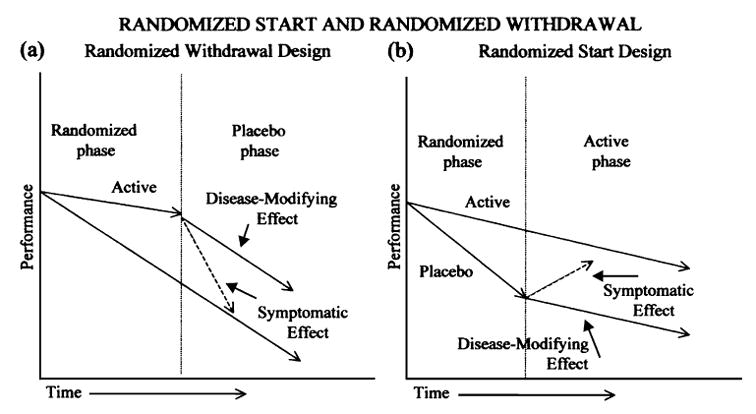
Study design summaries for disease modification.
While natural history studies can be important in the initial analysis of clinical or biomarker measures, they also have limitations. In these studies, subjects generally do not meet the same inclusion and exclusion criteria as they would in a randomized clinical trial, and thus have greater comorbidities and may be on medications that would otherwise be excluded. These limitations may lead to overestimates of the rate of decline or event rate, thus leading to underpowered and failed clinical studies. As illustrated in Table 1, rate of change in cognitive scores can also change over time, with placebo-treated patients showing slower rates of decline in more recent studies.
TABLE 1.
Rate of Decline in Alzheimer’s Disease Assessment Scale–Cognition (ADAS-cog) Scores in Five Studies87–91
| Study | N | Baseline MMSE | Mean ± SD |
|---|---|---|---|
| Thal et al, 1996 | 211 | 19.6 | 7.0 ± 7.8 |
| Thal et al, 2000 | 102 | 20.6 | 7.5 ± 8. |
| Aisen et al, 2000 | 69 | 22 | 6.3 ± 6.4 |
| Aisen et al, 2003 | 111 | 20.8 | 5.7 ± 8.2 |
| Reines et al, 2004 | 346 | 21 | 5.4 |
Each value in the far right column is the annual rate of decline in ADAS-cog score. MMSE, Mini-Mental State Examination.
Biomarkers may be used in phase 1 or 2 studies to show an effect of the investigational drug on its target. A potential caveat with this strategy is that some drugs require weeks to months to show such an effect (eg, antioxidants). In later phase studies, a biomarker may be used as a surrogate marker if the surrogate can be substituted for a clinical end point. In phase 2 studies, effects such as a reduction in CSF tau after 3 to 6 months or reduced isoprostanes in CSF can provide evidence of a biologic signal and may help with dose selection for phase 3. A potential caveat with this strategy is that biomarkers may be altered differently and may respond to treatment differently over the course of the disease. For phase 3 trials, primary outcome variables are likely to be clinical measures for the immediate future. Surrogate markers can be used in phase 3 trials to develop supportive evidence for efficacy and to support a claim of disease modification.
IMAGING TECHNIQUES USED IN THE STUDY OF CLINICALLY DIAGNOSED ALZHEIMER DISEASE
Changes in MR-based regional (hippocampus, entorhinal cortex, and corpus callosum) and global (whole brain and ventricles) brain volume measures have been demonstrated for patients with AD in a number of longitudinal studies.3,4,8–20 These studies consistently show loss of brain volumes in AD patients that are at least twice the rate of loss seen in age-matched control subjects. Longitudinal studies of vMRI in AD patients are listed in Table 2.
TABLE 2.
| Source | Region | N (Control/Alzheimer Disease [AD]) |
|---|---|---|
| Kaye et al, 1997 | Hippocampi | 18/12 (preclinical AD) |
| Parahippocampal gyri | ||
| Temporal lobes* | ||
| Intracranial volume | ||
| Jack et al, 1998, 2004 | Hippocampi* | 24/24 and 55/64 |
| Entorhinal cortex* | ||
| Temporal horns* | ||
| Whole brain* | ||
| Ventricle* | ||
| Fox et al, 2000 | Whole brain* | 18/18 |
| Laakso et al, 2000 | Hippocampi | 8/27 |
| Teipel et al, 2002 | Corpus callosum* | 10/21 |
| Bradley et al, 2002 | Whole brain* | 32/5 |
| Ventricle* | ||
| Ventricle/brain ratio* | ||
| Wang et al, 2002 | Cerebrum* | 14/14 |
| Lateral ventricles* | ||
| Temporal lobes* | ||
| Du et al, 2003, 2004 | Entorhinal cortex* | 23/21 and 25/21 |
| Hippocampus* | ||
| Schott et al, 2003 | Entorhinal cortex* | 20/5 (presymptomatic AD) |
| Hippocampus* | ||
| Temporal lobe* | ||
| Brain* | ||
| Thompson et al, 2004 | Hippocampus* | 14/12 |
| Ventricle* |
There were statistically significant differences between patients with Alzheimer disease and controls.
Additionally, patients with MCI, compared with control subjects with stable cognitive scores, have greater rates of volume loss for most brain areas regardless of whether they converted to AD. Further, MCI patients who did convert to AD have greater rates of change than those who are cognitively stable.21
An important consideration for imaging and other biomarker studies is whether results obtained at a single site can be replicated when the same measures are applied in a multiple site trial. The vMRI results from a 52-week study of milameline using 38 sites have been reported.21 Subjects were scanned at baseline and end point, and hippocampal and temporal horn volumes were obtained. Based on these results, sample size calculations for an AD trial designed to detect a 50% reduction in rate of progression would require 320 patients per arm based on change in ADAS-cog scores, compared with 21 patients per arm based on hippocampal volume and 54 patients per arm based on temporal horn volume. These estimates suggest that even considering the additional variability imposed by the use of multiple sites, vMRI can provide advantages in the number of subjects required in a clinical trial to demonstrate a statistically significant effect on a structural end point.
The fact that biomarker changes in observational studies do not always predict changes seen in therapeutic trials is illustrated by the apparent decrease in brain volumes seen in subjects actively immunized with Aβ1–42 (AN1792).22 Subjects who had measurable antibody titers in this trial also had improvements in cognitive scores using some instruments. While a number of potential explanations have been proposed for this surprising result (eg, loss of plaque volume or brain hydration), the dissociation between apparent response to active vaccination and expected change in brain volumes illustrates the need to examine biomarker changes in therapeutic as well as observational studies. The finding also illustrates the need for the further development and evaluation of additional promising therapeutic surrogates and the importance of considering the choice and number of putative surrogate end points to consider in a therapeutic trial, and the schedule in which these end points are assessed, such that the observed effect of a treatment on the end point(s) most likely reflects the treatment’s effect on disease progression.
FDG PET studies reveal characteristic and progressive reductions in regional measurements of the cerebral metabolic rate for glucose (CMRgl) in patients with AD5,6,23 and patients with MCI.24–27 In patients with AD, CMRgl reductions in the posterior cingulate, parietal, temporal, and prefrontal cortex are correlated with dementia severity6 and progression.5 In a retrospective study of patients with mild to moderate dementia, the pattern of hypometabolism was about 94% sensitive and 73% specific in predicting subsequent clinical decline and the histopathological diagnosis of AD.23 In patients with the diagnosis of amnestic MCI,24,25 isolated memory impairment,26 and nonamnestic MCI,27 regional CMRgl reductions helped distinguish subsequent AD converters from nonconverters, but with some overlap between groups. In a longitudinal study of amnestic MCI,25 the 1-year rate of CMRgl decline was greater in subsequent AD converters than in nonconverters.
Based on longitudinal CMRgl declines in AD patients, researchers have estimated the statistical power of FDG PET to detect the ability of a putative disease-modifying treatment to slow rates of regional CMRgl decline in randomized clinical trials.5 The estimated number of AD patients per treatment arm needed to detect an effect with FDG PET (Table 3) is roughly comparable to that needed to detect an effect with MRI and almost one-tenth the number of patients needed using clinical end points, suggesting the promise of these imaging techniques in proof-of-concept trials.
TABLE 3.
Sample Size Estimates Using FDG PET for Alzheimer Disease Trials5 Number of Alzheimer Disease Patients Per Treatment Group Needed to Detect an Effect With 80% Power in 1 Year
| Treatment Effect
|
||||
|---|---|---|---|---|
| 20% | 30% | 40% | 50% | |
| Frontal | 85 | 38 | 22 | 14 |
| Parietal | 217 | 97 | 55 | 36 |
| Temporal | 266 | 119 | 68 | 44 |
| Cingulate | 343 | 153 | 87 | 57 |
| Combined | 62 | 28 | 16 | 10 |
P = 0.01 (two-tailed).
No adjustment for normal aging effects or subject attrition.
Adapted from Alexander et al, 2002.
More recently, PET imaging studies using radioligands that bind directly to β-amyloid plaques have been performed.28,29 One of these ligands, Pittsburgh Compound-B (PIB), is a thioflavin derivative and appears to be relatively selective for β-amyloid plaques at the concentrations used for imaging studies. As shown in Figure 2, the binding of PIB to brain sections is highly correlated with total Aβ levels. Test/retest variability in clinical studies is less than 10% for most brain regions.30
FIGURE 2.
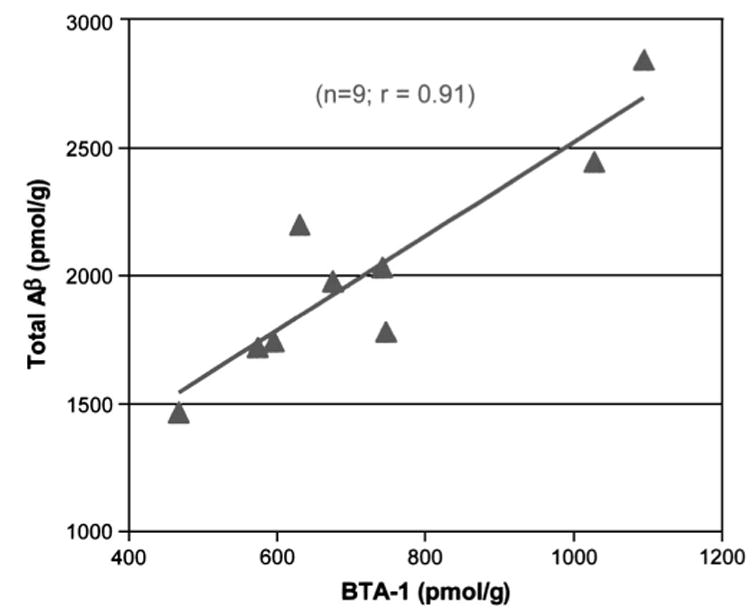
Correlation between PIB-derivative binding and β-amyloid levels in brain slices.28
Little longitudinal data have been accrued thus far for clinical PIB PET studies. A number of these studies have started recently or will begin in the near future. They will be crucial for determining sample size requirements for demonstrating a statistically significant effect of a given agent in clinical trials.
Newer imaging techniques may be available in the future that could be more sensitive to change or show qualitatively different effects of AD or effects of investigational drugs. For instance, automated algorithms are being used to deform MRIs into standard coordinates of a brain atlas, facilitating comparisons of the change in gray matter or white matter volume on a voxel-by-voxel basis.31–33 These differences offer promise in the differential diagnosis, early detection, and tracking of AD and in the evaluation of putative disease slowing treatments.
A strategy for using existing methods to determine brain volumes could entail the use of serial MRI images for several months prior to the initiation of an investigational drug. Such an approach would compare rate of decline within a subject before and after drug treatment, thus improving statistical power and reducing sample size requirements to demonstrate a statistically significant effect of a given agent.
Magnetic resonance spectroscopy has been used to assess concentrations of N-acetyl asparate (NAA), creatine, and choline in the brain; Alzheimer disease and MCI are associated with reduced NAA concentrations.34,35 While the ability to make biochemical measurements in brain parenchyma in vivo would be extremely valuable, these techniques currently suffer from lack of technical standardization and lack of correlation with clinical measures.
Pulsed arterial spin labeled (ASL) perfusion MRI may provide a means to determine regional cerebral blood flow without the use of radioactivity.36–38 Regional differences in blood flow may help to distinguish AD from frontotemporal dementia and vascular dementia. Continuous ASL techniques are also being investigated and may benefit from higher magnet strengths (ie, at least 4 Tesla).
BIOCHEMICAL MEASURES USED TO ASSESS RATE OF ALZHEIMER DISEASE PROGRESSION
Biochemical biomarkers may be assessed in different matrices or compartments, including CSF, blood, and urine. Many of the same considerations given to imaging techniques also apply to these biochemical measures (eg, the validity and accuracy of the analytical method and the variability among multiple sites). More specific to biochemical measures are the need to standardize sample-handling techniques and to standardize methods for obtaining and storing CSF.
Lumbar punctures and CSF analyses have been used routinely in the practice of neurology for decades, although with the advent of other diagnostic modalities, this procedure is now performed most frequently in research settings in the United States. Nevertheless, two large studies of lumbar punctures performed as part of an evaluation of possible AD biomarkers have shown that the procedure can be applied broadly and that it is well tolerated.39,40 The only recorded complication was post-lumbar puncture headache. With the use of a small diameter needle (0.7 mm), the rate of mild headache (duration less than 1 day, not affecting daily life) was less than 4%, and the rate of moderate or severe headache (duration more than 1one day and/or affecting daily life) was less than 1%.
While the initial pathogenic events in AD are not known with certainty, biochemical markers of the disease can be considered as more proximal or upstream, compared with more distal or downstream events. As shown in Figure 3, a number of potential biomarkers can be measured that may be proximal or distal in the pathogenic process.
FIGURE 3.
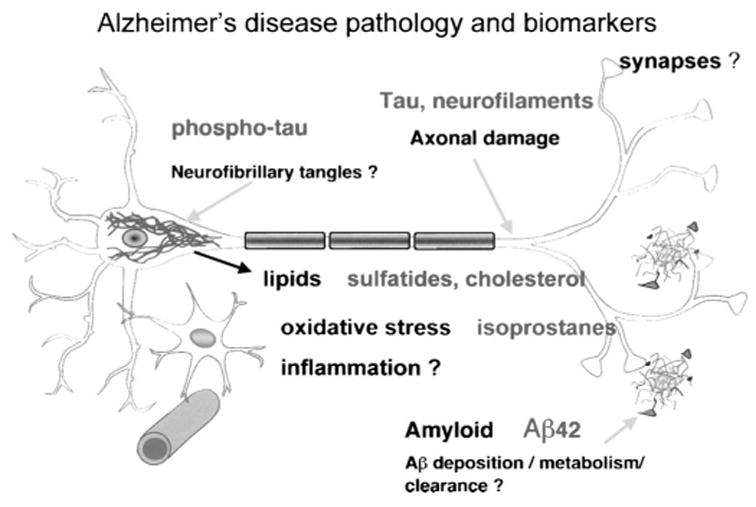
Potential biochemical biomarkers in AD. Question marks indicate processes or anatomic areas that may be proximal in the disease process. Possible biomarkers that can be considered given these postulated disease processes include tau, phospho-tau, sulfatides, cholesterol, isoprostanes, and Aβ42.
Each potential biomarker must have certain characteristics to be useful in multicenter trials. The assay must have excellent sensitivity and test/retest reliability. Sample handling requirements must be such that analyses have acceptable variability when samples are obtained at multiple sites. The biomarker analyte should reflect a key feature of AD pathology or a mechanism of disease. Finally, the pattern of change in the biomarker over time and variability of that change should be adequately described.
Aβ, and in particular Aβ1–42, has been studied frequently as a biomarker for AD. CSF concentrations of Aβ1–42 are reduced by 40% to 50%, whereas concentrations of Aβ1–40 or “Aβtotal” (using an ELISA that does not distinguish C-terminal length) are similar to those of age-matched controls. CSFAβ1–42 does correlate to an extent with dementia severity; however, in most studies concentrations are stable over intervals as long as 12 months.41
Plasma concentrations of Aβ1–42 do not correlate with those in CSF.42 Longitudinal studies have not shown a consistent change in plasma Aβ over time in AD patients,43 and cross-sectional differences between AD patients and controls that would allow plasma Aβ concentrations to be used as a diagnostic measure have not been identified.
Cerebrospinal fluid tau has also been studied as a potential biomarker in AD.44 Elevations of 2- to 3-fold of CSF total tau (T-tau) levels in patients with AD have been demonstrated in cross-sectional studies. In longitudinal studies, weak correlations are present with changes in cognitive scores, and CSF T-tau levels remain stably elevated in AD over time intervals of 12 months or longer. Tau may be phosphorylated at various sites, and forms of CSF tau reflecting specific sites of phosphorylation (P-tau 181, 199, 231, 235, 396, and 404) have been studied.
Three species of p-tau (p-thr231, p-ser199, and p-thr181) have been examined in detail in cross-sectional studies.45–49 All three species are elevated in the CSF of patients with AD, and concentrations of all three species appear to be linearly related. When assessed as diagnostic measures, these three measures have similar sensitivity, although p-thr231 may have somewhat greater specificity for AD versus other forms of dementia.45 Interestingly, p-thr231 tau, as well as other forms, is elevated in MCI patients compared with control subjects, but longitudinal studies of AD patients show a progressive decline in concentration with disease progression.50
There are several studies in which the diagnostic performance of the combination of CSF T-tau and Aβ1–42 has been evaluated.44 In most but not all studies,51 the sensitivity and specificity for the combination of these two biomarkers have been slightly higher (89% and 90%, respectively) than for T-tau (81% and 91%, respectively) or Aβ1–42 (86% and 89%, respectively) alone. Other combinations of CSF biomarkers have also resulted in slightly better diagnostic performance than the use of single markers. In a study on the combination of CSF p-tau181 and Aβ1–42, the sensitivity was 86% at a specificity of 97%,52 and in another study the combination of CSF T-tau and p-tau396/404 resulted in a sensitivity of 96% at a specificity of 100%.53 Further studies examining the value of combinations of biomarkers in larger series of patients and controls, and in particular in MCI, are needed.
As with imaging measures, sample size calculations for clinical trials can be made using Aβ and tau measures. As shown in Table 4, samples sizes using biochemical measures are similar to those achieved with imaging and are smaller than sample sizes based on clinical cognitive measures.
TABLE 4.
| Marker | Study | Number of Subjects Needed Per Group |
|---|---|---|
| CSF total tau in AD | Andreasen et al, 1999
Moriearty et al, 1999 |
40 |
| CSF Aβ42 in controls | Andreasen et al, 1999
Galasko et al, 1998 Prince et al, 2004 |
16 |
| CSF Aβ42 in AD | Andreasen et al, 1999
Moriearty et al, 1999 Simons et al, 2002 |
36 |
| Plasma Aβ | Hoglund et al, 2004 | 27 |
Assumptions: Two-arm, drug versus placebo; α = 0.05, β = 0.80; 25% effect size. AD, Alzheimer disease; CSF, cerebrospinal fluid.
Besides the pathologic hallmarks of the disease, which include amyloid plaques and neurofibrillary tangles, AD pathology is characterized by evidence of reactive-oxygen species (ROS)–mediated damage.54 ROS are formed under normal conditions, and although they are chemically unstable and highly reactive, their levels are kept relatively low by efficient antioxidant systems including catalase, glutathione, uric acid, and vitamins E and C. However, in some situations their generation can exceed the endogenous capacity to destroy them. As a consequence, the oxidant versus the antioxidant balance is altered and oxidative damage is the final result.55 Depending on the substrate attacked by ROS, oxidative damage will manifest as protein oxidation, DNA oxidation, or lipid peroxidation products, all of which have been described in AD brain (Fig. 4). In general, oxidative damage in the central nervous system predominantly manifests as lipid peroxidation because of its high content of polyunsaturated fatty acids that are easily susceptible to oxidation.56
FIGURE 4.
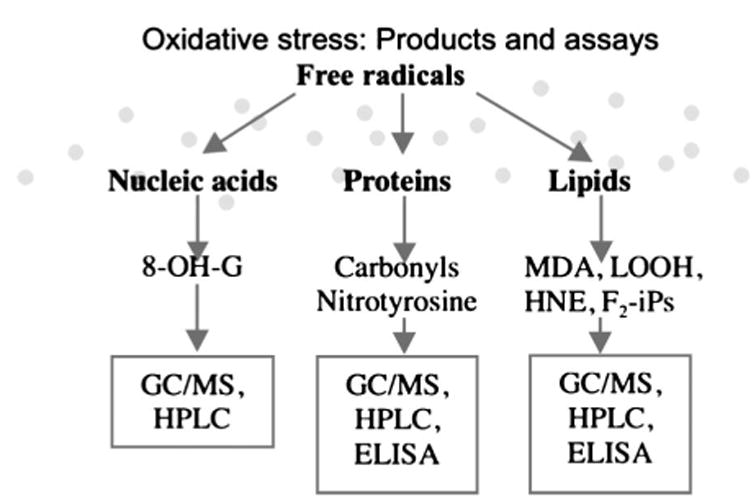
Products of reactive oxygen species–dependent attack of different substrates (nucleic acid, protein, lipid) and relative most employed analytical methods (GC/MS: gas chromatography/mass spectrometry; HPLC: high performance liquid chromatography; ELISA: enzyme-linked immuno-assay; 8-OH-G: 8-hydroxyguanosine; MDA: malondialdehyde; LOOH: lipid hydroperoxide; HNE: 4-hydroxynonenal).
Isoprostanes are members of a complex family of lipid oxidation products derived from an ROS-mediated attack on free or esterified fatty acids. One group of them, called F2-isoprostanes (F2-iPs) (Fig. 5), are present in detectable quantities in all normal biologic fluids and tissues. Assays for specific F2-iPs isomers using gas chromatography/mass spectrometry have identified 8,12-iso-iPF2α-VI (IPF2A) to be the most abundant F2-iP in human as well as in animals.57
FIGURE 5.
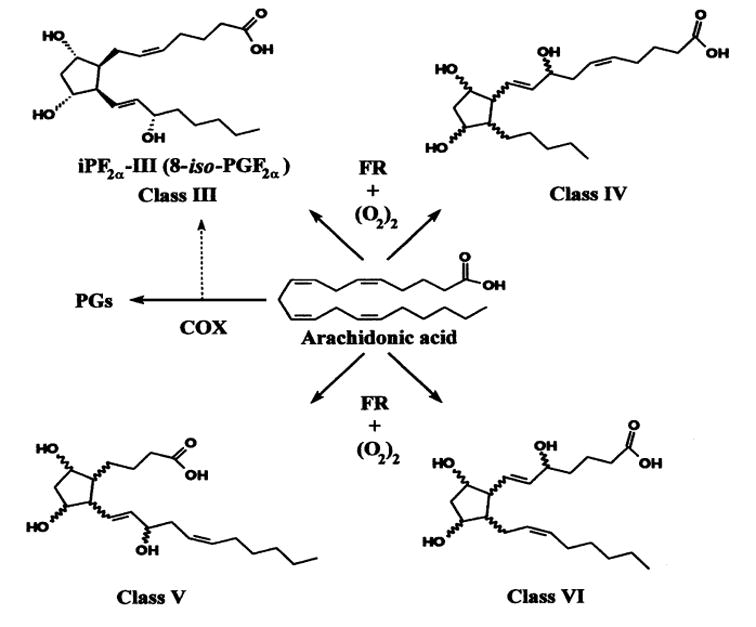
The four classes of F2-isoprostane deriving from the ROS-mediated oxidation of arachidonic acid.86
IPF2A concentrations are elevated in brain, CSF, and plasma of AD patients compared with controls.58,59 In cross-sectional studies, concentrations of IPF2A in CSF correlate directly with concentrations of total tau and inversely with Aβ1–42 levels.59 In patients with MCI, CSF concentrations of IPF2A are intermediate between those of AD patients and those of control subjects; interestingly, patients with MCI who progress to AD have higher concentrations than those who do not.59 Recent investigations were conducted to determine whether the increase in this marker of lipid peroxidation is present in neurodegenerative diseases other than AD. For this reason, histopathologically confirmed AD was compared with frontotemporal dementia (FTD) subjects, a heterogeneous group of dementing disorders with neurodegeneration. Levels of IPF2A were found to be markedly elevated in postmortem AD brains compared with corresponding areas of FTD and control brain tissues.60 This observation was also confirmed in CSF from living patients with clinical diagnosis of FTD.61
Longitudinal studies in MCI patients showed that CSF F2-iPs levels were elevated at both baseline (P < 0.001) and follow-up (P < 0.01) compared with controls. This resulted in an overall classification accuracy of 88%, both at baseline and follow-up. Moreover, a significant longitudinal change was seen in the MCI patients relative to controls. The longitudinal change yielded an overall classification accuracy of 76%, and post hoc examination showed a significant isoprostane increase restricted to the MCI group (de Leon M, DeSanti S, Zinkowski R, et al. Biomarkers for Alzheimer’s disease improve early diagnosis. Neurobiol Aging. 2005 [in press]).
Many, including Alzheimer himself, have observed enlarged (more recently referred to as activated) microglia and astrocytes in brain of Alzheimer patients. A 1989 report provided the first evidence of a neuropathogenic role for two of the principal cytokines derived from activated microglia and astrocytes, viz., IL-1, a potent pro-inflammatory cytokine, and S100B, a potent neurotogenic cytokine: (1) overexpression was shown to be already dramatic in neonates, children, and young adults with Down’s syndrome (a virtually certain risk for precocious development of AD)62 and (2) a similar overexpression was demonstrated in end-stage AD.63–65 The neuro-pathogenic role of these two cytokines has been further supported by findings that both S100B66 and IL-167 regulate production of the β-amyloid precursor protein (βAPP) as well as reports that the number of activated astrocytes and microglia overexpressing these two cytokines are related to the progression of β-amyloid plaques.68,69 Overexpression of these cytokines has been implicated in the pathogenesis of AD by their demonstrated influences on the genesis and formation of the two principal features in AD, neuritic β-amyloid plaques and neurofibrillary tangles, as well as synaptic loss, and neuronal dysfunction and loss.70–79 This strong evidence for cytokine involvement in such neurodegenerative processes, underscores the importance of astrocyte-derived and microglia-derived cytokines in the brain’s innate immune system and its role pathogenesis. In addition to overexpression of IL-1 and S100B, expression of IL-6,80 α1-ACT,81 and iNOS82 are increased in AD brain. Moreover, there is evidence that expression of each is up-regulated by IL-1 and that expression of each is decreased by suppression of IL-1.83–85 Findings such as these suggest that neuroinflammation, powered by glia-derived cytokines, drives a self-sustaining, self-amplifying cycle that leads to a vicious circle of inflammation and neuronal dysfunction and death. The way in which neuronal insults are implicated in AD neuropathogenesis is summarized in Figure 6.
FIGURE 6.
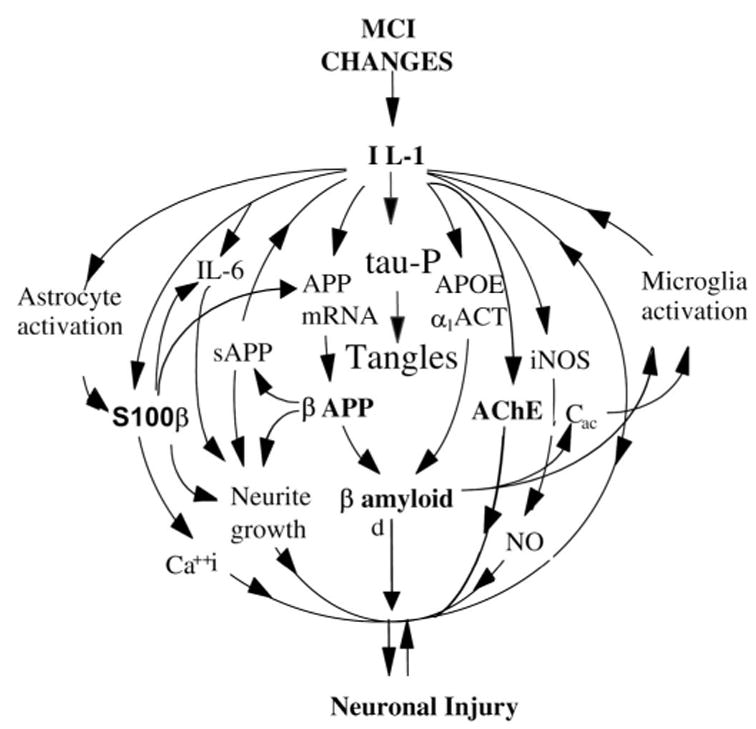
Inflammatory changes implicated in Alzheimer disease (AD). This conceptual scheme of the cytokine cycle illustrates the relationship of glial activation and inflammatory cytokine overexpression to neurodegenerative events in AD, with the effect of overexpression of IL-1 in the brain. IL-1 = interleukin-1; APOE = Apolipoprotein E; ACT = antichymotrypsin; sAPP = secreted amyloid precursor; iNOS = nitric oxide synthase; Cac = activated complement; MCI = mild cognitive impairment.
An important goal is to discover markers of conversion from a situation in which the brain can cope with a given level of insults and when the insults are sufficient to mildly impair brain function. Most of the studies cited above used brain tissue, but the need for studies of more accessible tissue has resulted in studies using CSF, some of which show tantalizing, but inconclusive, changes in α1-ACT and IL-6.2 Inflammatory markers have also been measured in serum, and early preliminary studies show that the relative levels of IL-1, IL-6, IL-8, IL-10, IL-12 and may be different in serum from patients who converted from control to MCI (Griffin et al, unpublished data). Measuring serum cytokine levels may serve as peripheral biomarkers of innate brain immune responses. At sufficient sensitivity, such biomarkers, although not likely to be specific for AD or other neurodegenerative condition, may be useful as peripheral indicators of progression of neural pathologies typical of AD. One way in which they might prove most useful is in testing the efficacy of therapeutic interventions. Not only the sensitivity but also the specificity of these putative biomarkers will need to be established in future trials.
PANEL DISCUSSION: BIOMARKERS USED IN THE STUDY OF CLINICALLY DIAGNOSED ALZHEIMER DISEASE
A panel (see Acknowledgments for participants) discussed a number of points related to the use of biomarkers for patients with AD. Several points of consensus were achieved, though a few points of disagreement were identified. Finally, a number of unmet needs for AD biomarker research were identified.
In the immediate future, biomarkers for AD are more likely to be used in clinical trials than in clinical practice. Use of CSF biomarkers as diagnostic measures in clinical practice is unlikely in the United States at this time; however, if a well-validated diagnostic marker and disease-modifying treatments were available, patients may be referred from primary care physicians to neurologists for diagnostic lumbar punctures. Diagnostic biomarkers in clinical practice also are more important for patients with earlier disease.
Imaging biomarkers may have greater face validity and are more well developed, but more data are needed to improve the understanding of both imaging and biochemical biomarkers and their expected behavior after treatment with investigational drugs. Of various volumes measured using vMRI, whole brain and ventricular volumes appear most sensitive to change. Whether concentrations of Aβ1–42 in CSF should increase or decrease with chronic dosing of a γ- or β-secretase inhibitor is unclear. The need for corrections in CSF concentrations based on ventricular volume has not been adequately assessed and more data are needed. More longitudinal studies with CSF measures are needed.
Biomarkers are likely to play an increasingly important role in the development of investigational drugs for AD. Lumbar punctures to obtain biochemical biomarkers can be performed safely, but may decrease recruitment rates in clinical trials. The choice of biomarker(s) in a clinical trial may depend on the mechanism of action of the investigational drug. There was not a clear consensus that a biomarker could be used as a primary outcome variable in a phase 2 clinical trial. Three potential uses for biomarkers in drug development were outlined: (1) for selection of homogeneous patient groups, (2) as an early indicator that an investigational drug is reaching its target and is having the intended effect, and (3) for indirect assessments of effects on disease progression. The validation of a biomarker as a surrogate marker for disease progression is likely to be iterative. Specifically, if a drug is identified that clearly changes clinical disease progression and that reduces or reverses an AD-specific biomarker, this would substantiate the use of such a biomarker for future therapeutic agents.
SUMMARY
Biomarkers are potentially very useful as tools in investigational drug trials of clinically diagnosed AD patients. Such markers could be used as indirect markers of disease severity, or might also be used as additional inclusion or exclusion criteria. When used as an indirect marker of disease severity, in almost all cases sample size estimates suggest that effects of putative disease-modifying drugs could be determined with fewer subjects using imaging or biochemical biomarkers than by using cognitive measures. Additional longitudinal multisite studies of these biomarkers, in particular FDG PET, amyloid-ligand PET, and CSF biochemical markers, would aid greatly in their applications to clinical trials. Such information will be obtained in a large observational study of patients with AD and MCI and elderly controls that will begin in mid-2005 (ADNI).
Acknowledgments
Biomarkers for AD progression trials panelists: Neil Buckholtz, PhD, Leon Thal, MD, John Growden, MD, John Morris, MD, Martin Farlow, MD, David Knopman, MD, Mony De Leon, EdD, and Howard Fillit, MD. The support of the Alzheimer’s Association and its Research Roundtable for this meeting is greatly appreciated. The assistance of Jay Lenn in the preparation of the manuscript is also greatly appreciated.
References
- 1.Matthews B, Siemers ER, Mozley PD. Imaging-based measures of disease progression in clinical trials of disease-modifying drugs for Alzheimer disease. Am J Geriatr Psychiatry. 2003;11:146–159. [PubMed] [Google Scholar]
- 2.Frank RA, Galasko D, Hampel H, et al. Biological markers for therapeutic trials in Alzheimer’s disease. Proceedings of the biological markers working group; NIA initiative on neuroimaging in Alzheimer’s disease. Neurobiol Aging. 2003;24:521–536. doi: 10.1016/s0197-4580(03)00002-2. [DOI] [PubMed] [Google Scholar]
- 3.Silbert LC, Quinn JF, Moore MM, et al. Changes in premorbid brain volume predict Alzheimer’s disease pathology. Neurology. 2003;61:487–492. doi: 10.1212/01.wnl.0000079053.77227.14. [DOI] [PubMed] [Google Scholar]
- 4.Fox NC, Cousens S, Scahill R, et al. Using serial registered brain magnetic resonance imaging to measure disease progression in Alzheimer disease: power calculations and estimates of sample size to detect treatment effects. Arch Neurol. 2000;57:339–344. doi: 10.1001/archneur.57.3.339. [DOI] [PubMed] [Google Scholar]
- 5.Alexander GE, Chen K, Pietrini P, et al. Longitudinal PET evaluation of cerebral metabolic decline in dementia: a potential outcome measure in Alzheimer’s disease treatment studies. Am J Psychiatry. 2002;159:738–745. doi: 10.1176/appi.ajp.159.5.738. [DOI] [PubMed] [Google Scholar]
- 6.Minoshima S, Frey KA, Koeppe RA, et al. A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. J Nucl Med. 1995;36:1238–1248. [PubMed] [Google Scholar]
- 7.Moriearty PL, Seubert P, Galasko D, et al. Effects of time and cholinesterase inhibitor treatment on multiple cerebrospinal fluid parameters in Alzheimer’s disease. Methods Find Exp Clin Pharmacol. 1999;21:549–554. doi: 10.1358/mf.1999.21.8.794837. [DOI] [PubMed] [Google Scholar]
- 8.Kaye JA, Swihart T, Howieson D, et al. Volume loss of the hippocampus and temporal lobe in healthy elderly persons destined to develop dementia. Neurology. 1997;48:1297–1304. doi: 10.1212/wnl.48.5.1297. [DOI] [PubMed] [Google Scholar]
- 9.Jack CR, Jr, Petersen RC, Xu Y, et al. Rate of medial temporal lobe atrophy in typical aging and Alzheimer’s disease. Neurology. 1998;51:993–999. doi: 10.1212/wnl.51.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laakso MP, Lehtovirta M, Partanen K, et al. Hippocampus in Alzheimer’s disease: a 3-year follow-up MRI study. Biol Psychiatry. 2000;47:557–561. doi: 10.1016/s0006-3223(99)00167-5. [DOI] [PubMed] [Google Scholar]
- 11.Bradley KM, Bydder GM, Budge MM, et al. Serial brain MRI at 3–6 month intervals as a surrogate marker for Alzheimer’s disease. Br J Radiol. 2002;75:506–513. doi: 10.1259/bjr.75.894.750506. [DOI] [PubMed] [Google Scholar]
- 12.Scahill RI, Schott JM, Stevens JM, et al. Mapping the evolution of regional atrophy in Alzheimer’s disease: unbiased analysis of fluid-registered serial MRI. Proc Natl Acad Sci USA. 2002;99:4703–4707. doi: 10.1073/pnas.052587399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith SM, Zhang Y, Jenkinson M, et al. Accurate, robust, and automated longitudinal and cross-sectional brain change analysis. Neuroimage. 2002;17:479–489. doi: 10.1006/nimg.2002.1040. [DOI] [PubMed] [Google Scholar]
- 14.Teipel SJ, Bayer W, Alexander GE, et al. Progression of corpus callosum atrophy in Alzheimer disease. Arch Neurol. 2002;59:243–248. doi: 10.1001/archneur.59.2.243. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Chalk JB, Rose SE, et al. MR image-based measurement of rates of change in volumes of brain structures. Part II: application to a study of Alzheimer’s disease and normal aging. Magn Reson Imaging. 2002;20:41–48. doi: 10.1016/s0730-725x(02)00472-1. [DOI] [PubMed] [Google Scholar]
- 16.Du AT, Schuff N, Zhu XP, et al. Atrophy rates of entorhinal cortex in AD and normal aging. Neurology. 2003;60:481–486. doi: 10.1212/01.wnl.0000044400.11317.ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schott JM, Fox NC, Frost C, et al. Assessing the onset of structural change in familial Alzheimer’s disease. Ann Neurol. 2003;53:181–188. doi: 10.1002/ana.10424. [DOI] [PubMed] [Google Scholar]
- 18.Du AT, Schuff N, Kramer JH, et al. Higher atrophy rate of entorhinal cortex than hippocampus in AD. Neurology. 2004;62:422–427. doi: 10.1212/01.wnl.0000106462.72282.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jack CR, Jr, Shiung MM, Gunter JL, et al. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology. 2004;62:591–600. doi: 10.1212/01.wnl.0000110315.26026.ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson PM, Hayashi KM, De Zubicaray GI, et al. Mapping hippocampal and ventricular change in Alzheimer disease. Neuroimage. 2004;22:1754–1766. doi: 10.1016/j.neuroimage.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 21.Jack CR, Jr, Slomkowski M, Gracon S, et al. MRI as a biomarker of disease progression in a therapeutic trial of milameline for AD. Neurology. 2003;60:253–260. doi: 10.1212/01.wnl.0000042480.86872.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fox NC, Black RS, Gilman S, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64:1563–1572. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- 23.Silverman DH, Small GW, Chang CY, et al. Positron emission tomography in evaluation of dementia: regional brain metabolism and long-term outcome. JAMA. 2001;286:2120–2127. doi: 10.1001/jama.286.17.2120. [DOI] [PubMed] [Google Scholar]
- 24.Arnaiz E, Jelic V, Almkvist O, et al. Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment. Neuroreport. 2001;12:851–855. doi: 10.1097/00001756-200103260-00045. [DOI] [PubMed] [Google Scholar]
- 25.Drzezga A, Lautenschlager N, Siebner H, et al. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: a PET follow-up study. Eur J Nucl Med Mol Imaging. 2003;30:1104–1113. doi: 10.1007/s00259-003-1194-1. [DOI] [PubMed] [Google Scholar]
- 26.Berent S, Giordani B, Foster N, et al. Neuropsychological function and cerebral glucose utilization in isolated memory impairment and Alzheimer’s disease. J Psychiatr Res. 1999;33:7–16. doi: 10.1016/s0022-3956(98)90048-6. [DOI] [PubMed] [Google Scholar]
- 27.Chetelat G, Desgranges B, de la Sayette V, et al. Mild cognitive impairment: can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology. 2003;60:1374–1377. doi: 10.1212/01.wnl.0000055847.17752.e6. [DOI] [PubMed] [Google Scholar]
- 28.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 29.Mathis CA, Klunk WE, Price JC, et al. Imaging technology for neuro-degenerative diseases: progress toward detection of specific pathologies. Arch Neurol. 2005;62:196–200. doi: 10.1001/archneur.62.2.196. [DOI] [PubMed] [Google Scholar]
- 30.Price JC, Klunk WE, Lopresti BJ, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab. 2005 doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- 31.Ashburner J, Csernansky JG, Davatzikos C, et al. Computer-assisted imaging to assess brain structure in healthy and diseased brains. Lancet Neurol. 2003;2:79–88. doi: 10.1016/s1474-4422(03)00304-1. [DOI] [PubMed] [Google Scholar]
- 32.Chételat G, Landeau B, Eustache F, et al. Using voxel-based morphometry to map the structural changes associated with rapid conversion in MCI: a longitudinal MRI study. Neuroimage. 2005;27:934–946. doi: 10.1016/j.neuroimage.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 33.Senjem ML, Gunter JL, Shiung MM, et al. Comparison of different methodological implementations of voxel-based morphometry in neuro-degenerative disease. Neuroimage. 2005;26:600–608. doi: 10.1016/j.neuroimage.2005.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyerhoff DJ, MacKay S, Constans JM, et al. Axonal injury and membrane alterations in Alzheimer’s disease suggested by in vivo proton magnetic resonance spectroscopic imaging. Ann Neurol. 1994;36:40–47. doi: 10.1002/ana.410360110. [DOI] [PubMed] [Google Scholar]
- 35.Chao LL, Schuff N, Kramer JH, et al. Reduced medial temporal lobe N-acetylaspartate in cognitively impaired but nondemented patients. Neurology. 2005;64:282–289. doi: 10.1212/01.WNL.0000149638.45635.FF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jahng GH, Zhu XP, Matson GB, et al. Improved perfusion-weighted MRI by a novel double inversion with proximal labeling of both tagged and control acquisitions. Magn Reson Med. 2003;49:307–314. doi: 10.1002/mrm.10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jahng GH, Song E, Zhu XP, et al. Human brain: reliability and reproducibility of pulsed arterial spin-labeling perfusion MR imaging. Radiology. 2005;234:909–916. doi: 10.1148/radiol.2343031499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson NA, Jahng GH, Weiner MW, et al. Pattern of cerebral hypo-perfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology. 2005;234:851–859. doi: 10.1148/radiol.2343040197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andreasen N, Minthon L, Davidsson P, et al. Evaluation of CSF-tau and CSF-Abeta42 as diagnostic markers for Alzheimer disease in clinical practice. Arch Neurol. 2001;58:373–379. doi: 10.1001/archneur.58.3.373. [DOI] [PubMed] [Google Scholar]
- 40.Blennow K, Wallin A, Hager O. Low frequency of post-lumbar puncture headache in demented patients. Acta Neurol Scand. 1993;88:221–223. doi: 10.1111/j.1600-0404.1993.tb04221.x. [DOI] [PubMed] [Google Scholar]
- 41.Andreasen N, Hesse C, Davidsson P, et al. Cerebrospinal fluid beta-amyloid(1–42) in Alzheimer disease: differences between early- and late-onset alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56:673–680. doi: 10.1001/archneur.56.6.673. [DOI] [PubMed] [Google Scholar]
- 42.Mehta PD, Pirttila T, Patrick BA, et al. Amyloid beta protein 1–40 and 1–42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci Lett. 2001;304:102–106. doi: 10.1016/s0304-3940(01)01754-2. [DOI] [PubMed] [Google Scholar]
- 43.Mayeux R, Honig LS, Tang MX, et al. Plasma A[beta]40 and A[beta]42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology. 2003;61:1185–1190. doi: 10.1212/01.wnl.0000091890.32140.8f. [DOI] [PubMed] [Google Scholar]
- 44.Blennow K. Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. Neurorx. 2004;1:213–225. doi: 10.1602/neurorx.1.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hampel H, Buerger K, Zinkowski R, et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry. 2004;61:95–102. doi: 10.1001/archpsyc.61.1.95. [DOI] [PubMed] [Google Scholar]
- 46.Ishiguro K, Ohno H, Arai H, et al. Phosphorylated tau in human cerebrospinal fluid is a diagnostic marker for Alzheimer’s disease. Neurosci Lett. 1999;270:91–94. doi: 10.1016/s0304-3940(99)00476-0. [DOI] [PubMed] [Google Scholar]
- 47.Kohnken R, Buerger K, Zinkowski R, et al. Detection of tau phosphorylated at threonine 231 in cerebrospinal fluid of Alzheimer’s disease patients. Neurosci Lett. 2000;287:187–190. doi: 10.1016/s0304-3940(00)01178-2. [DOI] [PubMed] [Google Scholar]
- 48.Vanmechelen E, Vanderstichele H, Davidsson P, et al. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett. 2000;285:49–52. doi: 10.1016/s0304-3940(00)01036-3. [DOI] [PubMed] [Google Scholar]
- 49.Zetterberg H, Wahlund LO, Blennow K. Cerebrospinal fluid markers for prediction of Alzheimer’s disease. Neurosci Lett. 2003;352:67–69. doi: 10.1016/j.neulet.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 50.Hampel H, Buerger K, Kohnken R, et al. Tracking of Alzheimer’s disease progression with cerebrospinal fluid tau protein phosphorylated at threonine 231. Ann Neurol. 2001;49:545–546. [PubMed] [Google Scholar]
- 51.Clark CM, Xie S, Chittams J, et al. Cerebrospinal fluid tau and beta-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol. 2003;60:1696–1702. doi: 10.1001/archneur.60.12.1696. [DOI] [PubMed] [Google Scholar]
- 52.Maddalena A, Papassotiropoulos A, Muller-Tillmanns B, et al. Biochemical diagnosis of Alzheimer disease by measuring the cerebrospinal fluid ratio of phosphorylated tau protein to beta-amyloid peptide42. Arch Neurol. 2003;60:1202–1206. doi: 10.1001/archneur.60.9.1202. [DOI] [PubMed] [Google Scholar]
- 53.Hu YY, He SS, Wang X, et al. Levels of nonphosphorylated and phosphorylated tau in cerebrospinal fluid of Alzheimer’s disease patients: an ultrasensitive bienzyme-substrate-recycle enzyme-linked immunosorbent assay. Am J Pathol. 2002;160:1269–1278. doi: 10.1016/S0002-9440(10)62554-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pratico D, Sung S. Lipid peroxidation and oxidative imbalance: early functional events in Alzheimer’s disease. J Alzheimers Dis. 2004;6:171–175. doi: 10.3233/jad-2004-6209. [DOI] [PubMed] [Google Scholar]
- 55.Pratico D, Delanty N. Oxidative injury in diseases of the central nervous system: focus on Alzheimer’s disease. Am J Med. 2000;109:577–585. doi: 10.1016/s0002-9343(00)00547-7. [DOI] [PubMed] [Google Scholar]
- 56.Pratico D. Alzheimer’s disease and oxygen radicals: new insights. Biochem Pharmacol. 2002;63:563–567. doi: 10.1016/s0006-2952(01)00919-4. [DOI] [PubMed] [Google Scholar]
- 57.Pratico D, Lawson JA, Rokach J, et al. The isoprostanes in biology and medicine. Trends Endocrinol Metab. 2001;12:243–247. doi: 10.1016/s1043-2760(01)00411-8. [DOI] [PubMed] [Google Scholar]
- 58.Pratico D, Lee VM, Trojanowski JQ, et al. Increased F2-isoprostanes in Alzheimer’s disease: evidence for enhanced lipid peroxidation in vivo. FASEB J. 1998;12:1777–1783. doi: 10.1096/fasebj.12.15.1777. [DOI] [PubMed] [Google Scholar]
- 59.Pratico D, Clark CM, Lee VM, et al. Increased 8,12-iso-iPF2alpha-VI in Alzheimer’s disease: correlation of a noninvasive index of lipid peroxidation with disease severity. Ann Neurol. 2000;48:809–812. [PubMed] [Google Scholar]
- 60.Yao Y, Zhukareva V, Sung S, et al. Enhanced brain levels of 8,12-iso-iPF2alpha-VI differentiate AD from frontotemporal dementia. Neurology. 2003;61:475–478. doi: 10.1212/01.wnl.0000070185.02546.5d. [DOI] [PubMed] [Google Scholar]
- 61.Grossman M, Farmer J, Leight S, et al. Cerebrospinal fluid profile in frontotemporal dementia and Alzheimer’s disease. Ann Neurol. 2005;57:721–729. doi: 10.1002/ana.20477. [DOI] [PubMed] [Google Scholar]
- 62.Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol. 1985;17:278–282. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]
- 63.Griffin WS, Stanley LC, Ling C, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Griffin WS, Sheng JG, McKenzie JE, et al. Life-long overexpression of S100beta in Down’s syndrome: implications for Alzheimer pathogenesis. Neurobiol Aging. 1998;19:401–405. doi: 10.1016/s0197-4580(98)00074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Royston MC, McKenzie JE, Gentleman SM, et al. Overexpression of S100beta in Down’s syndrome: correlation with patient age and with beta-amyloid deposition. Neuropathol Appl Neurobiol. 1999;25:387–393. doi: 10.1046/j.1365-2990.1999.00196.x. [DOI] [PubMed] [Google Scholar]
- 66.Li Y, Barger SW, Liu L, et al. S100beta induction of the proinflammatory cytokine interleukin-6 in neurons. J Neurochem. 2000;74:143–150. doi: 10.1046/j.1471-4159.2000.0740143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goldgaber D, Harris HW, Hla T, et al. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci USA. 1989;86:7606–7610. doi: 10.1073/pnas.86.19.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sheng JG, Mrak RE, Griffin WS. S100 beta protein expression in Alzheimer disease: potential role in the pathogenesis of neuritic plaques. J Neurosci Res. 1994;39:398–404. doi: 10.1002/jnr.490390406. [DOI] [PubMed] [Google Scholar]
- 69.Griffin WS, Sheng JG, Roberts GW, et al. Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. J Neuropathol Exp Neurol. 1995;54:276–281. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- 70.Mrak RE, Sheng JG, Griffin WS. Correlation of astrocytic S100 beta expression with dystrophic neurites in amyloid plaques of Alzheimer’s disease. J Neuropathol Exp Neurol. 1996;55:273–279. doi: 10.1097/00005072-199603000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheng JG, Mrak RE, Griffin WS. Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol. 1997;56:285–290. [PubMed] [Google Scholar]
- 72.Sheng JG, Mrak RE, Griffin WS. Neuritic plaque evolution in Alzheimer’s disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol (Berl) 1997;94:1–5. doi: 10.1007/s004010050664. [DOI] [PubMed] [Google Scholar]
- 73.Sheng JG, Zhou XQ, Mrak RE, et al. Progressive neuronal injury associated with amyloid plaque formation in Alzheimer disease. J Neuropathol Exp Neurol. 1998;57:714–717. doi: 10.1097/00005072-199807000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sheng JG, Griffin WS, Royston MC, et al. Distribution of interleukin-1-immunoreactive microglia in cerebral cortical layers: implications for neuritic plaque formation in Alzheimer’s disease. Neuropathol Appl Neurobiol. 1998;24:278–283. doi: 10.1046/j.1365-2990.1998.00122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sheng JG, Zhu SG, Jones RA, et al. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp Neurol. 2000;163:388–391. doi: 10.1006/exnr.2000.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sheng JG, Jones RA, Zhou XQ, et al. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer’s disease: potential significance for tau protein phosphorylation. Neurochem Int. 2001;39:341–348. doi: 10.1016/s0197-0186(01)00041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Y, Liu L, Barger SW, et al. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003;23:1605–1611. doi: 10.1523/JNEUROSCI.23-05-01605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu L, Li Y, Van Eldik LJ, et al. S100B-induced microglial and neuronal IL-1 expression is mediated by cell type-specific transcription factors. J Neurochem. 2005;92:546–553. doi: 10.1111/j.1471-4159.2004.02909.x. [DOI] [PubMed] [Google Scholar]
- 79.Liu DG, He SR, Zhang W, et al. Relationship between apoptosis of neurons and microglia activation in Alzheimer’s disease. Zhonghua Bing Li Xue Za Zhi. 2004;33:404–407. [PubMed] [Google Scholar]
- 80.Strauss S, Bauer J, Ganter U, et al. Detection of interleukin-6 and alpha 2-macroglobulin immunoreactivity in cortex and hippocampus of Alzheimer’s disease patients. Lab Invest. 1992;66:223–230. [PubMed] [Google Scholar]
- 81.Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine protease inhibitor alpha 1-antichymotrypsin in the brain amyloid deposits of Alzheimer’s disease. Cell. 1988;52:487–501. doi: 10.1016/0092-8674(88)90462-x. [DOI] [PubMed] [Google Scholar]
- 82.Lee SC, Zhao ML, Hirano A, et al. Inducible nitric oxide synthase immunoreactivity in the Alzheimer disease hippocampus: association with Hirano bodies, neurofibrillary tangles, and senile plaques. J Neuropathol Exp Neurol. 1999;58:1163–1169. doi: 10.1097/00005072-199911000-00006. [DOI] [PubMed] [Google Scholar]
- 83.Heneka MT, Sastre M, Dumitrescu-Ozimek L, et al. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 84.Morihara T, Teter B, Yang F, et al. Ibuprofen suppresses interleukin-1beta induction of pro-amyloidogenic alpha1-antichymotrypsin to ameliorate beta-amyloid (Abeta) pathology in Alzheimer’s models. Neuropsychopharmacology. 2005;30:1111–1120. doi: 10.1038/sj.npp.1300668. [DOI] [PubMed] [Google Scholar]
- 85.Richartz E, Stransky E, Batra A, et al. Decline of immune responsiveness: a pathogenetic factor in Alzheimer’s disease? J Psychiatr Res. 2005;39:535–543. doi: 10.1016/j.jpsychires.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 86.Pratico DF. (2)-isoprostanes: sensitive and specific non-invasive indices of lipid peroxidation in vivo. Atherosclerosis. 1999;147:1–10. doi: 10.1016/s0021-9150(99)00257-9. [DOI] [PubMed] [Google Scholar]
- 87.Thal LJ, Carta A, Clarke WR, et al. A 1-year multicenter placebo-controlled study of acetyl-L-carnitine in patients with Alzheimer’s disease. Neurology. 1996;47:705–711. doi: 10.1212/wnl.47.3.705. [DOI] [PubMed] [Google Scholar]
- 88.Thal LJ, Calvani M, Amato A, et al. A 1-year controlled trial of acetyl-l-carnitine in early-onset AD. Neurology. 2000;55:805–810. doi: 10.1212/wnl.55.6.805. [DOI] [PubMed] [Google Scholar]
- 89.Aisen PS, Davis KL, Berg JD, et al. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology. 2000;54:588–593. doi: 10.1212/wnl.54.3.588. [DOI] [PubMed] [Google Scholar]
- 90.Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289:2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 91.Reines SA, Block GA, Morris JC, et al. Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- 92.Fox NC, Cousens S, Scahill R, et al. Using serial registered brain magnetic resonance imaging to measure disease progression in Alzheimer disease: power calculations and estimates of sample size to detect treatment effects. Arch Neurol. 2000;57:339–344. doi: 10.1001/archneur.57.3.339. [DOI] [PubMed] [Google Scholar]
- 93.Prince JA, Zetterberg H, Andreasen N, et al. APOE epsilon4 allele is associated with reduced cerebrospinal fluid levels of Abeta42. Neurology. 2004;62:2116–2118. doi: 10.1212/01.wnl.0000128088.08695.05. [DOI] [PubMed] [Google Scholar]
- 94.Galasko D, Chang L, Motter R, et al. High cerebrospinal fluid tau and low amyloid beta42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype. Arch Neurol. 1998;55:937–945. doi: 10.1001/archneur.55.7.937. [DOI] [PubMed] [Google Scholar]
- 95.Hoglund K, Wiklund O, Vanderstichele H, et al. Plasma levels of beta-amyloid(1–40), beta-amyloid(1–42), and total beta-amyloid remain unaffected in adult patients with hypercholesterolemia after treatment with statins. Arch Neurol. 2004;61:333–337. doi: 10.1001/archneur.61.3.333. [DOI] [PubMed] [Google Scholar]
- 96.Simons M, Schwarzler F, Lutjohann D, et al. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease. Ann Neurol. 2002;52:346–350. doi: 10.1002/ana.10292. [DOI] [PubMed] [Google Scholar]