

Abstract
Background & Aims
Wnt/β-catenin activation is observed in normal liver development, regeneration, and liver cancer. Our aim was to elucidate the regulation and mechanism of this pathway in liver.
Methods
We report the generation and characterization of liver-specific nonmutated β-catenin–overexpressing transgenic mice. Transgenic livers were examined for their morphology and phenotype by histology, proliferation, apoptosis, and microarray analysis.
Results
Transgenic livers displayed a significant increase in cytoplasmic, membranous, and nuclear β-catenin in hepatocytes as compared with their wild-type littermates, which display a predominant membranous localization only. A 15%–20% increase in the liver weight–body weight ratio was evident in transgenic mice secondary to increased hepatocyte proliferation. Microarray analysis showed differential expression of approximately 400 genes in the transgenic livers. Epidermal growth factor receptor RNA and protein and increased levels of activated epidermal growth factor receptor and Stat3 were observed in the transgenic livers. Epidermal growth factor receptor promoter analysis showed a T-cell factor–binding site, and subsequent reporter assay confirmed epidermal growth factor receptor activation in response to Wnt-3A treatment that was abrogated by frizzled related protein 1, a known Wnt antagonist. Epidermal growth factor receptor inhibition successfully decreased liver size in transgenic mice. Next, 7 of 10 hepatoblastomas displayed simultaneous β-catenin and epidermal growth factor receptor up-regulation, thus suggesting a strong relationship between these 2 proteins in tumors.
Conclusions
β-Catenin transgenic mice show an in vivo hepatotrophic effect secondary to increased basal hepatocyte proliferation. Epidermal growth factor receptor seems to be a direct target of the pathway, and epidermal growth factor receptor activation might contribute toward some mitogenic effects of increased β-catenin in liver: epidermal growth factor receptor inhibition might be useful in such states.
The process of tumorigenesis is characterized by aberrant signal transduction.1 One such pathway that is reported to be abnormally active in many tumors, such as those of the colon, rectum, breast, and skin, is the Wnt/β-catenin pathway.2 This pathway is indispensable for normal development, as has been observed in numerous transgenic or loss-of-function studies that also elaborate and emphasize its ubiquitous distribution and functionality.3–5 The role of this pathway has been elucidated in key physiological phenomena, such as lineage specification, differentiation, stem cell renewal, epithelial–mesenchymal transition, proliferation, apoptosis, and cell–cell adhesion.3,6,7 Because of these roles, one can appreciate the implications of activation of this pathway during the process of tumorigenesis.
The Wnt/β-catenin pathway is normally turned “off” during the adult stage in many tissues.8 As shown previously, Ctnnb1 expression is low in hepatocytes, although significant levels of β-catenin protein are observed at the hepatocyte membrane, thus indicating its regulation at the level of degradation, as has been shown previously.9,10 In this phase, the key component of the pathway—β-catenin—is phosphorylated at specific serine and threonine residues by glycogen synthase kinase 3β and, with the aid of axin and adenomatous polyposis coli gene product (APC), is presented to β-transducin repeat-containing protein to undergo ubiquitin-mediated proteosomal degradation.11–13 This ensures minimal free cytosolic β-catenin levels for its nuclear signaling. During development in the presence of any of the Wnts or in cancers when either of the degradation complex components displays inactivating mutations or when the β-catenin gene itself displays mutations that alter the crucial serine and threonine residues, the pathway is active. This leads to nuclear translocation of β-catenin, where it, along with its cofactors T-cell factor family proteins (TCF), transactivate many target genes, such as c-myc, cyclin D1, urokinase plasminogen activator receptor, and others that act as ultimate effectors.14–16 Other important interactions of the β-catenin protein are at the cell membrane, where it complexes with E-cadherin and the hepatocyte growth factor receptor c-met, especially in hepatocytes.17,18
These interactions are regulated mainly by tyrosine phosphorylation of β-catenin at the C-terminal, and this further influences intercellular adhesion. However, we have also reported tyrosine phosphorylation–mediated nuclear translocation of β-catenin in hepatocytes, and this opens the possibility that various tyrosine kinases (such as hepatocyte growth factor) also influence downstream signaling of β-catenin via a noncanonical pathway.18 The role of β-catenin in liver pathology, especially in tumors such as hepatocellular carcinoma (HCC) and hepatoblastoma, is well established.19,20 Specific mutations in Ctnnb1 and axin have been shown to stabilize β-catenin protein in both of these tumor types in patients and in animal models.21–26 We have reported its activation during regulated liver growth in early liver regeneration after two-thirds partial hepatectomy.27 More recently, we have identified that a temporal expression of β-catenin during liver development and in vitro ablation of β-catenin in embryonic liver cultures at embryonic day 10 of mouse development affects proliferation, survival, and differentiation of the constituent cell population at that stage.9,28,29 In this study, we addressed the regulation of this pathway in liver by generating transgenic mice that overexpress nonmutated β-catenin under the transcriptional control of albumin promoter and enhancer. Simultaneously, we wanted to identify any novel target genes of the pathway that might be playing a noteworthy role in hepatic pathophysiology for possible therapeutic consequences.
Materials and Methods
Albumin/β-Catenin Expression Construct
An approximately 2.4-kilobase human β-catenin gene (Ctnnb1) consisting of its open reading frame (nucleotides 189–2554) was amplified from normal human liver by using primer 1 (CGGGATCCCAGCGTGGACAATGGCTA) and primer 2 (CCGGATCCAGGATGATTTACAGGTCA) containing BamHI sites. The fragment was inserted into the BamHI sites of an albumin promoter/enhancer-driven expression vector that also contained a polyadenylation cassette, kindly donated by Dr Zarnegar (University of Pittsburgh), to generate the transgenic plasmid.30
Generation of Albumin/β-Catenin Transgenic Mice
The previously described construct was linearized and microinjected into C57BL/6 × SJL hybrid mouse eggs to create transgenic mice by DNX Transgenic Sciences Corporation (Cranbury, New Jersey). The transgenic mice were identified by Southern blot and polymerase chain reaction (PCR) analysis by using tail genomic DNA digestion and primer 3 (ATGGGATCTGCATGCCCTCATCTAATG) and primer 4 (GTATGATTTTGTAATGGGGTAGGAACC). The transgenic founder mice were bred to C57BL/6 wild-type mice to produce F(1) hemizygous mice and bred further to obtain homozygous gene dosage. Out of the 11 lines generated initially, 3 lines were expanded on the basis of the robust protein expression of β-catenin, as described subsequently. All animal studies were performed in strict accordance with the institutional animal use and care committee at the University of Pittsburgh School of Medicine and Institutes of Health guidelines.
Protein Extraction and Western Blots
Transgenic and wild-type livers (n > 5) were used for whole-cell lysate preparation by using RIPA buffer (9.1 mmol/L dibasic sodium phosphate, 1.7 mmol/L monobasic sodium phosphate, 150 mmol/L sodium chloride, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl-sulfate [pH adjusted to 7.4]) containing fresh protease- and phosphatase-inhibitor cocktails (Sigma, St Louis, MO), as described previously.27 Protein concentration was determined by bicinchoninic acid protein assay. The lysates from transgenic and wild-type livers were kept separate for analysis (n > 5) unless mentioned specifically. All experiments were performed in triplicate, and the data shown are representative of the 3 sets. The transgenic and wild-type mice were matched for age and sex for all analyses. The protein analysis presented is from 1- or 3-month-old transgenic and wild-type livers and has been specified in Results. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis was performed by using 20 μg of protein on 7.5% ready gels by using the mini–PROTEIN 3 electrophoresis module assembly (Bio-Rad, Hercules, CA). After overnight transfer to Immobilon polyvinylidene difluoride membranes (Millipore, Bedford, MA), the Western blot was performed as discussed elsewhere.27 The blots were subjected to fresh Super-Signal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL) and visualized by autoradiography. Blots were stripped in immunoglobulin G elution buffer (Pierce) for reprobing.
Primary antibodies used in this study were against β-catenin, epidermal growth factor receptor (EGFR), cyclin D1, and c-myc (Santa Cruz Biotechnology, Santa Cruz, CA); anti–serine 45/threonine 41 β-catenin, anti–serine 33 and 37/threonine 41 β-catenin, anti–tyrosine 992 EGFR, anti–serine 217 and 221 MEK/ERK, and anti–phospho-Stat3 (tyrosine 705; Cell Signaling, Beverly, MA); anti–Ki-67 (Dako, Carpinteria, CA); anti–proliferating cell nuclear antigen (PCNA; Signet Laboratories, Dedham, MA); and anti-actin (Chemicon, Temecula, CA). Horseradish peroxidase–conjugated secondary antibodies (Chemicon) were used at 1:50,000 dilution.
Nuclear Protein Isolation and Electrophoretic Mobilization Shift Assay for β-Catenin Activation
Liver tissue (0.2–0.5 g) from 3-month-old transgenic (n = 4) or wild-type (n = 3) males was finely chopped and then homogenized in a glass Dounce homogenizer (50–100 strokes) in 1 mL of hypotonic buffer (10 mmol/L HEPES, pH 7.9, 10 mmol/L NaH2PO4, 1.5 mmol/L MgCl2, 1 mmol/L dithiothreitol [DTT], 0.5 mmol/L spermidine, and 1 mol/L NaF, with protease- and phosphatase-inhibitor cocktails [Sigma; P8340, P5726, and P2850] used at 1:100 dilution). Homogenization was monitored under a microscope by using trypan blue dye to control for release of nuclei. The homogenates were centrifuged for 5 minutes at 800g. The pelleted nuclei were washed and repelleted 2 times in 2 mL of hypotonic buffer. Nuclei pellets were snap-frozen in liquid nitrogen.
Nuclear proteins were extracted in 25–50 μL of hypertonic buffer (30 mmol/L HEPES, pH 7.9, 25% glycerol, 450 mmol/L NaCl, 12 mmol/L MgCl2, 1 mmol/L DTT, and 0.1 mmol/L ethylenediaminetetraacetic acid [EDTA], with protease- and phosphatase-inhibitor cocktails [Sigma; P8340, P5726, and P2850] used at 1:200 dilution) for 30–45 minutes at 4°C with continuous agitation. Extracts were centrifuged at 30,000g for 30 minutes. The supernatants were collected, and protein concentration was determined by bicinchoninic acid assay with bovine serum albumin as a standard.
Electrophoretic mobility shift assay was performed by incubating 5 μg of nuclear protein extracts with a 32P end-labeled oligonucleotide containing the core TCF/lymphoid enhancement factor (LEF) binding site (5′-CTCTGCCGGGCTTTGATCTTTGCTTAACAACA-3′) at room temperature for 20 minutes.31 The binding reaction conditions were 15 mmol/L HEPES, pH 7.9, 75 mmol/L NaCl, 6 mmol/L MgCl2, 0.025 mmol/L EDTA, 2.5 mmol/L Tris, pH 7.6, 12.5% glycerol, and 1 mmol/L DTT, with protease- and phosphatase-inhibitor cocktails (Sigma; P8340, P5726, and P2850) at 1:400 dilution. Reaction products were analyzed on a 5% nondenaturing polyacrylamide gel with 0.5% Tris/borate/EDTA buffer. The specificity of the DNA binding complex was determined by the addition of 20× cold TCF/LEF oligonucleotide as a competitor. Gels were dried and exposed to BioMax MR film (Eastman Kodak, Rochester, NY).
Histology and Immunohistochemistry
Sections from the age- and sex-matched transgenic and wild-type littermates’ livers were subjected to immunohistochemical analysis for β-catenin, EGFR, PCNA, and Ki-67 to determine their expression, localization, or both by using the indirect immunoperoxidase technique as discussed previously.9 Briefly, 4-μm-thick paraffin sections were passed through xylene and graded alcohol and rinsed in phosphate-buffered saline. Endogenous peroxide was inactivated by using 3% hydrogen peroxide (Sigma). Slides were microwaved in citrate buffer for 20 minutes, followed by blocking in the blue blocker (Shandon Lipshaw, Pittsburgh, PA) and overnight incubation at 4°C in the anti–β-catenin antibody (Santa Cruz Biotechnology). After washing, the sections were incubated in the secondary anti-mouse horseradish peroxidase–conjugated antibody (Chemicon) for 1 hour at room temperature, and signal was detected by using an ABC Elite kit (Vector Laboratories, Burlingame, CA) according to the manufacturer’s instructions. Sections were counterstained with Harris hematoxylin solution (Sigma) and passed through a dehydration process, followed by coverslipping and mounting by using DPX (Fluka Labs, St Louis, MO). For negative control, the sections were incubated with secondary antibodies only. Slides were viewed under an Axioskop 40 (Zeiss) upright research microscope, and digital images were obtained by a Nikon Coolpix camera. Collages were prepared with Adobe Photoshop 5.0 software.
For proliferation assay in the age- and sex-matched transgenic and wild-type livers, immunohistochemistry was performed for PCNA that recognizes cells in the cell cycle. To detect apoptosis, we used terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling staining with the ApopTag Peroxidase kit (Intergen Company). The apoptotic nuclei were detected by the presence of positive brown staining.
RNA Isolation and Affymetrix Microarray
Three male transgenic livers and 3 male wild-type littermate livers from 3-month-old mice were used for isolating and purifying RNA by Qiagen RNeasy kit (Qiagen, San Diego, CA). Increased protein levels (2.5-fold) of β-catenin in transgenic mice were confirmed by Western blots, as described previously. RNA was used for chromosomal RNA (cRNA) preparation and for generating a biotinylated cRNA probe from transgenic and wild-type livers; further steps were performed according to the Affymetrix protocols and as described previously.9 Briefly, 5 μg of total RNA was used in the first-strand complementary DNA (cDNA) synthesis with T7-d(T)24 primer Superscript II (GIBCO-BRL, Rockville, MD). The second-strand cDNA synthesis was performed at 16°C by adding Escherichia coli DNA ligase, E coli DNA polymerase I, and ribonuclease H to the reaction. The newly synthesized cDNA was purified and incubated at 37°C for 4 hours in an in vitro transcription reaction to produce cRNA labeled with biotin by using a MEGAscript system (Ambion, Inc, Austin, TX). This probe was further used for Affymetrix chip hybridization (U74A) according to the manufacturer’s instructions. After hybridization, the chips were washed in a fluidic station and stained with streptavidin phycoerythrin, followed by incubation with biotinylated mouse anti-avidin antibody and restaining with streptavidin phycoerythrin. The chips were scanned in an HP ChipScanner (Affymetrix Inc, Santa Clara, CA) to detect hybridization signals. Final analysis was performed by using Affymetrix Microarray Suite 5.0 software, and data were exported and organized in an Excel spreadsheet (Microsoft Corp, Redmond, WA). The signals from the transgenic and wild-type livers were averaged and compared and are presented as -fold change. The cutoff was 2-fold or higher, representing the genes that were up-regulated in the transgenic livers, and 0.5-fold or lower, representing the down-regulated genes in the transgenic livers. The data were specifically examined for some of the more relevant and liver-specific pathways.
Cell Culture and Luciferase Assay
Wnt-3A–conditioned medium containing biologically active Wnt-3A was obtained after the culture of the L Wnt-3A (CRL-2647, American Type Culture Collection).32 Control conditioned medium was generated from the recommended parental cell line (CRL-2648, American Type Culture Collection). Human recombinant soluble frizzled related protein (sFRP)-1 to also confirm the specificity of the Wnt-conditioned media was a kind gift from Dr Jeffery S. Rubin (Laboratory of Cellular and Molecular Biology, Institutes of Health, Bethesda, MD). These media were used to evaluate luciferase activity in the MCF7 cells stably transfected with a full-length EGFR promoter and a luciferase reporter construct (EGFR-luc), which were a kind gift from Dr Alfred Johnson (Laboratory of Molecular Biology, Center for Cancer Research, National Cancer Institute, Institutes of Health). The cells were cultured to confluence and treated with control media or Wnt-conditioned media alone or in the presence of sFRP-1 for 3 hours, and cells were lysed and prepared by using the luciferase reporter assay system (Promega, Madison, WI). The activity was read on a luminometer (Lumat; EG & G Berthold).
Drug Treatment of Transgenic Mice
Anti-EGFR compound (AG1478) was purchased from Tocris Cookson Inc (Ellisville, MO). It was dissolved in dimethyl sulfoxide (DMSO; Sigma) to prepare a stock solution. Six transgenic mice, aged 3 months, were divided into a control or treatment group. The control group (1 male and 2 females) received intraperitoneal DMSO twice a week for 5 weeks. The treatment group received intraperitoneal AG1478 (1 μg per mouse) twice a week for 5 weeks. The animals were killed according to institutional animal care and utilization committee guidelines, and livers were weighed and stored.
Examination of Patient Tissues
Paraffin blocks from 10 hepatoblastoma patients were obtained from the Department of Pathology at Children’s Hospital at the University of Pittsburgh School of Medicine under an approved institutional review board protocol. Four-micrometer-thick sections were examined for β-catenin and EGFR expression and localization by indirect immunohistochemistry as described previously.
Statistical Methods
For comparing the liver weight–body weight ratios, averages ± SEM were calculated for the age-matched transgenic and wild-type mice and were compared for statistical significance by Student t tests. For quantitative assessment, Western blots were scanned and subjected to densitometry by NIH Image software. The mean integrated optical density was compared in the wild-type and transgenic mice for statistical significance by the Student t test (InStat 2.01; GraphPad Software), and P < .001 was considered significant.
The proliferation labeling index was calculated by counting the number of PCNA-positive cells per the total number of cells in 5 fields from 3 transgenic and 3 wild-type 3-month-old male mice. The index (mean ± SEM) was compared for statistical significance by using the Student t test.
For the EGFR-inhibitor effect on liver weights, the livers harvested from the control and experimental groups were weighed. The weight comparisons were performed by paired tests (InStat 2.01), and 2-tailed P values of <.05 were considered significant. The values were exported to Kaleidagraph (Synergy Software) for generating bar graphs.
Results
Increased β-Catenin Protein in Transgenic Mice
By using the albumin/β-catenin construct injection into the blastocysts, transgenic mice were generated (Figure 1A). Out of the 11 founder lines obtained that expressed the transgene as shown by the PCR analysis (Figure 1B), 3 were chosen for expansion on the basis of the robust protein expression of β-catenin. One such representative analysis in 1-month-old (upper panel) and 3-month old (lower panel) mice shows the presence of transgene and increased levels of hepatic β-catenin protein in transgenic mice only (Figure 1C). On average, the transgenic livers (n = 20) displayed 2.4-fold higher protein levels of β-catenin than their sex- and age-matched wild-type littermates (n = 10), and this was statistically significant (P < .001; Figure 1D). The β-catenin protein levels were proportional to the gene copy numbers on the basis of hemizygous or homozygous gene dosage. On average, the gene copy was >25 gene copies per cell equivalent (not shown).
Figure 1.

β-Catenin transgenic mice display successful transgene integration and increased levels of cytoplasmic and nuclear β-catenin protein. (A) A 2.4-kilobase human β-catenin gene (Ctnnb1; open box) was amplified from adult human liver by using primers 1 and 2 and was inserted into the BamH1 site of human growth hormone (h-GH; black boxes) exon 1, regulated by albumin promoter/enhancer (gray box and vertical hatches), and a human growth hormone polyadenylation site (black horizontal hatched box). (B) Eleven founders were confirmed for the transgene by using primers 3 and 4 as compared with the positive controls with purified construct as a template for PCR confirmation (C1 to C4). C1, 1 gene copy per cell equivalent; C2, 5 gene copies per cell equivalent; C3, same as C1 with mouse genomic DNA; C4, same as C2 with mouse genomic DNA. bp, base pairs. (C) A representative analysis showing transgenic status confirmation by PCR in the upper panel and Western blot showing increased total β-catenin protein (1-month-old mice) and actin loading control in the middle panel. The lower panel is a Western blot from additional mice at 3 months of age showing variation in the increased β-catenin levels in transgenic mice. (D) Bar graph displays an overall increase of more than 2-fold in total β-catenin protein in the transgenic livers (n = 20) as compared with wild-type littermates (n = 10) from approximately 1-month-old mice; this is statistically significant (P <.001). IOD, integrated optical density. (E) A representative section from wild-type liver at 3 months of age displays predominantly membranous localization of β-catenin (original magnification, 40×). (F) A representative section from transgenic liver at 3 months of age displays membranous, cytoplasmic (arrow), and nuclear (arrowhead) localization of β-catenin (original magnification, 40×). (G) A representative Western blot using nuclear extracts from livers of 3-month-old male transgenic mice (T1 and T2) and matched control (W) shows increased β-catenin protein in transgenic livers only. (H) A representative electrophoretic mobility shift assay using nuclear lysates from livers of 3-month-old transgenic (T1) and wild-type (W) liver displays β-catenin/TCF binding in the transgenic liver only. F, free probe; Wc, competition with cold probe in wild-type liver nuclear lysate; Tc, competition with cold probe in nuclear lysate from transgenic liver; ATG, methionine start site; L, ladder; P, positive controls; WB, Western blot.
Next, transgenic and wild-type livers were examined for β-catenin distribution by immunohistochemistry. In wild-type littermate livers, β-catenin is con-fined to the hepatocyte membrane (Figure 1E). The transgenic livers show cytoplasmic staining, in addition to its physiological membranous localization (Figure 1F). In addition, several hepatocytes also show nuclear distribution of β-catenin. Representative transgenic and matched controls were used for nuclear protein isolation that confirmed the presence of increased β-catenin protein in most transgenic livers (Figure 1G). To further confirm the activity of β-catenin, nuclear protein was examined for β-catenin/TCF binding by electrophoretic mobility shift assay. Enhanced β-catenin/TCF binding was evident in transgenic livers, as shown in a representative analysis of 3-month-old transgenic and wild-type liver (Figure 1H). Most transgenic livers that displayed an increase in total β-catenin protein showed enhanced cytoplasmic and nuclear localization of β-catenin, as well as enhanced β-catenin/TCF binding, thus showing its activation in these livers.
β-Catenin Transgenic Mice Display Hepatomegaly but No Tumors
β-Catenin transgenic mice were examined for any obvious phenotypes. The mice were followed up to 18–24 months. There was no difference in survival of these mice as compared with their wild-type littermates. On death at different ages, no unusual lesions or tumors were observed in the transgenic or wild-type mice. However, the transgenic mice displayed larger livers as early as 30 days after birth. Approximately 70% of all transgenic mice consistently display hepatomegaly. A representative analysis from 1-month-old transgenic (n = 20) and wild-type (n = 10) mice, along with their liver weight–body weight ratios, is provided in Table 1. The liver weight–body weight ratio (liver weight/body weight × 100) for most transgenic mice ranged from 5.42 to 5.91 (mean, 5.50) as compared with their wild-type littermates, which displayed a ratio range from 4.21 to 5.0 (mean, 4.65). The difference in these ratios is approximately 15% and is statistically significant (P < .0001; Figure 2). This analysis clearly emphasizes a hepatotrophic effect of excess β-catenin in most transgenic mice as compared with their wild-type littermates.
Table 1.
A Representative Analysis of 20 Transgenic Mice and 10 Wild-type Littermates Displaying an Increase in the Liver Weight–Body Weight Ratio at or Around 1 Month of Age
| No. | TG/WT | Sex | Body weight (g) | Liver weight (g) | Ratio (×100) |
|---|---|---|---|---|---|
| 1 | TG | F | 18 | 1.0 | 5.56 |
| 2 | TG | F | 16 | 0.9 | 5.62 |
| 3 | TG | F | 20 | 1.1 | 5.50 |
| 4 | TG | F | 21 | 1.2 | 5.71 |
| 5 | TG | F | 19 | 1 | 5.26a |
| 6 | TG | F | 18 | 1 | 5.56 |
| 7 | TG | F | 17 | 1.0 | 5.88 |
| 8 | TG | F | 20 | 1.1 | 5.50 |
| 9 | TG | F | 19 | 1 | 5.26a |
| 10 | TG | F | 18 | 1 | 5.56 |
| 11 | TG | M | 22 | 1.3 | 5.91 |
| 12 | TG | M | 22 | 1.2 | 5.45 |
| 13 | TG | M | 23 | 1.2 | 5.22a |
| 14 | TG | M | 21 | 1.2 | 5.71 |
| 15 | TG | M | 24 | 1.2 | 5.00a |
| 16 | TG | M | 24 | 1.3 | 5.42 |
| 17 | TG | M | 22 | 1.1 | 5.00a |
| 18 | TG | M | 21 | 1.2 | 5.71 |
| 19 | TG | M | 20 | 1 | 5.00a |
| 20 | TG | M | 22 | 1.2 | 5.45 |
| 21 | WT | F | 20 | 0.9 | 4.50 |
| 22 | WT | F | 19 | 0.8 | 4.21 |
| 23 | WT | F | 17 | 0.8 | 4.71 |
| 24 | WT | F | 21 | 1 | 4.76 |
| 25 | WT | F | 19 | 0.8 | 4.74 |
| 26 | WT | M | 22 | 1 | 4.55 |
| 27 | WT | M | 21 | 1 | 4.76 |
| 28 | WT | M | 23 | 1.1 | 4.78 |
| 29 | WT | M | 20 | 1 | 5.00 |
| 30 | WT | M | 20 | 0.9 | 4.50 |
TG, transgenic; WT, wild type.
Outliers: transgenic mice that displayed relatively lower liver weight–body weight ratios (≤5.26).
Figure 2.

Column comparison by analysis of variance shows a significant increase in the liver weight–body weight ratio by approximately 15% in this representative analysis from 20 transgenic and 10 wild-type littermates (P < .0001). The mean increase was more than 20% if the outliers (liver weight–body weight ratio ≤5.26) were excluded from the analysis. WT, wild type; TG, transgenic.
Increased Basal Proliferation in Transgenic Livers: Apoptosis Unaffected
To comment on the increased liver weight–body weight ratios in the transgenic mice, we examined the histology of the transgenic livers. Representative analysis from 3-month-old male transgenic and wild-type livers is shown in this section. Routine H&E stains for the transgenic livers were unremarkable as compared with those for the wild-type livers (Figure 3A and B). To elucidate the mechanism of increased liver size, we next examined the livers for any decrease in apoptosis as being the cause of liver enlargement by examining the numbers of apoptotic nuclei after terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling staining. This analysis was also unremarkable, with no differences between transgenic and wild-type livers (Figure 3C and D). Next, by immunohistochemistry for PCNA, we examined whether the change in proliferation was contributing to the enlarged livers. The 3-month-old transgenic livers displayed more PCNA-positive cells than their wild-type littermates, thus indicating increased basal proliferation in β-catenin transgenic livers (Figure 3E–H).
Figure 3.

β-Catenin transgenic mice display increased proliferation, whereas histology and apoptosis remain unaffected. (A) A representative H&E-stained section from a wild-type liver at 3 months of age displays normal hepatic architecture (original magnification, 40×). Inset shows normal hepatocytes (original magnification, 60×). (B) A representative H&E-stained section from a transgenic liver at 3 months of age also displays normal hepatic architecture (original magnification, 40×). Inset also displays normal hepatocytes (original magnification, 60×). (C) TUNEL staining shows minimal apoptosis as seen by the presence of a single apoptotic nucleus (arrowhead) in this field in a wild-type liver. (D) Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL) staining in a representative transgenic liver also shows comparable numbers of apoptotic nuclei (arrowhead). (E) Representative PCNA staining shows only 1 cell to be positive (arrowhead) in this low-power field (original magnification, 20×) in a 3-month-old wild-type liver. (F) Representative PCNA staining shows many cells to be positive (arrowheads) in this low-power field (original magnification, 20×) in a 3-month-old transgenic liver. (G) High-power magnification of wild-type liver shows no PCNA-positive cells (original magnification, 60×). (H) High-power magnification (original magnification, 60×) of transgenic liver shows 2 PCNA-positive cells (arrowheads).
The proliferation index was determined and compared for statistical significance between the transgenic and wild-type mice as described in Materials and Methods. On average, there was an approximately 67% increase in proliferation index in the transgenic livers as compared with their wild-type littermates, and this was statistically significant (P < .001), thus implying that increased basal hepatocyte proliferation could be the contributing factor for the enlarged hepatic phenotype observed in the β-catenin overexpressers (Figure 4).
Figure 4.

Bar graph shows a significantly higher proliferation index in transgenic mice as compared with wild-type mice (P < .001). Five fields from 3 transgenic and wild-type livers, aged 3 months, were used for this analysis.
Distinct Gene Expression Changes in Transgenic Livers
To elucidate the molecular mechanism of hepatomegaly in the β-catenin transgenic mice, 3 livers from 3-month-old male transgenic mice (showing at least a 2.5-fold increase in total β-catenin protein by Western blots) and their 3 age- and sex-matched wild-type mice were used for messenger RNA (mRNA) extraction and Affymetrix gene array by using the U74A mouse gene chip. The analysis showed at least 210 genes that displayed ≥2.0-fold expression as compared with their wild-type littermate livers (additional information available online at http://www.pathology.pitt.edu/lectures/monga/onlineSupplement.pdf). Also, another 189 genes showed a ≥2.0-fold decrease in the transgenic livers. A list of some of the more relevant liver genes, along with their -fold change in expression, is shown in Table 2.
Table 2.
Selected List of Genes That Were Up-regulated or Down-regulated in Transgenic Livers as Compared with the -Wild-type Livers (Additional Information Available Online at http://www.pathology.pitt.edu/lectures/monga/onlineSupplement.pdf)
| Gene | -Fold change |
|---|---|
| Up-regulated | |
| ZT2 gene encoding zinc finger protein 125 | 3.8 |
| Basic-helix-loop-helix protein (Hey1 gene) | 3.7 |
| Cytochrome P450, 7b1 | 3.7 |
| Creb-rp, Tnx, and Cyp21 genes for cAMP response element–binding protein-related protein, tenascin X, and steroid 21-hydroxylase | 3.3 |
| Cyclin-dependent kinase inhibitor 1A (P21) | 3.1 |
| Calcium-modulating ligand/cds | 3.1 |
| Tubby | 3.1 |
| Fibroblast growth factor 4 | 2.7 |
| Aquaporin 1 | 2.7 |
| Glycosylphosphatidylinositol anchor attachment 1(GPAA1) | 2.6 |
| Cyclin A2 | 2.6 |
| Amino levulinate synthase (ALAS-H) | 2.6 |
| Frizzled homolog 7 | 2.6 |
| Annexin VII | 2.5 |
| Ring finger protein 3 mRNA | 2.5 |
| Inositol polyphosphate-5-phosphatase | 2.5 |
| Eph-and Elk-related kinase (eek) | 2.3 |
| Epidermal growth factor receptor (EGFR) | 2.2 |
| NEDD8-conjugating enzyme (Uba3) | 2.2 |
| Sterol 12α hydroxylase CYP8B1 (Cyp8b1) | 2.2 |
| Catenin α2 | 2.2 |
| Mouse glyceraldehyde-3-phosphate dehydrogenase mRNA | 2.1 |
| Pyruvate kinase | 2.1 |
| Peroxisome membrane protein PEX2 | 2.1 |
| Adrenergic receptor 31 | 2.1 |
| Down-regulated | |
| Protein kinase inhibitor p58 | 0.5 |
| Dutt1 mRNA | 0.5 |
| Calpain large subunit (nCL-4) mRNA | 0.5 |
| Adrenergic receptor β3 | 0.5 |
| Stromal cell–derived factor 1 | 0.5 |
| Glutamate receptor, ionotropic, δ1 | 0.4 |
| MAdCAM-1 mRNA | 0.4 |
| Cdk4 and Cdk6 inhibitor p19 protein mRNA | 0.4 |
| TRA1 mRNA | 0.4 |
| HNF-1β mRNA for hepatic nuclear factor 1β long form | 0.4 |
| Growth arrest–specific 1 protein mRNA | 0.4 |
| Plexin 2 mRNA | 0.4 |
| Plasminogen activator, tissue | 0.4 |
| HNF-3/forkhead homolog 1 | 0.4 |
| Mouse liver receptor homologous protein (LRH-1)mRNA | 0.3 |
| Notch gene homolog 3 | 0.3 |
| Casein kinase II α subunit mRNA | 0.3 |
| Glutamate receptor, ionotropic, δ2 | 0.3 |
| Apoptosis associated tyrosine kinase (AATYK) mRNA | 0.3 |
| TIE1 mRNA | 0.3 |
| Metallothionein 2 | 0.2 |
| Metallothionein 3 | 0.2 |
cAMP, adenosine 3′, 5′-cyclic monophosphate.
This gene-array analysis provides insight into some of the genes that seem to be modulated by increased expression of β-catenin and thus might also be among the molecular basis for the observed changes in the transgenic livers, including a positive effect on cell proliferation. In general, several of the up-regulated genes are known to have a metabolic role. More importantly, by regulating some of the known transcription factors such as ZT2, Hey1, and ring finger protein 3, β-catenin might be influencing multiple other effectors and affectors. Also, other stimulators of growth in terms of proliferation, such as adrenergic receptor β1 and fibroblast growth factor (FGF)-4 (an established target), might be contributing to the increased liver size as well. However, a more crucial and relevant observation seems to be an in crease in the expression of EGFR that clearly possess liver mitogenic activity. The transcription factors hepatocyte nuclear factor (HNF)-1 βand HNF-3/forkhead homolog 1 showed a decrease in their mRNA expression. Additional negative regulators of cell proliferation that are significantly down-regulated in transgenic livers are the protein kinase inhibitors p58, cyclin-dependent kinase (Cdk)4, and Cdk6 inhibitor p19 protein and the growth arrest–specific 1 protein. Thus, there seems to be a global trend of an up-regulation of the stimulatory genes that is supplemented by a concomitant decrease in some of the growth-inhibitory genes, thus giving an overall growth advantage to the transgenic livers in terms of their cell proliferation.
Epidermal Growth Factor Receptor Up-regulation and Activation in Transgenic Livers
One of the more established stimulatory pathways for hepatocytes is the epidermal growth factor (EGF). Gene-array analysis also showed a 2.2-fold higher expression of EGFR in the transgenic livers as compared with their littermates (Table 2). A representative Western blot shows increased total EGFR protein in transgenic mice as compared with their age- and sex-matched wild-type littermates at 1 and 3 months of age (Figure 5A). These corresponded to their respective β-catenin levels. Quantitative assessment after densitometry showed an approximately 2-fold increase that was significantly higher than that in the wild-type littermates (P < .001; Figure 5B).
Figure 5.
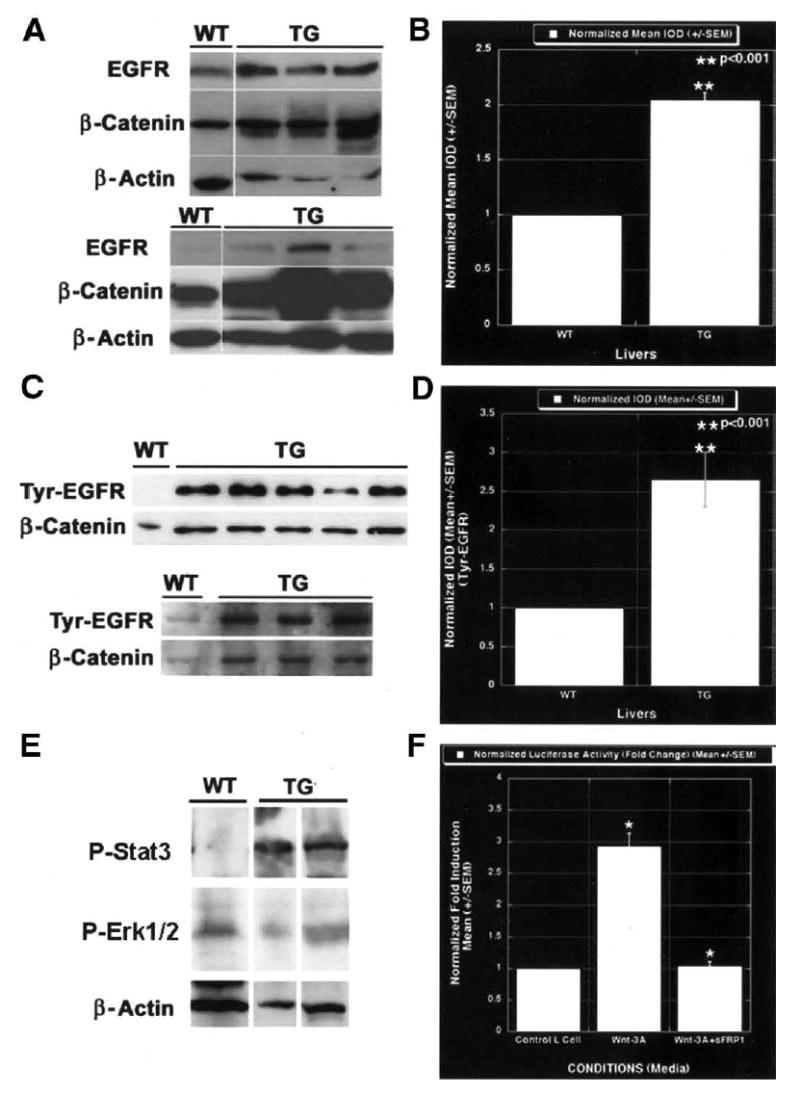
EGFR is regulated by β-catenin in liver and is a direct target of the Wnt/β-catenin pathway. (A) Western blot shows an increase in total EGFR protein in livers from three 1-month-old transgenic male mice as compared with their wild-type male littermates; this was proportional to their β-catenin levels. The lower set of Western blots shows a representative analysis from 3-month-old female transgenic and wild-type littermates showing increased levels of EGFR protein in transgenic livers only. These were also comparable to the total β-catenin levels. The lower EGFR autoradiograph was a very short exposure to highlight the difference between the wild-type and transgenic EGFR content. WT, wild type; TG, transgenic. (B) Normalized mean integrated optical density (IOD) obtained from densitometric analysis of the blots shows more than a 2-fold increase in total EGFR protein in the transgenic livers; this was statistically significant (P < .001). (C) Representative Western blots show increased levels of tyrosine-phosphorylated EGFR in transgenic livers, with minimal levels in wild-type littermates. The upper panel is a Western blot showing tyrosine-phosphorylated EGFR in liver lysates from 3-month-old wild-type males, 3 transgenic males, and 2 transgenic females, along with their respective total β-catenin levels (from left to right). The lower panel shows tyrosine-phosphorylated EGFR in 1 wild-type and 3 transgenic livers from 1-month-old males. Tyr, tyrosine. (D) Column comparison shows a 2.5-fold increase in tyrosine 992/EGFR levels in transgenic livers as compared with the wild-type littermate livers; this is statistically significant (P < .001). (E) Representative Western blots show increased levels of phospho-Stat3 in 3-month-old transgenic livers as compared with control (top). No change was evident in phospho-Erk1/2 levels (middle). β-Actin was used as a loading control (bottom). (F) Wnt-3A–conditioned medium induced approximately 3.0-fold EGFR reporter activity as compared with the control medium that was abrogated by the addition of sFRP-1, a Wnt antagonist. The luciferase activity was normalized to the negative control media.
Next we wanted to examine the status of the EGF pathway. Normal, essential amounts of EGF are present in the portal circulation. EGFR up-regulation would thus imply more downstream effector availability for the ligand and, hence, the activity of that pathway. This led us to investigate and compare the tyrosine phosphorylation status of EGFR by using a phosphospecific antibody against the same (tyrosine 992). Again, increased levels of tyrosine-phosphorylated or active EGFR were apparent in the transgenic mice as compared with their age-and sex-matched wild-type littermates at 1 and 3 months of age, as shown in Figure 5C. The increase was >2.5-fold in the transgenic livers and was statistically significant (P < .001; Figure 5D).
To further comment on the downstream effectors of EGFR in response to increased activation of EGFR in transgenic livers, we examined the status of Stat3 and Erk1/2. Whereas an increase in activated Stat3 was observed in the transgenic livers as compared with controls, the phospho-Erk1/2 levels remained unchanged, as shown in a representative Western blot in Figure 5E. This suggests preferential activation of Stat3 in response to increased β-catenin and EGFR activation. Thus, transgenic livers show activation of the EGF/EGFR/Stat3 axis that is among the more prominent mitogenic pathways for liver and thus may be a crucial basis for liver enlargement in transgenic mice.
EGFR is a direct target of the Wnt/β-catenin pathway. To further our understanding of regulation of EGFR by the Wnt/β-catenin pathway, we examined the promoter of EGFR for any potential β-catenin or TCF binding sites. We observed a single consensus sequence for TCF binding (5′-WWCAAWG-3′) that was located at nucleotides −79 to −86 (GCCAACG) in the EGFR promoter. Next, we examined the reporter activity by using the EGFR/luciferase construct stably transfected in the MCF7 cells. The MCF7 cells with this construct and the control plasmid were grown to confluence and cultured in the Wnt-3A–negative control media and Wnt-3A–conditioned media with or without sFRP-1. An approximately 3-fold increase in the luciferase activity was observed in the presence of the Wnt-3A–conditioned media as compared with the negative control media (Figure 5F). In addition, the presence of sFRP-1 in the Wnt-3A–conditioned media, which sequesters Wnt-3A and makes it unavailable to transduce β-catenin stabilization, blunted the luciferase activity to the one observed in the presence of negative control media (Figure 5F). This analysis shows EGFR as a novel target of the Wnt/β-catenin pathway.
Concomitant β-Catenin and Epidermal Growth Factor Receptor Increase in Hepatoblastomas
Next, to establish any relationship between the EGFR status and β-catenin in clinical samples, we examined 10 hepatoblastoma liver samples that were available to us. Immunohistochemistry showed aberrant β-catenin localization in all 10 samples, clearly displaying nuclear and cytoplasmic abundance in the tumors (Figure 6A, C, E, and G). This shows activation of the Wnt/β-catenin pathway in all the examined hepatoblastomas. The same samples were next analyzed for EGFR status in consecutive sections. Seven of the 10 samples showed a concomitantly increased immunohistochemical localization of EGFR in the tumors. Also, the tissue distribution was identical to that of the β-catenin in the tumors (Figure 6B, D, and F). However, the remaining 3 samples did not show any EGFR up-regulation, especially in the areas of tumors that displayed β-catenin activation (Figure 6G and H). These results indicate a concurrent β-catenin and EGFR up-regulation in a significant number of patient tumors and thus strengthen the hypothesis that EGFR is regulated by β-catenin in these tumors.
Figure 6.

Concomitant β-catenin and EGFR increase in pediatric hepatoblastomas. (A) Low-power view (original magnification, 5×) shows β-catenin distribution in a pediatric liver with hepatoblastoma. (B) An adjacent section displays a similar distribution of EGFR in the same tumor (original magnification, 5×). (C) Another hepatoblastoma displays aberrant nuclear β-catenin localization in the tumor (original magnification, 20×). (D) An adjacent section shows abnormal EGFR up-regulation in the same sample (original magnification, 20×). (E) Another patient also displays abnormal β-catenin localization in the hepatoblastoma (original magnification, 20×). (F) A consecutive section displays a similar anomalous increase in EGFR (original magnification, 20×). (G) Another hepatoblastoma sample shows abnormal cytoplasmic and nuclear 3-catenin (original magnification, 20×). (H) No EGFR was observed in the adjacent section in this tumor (original magnification, 20×).
Epidermal Growth Factor Receptor Inhibition Normalized Liver Weights in Transgenic Mice
To examine the functional relevance of EGFR up-regulation in the transgenic mice, we initiated intra-peritoneal injections of AG1478 or DMSO only twice a week for 5 weeks in 3-month-old transgenic mice (n = 6). There was a consistent decrease in liver weights after 5 weeks in the age- and sex-matched controls (Figure 7A). This was statistically significant and amounted to a 15% decrease in liver weights as compared with the transgenic mice that received vehicle injection only (Figure 7B). The harvested livers from all mice showed increased β-catenin protein by Western blot (not shown). This study indicates the importance of EGFR inhibition in increased– β-catenin protein states.
Figure 7.
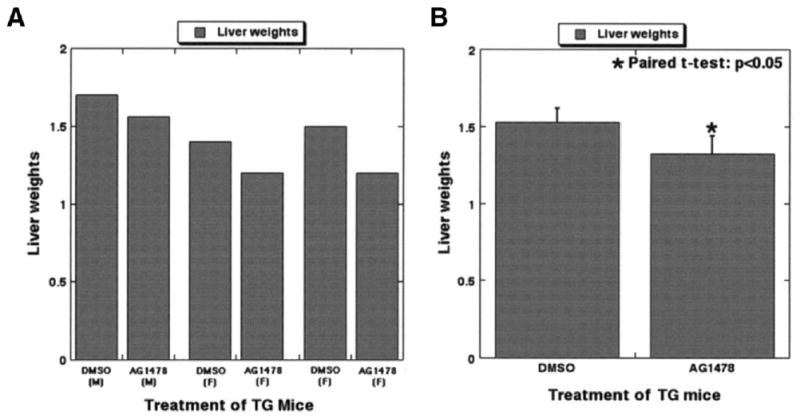
EGFR inhibition affected liver weights in transgenic mice. (A) Paired liver weights (sex matched) from 3-month-old transgenic mice after AG1478 or DMSO treatment for 5 weeks are displayed in this bar graph, which shows a consistent decrease in the experimental group. (B) Paired t tests showed a significant decrease in liver weights in the experimental group as compared with the controls (P < .05), thus confirming the role of EGFR in β-catenin–induced increase in liver size.
A Subset of Transgenic Mice Maintain Normal β-Catenin, Epidermal Growth Factor Receptor, and Liver Size
Another interesting observation was the presence of normal β-catenin protein levels in approximately 30% of all transgenic mice examined. The livers in these transgenic mice retained normal β-catenin levels that were comparable to those of their wild-type littermate livers, as shown in this representative analysis in 1-month-old transgenic mice (Figure 8A). In addition, most of these mice preserved their normal liver weight–body weight ratio, which was akin to that of their wild-type littermates. When investigated for their total EGFR levels, these animals retained normal EGFR levels in their livers, which were again comparable to those of their littermates (not shown).
Figure 8.
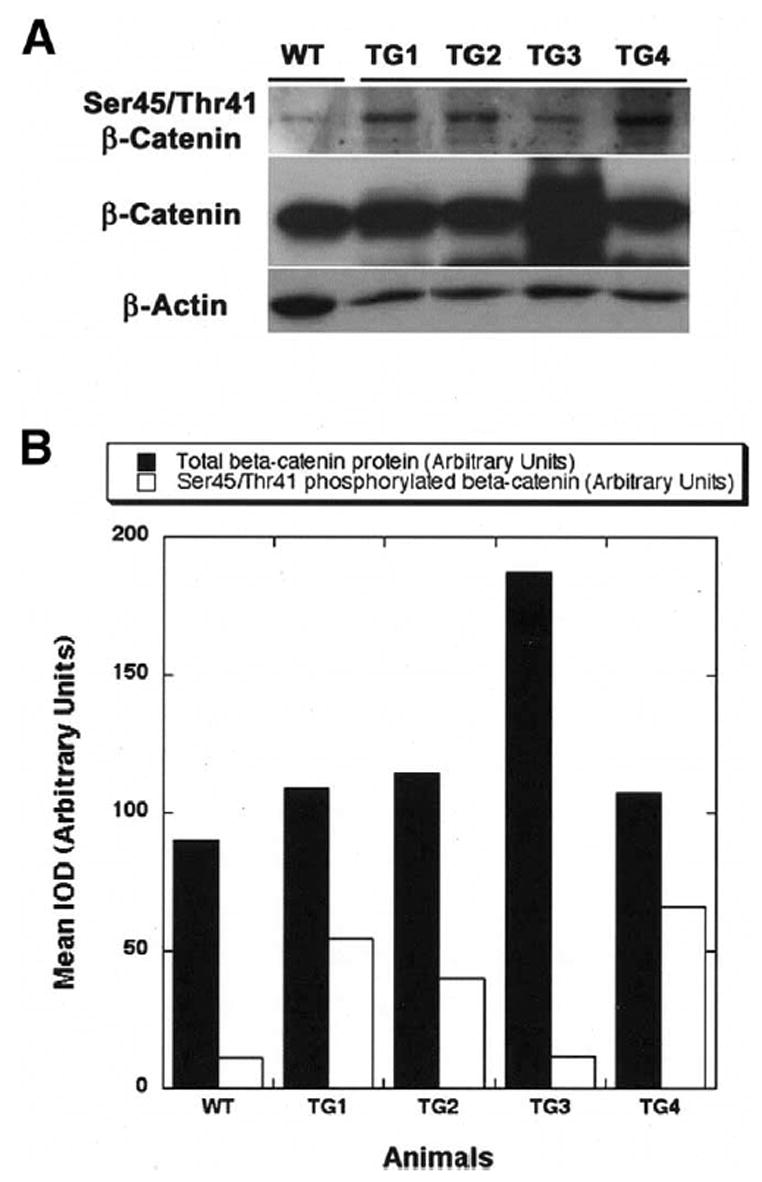
Increased degradation of β-catenin enables maintenance of its normal levels in a subset of transgenic mice. (A) A representative Western blot analysis shows increased levels of serine 45/threonine 41–phosphorylated β-catenin, corresponding to lower levels of total β-catenin in livers (TG1, TG2, and TG4) of 1-month-old transgenic mice. The higher levels of total β-catenin protein corresponded to less phosphorylation (TG3) and more cytoplasmic and nuclear localization (not shown). (B) A bar graph depicts a clear inverse relationship between total β-catenin levels in arbitrary units (closed box) and serine 45/threonine 41–phosphorylated β-catenin (open box), thus indicating some transgenic animals can regulate β-catenin levels by enhancing its degradation. Wild-type livers display reasonable levels of β-catenin and less phosphorylation; however, the gene expression is also very low as compared with all transgenic mice (not shown). WT, wild type; TG, transgenic; IOD, integrated optical density.
To further investigate the mechanism of maintenance of the normal β-catenin levels in such a subset, especially for epigenetic regulation, because these mice still had high gene copies per cell equivalent, we studied its degradation by examining the serine/threonine phosphorylation status of β-catenin. Using phosphospecific antibody against serine 45/threonine 41, we detected increased levels in the fraction of transgenic livers that retain normal total β-catenin levels (Figure 8A). This indicates a spontaneous activation of β-catenin phosphorylation and degradation that is able to preserve normal β-catenin levels and normal liver weight–body weight ratios in a subset of transgenic mice. However, how these mice differ from the remaining transgenic mice that display increased levels of total β-catenin and continued decreased phosphorylation remains undetermined (Figure 8A). The inverse relationship of total β-catenin and the percentage of serine 45/threonine 41–phosphorylated β-catenin is better shown in a densitometric analysis from the same representative blot, which suggests the mechanism of β-catenin regulation in the transgenic mice (Figure 8B).
Discussion
The molecular basis of tumors has long been studied; nevertheless, its importance has been restricted to the pathogenesis of these disorders. More recently, aberrant signal transduction has been increasingly studied from a therapeutic viewpoint.33,34 However, such studies have shown several new issues, such as the differences in involved molecular pathways in patients with a similar histological tumor type. Another closely associated issue is the differences in tissue specificity of transcriptional targets of a pathway. These are among a few challenges that daunt custom chemotherapy. In our effort to address the tissue specificity of a pathway that has been shown to be aberrantly activated in multiple tumors and for which no specific therapy is available, we generated a liver-specific transgenic mouse that overexpresses β-catenin, a component of the Wnt pathway.
Activation of the Wnt/β-catenin pathway has been observed during early liver development and during liver regeneration.9,27–29 These events are characterized by nuclear translocation of β-catenin, as well as some stabilization and increased transcription of this protein. Although mutations in ctnnb1 or in other components of this pathway have been shown to occur in certain subsets of HCCs and hepatoblastomas, transgenic mice that have been generated by using such mutated β-catenin have not displayed any liver tumors.35 Our results from the transgenic mice that overexpress wild-type β-catenin (nonmutated) in their livers are also in agreement because of the absence of any spontaneous hepatic tumorigenicity in these animals. The lack of tumors in our transgenic mice could be multifactorial. One prominent reason could be the strain of mice used for generating these mice. C57BL/6 mice are known to be resistant to hepatocarcinogenesis, and we used this model in an effort to reproduce the patient scenario.36–38 This might be supplemented by the successful degradation of overexpressed wild-type β-catenin protein, which kept the total β-catenin levels regulated enough that they were unable to induce spontaneous tumors. This is supported by a recent study that shows spontaneous hepatomas in APC conditional knockout mice secondary to increased and activated β-catenin, although most HCC patients with β-catenin stabilization have not shown any mutations in the APC gene.39–41 As shown in another study, it is possible that β-catenin alone is insufficient to initiate HCC and that it acts in concert with other pathways, such as H-Ras.42
However, an overall similar (although milder) phenotype that consists of liver enlargement was observed in our transgenic mice. This could clearly be the influence of the nondegradable form of β-catenin or might be due to a dissimilar promoter/enhancer system used by other investigators. We observed an increase in total β-catenin protein, its cytoplasmic stabilization, and its nuclear translocation and activation in most transgenic mice. These events resulted in stimulating liver growth secondary to increased basal proliferation of hepatocytes in the nonchallenged transgenic livers. It is interesting to note that we did not observe any major difference in apoptosis as could have been expected according to our earlier results in ex vivo liver cultures, in which a decrease in β-catenin accelerated apoptosis in developmental hepatocytes and biliary epithelial cells.28 In light of these differences, β-catenin might be playing a role in regulating apoptosis during prenatal liver development, in which more apoptotic nuclei are normally evident as compared with normal adult livers, which show minimal apoptosis (unpublished data).
Although most of the transgenic mice displayed hepatomegaly secondary to increased proliferation, β-catenin, and EGFR, a subset of these mice were able to regulate their β-catenin levels. This was sufficient to normalize their liver weights, as was evident by an increase in the serine 45/threonine 41–phosphorylated fraction of β-catenin in these animals. We do not have an adequate explanation for the molecular basis of this observed difference in the ability of some of these transgenic mice to be able to successfully down-regulate β-catenin by enhancing its phosphorylation and, hence, degradation, and it might be due to the existence of some additional autocrine feedback loops. Several such loops are known to exist in this pathway at many different levels. One such example is that many of the positive and negative regulators of this pathway, such as sFRP-2, axin 2, and TCF-1, are in fact downstream targets of this pathway.43–45 Studies relating the qualitative and quantitative changes in these proteins and other crucial components of the pathway are ongoing.
Gene-array studies were aimed at giving a global molecular mechanism of the hepatomegaly observed in the β-catenin transgenic mice. Some of the genes that are showing increased expression in the transgenic livers are related to the synthetic and metabolic functions of the hepatocytes. These are genes such as cytochrome P450, 7b1, amino levulinate synthase, inositol polyphosphate-5-phosphatase, sterol 12-α hydroxylase cytochrome P450 8B1 (Cyp8b1), glyceraldehyde-3-phosphate dehydrogenase, and pyruvate kinase. However, although these do define a metabolically heightened environment, the specific genes involved in glutamine metabolism that have been previously shown to be regulated by mutated β-catenin, including glutamate transporter 1, glutamine synthetase, and ornithine aminotransferase, remained unchanged.46 These differences might be due to differences in the transcriptional activation capability and specificity of mutated vs wild-type β-catenin. However, we believe that varied expression of these genes might be a function of multiple other factors, such as diet, time of the day, species, age, and other such factors.
The transgenic livers also displayed up-regulation of some factors, such as ZT2, Hey1, and ring finger protein 3, which have not yet been investigated for their importance in the liver.47,48 Hey1 might be of specific interest because of its involvement in proliferation and because it is a direct target of the Notch pathway.49 Others, such as FGF-4, are known targets of this pathway.50 We have also previously defined changes in β-catenin distribution secondary to exogenous FGF application, and, thus, interactions between the 2 pathways might be at multiple levels.51 However, some of the more prominent targets, such as c-myc and cyclin D1, remained unaffected in the transgenic livers, thus indicating tissue or stage specificity of the β-catenin targets.14,15 These results are also in accordance with the mutated β-catenin transgenic mice.35 Other relevant genes included frizzled 7 (Wnt receptor) and Nedd8-conjugating enzyme (ubiquitination), which might be part of the autoregulatory loop within the Wnt/β-catenin pathway. Some of the other genes showing an up-regulation had a more specific function, such as aquaporin 1 (expressed in bile ducts and intrahepatic cholangiocytes and required for fat absorption), annexin VII (tumor-suppressor gene in prostate carcinoma and HCC), and adrenergic receptor β1 (nor-epinephrine-mediated comitogen effect on liver).52–55 Some of the genes that display a decrease in their expression are the liver-specific transcription factors HNF-1 β and HNF-3 (forkhead homolog 1), which have been shown to correlate with decreased proliferation and increased differentiation of hepatocytes.56,57 Other specific genes that are more defined growth inhibitors, such as Cdk4 and Cdk6 inhibitor p19 protein and the growth arrest–specific 1 protein, also show a significant decrease in transgenic livers.58,59 Other genes that show a decreased expression possess developmental and/or tumor-suppressor properties, such as Dutt1 mRNA, SDF-1, and Notch gene.60–64 Other down-regulated genes with specific functions were Tie1 (angiogenesis) and metallothionein (cytoprotective against heavy metals and metabolism of trace elements).65,66 The collective premise that emerges from the microarray is an up-regulation of pro-mitogenic factors and a down-regulation of antimitogenic factors, thus giving a basis of the observed phenotype in the transgenic livers.
Although enhanced proliferation and liver size may be multifactorial, we attempted to examine the EGFR/β-catenin interaction in depth. EGFR up-regulation and its therapeutic inhibition have been shown to be effective in many cancers, such as those of the lung, breast, and prostate.67,68 The availability of EGFR inhibitors has greatly influenced the prognosis of these malignancies. More importantly, the availability of such drugs that affect individual signal transduction pathways at various levels provides an opportunity to customize therapies in many of the cancers.33 In our study, we show higher levels of EGFR mRNA and protein in β-catenin–overexpressing livers. The functionality of this interaction was further substantiated by the presence of activated EGFR in the transgenic livers that is probably secondary to the presence of normally higher levels of EGF that are synthesized by the Brunner’s glands and make their way into the portal circulation. Thus, EGFR up-regulation is sufficient to increase the sensitivity of hepatocytes to the available EGF to further prove to be mitogenic. This was further strengthened by the observed Stat3 activation in these livers. Stat3 activation is responsible for inducing proliferation in various tumors, such as those of the breast and skin.69,70
More than 80 target genes are known to be functioning in response to the Wnt/β-catenin pathway, and their tissue and stage specificity are becoming increasingly evident (http://www.stanford.edu/~rnusse/pathways/targets.html). As mentioned, we did not find any changes in c-myc or cyclin D1 levels, similar to the mutated β-catenin transgenic mice.35 Also, because of the unavailability of molecular therapies against β-catenin, it is vital to define any newer downstream effectors of this pathway that might be targeted during the activation of this pathway in an organ-specific manner. The EGFR promoter showed a TCF-binding site that is a binding cofactor of β-catenin to support transcriptional activation effectively. The relevance of this site is clear from the EGFR/luciferase reporter activation in the presence of Wnt-3A–conditioned media that is abrogated by the presence of sFRP-1 in the same.29,32,71 Nuclear translocation of β-catenin in response to Wnt-3A–conditioned media has been described extensively.10,32 This observation was also extended to hepatoblastoma tumors. These pediatric tumors are known to harbor mutations in the Ctnnb1 gene, thus resulting in activation of the β-catenin pathway.20,24 Upon examination, we detected a concomitant increase and nuclear translocation of β-catenin and EGFR in a significant number of these tumors. Nuclear EGFR has been reported previously and indicates a positive correlation with cell proliferation.72–74 This observation suggests that EGFR up-regulation might be one of the downstream signaling events secondary to β-catenin activation and that it might be relevant to interfere with EGFR signaling in β-catenin–activated states. Indeed, when EGFR inhibitor was administered to the transgenic mice, there was an approximately 15% decrease in their liver weights to normal levels as compared with the control-treated animals. Again, we cannot rule out the possibility of interaction of the Wnt/β-catenin and EGF pathway at more than 1 level, as has been shown previously.75,76 Our study thus shows a direct regulation of EGFR expression by β-catenin, thus highlighting the relevance of therapeutic EGFR inhibition in β-catenin–increased states. Thus, although these distinct mitogenic pathways might have cooperative detrimental pathogenic implications, there might also be exploitable therapeutic benefits of such a relationship.
Supplementary Material
Acknowledgments
The authors thank Dr Jeff Rubin (National Cancer Institute [NCI] and National Institutes of Health [NIH]) for providing the sFRP-1 protein and Dr Alfred Johnson (NCI and NIH) for providing the EGFR/luciferase or control plasmid–transfected MCF7 cells for this study.
Abbreviations used in this paper
- APC
adenomatous polyposis coli gene product
- cdk
cyclin-dependent kinase
- cRNA
chromosomal RNA
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- EGF
epidermal growth factor
- GFR
epidermal growth factor receptor
- FGF
fibroblast growth factor
- HCC
hepatocellular carcinoma
- HNF
hepatocyte nuclear factor
- PCNA
proliferating cell nuclear antigen
- PCR
polymerase chain reaction
- sFRP
soluble frizzled related protein
- TCF
T-cell factor family protein
Footnotes
George K. Michalopoulos is a consultant for Kytaron, Corp. There are no other conflicts of interest to be disclosed.
Funded by American Cancer Society Grant RSG-03-141-01-CNE and National Institutes of Health Grant 1RO1DK62277 (S.P.S.M.).
References
- 1.Pennisi E. How a growth control path takes a wrong turn to cancer (published erratum appears in Science 1998;281:1809) Science. 1998;281:1438–1439. doi: 10.1126/science.281.5382.1438. 1441. [DOI] [PubMed] [Google Scholar]
- 2.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 3.Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 4.Huelsken J, Vogel R, Brinkmann V, Erdmann B, Birchmeier C, Birchmeier W. Requirement for beta-catenin in anterior-posterior axis formation in mice. J Cell Biol. 2000;148:567–578. doi: 10.1083/jcb.148.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development. 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- 6.Arias AM. Epithelial mesenchymal interactions in cancer and development. Cell. 2001;105:425–431. doi: 10.1016/s0092-8674(01)00365-8. [DOI] [PubMed] [Google Scholar]
- 7.Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19:379–383. doi: 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- 8.Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science. 2000;287:1606–1609. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- 9.Micsenyi A, Tan X, Sneddon T, Luo JH, Michalopoulos GK, Monga SP. Beta-catenin is temporally regulated during normal liver development. Gastroenterology. 2004;126:1134–1146. doi: 10.1053/j.gastro.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 10.Willert K, Shibamoto S, Nusse R. Wnt-induced dephosphorylation of axin releases beta-catenin from the axin complex. Genes Dev. 1999;13:1768–1773. doi: 10.1101/gad.13.14.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 12.Nakamura T, Hamada F, Ishidate T, Anai K, Kawahara K, Toyoshima K, Akiyama T. Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level. Genes Cells. 1998;3:395–403. doi: 10.1046/j.1365-2443.1998.00198.x. [DOI] [PubMed] [Google Scholar]
- 13.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 15.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 16.Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci U S A. 1999;96:1603–1608. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huber AH, Weis WI. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105:391–402. doi: 10.1016/s0092-8674(01)00330-0. [DOI] [PubMed] [Google Scholar]
- 18.Monga SP, Mars WM, Pediaditakis P, Bell A, Mule K, Bowen WC, Wang X, Zarnegar R, Michalopoulos GK. Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer Res. 2002;62:2064–2071. [PubMed] [Google Scholar]
- 19.Legoix P, Bluteau O, Bayer J, Perret C, Balabaud C, Belghiti J, Franco D, Thomas G, Laurent-Puig P, Zucman-Rossi J. Beta-catenin mutations in hepatocellular carcinoma correlate with a low rate of loss of heterozygosity. Oncogene. 1999;18:4044–4046. doi: 10.1038/sj.onc.1202800. [DOI] [PubMed] [Google Scholar]
- 20.Wei Y, Fabre M, Branchereau S, Gauthier F, Perilongo G, Buendia MA. Activation of beta-catenin in epithelial and mesenchymal hepatoblastomas. Oncogene. 2000;19:498–504. doi: 10.1038/sj.onc.1203356. [DOI] [PubMed] [Google Scholar]
- 21.de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847–8851. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devereux TR, Anna CH, Foley JF, White CM, Sills RC, Barrett JC. Mutation of beta-catenin is an early event in chemically induced mouse hepatocellular carcinogenesis. Oncogene. 1999;18:4726–4733. doi: 10.1038/sj.onc.1202858. [DOI] [PubMed] [Google Scholar]
- 23.Huang H, Fujii H, Sankila A, Mahler-Araujo BM, Matsuda M, Cathomas G, Ohgaki H. Beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am J Pathol. 1999;155:1795–1801. doi: 10.1016/s0002-9440(10)65496-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koch A, Denkhaus D, Albrecht S, Leuschner I, von Schweinitz D, Pietsch T. Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res. 1999;59:269–273. [PubMed] [Google Scholar]
- 25.Nhieu JT, Renard CA, Wei Y, Cherqui D, Zafrani ES, Buendia MA. Nuclear accumulation of mutated beta-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am J Pathol. 1999;155:703–710. doi: 10.1016/s0002-9440(10)65168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, Sasaki Y, Imaoka S, Murata M, Shimano T, Yamaoka Y, Nakamura Y. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245–250. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- 27.Monga SP, Pediaditakis P, Mule K, Stolz DB, Michalopoulos GK. Changes in WNT/beta-catenin pathway during regulated growth in rat liver regeneration. Hepatology. 2001;33:1098–1109. doi: 10.1053/jhep.2001.23786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monga SP, Monga HK, Tan X, Mule K, Pediaditakis P, Michalopoulos GK. Beta-catenin antisense studies in embryonic liver cultures: role in proliferation, apoptosis, and lineage specification. Gastroenterology. 2003;124:202–216. doi: 10.1053/gast.2003.50000. [DOI] [PubMed] [Google Scholar]
- 29.Hussain SZ, Sneddon T, Tan X, Micsenyi A, Michalopoulos GK, Monga SP. Wnt impacts growth and differentiation in ex vivo liver development. Exp Cell Res. 2004;292:157–169. doi: 10.1016/j.yexcr.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 30.Bell A, Chen Q, DeFrances MC, Michalopoulos GK, Zarnegar R. The five amino acid-deleted isoform of hepatocyte growth factor promotes carcinogenesis in transgenic mice. Oncogene. 1999;18:887–895. doi: 10.1038/sj.onc.1202379. [DOI] [PubMed] [Google Scholar]
- 31.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shibamoto S, Higano K, Takada R, Ito F, Takeichi M, Takada S. Cytoskeletal reorganization by soluble Wnt-3a protein signalling. Genes Cells. 1998;3:659–670. doi: 10.1046/j.1365-2443.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- 33.Summerton JE. Custom cancer therapies: safe and effective treatments for most or all cancers. Ann N Y Acad Sci. 2003;1002:189–196. doi: 10.1196/annals.1281.016. [DOI] [PubMed] [Google Scholar]
- 34.Arteaga CL. Selecting the right patient for tumor therapy. Nat Med. 2004;10:577–578. doi: 10.1038/nm0604-577. [DOI] [PubMed] [Google Scholar]
- 35.Cadoret A, Ovejero C, Saadi-Kheddouci S, Souil E, Fabre M, Romagnolo B, Kahn A, Perret C. Hepatomegaly in transgenic mice expressing an oncogenic form of beta-catenin. Cancer Res. 2001;61:3245–3249. [PubMed] [Google Scholar]
- 36.Diwan BA, Rice JM, Ward JM. Strain-dependent effects of phenobarbital on liver tumor promotion in inbred mice. Prog Clin Biol Res. 1990;331:69–83. [PubMed] [Google Scholar]
- 37.Kemp CJ, Drinkwater NR. Genetic variation in liver tumor susceptibility, plasma testosterone levels, and androgen receptor binding in six inbred strains of mice. Cancer Res. 1989;49:5044–5047. [PubMed] [Google Scholar]
- 38.Stanley LA, Devereux TR, Foley J, Lord PG, Maronpot RR, Orton TC, Anderson MW. Proto-oncogene activation in liver tumors of hepatocarcinogenesis-resistant strains of mice. Carcinogenesis. 1992;13:2427–2433. doi: 10.1093/carcin/13.12.2427. [DOI] [PubMed] [Google Scholar]
- 39.Chen TC, Hsieh LL, Ng KF, Jeng LB, Chen MF. Absence of APC gene mutation in the mutation cluster region in hepatocellular carcinoma. Cancer Lett. 1998;134:23–28. doi: 10.1016/s0304-3835(98)00238-9. [DOI] [PubMed] [Google Scholar]
- 40.Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, Giovannini M, Perret C. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci U S A. 2004;101:17216–17221. doi: 10.1073/pnas.0404761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishizaki Y, Ikeda S, Fujimori M, Shimizu Y, Kurihara T, Itamoto T, Kikuchi A, Okajima M, Asahara T. Immunohistochemical analysis and mutational analyses of beta-catenin, Axin family and APC genes in hepatocellular carcinomas. Int J Oncol. 2004;24:1077–1083. [PubMed] [Google Scholar]
- 42.Harada N, Oshima H, Katoh M, Tamai Y, Oshima M, Taketo MM. Hepatocarcinogenesis in mice with beta-catenin and Ha-ras gene mutations. Cancer Res. 2004;64:48–54. doi: 10.1158/0008-5472.can-03-2123. [DOI] [PubMed] [Google Scholar]
- 43.Roose J, Clevers H. TCF transcription factors: molecular switches in carcinogenesis. Biochim Biophys Acta. 1999;1424:M23–M37. doi: 10.1016/s0304-419x(99)00026-8. [DOI] [PubMed] [Google Scholar]
- 44.Yan D, Wiesmann M, Rohan M, Chan V, Jefferson AB, Guo L, Sakamoto D, Caothien RH, Fuller JH, Reinhard C, Garcia PD, Randazzo FM, Escobedo J, Fantl WJ, Williams LT. Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/beta-catenin signaling is activated in human colon tumors. Proc Natl Acad Sci U S A. 2001;98:14973–14978. doi: 10.1073/pnas.261574498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lescher B, Haenig B, Kispert A. sFRP-2 is a target of the Wnt-4 signaling pathway in the developing metanephric kidney. Dev Dyn. 1998;213:440–451. doi: 10.1002/(SICI)1097-0177(199812)213:4<440::AID-AJA9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 46.Cadoret A, Ovejero C, Terris B, Souil E, Levy L, Lamers WH, Kitajewski J, Kahn A, Perret C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene. 2002;21:8293–8301. doi: 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- 47.Giorgi S, Polimeni M, Senni MI, De Gregorio L, Dragani TA, Molinaro M, Bouche M. Isolation and characterization of the murine zinc finger coding gene, ZT2: expression in normal and transformed myogenic cells. Gene. 1999;230:81–90. doi: 10.1016/s0378-1119(99)00044-x. [DOI] [PubMed] [Google Scholar]
- 48.Maier MM, Gessler M. Comparative analysis of the human and mouse Hey1 promoter: Hey genes are new Notch target genes. Biochem Biophys Res Commun. 2000;275:652–660. doi: 10.1006/bbrc.2000.3354. [DOI] [PubMed] [Google Scholar]
- 49.Zamurovic N, Cappellen D, Rohner D, Susa M. Coordinated activation of notch, Wnt, and transforming growth factor-beta signaling pathways in bone morphogenic protein 2-induced osteogenesis. Notch target gene Hey1 inhibits mineralization and Runx2 transcriptional activity. J Biol Chem. 2004;279:37704–37715. doi: 10.1074/jbc.M403813200. [DOI] [PubMed] [Google Scholar]
- 50.Kratochwil K, Galceran J, Tontsch S, Roth W, Grosschedl R. FGF4, a direct target of LEF1 and Wnt signaling, can rescue the arrest of tooth organogenesis in Lef1(−/−) mice. Genes Dev. 2002;16:3173–3185. doi: 10.1101/gad.1035602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sekhon SS, Tan X, Micsenyi A, Bowen WC, Monga SP. Fibroblast growth factor enriches the embryonic liver cultures for hepatic progenitors. Am J Pathol. 2004;164:2229–2240. doi: 10.1016/S0002-9440(10)63779-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Broten J, Michalopoulos G, Petersen B, Cruise J. Adrenergic stimulation of hepatocyte growth factor expression. Biochem Biophys Res Commun. 1999;262:76–79. doi: 10.1006/bbrc.1999.1183. [DOI] [PubMed] [Google Scholar]
- 53.Srivastava M, Montagna C, Leighton X, Glasman M, Naga S, Eidelman O, Ried T, Pollard HB. Haploinsufficiency of Anx7 tumor suppressor gene and consequent genomic instability promotes tumorigenesis in the Anx7(+/−) mouse. Proc Natl Acad Sci U S A. 2003;100:14287–14292. doi: 10.1073/pnas.2235927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma T, Jayaraman S, Wang KS, Song Y, Yang B, Li J, Bastidas JA, Verkman AS. Defective dietary fat processing in transgenic mice lacking aquaporin-1 water channels. Am J Physiol Cell Physiol. 2001;280:C126–C134. doi: 10.1152/ajpcell.2001.280.1.C126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Couchie D, Holic N, Chobert MN, Corlu A, Laperche Y. In vitro differentiation of WB-F344 rat liver epithelial cells into the biliary lineage. Differentiation. 2002;69:209–215. doi: 10.1046/j.1432-0436.2002.690414.x. [DOI] [PubMed] [Google Scholar]
- 56.Halmos B, Basseres DS, Monti S, D’Alo F, Dayaram T, Ferenczi K, Wouters BJ, Huettner CS, Golub TR, Tenen DG. A transcriptional profiling study of CCAAT/enhancer binding protein targets identifies hepatocyte nuclear factor 3 beta as a novel tumor suppressor in lung cancer. Cancer Res. 2004;64:4137–4147. doi: 10.1158/0008-5472.CAN-03-4052. [DOI] [PubMed] [Google Scholar]
- 57.Mizuguchi T, Mitaka T, Hirata K, Oda H, Mochizuki Y. Alteration of expression of liver-enriched transcription factors in the transition between growth and differentiation of primary cultured rat hepatocytes. J Cell Physiol. 1998;174:273–284. doi: 10.1002/(SICI)1097-4652(199803)174:3<273::AID-JCP1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 58.Weitzman JB, Fiette L, Matsuo K, Yaniv M. JunD protects cells from p53-dependent senescence and apoptosis. Mol Cell. 2000;6:1109–1119. doi: 10.1016/s1097-2765(00)00109-x. [DOI] [PubMed] [Google Scholar]
- 59.Del Sal G, Ruaro ME, Philipson L, Schneider C. The growth arrest-specific gene, gas1, is involved in growth suppression. Cell. 1992;70:595–607. doi: 10.1016/0092-8674(92)90429-g. [DOI] [PubMed] [Google Scholar]
- 60.Varnum-Finney B, Purton LE, Yu M, Brashem-Stein C, Flowers D, Staats S, Moore KA, Le Roux I, Mann R, Gray G, Artavanis-Tsakonas S, Bernstein ID. The Notch ligand, Jagged-1, influences the development of primitive hematopoietic precursor cells. Blood. 1998;91:4084–4091. [PubMed] [Google Scholar]
- 61.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 62.Qi R, An H, Yu Y, Zhang M, Liu S, Xu H, Guo Z, Cheng T, Cao X. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003;63:8323–8329. [PubMed] [Google Scholar]
- 63.David NB, Sapede D, Saint-Etienne L, Thisse C, Thisse B, Dambly-Chaudiere C, Rosa FM, Ghysen A. Molecular basis of cell migration in the fish lateral line: role of the chemokine receptor CXCR4 and of its ligand, SDF1. Proc Natl Acad Sci U S A. 2002;99:16297–16302. doi: 10.1073/pnas.252339399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xian J, Clark KJ, Fordham R, Pannell R, Rabbitts TH, Rabbitts PH. Inadequate lung development and bronchial hyperplasia in mice with a targeted deletion in the Dutt1/Robo1 gene. Proc Natl Acad Sci U S A. 2001;98:15062–15066. doi: 10.1073/pnas.251407098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coyle P, Philcox JC, Carey LC, Rofe AM. Metallothionein: the multipurpose protein. Cell Mol Life Sci. 2002;59:627–647. doi: 10.1007/s00018-002-8454-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rodewald HR, Sato TN. Tie1, a receptor tyrosine kinase essential for vascular endothelial cell integrity, is not critical for the development of hematopoietic cells. Oncogene. 1996;12:397–404. [PubMed] [Google Scholar]
- 67.Roskoski R., Jr The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun. 2004;319:1–11. doi: 10.1016/j.bbrc.2004.04.150. [DOI] [PubMed] [Google Scholar]
- 68.Schiller JH. New directions for ZD1839 in the treatment of solid tumors. Semin Oncol. 2003;30:49–55. doi: 10.1053/sonc.2003.50032. [DOI] [PubMed] [Google Scholar]
- 69.Chan KS, Carbajal S, Kiguchi K, Clifford J, Sano S, DiGiovanni J. Epidermal growth factor receptor-mediated activation of Stat3 during multistage skin carcinogenesis. Cancer Res. 2004;64:2382–2389. doi: 10.1158/0008-5472.can-03-3197. [DOI] [PubMed] [Google Scholar]
- 70.Sartor CI, Dziubinski ML, Yu CL, Jove R, Ethier SP. Role of epidermal growth factor receptor and STAT-3 activation in autonomous proliferation of SUM-102PT human breast cancer cells. Cancer Res. 1997;57:978–987. [PubMed] [Google Scholar]
- 71.Finch PW, He X, Kelley MJ, Uren A, Schaudies RP, Popescu NC, Rudikoff S, Aaronson SA, Varmus HE, Rubin JS. Purification and molecular cloning of a secreted, frizzled-related antagonist of Wnt action. Proc Natl Acad Sci U S A. 1997;94:6770–6775. doi: 10.1073/pnas.94.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 73.Marti U, Wells A. The nuclear accumulation of a variant epidermal growth factor receptor (EGFR) lacking the transmembrane domain requires coexpression of a full-length EGFR. Mol Cell Biol Res Commun. 2000;3:8–14. doi: 10.1006/mcbr.2000.0177. [DOI] [PubMed] [Google Scholar]
- 74.Tao Y, Song X, Deng X, Xie D, Lee LM, Liu Y, Li W, Li L, Deng L, Wu Q, Gong J, Cao Y. Nuclear accumulation of epidermal growth factor receptor and acceleration of G1/S stage by Epstein-Barr-encoded oncoprotein latent membrane protein 1. Exp Cell Res. 2005;303:240–251. doi: 10.1016/j.yexcr.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 75.Li Y, Ren J, Yu W, Li Q, Kuwahara H, Yin L, Carraway KL, III, Kufe D. The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and beta-catenin. J Biol Chem. 2001;276:35239–35242. doi: 10.1074/jbc.C100359200. [DOI] [PubMed] [Google Scholar]
- 76.Lu Z, Ghosh S, Wang Z, Hunter T. Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin, increased transcriptional activity of beta-catenin, and enhanced tumor cell invasion. Cancer Cell. 2003;4:499–515. doi: 10.1016/s1535-6108(03)00304-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.