

Abstract
HRG4(UNC119) is a photoreceptor protein predominantly localized to the photoreceptor synapses and to the inner segments to a lesser degree. A heterozygous truncation mutation in HRG4 was found in a patient with late onset cone-rod dystrophy, and a transgenic (TG) mouse expressing the identical mutant protein developed late onset retinal degeneration, confirming the pathogenic potential of HRG4. Recently, the dominant negative pathogenic mechanism in the TG model was shown to involve increased affinity of the truncated mutant HRG4 for its target, ARL2, which leads to a delayed decrease in its downstream target, mitochondrial ANT1, mitochondrial stress, synaptic degeneration, trans-synaptic degeneration, and whole photoreceptor degeneration by apoptosis. In this study, the mouse HRG4 (MRG4) gene was cloned and targeted to construct a knock-out (KO) mouse model of HRG4 in order to study the effects of completely inactivating this protein. The KO model was examined by genomic Southern blotting, western blotting, immunofluorescence, funduscopy, LM and EM histopathology, ERG, and TUNEL analyses. The KO model developed a slowly progressive retinal degeneration, characterized by mottling in the fundus, mild thinning of the photoreceptor layer, and increase in apoptosis as early as 6 months, dramatic acceleration at ~17 months, and virtual obliteration of the photoreceptors by 20 months. When compared to retinal degeneration in the TG model, significant differences existed in the KO consisting of more severe and early photoreceptor death without evidence of early synaptic and trans-synaptic degeneration as seen in the TG, confirmed by LM and EM histopathology, ERG, and western blotting of synaptic proteins. The results indicated a dysfunction in the KO outside the synapses in the distal end of photoreceptors where MRG4 is also localized. Differences in the phenotypes of retinal degeneration in the KO and TG models reflect a dysfunction in the two opposite ends of photoreceptors, i.e., the distal inner/outer segments and proximal synapses, respectively, indicating a second function of MRG4 in the distal photoreceptor and dual functionality of MRG4. Thus, inactivation of MRG4 by gene targeting resulted in a retinal degeneration phenotype quite different from that previously seen in the TG, attesting to the multiplicity of MRG4 function, in addition to the importance of this protein for normal retinal function. These models will be useful in elucidating the functions of HRG4/MRG4 and the mechanism of slow retinal degeneration.
Keywords: retinal degeneration, knock-out model, transgenic model, photoreceptor, synapse, inner segments, outer segments
Introduction
Retinal degeneration is a major cause of blindness for which there is no cure or effective treatment. "Over nine million Americans of every age and race suffer vision loss from these blinding diseases" (Foundation Fighting Blindness web page (blindness.org)). The first step in moving towards understanding of these diseases is to identify the causative genes. Once the genes are identified, the functional defect present in the disease can be elucidated. Animal models can be constructed to investigate the actual mechanism of pathogenesis that occurs in the disease. Information gained from these studies will aid in finding a cure or the best treatment for the retinal degeneration. Along this line, very encouraging results were obtained from gene therapy of a dog model of Leber congenital amaurosis (Acland et al., 2001).
HRG4 (UNC119) is a photoreceptor synaptic protein that was cloned in our laboratory through a subtractive cloning strategy to isolate novel retinal genes that may be candidate retinal degeneration genes (Higashide et al., 1996). HRG4 is homologous to C. elegans neuroprotein UNC119, loss of which causes disorganized neural architecture and paralysis in the worm (Maduro and Pilgrim, 1995). UNC119 has also been shown recently to be required for normal development of the zebrafish nervous system (Manning et al., 2004). HRG4 is one of the most abundant proteins in the retina, consistent with its functional importance in the retina (Wistow et al., 2002). It is present in the presynaptic space of rod and cone photoreceptors, predominantly associated with synaptic vesicles and also in the inner segments (IS) of photoreceptors to a degree (Higashide et al., 1998). Although the precise function of HRG4 is not known yet, it has been shown to interact with the ARF-like protein 2 (ARL-2) by the yeast two-hybrid strategy, opening up several possibilities for function (Kobayashi et al. 2003). The HRG4 gene consists of 5 exons, mapping to chromosome 17q11.2 (Higashide and Inana, 1999).
A heterozygous truncation mutation of HRG4 was demonstrated in a patient with late onset cone-rod dystrophy, and a transgenic model (TG) expressing the same mutant protein was shown to also develop late onset retinal degeneration, confirming the pathogenic potential of a defect in this protein (Kobayashi et al. 2000). The retinal degeneration in the TG model was marked by slow progression, selective reduction in eletroretinogram (ERG) b-wave, severe synaptic degeneration, and significant trans-synaptic degeneration in the inner nuclear layer (INL) in addition to outer nuclear layer (ONL) thinning. Consistent with the localization of HRG4 in the photoreceptor synapse, specific decrease in the levels of several synaptic vesicle membrane proteins was observed in the transgenic retina, indicative of synaptic dysfunction (Kubota et al., 2002). More recently, we have demonstrated a dominant negative mechanism of pathogenesis in the TG to be based on the increased affinity of the mutant truncated HRG4 for its target, ARL2, resulting in perturbation of the formation of a complex between ARL2 and Binder-of-ARL2 (BART) and a decrease in its downstream target, the mitochondrial adenine nucleotide transporter 1 (ANT1) in the synapses (Mori et al. 2006). The deficit in ANT1, a key regulator of cellular energetics that exchanges mitochondrial ATP for cytoplasmic ADP, leads to mitochondrial stress, cytochrome C release, and caspase 3 activation in the synapses. As a result, the synapses degenerate, inducing trans-synaptic degeneration and photoreceptor cell body degeneration by apoptosis. Thus, the TG model elucidated a function of HRG4 related to mitochondrial ANT1 and cellular energetics, and demonstrated the mutant truncated HRG4 to cause a retinal degeneration that initiates in the photoreceptor synapses.
To further advance the elucidation of the function and pathogenic mechanism of HRG4, we have constructed a knock-out (KO) model of HRG4 by gene targeting. In this paper, we describe its initial analysis which has revealed that inactivating the HRG4 gene results in severe retinal degeneration based on a dysfunction in the inner/outer segments as opposed to synapses in the model, demonstrating the essential requirement and dual functions of this protein in the normal retina.
Materials and Methods
Cloning of the mouse HRG4 (MRG4) gene
Two million lambda Fix II clones from the 129 SvJ mouse genomic library (Stratagene, La Jolla, CA) were plated out and screened by hybridization with a rat HRG4 (RRG4) cDNA probe using standard procedures (Higashide et al., 1996; Sambrook and Fritsch, 1989). Positive clones were purified through multiple picking and hybridization screening, and characterized by restriction mapping and DNA sequencing. A genomic library made from a 129 SvJ mouse was used to minimize the difference between the sequence of a gene clone and its intended target, the genomic sequence of the MRG4 gene in the embryonic stem (ES) cell derived from 129 SvJ mouse.
Construction of the MRG4 targeting vector
The pPNT targeting vector containing a neomycin resistance gene and a herpes virus thymidine kinase at the 3' end was used for targeting the MRG4 gene (Tybulewicz et al., 1991). A 2.6 kbp Pst I-Pst I fragment containing most of exon 1 and its upstream sequence and a 3.0 kbp Hind III-Hind III fragment containing exons 2, 3, and part of 4 were isolated from the MRG4 gene clone λmrg4gene14-2. The two fragments were subcloned into the upstream and downstream regions of the 1.8 kbp region containing the neomycin resistance gene, respectively, in the pPNT vector, such that a contiguous region of ~7 kbp with 80% homology to the MRG4 gene was created.
Construction of the KO mouse
All procedures using animals were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The pPNT targeting vector was constructed in our laboratory, and electroporated into RW4 ES cells grown on murine embryonic fibroblast feeder cells at Genome Systems (St. Louis, MO). Neomycin-resistance cell lines were isolated after selection with G418 (300–400 μg/ml) for ~5 days and frozen. DNA was prepared from the isolated cell lines and used to confirm successful targeting of the MRG4 gene by genomic Southern blot analysis in our laboratory. The ES cell lines determined to be successfully targeted were injected into C57Bl/6 blastocysts at Genome Systems and chimeras were produced after transplantation into female C57Bl/6 mice. The degree of chimerism was approximated by the amount of agouti coloring of the coat. Chimeras were mated with C57Bl/6 partners, and germ line transmission in pups was determined by the agouti phenotype. The presence of a targeted MRG4 allele was confirmed in agouti pups by DNA isolation from the tail and genomic Southern blot analysis. Targeted agouti F1s were crossed with each other to produce homozygous targeted F2s. Homozygosity of the targeted MRG4 alleles was confirmed by DNA isolation and genomic Southern blot analysis or polymerase chain reaction (PCR) amplification.
Western blot analysis
Mouse retinas were homogenized in PBS containing 1 μg/mL aprotinin and 100 μg/mL phenylmethylsulfonylfluoride (PMSF) and centrifuged at 15,000 xg. Protein concentration of the supernatant was measured using the Protein Determination Kit (Sigma, St. Louis, MO). Equal amounts of retinal proteins were boiled and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the proteins were electroblotted onto a nitrocellulose membrane (Immobilon-P; Millipore, Bedford, MA). The blot was incubated at 4°C overnight in a blocking buffer containing 5% (wt/vol) nonfat dried milk in 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, and 0.05% Tween20. After the blocking of nonspecific binding sites, the blot was incubated with appropriate primary antibody (1:200 affinity-purified polyclonal antibody against rat HRG4 (RRG4) (Higashide et al., 1998), 1:1000 monoclonal anti-rat synaptotagmin antibody, 1:200 monoclonal anti-rat synaptogyrin antibody, and 1:3000 monoclonal anti-rat syntaxin antibody (Transduction Laboratories, Lexington, KY)) at room temperature for one hour. After three washes with TBS-T (0.1% Tween 20 in TBS), the membranes were incubated with the secondary antibody (peroxidase-conjugated anti-rabbit or anti-mouse IgG antibody; Amersham Pharmacia Biotech, Piscataway, NJ) at a dilution of 1:1000 in TBS-T at room temperature for one hour. Finally, the membranes were washed five times in TBS-T and processed for detection by enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech). Signals on exposed film were quantitated by densitometry, and statistical significance of differences in signals was determined by the Student t-test.
Immunofluorescence
Age-matched KO and control (sibling) mice were examined. Mouse eyes were enucleated, fixed in 4% paraformaldehyde in 0.1 M phosphate buffer for 2 hours, and immersed in 20% sucrose in 0.1M phosphate buffer overnight. Before sectioning, the eyes were embedded in OCT (Miles, Elkhart, IN) and frozen in liquid nitrogen. Six μm thick sections were cut on a cryostat and stored at −20° C until used. The frozen sections were warmed to room temperature, blocked for 60 minutes with blocking buffer (10% goat serum, 1% BSA, 0.1% Triton-X 100 in PBS, pH 7.4), and incubated with the primary antibody against rat HRG4 (RRG4, 1:100) in PBS for 90 minutes (Higashide et al., 1998). After three washes in PBS, sections were incubated with the secondary antibody (Alexa Fluor 488 goat anti-rabbit IgG, Molecular Probes/Invitrogen, Carlsbad, CA) at a dilution of 1:50 for 30 minutes, followed by three final washes in PBS. The sections were mounted in Fluoromount-G (Southern Biotechnology Associates, Inc, Birmingham, AL), analyzed, and digitally photographed with Zeiss photomicroscope III or Zeiss Laser Scanning System LSM 510 (Carl Zeiss, Oberkochen, Germany).
Fundus examination
Mice were anesthetized with a mixture of ketamine, xylazine, and urethane, pupils were dilated with phenylephrine and atropine, and fundus photographs were taken with a Kowa RC2 camera (Kowa, Tokyo, Japan).
Electroretinogram (ERG) study
Knock-out and age-matched control (sibling) mice of various ages (2 (2 KO, 1 N), 4 (1 KO, 1 N), 6 (1 KO, 1 N), 12 (2 KO, 1 N), 14 (2 KO, 1 N), 15 (2 KO, 1 N), 18 (1 KO, 1 N), and 19 months (1 KO, 1 N)) were subjected to ERG study. By the time of the experiment the animals had been dark adapted for 14–16 hours; all the subsequent steps in preparation for recording were performed under dim red light. Mice were anesthetized with an intraperitoneal injection of ketamine (60.0–75.0 mg/kg) and xylazine (6.7–8.3 mg/kg). After pupils were dilated (tropicamide 1%, phenylephrine HCl 2.5%; Bausch and Lomb Pharmaceuticals, Inc., Tampa, FL), the corneal surface was anesthetized with proparacaine HCl 0.5% (Alcon Laboratories, Inc., Fort Worth, TX) before the animal was immobilized in a holder. ERGs were recorded simultaneously from both eyes using an Espion visual electrophysiology system (Diagnosys LCC, Littleton, MA) as previously described (Sokolov et al., 2004). The recording electrodes were platinum wires enclosed inside plastic armatures that completely covered the mouse pupil. Electrical contact between the cornea and the electrode was achieved through a hypromellose solution (Gonak; Akorn, Inc., Buffalo Grove, IL) which also protected the eyes from drying during the experiment. The reference electrode was clipped at the cheek and electrical contact was established through Gonak; the ground electrode was clipped at the tail. Mouse body temperature was maintained at 37 °C using a homeothermic blanket connected to its control unit (Harvard Apparatus, Holliston, MA). Signals were bandpass-filtered at 0.15–500 Hz and sampled at 1 kHz. Flash intensity ranged from −3.52 to 1.76 log cd s/m2. Data analysis was performed with Igor Pro 5.0 software (Wavemetrics, Lake Oswego, OR).
Histopathology
Frozen and paraffin sections of retina from normal and KO mice of different ages were stained with hematoxylin and eosin (H&E), and examined microscopically. For determination of outer nuclear layer thickness, retinal sections were obtained along the vertical meridian through the optic nerve head (ONH) from the superior to inferior periphery. The entire span was divided into zones 4 mm (peripheral), 2 mm (mid), and 0.5 mm (central) away from the ONH, and the thickness of the outer nuclear layer (ONL) was measured in each zone in the digitized images. 4–5 readings were taken for each zone and averages were calculated. Statistical significance of differences in nuclear layer thickness in each zone of the retina for a given age in KO and normal (sibling) mice was determined by the Student’s t-test. Photomicrographs were taken to document the histopathological changes.
Ultrastructural analysis by electron microscopy (EM)
Age-matched KO and normal control mice (sibling) were examined at ages 7, 12, and 18 months. Mice were euthanized and eyes were enucleated and immersed in fixative containing 2.0% paraformaldehyde and 2.5% glutaraldehyde in 0.1M phosphate buffer, pH 7.4. For EM, blocks of tissue, fixed as above, were prepared from affected retinal regions of KO mice and comparable regions in controls, then post-fixed in buffered 1% OsO4, dehydrated through a graded series of alcohols and embedded in epoxy resin following standard procedures. Ultrathin sections stained with uranyl acetate and lead citrate were examined in a JEOL electron microscope.
TUNEL staining
Fragmentation of DNA in apoptotic cells was detected by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end-labeling (TUNEL) assay, using the ApoAlert DNA Fragmentation Assay Kit (Clontech Laboratories, Palo Alto, CA) with fluorescence microscopy, or with the DeadEnd Colorimetric TUNEL System (Promega, Madison, WI) and light microscopy according to the manufacturer’s protocol. For quantitative assessment of apoptosis, in order to increase the signal-to-noise ratio, the DeadEnd Colorimetric TUNEL System was converted into Alexa Fluor-based fluorescence detection system by using the biotinylated dUTP for streptavidin-conjugated Alexa Fluor 488 binding (Molecular Probes/Invitrogen, Carlsbad, CA). Numbers of apoptotic cells were determined by counting TUNEL-positive cells in the entire retina, superior to inferior, of vertical meridian sections of the eye from age-matched KO and normal (sibling) mice. Statistical significance was determined by the Student's t-test.
Results
Cloning of the mouse HRG4 gene
A genomic clone containing the murine orthologue of HRG4 gene was isolated from a 129 SvJ mouse genomic library (Stratagene, La Jolla, CA) by hybridization screening using a rat HRG4 (RRG4) cDNA as probe (Higashide et al., 1996). The 129 SvJ library was used to obtain a mouse gene clone with minimal difference to the target gene of homologous recombination in the embryonic stem (ES) cells of 129 SvJ lineage. A lambda Fix II clone (λmrg4gene14-2) with a ~20 kbp insert containing the mouse HRG4 gene (MRG4) was obtained. Restriction digestion and sequencing analyses demonstrated that the mouse gene is similar to the human gene in size and exon/intron structure (Fig. 1A) (Fig. 1A here) (Higashide et al., 1999). The MRG4 gene, like the HRG4 gene, is made up of 5 exons spread over a ~5 kbp genomic region with the translational initiation codon in exon 1.
Figure 1.
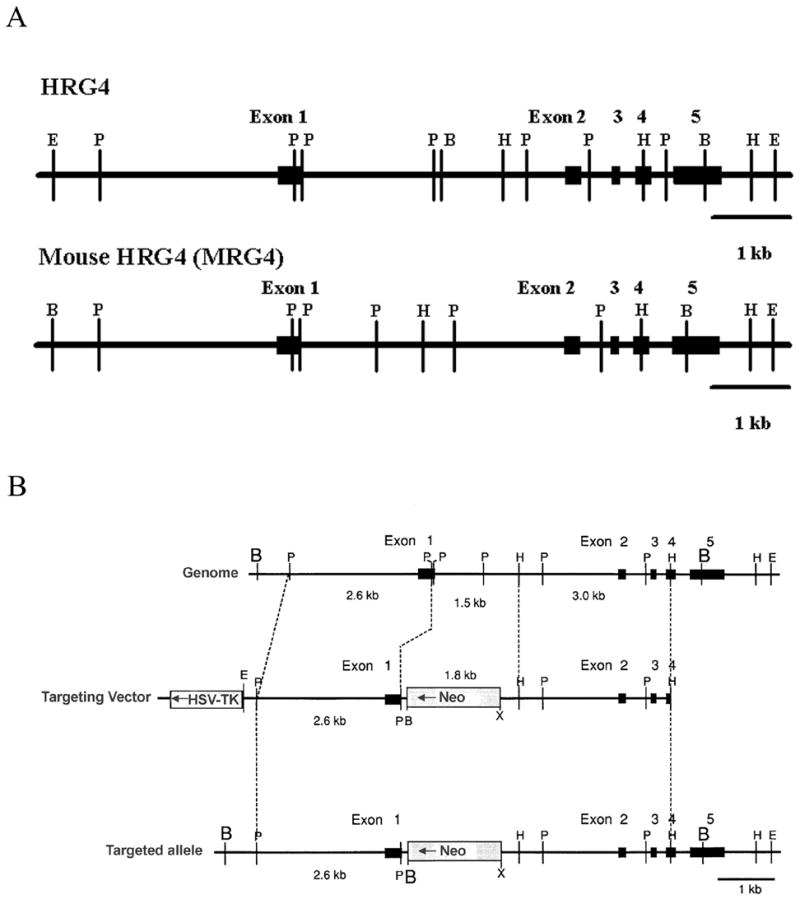
Structure and targeting of the mouse HRG4 (MRG4) gene. A. Exon/intron sturcture of the MRG4 gene (bottom) is compared to that of the human HRG4 gene (top). The mouse gene, like the human gene, consists of 5 exons encompassing ~5 kbp of genomic sequence. Exons are represented by black boxes and labeled. Restriction enzyme sites are shown by letters. B, Bam HI; P, Pst I; H, Hind III; E, Eco RI. B. Targeting of the MRG4 gene. Targeting of the MRG4 gene (top) by the pPNT targeting vector (middle) is shown. The resulting targeted gene is shown at the bottom. The targeting vector contains a neomycin resistance gene in a ~7 kbp sequence homologous to the mouse genomic region containing the upstream region and exons 1 to 4 of the MRG4 gene. A herpes simplex virus thymidine kinase (HSV-TK) gene is also attached at one end of the homologous region in the vector to facilitate the selection of successfully targeted cells. Correct targeting of the vector should result in elimination of this marker. Successful targeting results in replacement of part of exon 1 and intron 1 with the neomycin resistance gene-containing sequence (bottom). A termination codon is built into the sequence downstream of exon 1 to prevent run-on translation. Restriction enzyme sites: B, Bam HI; P, Pst I; H, Hind III; E, Eco RI; X, Xho I. Neo, neomycin resistance gene. HSV-TK, herpes simplex virus thymidine kinase gene. Gene exons are represented by black boxes.
Targeting of the MRG4 gene
The pPNT targeting vector containing a neomycin resistance gene and a herpes virus thymidine kinase at the 3' end and the λmrg4gene14-2 clone were used to construct a MRG4 targeting construct (Tybulewicz et al., 1991). The targeting construct contained a ~7 kbp fragment homologous to the MRG4 gene designed to replace the distal region of exon 1 and intron 1 of the endogenous MRG4 gene with the neomycin resistance gene (Fig. 1B) (Fig. 1B here). A termination codon was incorporated at the junction to prevent run-on translation. A ~6 kbp region of identity to the endogenous MRG4 gene was included in the construct to assure homologous recombination at the correct site.
Construction of the KO mouse
The targeting construct was electroporated into ES cells (Genome Systems, St. Louis, MO), and neomycin-resistant lines were isolated after selection. The ES cell lines were screened for successful targeting of the MRG4 gene by genomic Southern blot analysis of their DNA. Two different ES cell lines demonstrated targeting by the presence of a 5.3 kbp Bam HI fragment produced by the replacement of the gene by the ~7 kbp homologous sequence (Fig. 1B) (data not shown). After injection into mouse blastocysts (C57B6), one of the targeted ES cell lines, #95, produced chimeras with high degree of chimerism (95%). After mating with normal female (Black Swiss Webster), one of the chimeras produced an Agouti pup, indicating germ line transmission. The targeted MRG4 allele was confirmed in the pup, which should be heterozygous, by the presence of the 5.3 kbp Bam HI fragment by genomic Southern blot analysis (data not shown). Back crossing of the Agouti pups produced offspring homozygous for the targeted gene, indicated by the unique pattern of Bam HI fragments on genomic Southern blot analysis (data not shown).
Inactivation of the MRG4 gene by targeting was confirmed by isolation of retinal proteins from 2 months old normal, heterozygous, and homozygous knock-out mice and western blot analysis with the rat HRG4 (RRG4) antibody (Higashide et al., 1998). The presence of MRG4, shown to be enriched in photoreceptors (Higashide et al., 1998), was demonstrated by a strongly reactive band in the normal mouse retina, while a band approximately half as reactive was present in the heterozygous retina, consistent with expression from only one allele (Fig. 2A) (Fig. 2 here). The homozygous retina showed complete absence of MRG4, consistent with the targeted inactivation of both alleles of the MRG4 gene (Fig. 2A). Immunofluorescence microscopic analysis of retinas from age-matched normal and homozygous KO mice demonstrated as expected the presence of MRG4 mostly in the outer plexiform layer (site of photoreceptor synapses) and in the IS in the normal retina as previously demonstrated (Higashide et al., 1998). By contrast, a complete absence of MRG4 immunofluorescent staining was observed in the knock-out retina, consistent with the western blot result (Fig. 2B).
Figure 2.
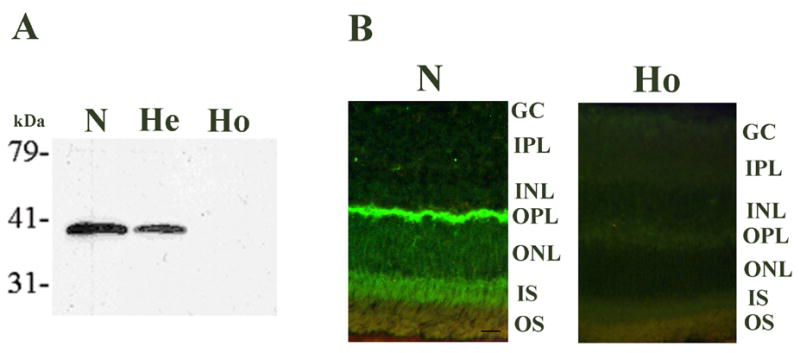
Confirmation of the MRG4 gene targeting in the knock-out mouse. A. Western blot analysis of retinal proteins from normal (N), heterozygous (He), and homozygous (Ho) knockout mouse with the rat HRG4 (RRG4) antibody (1:200). A strongly reactive MRG4 protein band is present in the normal retina (N), whereas a band approximately half as reactive is present in the heterozygous retina (He), consistent with one allele being knocked out by targeting, and a complete absence of the protein band in the homozygous retina (Ho), consistent with both alleles being knocked out. Protein size markers are shown on the left. kDa, kilo Daltons. B. MRG4 immunofluorescence (polyclonal anti-rat HRG4 (RRG4) antibody, 1:100) in 12 months old normal (N) and homozygous knock-out mouse (Ho) retinas. The presence of MRG4, predominantly in the outer plexiform layer and less in the inner segments of the retina, is shown in the normal retina (N). There is complete absence of MRG4 signal in the homozygous knockout mouse retina (Ho). There is evidence of retinal degeneration visible in the homozygous knock-out mouse, i.e., thinning of the outer nuclear layer compared to normal thickness. GC, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; IS, inner segments; OS, outer segments. Scale bar in N = 10 micrometer.
Phenotype of the knock-out mouse
The homozygous KO mice did not show any abnormality in general breeding or developmental characteristics. No gross physical abnormality could be detected. Funduscopic examination of homozygous KO mice of different ages, along with selected normals, demonstrated a progressive pattern of abnormality consistent with retinal degeneration. Figure 3A (Fig. 3 here) shows representative fundus photographs of 2, 5, 10, 17, 20, and 26 month old homozygous KO mice compared with 17 and 19 month old normals. Mottling of retinal pigment epithelium (RPE) was visible in the fundus as early as 5 months of age in the KO mice. The mottling appeared to be randomly distributed throughout the retina with no specific pattern. An increase in the areas of mottling with regions of coalescence was observed by 10 months of age. Thinning of blood vessels was also present. The whole retina appeared to be involved by 17 months. Evidence of severe retinal degeneration was present at 20 months with areas of photoreceptor thinning. Severe end-stage retinal degeneration was present at 26 months.
Figure 3.
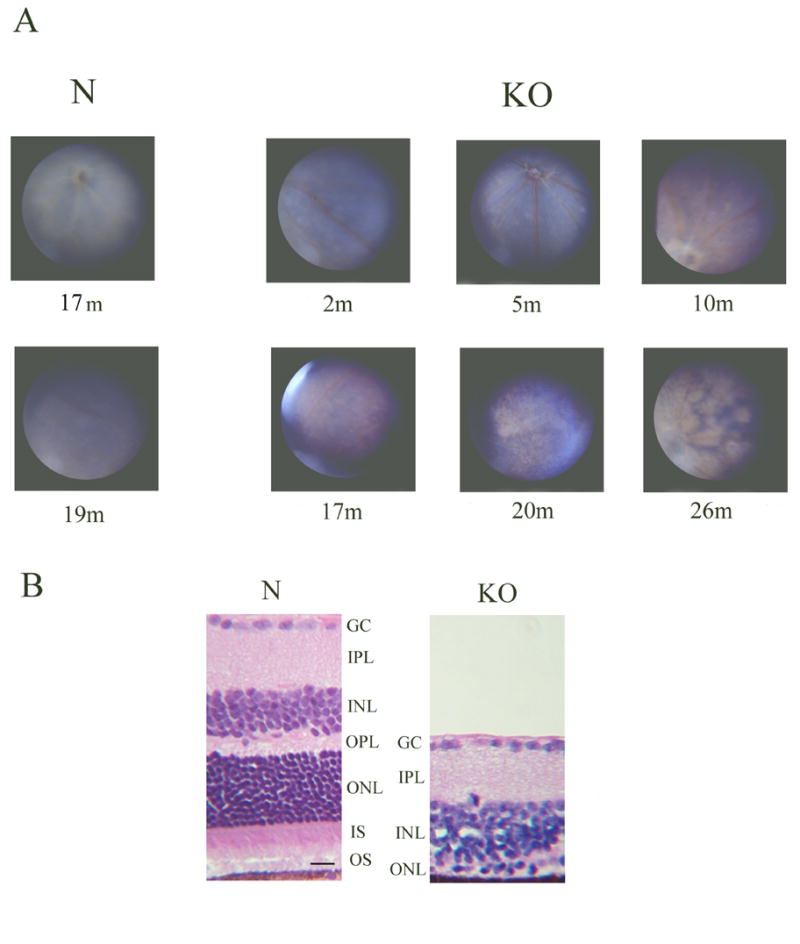
Fundus images of the homozygous knock-out mouse and histologic confirmation of retinal degeneration. A. Fundus photographs of 17 and 19 months old normal mice (N) and 2, 5, 10, 17, 20, and 26 months old homozygous KO mice are shown. The fundus photographs were taken with the Kowa RC2 camera. No significant abnormality is present in the KO fundus at 2 months of age, but mottling of the RPE begins to appear in all areas of the retina at 5 months of age. The mottling increases in area and begins to coalesce, and blood vessels begin to narrow by 10 months. The entire retina is involved and areas of retinal thinning are visible by 17 months of age. By 20 to 26 months, severe retinal degeneration affecting the entire retina is seen. No abnormality is seen in the fundus of 17 and 19 months old normal mice (N). B. Histopathology of normal (N) and homozygous KO mouse retina at 20 months of age. H&E stained sections of mid retina are shown. Severe retinal degeneration is present in the KO retina with barely a single row of photoreceptor nuclei left (ONL), consistent with the fundus image obtained. GC, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; IS, inner segments; OS, outer segments. Scale bar in N = 10 micrometer.
The presence of severe retinal degeneration in this model is illustrated by hematoxylin-eosin (H&E) staining of retinal sections from a 20 month old homozygous KO mouse compared to normal (Fig. 3B). As can be seen, the retina in the knock-out model is significantly thinned with most of the outer nuclear layer gone except for one partial row of nuclei.
Morphometric analysis of the retina from normal and KO mice at different ages was performed. The thickness of the outer nuclear layer comprising photoreceptor nuclei was determined in the retinal sections of 6, 10, 13, 17, 20, and 26 month old normal and KO mice (Fig. 4) (Fig. 4 here). A clear pattern of progressive retinal degeneration was observed. A significant thinning of the ONL was present as early as 6 months, in agreement with the funduscopic analysis (Fig. 4). The progress of the degeneration, as gauged by the thinning of the ONL, was relatively slow in the first 13 months, but accelerated at 17 months. Although the superior retina appeared to be more affected at 6 months, the degeneration seemed to affect superior and inferior regions equally overall, consistent again with the funduscopic analysis. Severe retinal degeneration in this model was confirmed by the thinning of the ONL down to almost nothing by 26 months of age (Fig. 4).
Figure 4.
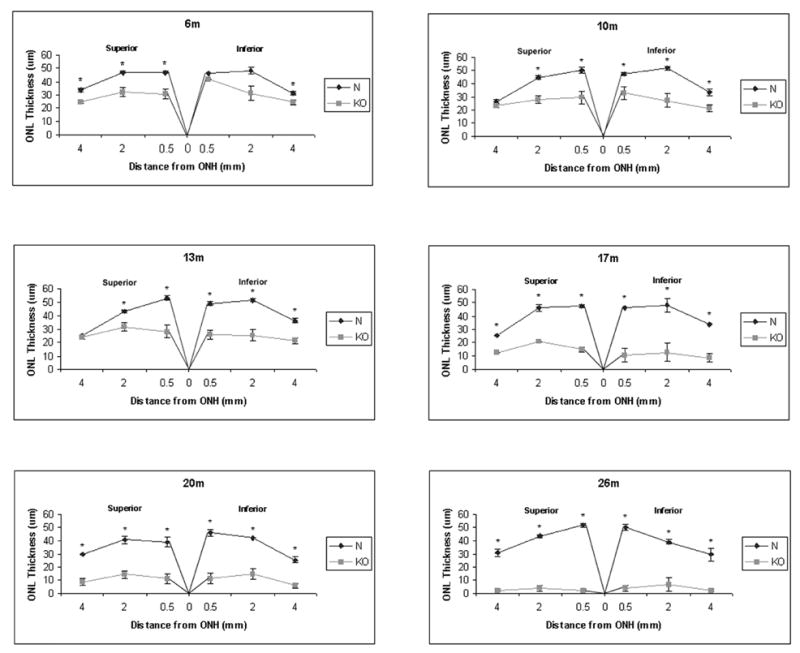
Morphometric analysis of normal and KO retinas at different ages. Thickness in μm of the outer nuclear layer (ONL) containing the photoreceptors is shown for normal (N) and homozygous KO retinas at 6, 10, 13, 17, 20, and 26 months of age. For each measurement, ONL thickness was determined at a point 0.5, 2, and 4 mm from the optic nerve head (ONH) superior or inferior in a vertical meridian section of the retina stained with H&E. Each point on the graph represents an average of measurements from 3–4 animals, and error bars represent standard error of means. Asterisk indicates statistically significant difference between the normal and KO ONL thickness at p<0.05. The results demonstrate a pattern of progressive retinal degeneration in the KO retina, consisting of thinning of the ONL which follows a relatively slow and steady pace until 17 months of age when it accelerates dramatically to result in barely a single row of photoreceptor nuclei left by 20 months of age. No difference in the severity of degeneration is observed between the superior and inferior retina.
The level of apoptosis was measured in KO and normal (sibling) retinas at different ages by counting the number of TUNEL-positive cells present in the ONL in the whole 6 μm vertical meridian sections of the retina (Fig. 5A) (Fig. 5 here). A graph of the data (Fig. 5B) demonstrates that as early as 6 months of age, a higher number of apoptotic cells is present in the KO retina compared to normal. A burst of apoptosis appears to occur in the KO retina starting around 17 months and continuing through 20 months, consistent with the results of both the funduscopic and morphometric analysis. The number of TUNEL-positive cells subsequently decreases to almost none by 26 months of age. In the normal ONL, the number of apoptotic cells is relatively constant, at 2 to 3 per section, at all ages examined (6–26 months).
Figure 5.
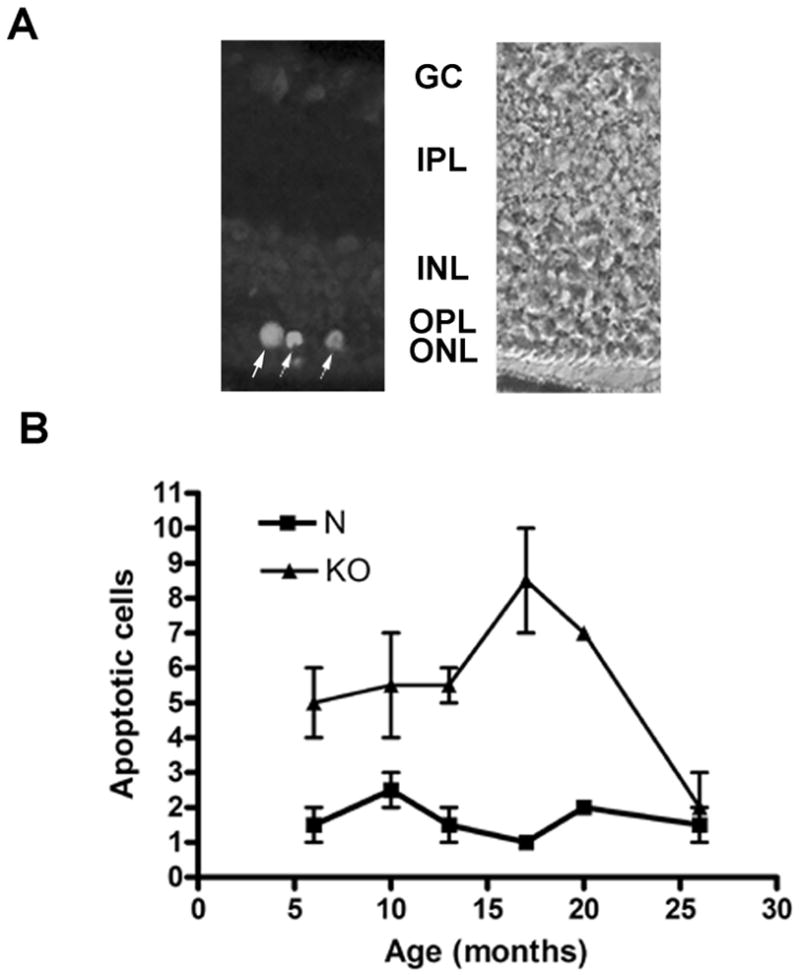
Apoptosis in normal and KO retinas at different ages. A. Immunofluorescence (Alexa Fluor 488) of TUNEL-positive photoreceptor cells (arrows) is shown in 20 months old KO retina (left) with corresponding phase image (right). Most of the photoreceptors in the outer nuclear layer is degenerated except for 1–2 rows of nuclei. GC, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer. B. A graph of number of TUNEL-positive apoptotic cells in KO and normal (N) (sibling) retinas at different ages is shown. Each point represents the average number of TUNEL-positive photoreceptors present in a microscopic section (vertical meridian) spanning the whole retina from three normal or homozygous KO mice of specified age. An increase in number of apoptotic cells is seen in the KO retina compared to normal as early as 6 months. An abrupt increase in apoptosis is seen in the KO retinas at 17–20 months, consistent with the acceleration of the retinal degeneration after 17 months demonstrated by the morphometric analysis. Error bars = standard error of means. The differences in the number of apoptotic cells between the normal and KO retinas were statistically significant (p<0.05) by the Student's t-test for all ages except 10 months.
Histopathologic analysis of the normal and KO retina at various ages was performed. Figure 6 (Fig. 6 here) shows representative H&E sections of mid retina of normal and KO mice at 6, 10, 13, 17, 20, and 26 months of age. A pattern of progressive retinal degeneration is observed which progresses relatively slowly in the first 13 months, and accelerates at 17 months, to result in complete obliteration of the ONL by 26 months of age. The architecture of the ONL, however, appears to remain relatively intact during the process of gradual thinning, in contrast to significant disorganization that was observed in the TG model (Kobayashi et al., 2000). The inner nuclear layer (INL) also does not appear to show significant thinning in the KO retina compared to normal until the terminal stage of retinal degeneration, again in contrast to prominent early thinning of the INL, reflecting significant trans-synaptic degeneration, that was observed in the TG model (Mori et al., 2006).
Figure 6.
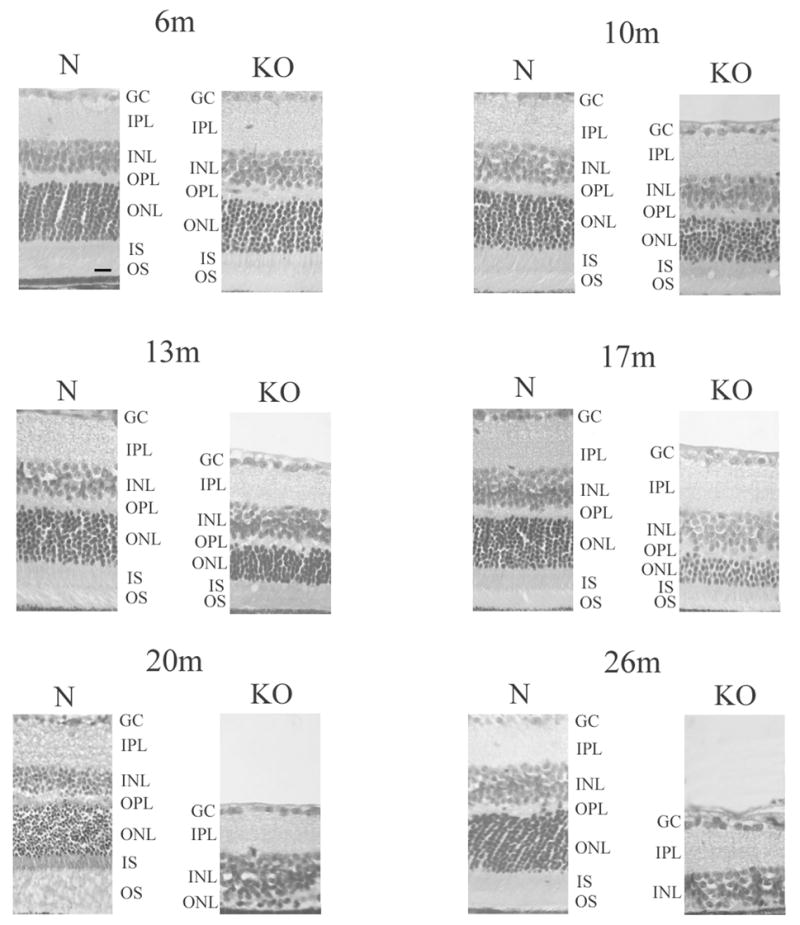
Histopathology of the KO retina. H&E stained sections of retinas from normal (N) and homozygous KO mice at 6, 10, 13, 17, 20, and 26 months of age are shown. Progressive retinal degeneration is evident with gradual thinning of the outer nuclear layer containing the photoreceptors until 17 months of age when there is an acceleration of the degeneration, resulting in virtual disappearance of the photoreceptors by 20 and 26 months of age. The thickness of the inner nuclear layer does not appear to decrease much in the KO retina over time until the terminal stage at 26 months. The architecture of the outer nuclear layer appears to be maintained while it undergoes thinning in the KO retina. The histopathologic time course of retinal degeneration in the KO retina is consistent with the time course of changes seen in the funduscopic, morphometric, and apoptosis analyses. GC, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; IS, inner segments; OS, outer segments. Scale bar in 6m N = 10 micrometer.
Analysis of the photoreceptors at the ultrastructural level by EM of the KO and age-matched normal control retina at 7, 12, and 18 months of age demonstrated evidence of degenerating photoreceptors and synapses, especially at the older ages (data not shown). No evidence of early synaptic degeneration was present without photoreceptor degeneration. Significantly, synapses filled with dark granular deposits consistent with "dark degeneration" of terminals (Ghetti et al., 1972) that were commonly observed in the TG (Kobayashi et al., 2000) were not present in the KO, regardless of age, indicating a different mechanism of synaptic degeneration in the KO.
ERG study of KO and age-matched control (sibling) mice of various ages (2, 4, 6, 12, 14, 15, 18, and 19 months) demonstrated that mice 12 months and younger had virtually normal ERG. Even though statistical analysis was not possible based on only one or two animals available at each age, the ERG of all six mice of ages 2 to 12 months were consistently within the normal range. However, the ERG of six older animals was clearly abnormal. Examples from a 14-month-old and a 15-month-old mouse are shown in Figure 7 (Fig. 7 here) with the morphology of their retinas. As the histology from the same animal shows, the ONL had thinned to 5–8 rows and 3–5 rows at 14 and 15 months, respectively. A reduction in both a- and b-wave was preliminarily noted at high light intensity which did not normalize with increase in interstimulus intervals. Animals at more advanced stages of retinal degeneration had severely diminished or no recordable ERG responses. In contrast to the previously reported ERG of the TG mice, none of the KO animals showed any sign of a drastically reduced b-wave concomitantly with a substantially preserved a-wave. Furthermore, oscillatory potentials (OP) superimposed on the slower waveform in the 14 months KO were similar to those of normal, at least in the mid-range of light intensities. These OP are contributions to the ERG responses by neurons in the inner retina (Wachtmeister, 1998). The appearances of the residual b-waves and OP in the 15 months KO suggested that in spite of the severe thinning of the ONL, the transmission of visual signal from the remaining photoreceptors to the inner retina remained relatively intact.
Figure 7.
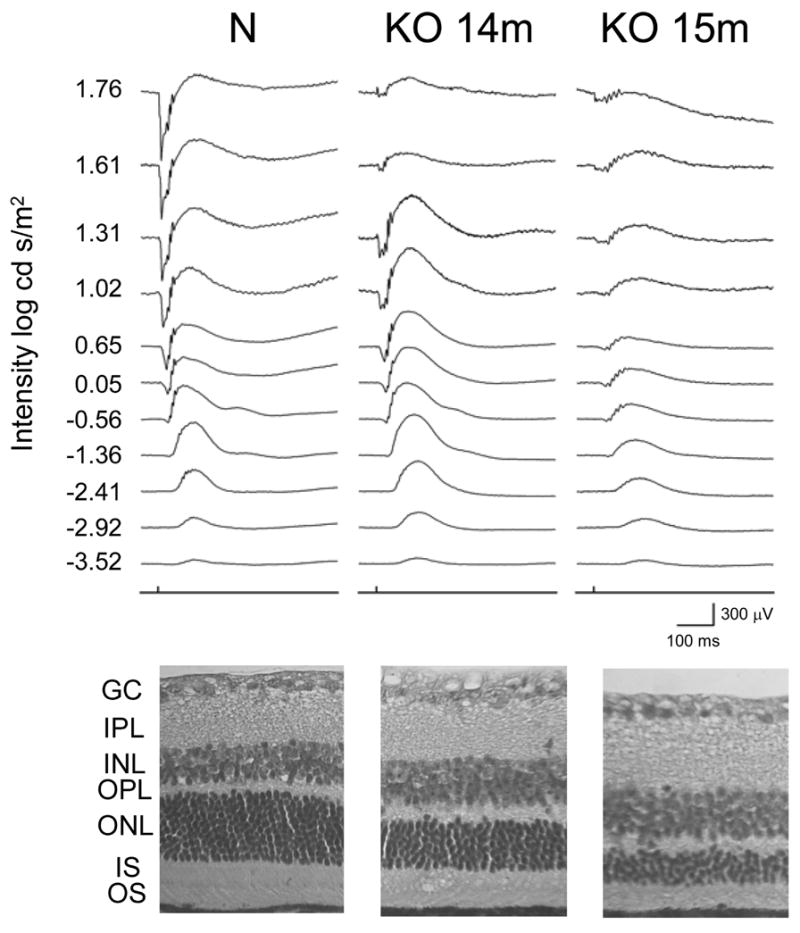
ERG study of KO and control mice. The ERG of 14-month-old KO, 15-month-old KO (2 mice of each age), and control mice (one of each age) are shown with retinal morphology of the same mice. Abnormal ERG characterized by a reduction in both a- and b-wave at high light intensity is shown for the KO mice which worsens with age and correlates with the thinning of the photoreceptor layer (ONL). The ONL of the 14-month-old and 15-month-old KO is thinned to 5–8 rows and 3–5 rows of nuclei, respectively, compared to 10–12 rows for the normal control. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; IS, inner segments; OS, outer segments.
In view of the morphological evidence by light microscopy and EM and functional evidence by ERG indicating absence of early photoreceptor synaptic degeneration in the KO, the functional status of synapses in the KO retina was investigated further by analysis of synaptic proteins by western blotting. A similar analysis of the TG model, which showed early photoreceptor synaptic degeneration, demonstrated changes in the levels of various synaptic proteins by 13 months of age (Kubota et al. 2002). Status of synaptotagmin and synaptogyrin which are synaptic vesicle proteins, and syntaxin which is a pre-synaptic membrane protein (Sudof 1995) was examined. Western blotting of retinal proteins from 3, 6, 10, and 20 months old KO and normal siblings for synaptotagmin, synatptogyrin, and syntaxin demonstrated no significant difference between the KO and control, consistent with the absence of early synaptic degeneration in the KO (Fig. 8) (Fig. 8 here).
Figure 8.
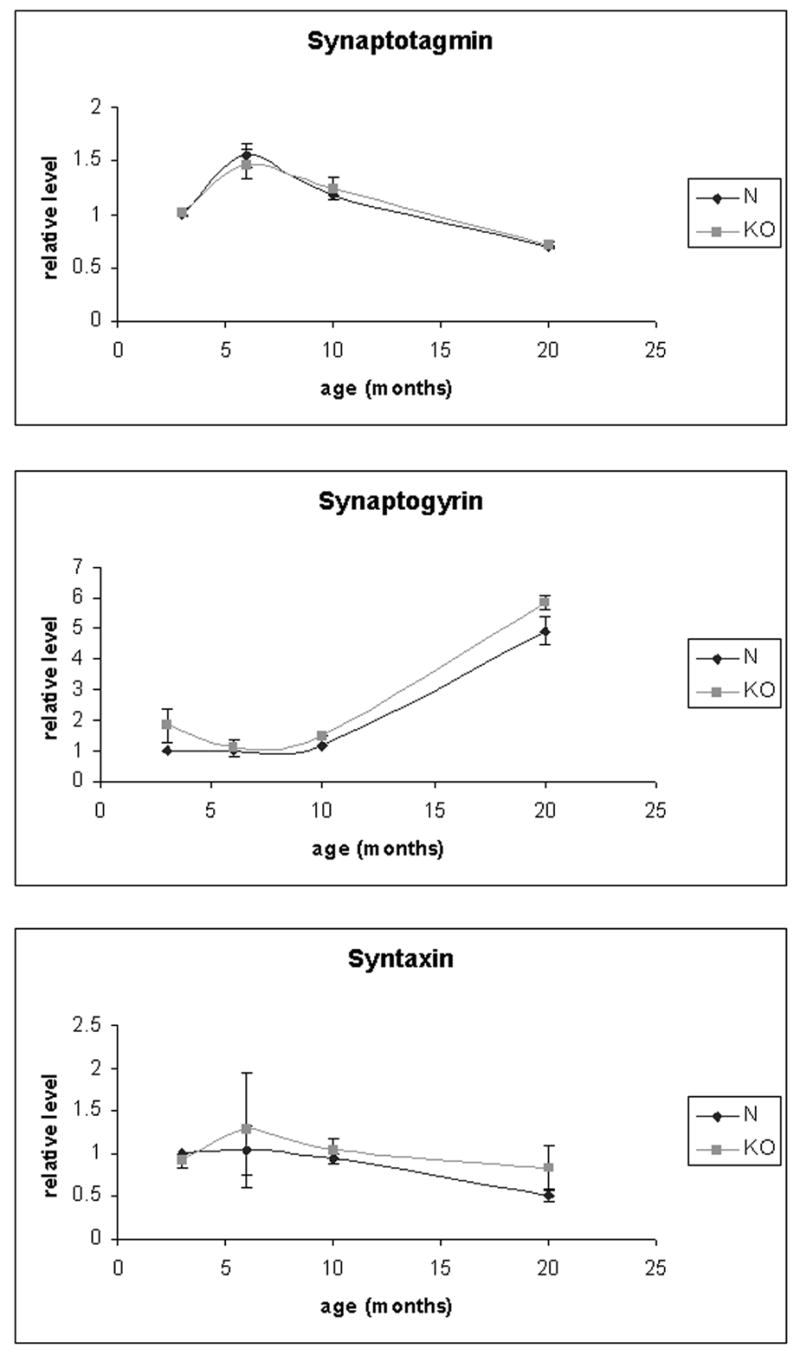
Western blot analysis of synaptic proteins in the KO and control retina. The protein levels of synaptotagmin, synaptogyrin, and syntaxin in the KO and normal control retinas at 3, 6, 10, and 20 months of age are shown. The levels of proteins were obtained by western blot analysis of total retinal proteins with the corresponding antibodies (duplicates), quantitated by densitometry of the ECL bands, and normalized to the average level of 3 months normal control as 1 for each protein. The error bars are standard error of means. There was no statistically significant difference between the KO and control for any of the tested age and protein.
Preliminary histopathologic analysis of heterozygous KO mice showed a variable pattern of retinal phenotype. Most of the heterozygotes had a normal retina regardless of age, but up to a third of mice older than 17 months showed mild thinning of ONL. The morphology was identical to that seen in the homozygous KO, except the ONL thinning was partial (up to 40–50%) instead of complete as in the homozygotes.
Discussion
Inactivation of mouse HRG4 (MRG4) by gene targeting resulted in progressive degeneration of the retina. Thus, both dominant negative mutation of HRG4 in the TG model (Kobayashi et al., 2000) and elimination of HRG4 by gene targeting in the KO model resulted in retinal degeneration, attesting to the importance of HRG4 for normal function of the retina. Although both gene manipulations resulted in slow retinal degeneration, there were significant differences in the phenotypes produced in the two models, reflecting important differences in the predominant pathogenic mechanism involved in each.
First, the TG and KO models differed in the timing and severity of the retinal degeneration. Evidence of photoreceptor degeneration, indicated by increased apoptosis and thinning of the ONL, was present as early as 6 months in the homozygous KO, whereas clear evidence of ONL thinning was not present in the TG until they were >12 months old (Kobayashi et al., 2000). There was a clear difference in the severity of the retinal degeneration at the terminal stage in the two models with barely a single row of photoreceptor nuclei, most likely cones, remaining in the KO by 20 months compared to 4–5 rows of nuclei present at 24 months in the TG (Kobayashi et al., 2000; Mori et al., 2006). The heterozygous KO demonstrated a variable phenotype of much milder retinal degeneration, suggesting partial haploinsufficiency, but this requires further investigation for confirmation.
Second, while significant evidence of trans-synaptic degeneration with thinning of the INL could be appreciated in the TG even at relatively early stage (12–18 months) of retinal degeneration in the TG model (Kobayashi et al., 2000; Mori et al., 2006), there was essentially no evidence of trans-synaptic degeneration in the KO until the terminal stage (>20 months) of retinal degeneration when barely a single row of photoreceptors was present. The absence of trans-synaptic degeneration was also confirmed by normal OP in the ERG of 14 and 15 months KO, reflecting normal function of the inner retina. This is a very important point that reflects the presence or absence of early synaptic degeneration. We have recently demonstrated in the TG model that trans-synaptic degeneration of a secondary neuron such as bipolar cell is directly caused by the degeneration that occurs in the synapse of the primary neuron (photoreceptor) associated with the secondary neuron and that this can occur even before the death of the whole photoreceptor (apoptosis in the cell body) is evident in the early stage of retinal degeneration (Mori et al., 2006). Thus, the absence of trans-synaptic degeneration in the KO strongly suggests that early synaptic degeneration does not occur in the KO as does in the TG, and a different mechanism of photoreceptor degeneration is operating.
Third, the ultrastructural analysis of photoreceptor synapses in the KO by EM showed evidence of degenerating synapses at ages, especially older ages, when photoreceptor degeneration was present (7, 12, and 18 months), as expected. However, there was no evidence at any of the ages examined of dark degeneration of synaptic termini, i.e., degenerating synapses filled with dark granules (Ghetti et al., 1972), in the KO as was observed in the TG. In the TG these dark degenerating synapses were commonly observed in the outer plexiform layer (OPL) where photoreceptor synapses reside (Kobayashi et al., 2000), and photoreceptor synaptic degeneration has been shown to be the initial event, followed by trans-synaptic degeneration and whole photoreceptor degeneration by apoptosis in the cell body (Mori et al., 2006). Thus, the dark degeneration of the synapses was a signature characteristic of the early synaptic degeneration that occurs in the TG. The absence of this phenomenon in the KO again strongly supports the absence of early synaptic degeneration in the KO and a different mechanism of cell death in the KO in which the whole photoreceptor dies instead of a sequential process of synaptic degeneration, followed by whole photoreceptor death.
The result of the ERG analysis of the KO clearly confirmed the absence of early synaptic degeneration in the KO and firmly supported a different mechanism by which whole photoreceptors degenerate and die in the KO. When ERG dysfunction was present, reduction in both the a-wave, reflecting phototransduction in the outer segments (OS), and b-wave, reflecting synaptic transmission, was present. There was preliminary evidence of reduction of both a- and b-wave at high light intensity, perhaps not corrected by lengthening of interstimulus intervals, suggesting that recovery may not be the problem. This preliminary observation may reflect a unique effect of the lack of HRG4 and is a subject of future investigation. Importantly, there was no evidence of early reduction in the b-wave while the a-wave was preserved, as seen in the TG. Thus, the ERG defect seen in the KO is consistent with a dysfunction of whole photoreceptors without evidence of preceding synaptic degeneration. This is dramatically different from the TG which showed early b-wave reduction, reflecting synaptic dysfunction, before real evidence of OS dysfunction and whole photoreceptor death was observed (Kobayashi et al., 2000). Thus, the result of the ERG study agreed with the absence of trans-synaptic degeneration and the result of the EM analysis in demonstrating a mechanism of pathogenesis in the KO that is quite different from that in the TG, i.e., a mechanism that operates outside the synapses, namely in the distal end of photoreceptors (inner segments/outer segments, IS/OS) where HRG4 is also localized (Higashide et al., 1998).
Finally, the results of the western blot analyses of synaptic proteins in the KO and normal retina at various ages starting at 3 months confirmed the absence of early synaptic degeneration in the KO. The levels of proteins associated with synaptic vesicles (synaptotagmin, synaptogyrin) and pre-synaptic membrane (syntaxin) were all essentially unchanged in the KO compared to normal. The levels of these proteins may be expected to show a decrease in the older KO due to a significant degeneration of photoreceptors. The lack of significant decrease must be due to the persistent intactness of the inner nuclear and inner plexiform layer which contains many synapses, which again argues against early synaptic degeneration which would have induced trans-synaptic degeneration and inner nuclear layer dysfunction. The TG model which showed early synaptic degeneration, however, demonstrated alterations in synaptic proteins, including synaptotagmin and syntaxin, even at 13 months of age (Kubota et al. 2002). The absence of such alteration in the synaptic proteins in the KO clearly supports the absence of early synaptic degeneration in this model.
The remarkable difference in the phenotype of the retinal degeneration in the TG and KO, one involving early synaptic degeneration at one end and the other involving IS/OS dysfunction at the other end of photoreceptors, respectively, point to a dual function of HRG4. The dominant negative truncated mutant HRG4 in the TG was shown to sequester its target, ARL2, three times as much as the normal HRG4, leading to a decrease in the downstream target, synaptic mitochondrial ANT1, mitochondrial stress, and synaptic degeneration which triggered trans-synaptic degeneration and whole photoreceptor death by apoptosis (Mori et al. 2006). This process was slowly progressive and was unique to the photoreceptor synapses, reflecting the importance of this mechanism in the synapses. Absence of HRG4 in the KO may be expected to affect the same mechanism in the synapses, yet synaptic degeneration was not present as the first initiating event in the KO. Instead, there was preliminary evidence of a unique photoreceptor dysfunction with reduction of both a-wave and b-wave whenever ERG abnormality and photoreceptor degeneration was present. This indicated the presence of a second function of HRG4, not related to synapse, in the IS/OS region of photoreceptors where HRG4 also localizes which is defective in the KO and leads to cell dysfunction and death. Defects in phototransduction proteins in the OS accompanied by phototransduction abnormalities have been well documented to cause photoreceptor death, as in the case for rhodopsin, cGMP-phosphodiesterase, and transducin (Dryja et al., 1990, 1996; Bowes et al., 1990; Pittler and Baehr, 1991; Michaelides et al., 2004). A defect in the putative second function of HRG4 may lead to abnormality in a phototransduction protein, causing photoreceptor death. The photoreceptor death occurs much earlier in the KO compared to the TG, indicating that this IS/OS dysfunction is serious enough to kill the photoreceptors more quickly. Thus, the absence of initial synaptic degeneration in the KO may be due to the quicker demise of photoreceptors from the IS/OS dysfunction even before the absence of HRG4 has a chance to cause synaptic degeneration. The IS/OS function affected in the KO is unlikely to be the mitochondrial ANT-1-mediated mechanism, elucidated in the TG, since this mechanism has been shown to operate predominantly in the synapse by the TG model. Thus, the findings point to a second function of HRG4 which is important in the IS/OS region and which is defective in the KO, leading to photoreceptor death. In this regard, in contrast to the TG model, the phenotype of the KO model is somewhat similar to other mouse models of retinal degeneration based on abnormalities of OS proteins, such as rhodopsin, rds/peripherin, and rom-1, in terms of rate of photoreceptor degeneration and absence of early synaptic degeneration (Kedzierski et al. 2001).
A function of HRG4 in the IS/OS region is consistent with the presence of HRG4 in the IS which has always been noted (Higashide et al., 1998). Although the precise function of HRG4 in the IS is not known at this time, it is plausible that absence of this function in the IS in the KO leads to a serious defect in the distal end of photoreceptors and photoreceptor degeneration as observed. Thus, the KO model of HRG4 has revealed a likely second, new function of HRG4, present and important in the IS/OS region and uniquely highlighted because of the specific phenotype its absence produces in the KO. The KO and TG models of various retina-expressed proteins, such as rhodsopsin, cGMP-phosphodiesterase gamma, arrestin, transducin alpha, RPE65, peripherin/rds, retinol dehydrogenase, TrkB receptor, and interphotoreceptor retinoid-binding protein (IRBP), have been invaluable in understanding their function and pathogenic mechanism of retinal dysfunction they can cause (Lee et al., 2006; Burns et al., 2006; Naash et al., 2004; Maeda et al., 2005; Fan et al., 2003; Jones et al., 2003; Rohrer, 2001; Calvert et al., 2000, 2001; Frederick et al., 2001; Ripps et al., 2000; Palczewski et al., 1999; Lem et al., 1999; Tsang et al., 1998; Humphries et al., 1997). Similarly, the KO model should be invaluable in elucidating the putative new function of HRG4 in the distal end of photoreceptors, and in combination with the TG model which shows synaptic degeneration at the proximal end of photoreceptors (Kobayashi et al., 2000; Mori et al., 2006), should help us to understand the dual function of HRG4.
Acknowledgments
Supported by NIH (EY10848, P30 EY014801, RO1EY14209, P30EY05722), Foundation Fighting Blindness, Inc., and Research to Prevent Blindness, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, Jacobson SG, Hauswirth WW, Bennett J. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- Burns ME, Mendez A, Chen CK, Almuete A, Quillinan N, Simon MI, Baylor DA, Chen J. Deactivation of phosphorylated and nonphosphorylated rhodopsin by arrestin splice variants. J Neurosci. 2006;26:1036–1044. doi: 10.1523/JNEUROSCI.3301-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert PD, Krasnoperova NV, Lyubarsky AL, Isayama T, Nicolo M, Kosaras B, Wong G, Gannon KS, Margolskee RF, Sidman RL, Pugh EN, Jr, Makino CL, Lem J. Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha -subunit. Proc Natl Acad Sci U S A. 2000;97:13913–13918. doi: 10.1073/pnas.250478897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert PD, Govardovskii VI, Krasnoperova N, Anderson RE, Lem J, Makino CL. Membrane protein diffusion sets the speed of rod phototransduction. Nature. 2001;411:90–94. doi: 10.1038/35075083. [DOI] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, Sandberg MA, Berson EL. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- Dryja TP, Hahn LB, Reboul T, Arnaud B. Missense mutation in the gene encoding the alpha subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat Genet. 1996;13:358–360. doi: 10.1038/ng0796-358. [DOI] [PubMed] [Google Scholar]
- Fan J, Rohrer B, Moiseyev G, Ma JX, Crouch RK. Isorhodopsin rather than rhodopsin mediates rod function in RPE65 knock-out mice. Proc Natl Acad Sci U S A. 2003;100:13662–13667. doi: 10.1073/pnas.2234461100. Epub 2003 Oct 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick JM, Krasnoperova NV, Hoffmann K, Church-Kopish J, Ruther K, Howes K, Lem J, Baehr W. Mutant rhodopsin transgene expression on a null background. Invest Ophthalmol Vis Sci. 2001;42:826–833. [PubMed] [Google Scholar]
- Ghetti B, Horoupian DS, Wisniewski HM. Transsynaptic response of the lateral geniculate nucleus and the pattern of degeneration of the nerve terminals in the rhesus monkey after eye enucleation. Brain Research. 1972;45:31–48. doi: 10.1016/0006-8993(72)90214-4. [DOI] [PubMed] [Google Scholar]
- Higashide T, Murakami A, McLaren MJ, Inana G. Cloning of the cDNA for a novel photoreceptor protein. J Biol Chem. 1996;271:1797–1804. doi: 10.1074/jbc.271.3.1797. [DOI] [PubMed] [Google Scholar]
- Higashide T, McLaren MJ, Inana G. Localization of HRG4, a photoreceptor protein homologous to Unc-119, in ribbon synapse. Invest Ophthalmol Vis Sci. 1998;39:690–698. [PubMed] [Google Scholar]
- Higashide T, Inana G. Characterization of the gene for HRG4 (UNC119), a novel photoreceptor synaptic protein homologous to unc-119. Genomics. 1999;57:446–450. doi: 10.1006/geno.1999.5791. [DOI] [PubMed] [Google Scholar]
- Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, Sieving PA, Sheils DM, McNally N, Creighton P, Erven A, Boros A, Gulya K, Capecchi MR, Humphries P. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–219. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]
- Jones BW, Watt CB, Frederick JM, Baehr W, Chen CK, Levine EM, Milam AH, Lavail MM, Marc RE. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003;464:1–16. doi: 10.1002/cne.10703. [DOI] [PubMed] [Google Scholar]
- Kedzierski W, Nusinowitz S, Birch D, Clarke G, McInnes RR, Bok D, Travis GH. Deficiency of rds/peripherin causes photoreceptor death in mouse models of digenic and dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 2001;98:7718–7723. doi: 10.1073/pnas.141124198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Higashide T, Hamasaki D, Kubota S, Sakuma H, An W, Fujimaki T, McLaren MJ, Weleber RG, Inana G. HRG4 (UNC119) mutation found in cone-rod dystrophy causes retinal degeneration in a transgenic model. Invest Ophthalmol Vis Sci. 2000;41:3268–3277. [PubMed] [Google Scholar]
- Kobayashi A, Kubota S, Mori N, McLaren MJ, Inana G. Photoreceptor synaptic protein HRG4 (UNC119) interacts with ARL2 via a putative conserved domain. FEBS Lett. 2003;534:26–32. doi: 10.1016/s0014-5793(02)03766-3. [DOI] [PubMed] [Google Scholar]
- Kubota S, Kobayashi A, Mori N, Higashide T, McLaren MJ, Inana G. Changes in retinal synaptic proteins in the transgenic model expressing a mutant HRG4 (UNC119) Invest Ophthalmol Vis Sci. 2002;43:308–313. [PubMed] [Google Scholar]
- Lee ES, Burnside B, Flannery JG. Characterization of peripherin/rds and rom-1 transport in rod photoreceptors of transgenic and knockout animals. Invest Ophthalmol Vis Sci. 2006;47:2150–2160. doi: 10.1167/iovs.05-0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, Nicolo M, Makino CL, Sidman RL. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci U S A. 1999;96:736–741. doi: 10.1073/pnas.96.2.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro M, Pilgrim D. Identification and cloning of unc-119, a gene expressed in the Caenorhabditis elegans nervous system. Genetics. 1995;141:977–988. doi: 10.1093/genetics/141.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Maeda T, Imanishi Y, Kuksa V, Alekseev A, Bronson JD, Zhang H, Zhu L, Sun W, Saperstein DA, Rieke F, Baehr W, Palczewski K. Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J Biol Chem. 2005;280:18822–18832. doi: 10.1074/jbc.M501757200. Epub 2005 Mar 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AG, Crawford BD, Waskiewicz AJ, Pilgrim DB. unc-119 homolog required for normal development of the zebrafish nervous system. Genesis. 2004;40:223–230. doi: 10.1002/gene.20089. [DOI] [PubMed] [Google Scholar]
- Michaelides M, Aligianis IA, Holder GE, Simunovic M, Mollon JD, Maher ER, Hunt DM, Moore AT. Cone dystrophy phenotype associated with a frameshift mutation M280fsX291) in the alpha-subunit of cone specific transducin (GNAT2) Br J Ophthalmol 87, 1317–1320 Erratum in: Br J Ophthalmol 2004. 2003;88:314. doi: 10.1136/bjo.87.11.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori N, Ishiba Y, Kubota S, Kobayashi A, Higashide T, McLaren MJ, Inana G. Truncation mutation in HRG4(UNC119) leads to mitochondrial ANT-1-mediated photoreceptor synaptic and retinal degeneration by apoptosis. Invest Ophthalmol Vis Sci. 2006;47:1281–1292. doi: 10.1167/iovs.05-0493. [DOI] [PubMed] [Google Scholar]
- Naash MI, Wu TH, Chakraborty D, Fliesler SJ, Ding XQ, Nour M, Peachey NS, Lem J, Qtaishat N, Al-Ubaidi MR, Ripps H. Retinal abnormalities associated with the G90D mutation in opsin. J Comp Neurol. 2004;478:149–163. doi: 10.1002/cne.20283. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Van Hooser JP, Garwin GG, Chen J, Liou GI, Saari JC. Kinetics of visual pigment regeneration in excised mouse eyes and in mice with a targeted disruption of the gene encoding interphotoreceptor retinoid-binding protein or arrestin. Biochemistry. 1999;38:12012–12019. doi: 10.1021/bi990504d. [DOI] [PubMed] [Google Scholar]
- Pittler SJ, Baehr W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc Natl Acad Sci U S A. 1991;88:8322–8326. doi: 10.1073/pnas.88.19.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripps H, Peachey NS, Xu X, Nozell SE, Smith SB, Liou GI. The rhodopsin cycle is preserved in IRBP "knockout" mice despite abnormalities in retinal structure and function. Vis Neurosci. 2000;17:97–105. doi: 10.1017/s095252380017110x. [DOI] [PubMed] [Google Scholar]
- Rohrer B. Gene dosage effect of the TrkB receptor on rod physiology and biochemistry in juvenile mouse retina. Mol Vis. 2001;7:288–296. [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2. Cold Spring Harbor; New York: 1989. [Google Scholar]
- Sokolov M, Strissel KJ, Leskov IB, Michaud NA, Govardovskii VI, Arshavsky VY. Phosducin facilitates light-driven transducin translocation in rod photoreceptors. Evidence from the phosducin knockout mouse. J Biol Chem. 2004;279:19149–19156. doi: 10.1074/jbc.M311058200. Epub 2004 Feb 18. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Tsang SH, Burns ME, Calvert PD, Gouras P, Baylor DA, Goff SP, Arshavsky VY. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998;282:117–121. doi: 10.1126/science.282.5386.117. [DOI] [PubMed] [Google Scholar]
- Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Wachtmeister L. Oscillatory potentials in the retina: what do they reveal? Prog Ret Eye Res. 1998;17:485–521. doi: 10.1016/s1350-9462(98)00006-8. [DOI] [PubMed] [Google Scholar]
- Wistow G, Bernstein SL, Wyatt K, Ray S, Behal A, Touchman JW, Bouffard G, Smith D, Peterson K. Expressed sequence tag analysis of human retina for the NEIBank Project: retbindin, an abundant, novel retinal cDNA and alternative splicing of other retina-preferred gene transcripts. Mol Vis. 2002;8:196–204. [PubMed] [Google Scholar]