

Abstract
Hepatocyte growth factor/scatter factor (HGF/SF) is a multifunctional cytokine that is involved in many normal as well as pathological conditions. HGF/NK1, a splice variant of HGF/SF, has been reported to have either antagonistic or agonistic effects with regard to c-Met signaling depending on the cell type. In these experiments, we have determined that HGF/NK1 is a potent mitogen for rat hepatocytes in culture. Furthermore, we have found that coagulation factor Xa (fXa) is capable of cleaving HGF/NK1 and single chain HGF/SF (scHGF/SF). The products resulting from cleavage of HGF/NK1 or scHGF/SF by fXa appear as single bands under non-reducing conditions. The reaction products from the digestion of HGF/NK1 by fXa were separated under reducing conditions, and the cleavage site, as determined by N-terminal sequencing, was located C-terminal to arginine 134. Previous work established that the heparin-binding domain for HGF/SF is located in the N domain of HGF/SF. Additionally, the dimerization of the HGF/SF receptor (c-Met) by the ligand HGF/NK1 is facilitated by heparin and related sulfonated sugars on the cell surface, whereas heparin is not required for HGF/SF-mediated dimerization. Cleavage of single chain HGF/SF or HGF/NK1 by factor Xa does not alter the affinity of the respective molecules for heparin, but it did variably affect the associated mitogenic activity of these factors. The associated mitogenic activity of HGF/NK1 was reduced by more than 90%, whereas the mitogenic activity of scHGF/SF was unaffected. This suggests mandatory maintenance of a steric interaction of the N domain and the first kringle domain for HGF/NK1 to act as an agonist for rat hepatocyte growth but is not required by full-length HGF/SF.
Hepatocyte growth factor/scatter factor (HGF/SF)1 (1) was first discovered by its ability to induce a scatter response with Madin-Darby canine kidney cells (2) as well as by its ability to induce a proliferative response with primary cultures of hepatocytes (3). HGF/SF is involved in many normal as well as pathological biological processes (4). During embryonic development, HGF/SF is vital for organ and tissue morphogenesis and growth (5–7). In injury models of the kidney or liver, HGF/SF is a key mediator in the repair process (8, 9).
HGF/SF acts through its tyrosine kinase-endowed receptor, c-Met (10). Because mesenchymal cells produce HGF/SF and epithelial cells express c-Met (11–13), HGF/SF is an important mediator of mesenchymal-epithelial interaction (14, 15). Furthermore, in pathologic conditions, sustained activation of c-Met occurs in a wide variety of tumors (16–21).
HGF/SF is a member of the plasminogen-related growth factor family. In addition to sharing several structural motifs, HGF/SF shares approximately a 40% overall homology to plasminogen (22). The common structural motifs are best defined at the gene sequence level and include an N-terminal N domain (Glu30–Asn121), which is similar to the activation peptide of plasminogen; four kringle domains: Cys128–Cys206, Cys211–Cys288, Cys305–Cys383, an Cys391–Cys469; and in the case of HGF/SF, an inactive serine protease domain (Val495–Ser728). This domain is inactive because of non-conservative mutations within the catalytic triad (His534 → Gln and Ser673 → Tyr) (1). The kringle domain, first identified in prothrombin (23), is a stretch of 78 largely hydrophobic amino acids that form a globular structure that is stabilized by three highly conserved disulfide bonds. In order for the pro-form of HGF/SF, single chain HGF/SF (scHGF/SF), to exhibit biological activity, cleavage must occur at the trypsin-like site Arg494–Val495 (24). The resulting product is an active two-chain HGF/SF molecule (tcHFGF/SF), which is held together by a single disulfide bond (Cys487–Cys604) (1). Two proteases have been identified that can cleave scHGF/SF at Arg494, urokinase plasminogen activator (uPA) (25) and HGF activator (26).
In addition to the full-length isoform of HGF/SF, three additional splice variants of HGF/SF exist in vivo. The first is referred to as deleted HGF/SF (delHGF/SF) and is identical to full-length HGF/SF except for a 5-amino acid deletion within the first kringle domain (27). To date, both the full-length and the deleted isoforms of HGF/SF have been shown to have comparable mitogenic, cytotoxic, and scatter activities (27, 28). The remaining splice variants result in much smaller protein products and consist of the N-terminal N domain followed by either the first kringle (HGF/NK1) (29) or the first two kringles (HGF/NK2) (30). These two splice variants have been described as having agonistic as well as antagonistic activities. The phenotype of mice transgenic for HGF/NK1 was similar to that of transgenic mice that overexpressed HGF/SF, thus demonstrating that HGF/NK1 can act as an agonist in vivo (31).
Analysis of function of full-length HGF/SF by selective deletion of specific domains has been very useful in clarifying the roles of the various domains with regard to heparin and receptor binding activity (22, 24, 28, 32). The heparin binding activity is largely confined to two separate domains within the HGF/SF molecule, the N-terminal N domain and kringle 2 (32, 33). Recent work utilizing NMR spectroscopy with the N domain alone or in the molecule HGF/NK1 shows that the primary heparin-binding site in the N-terminal N domain primarily involves the amino acids, Lys60, Lys62, and Arg73, as well as several other nearby basic amino acids and Asn77–Leu80 (34). The receptor-binding domain of HGF/SF lies in kringle 1 (24, 35). The most significant amino acids with regard to receptor binding as identified by site-directed mutagenesis are Glu159, Ser161, Glu195, and Arg197 (36). Receptor binding and biological activity may be reduced by as much as 50-fold if some of these amino acids are altered (36).
Virtually all tyrosine kinase receptors with the exception of the insulin receptor undergo a dimerization event following ligand binding. With regard to HGF/NK1, the dimerization of c-Met is facilitated by heparin binding to the N-terminal hairpin loop which in turn brings the two HGF/NK1 molecules together while exposing two receptor binding domains (34). If heparin or cell surface heparan sulfate proteoglycan (HSPG) is not present, then HGF/NK1 does not function as an agonist but rather as an antagonist (37). With full-length HGF/SF, this is not the case. HGF/SF remains fully active with cells that are devoid of HSPG (35). Yet deletion of the N-terminal N-domain does significantly diminish the activity of HGF/SF suggesting that there may be another role for the N-terminal hairpin loop in the action of full-length HGF/SF (32). An engineered variant consisting of the N-terminal N domain and all four kringles, HGF/NK4, is considered to be the only true HGF/SF antagonist (38). Not only does it compete with full-length HGF/SF for receptor binding, it does not stimulate any detectable phosphorylation of c-Met which is seen with HGF/NK1 and HGF/NK2.
In the current experiments described herein, we have determined that HGF/NK1 is a potent mitogen for primary cultures of rat hepatocytes. Furthermore, we have found that coagulation factor Xa (fXa) is capable of cleaving HGF/NK1 and scHGF/SF at a previously described secondary cleavage site of fXa, while differentially affecting the associated mitogenic activities of HGF/NK1 and scHGF/SF. The mitogenic activity of HGF/NK1 was largely abolished following cleavage by fXa, whereas the mitogenic activity of scHGF/SF was unaffected. The primary cleavage site by fXa as determined by N-terminal sequencing of the HGF/NK1 cleavage products is located C-terminal to Arg134. Addition of heparin to the fXa cleavage reaction of HGF/NK1 did not stabilize HGF/NK1 in such a way as to preserve its mitogenic activity. Additionally, the fXa processing of HGF/NK1 or scHGF/SF did not alter the heparin affinity of either molecule as determined by high pressure liquid chromatography analysis utilizing a heparin column.
MATERIALS AND METHODS
HGF/NK1 and HGF/NK2 were gifts from Dr. Jeffery Rubin at the National Institutes of Health, and scHGF/SF was a gift of Toyobo (Tokyo, JP). All other chemicals were obtained from Sigma unless otherwise stated.
Hepatocyte Isolation, Culture, and Harvesting
All animals were humanely treated and housed in accordance with the guidelines set by the Institutional Animal Use and Care Committee of the University of Pittsburgh (protocol number 0699068A-1). Male Fisher 344 rats (NCI, National Institutes of Health, Frederick, MD) were housed in a 12-h dark cycle and allowed water and food ad libitum. The rats were anesthetized for surgery with Nembutal (0.5 μg/g body weight), and the hepatocytes were isolated following collagenase perfusion of the liver (39). The resulting isolated hepatocytes were suspended and plated at a concentration of 100,000 cells/well on 6-well 35-mm tissue culture plates (BD PharMingen). Prior to use, the 6-well plates were coated with 1 ml of 10% Vitrogen (Cohesion, Palo Alto, CA) and allowed to dry. The isolated hepatocyte viability was determined prior to use by trypan blue exclusion and was consistently greater than 90%. The composition of the media used was minimum Eagle’s medium with nonessential amino acids (Invitrogen) supplemented with bovine insulin (5 μg/ml), pyruvate (1 mmol/liter), and gentamicin (10 μg/ml). Following plating, the cells were incubated at 37 °C in 5% CO2 for 2–4 h until cells attached. Following cell attachment, the media was changed with 1 ml of the previously described minimum Eagle’s medium supplemented with 2.5 μCi/ml of [3H]thymidine (ICN, Irvine, CA). At this point, growth factors were added to the cell cultures as described. After 48 h in culture at 37 °C in 5% CO2, the feeding media were removed, and 1.5 ml of cold 5% (w/v) trichloroacetic acid was added to each well. The plates were placed at 4 °C for 1 h and then were washed and allowed to air-dry. The precipitate was solubilized by the addition of 1 ml of 0.33 m NaOH, and an aliquot was added to Universol (ICN, Irvine, CA). The counts/min of the solubilized precipitates were determined with a scintillation counter (Beckman Instruments, Palo Alto, CA).
Digestion of HGF/SF Isoforms with Factor Xa
Factor Xa (American Diagnostica, Greenwich, CT) was used at a concentration of 1–3 μm. The reaction buffer was Tris-buffered saline (TBS: 147 mm NaCl, 3 mm KCl, 25 mm Tris, pH 7.4) supplemented with CaCl2 to a final concentration of 5 mm. The reaction was stopped by either heating the reaction to 60 °C for 5 min or by adding the sample to a 2× gel-loading buffer (62 mm Tris, 2% SDS, 10% glycerol, 0.14 mm bromphenol blue, and 10 mm dithiothreitol).
Electrophoresis and N-terminal Sequencing
Samples were subjected to continuous 10% Tris-Tricine SDS-PAGE (40) unless otherwise stated. This was followed by electroblotting of the protein to an Immobilon-Psq polyvinylidene difluoride membrane (Millipore, Bedford, MA). The buffer used for the transfer was 10 mm CAPS, pH 11.0, containing 10% (v/v) methanol. The blots were visualized with Coomassie Blue (Bio-Rad). The visible bands were excised from the blot, and a 5-amino acid N-terminal sequence of the band was obtained by automated Edman degradation utilizing an Applied Biosystems model 477A Protein Sequencer (Foster City, CA).
Protein Iodination
Iodo-BEADS (Pierce) were prepared as per the instructions of the manufacturer. Between 2 and 5 μg of protein were placed in a tube, which had been treated with Sigmacote (Sigma), with 100 mm sodium phosphate reaction buffer, pH 6.8, and 0.5 mCi of sodium iodide (Amersham Biosciences). The final reaction volume was 100 μl. The reaction was allowed to proceed for 10 min and was stopped by removal of the reaction solution from the Iodo-BEADS. The excess reactive free iodine was quenched by the addition of tyrosine to a final concentration of 5 mm. The tyrosine-containing reaction mixture was loaded onto a 5-ml G-25 desalting column (Pierce), which was previously equilibrated in TBS, pH 7.4, containing protease-free albumin (1 mg/ml). The iodinated protein was evaluated by gel chromatography to ensure that the HGF/SF remained intact. Specific activity was 17,000–25,000 cpm/ng (HGF/SF) and 4,000–8,000 cpm/ng (HGF/NK1).
High Performance Liquid Chromatography
The solvent delivery system (Eldex, San Carlos, CA) was attached to a TSK-heparin column (Toyo, Tokyo, JP) and the following gradient was used. From 0 to 10 min following sample injection, the mobile phase was isocratic (0.5 m NaCl). From 10 to 40 min, the mobile phase was a linear gradient from 0.5 to 2 m NaCl. The flow rate for the entire run was 1.0 ml/min. Elution was monitored by the absorbance at 280 nm with a spectrophotometer (Linear, Reno, NV).
RESULTS
Stimulation of DNA Synthesis in Hepatocyte Cultures by HGF/NK1 and scHGF/SF
The HGF/NK1 utilized in these experiments has been well characterized with regard to its structure and its associated activities and is equivalent to HGF/NK1 produced in eukaryotes (41). Hepatocytes were isolated as described under “Materials and Methods,” and either scHGF/SF or HGF/NK1 was administered to the cultures as described. The concentration of growth factor used ranged between 0 and 210 ng/ml. To ensure that the buffer (TBS) used to dilute the growth factors was not toxic or stimulatory to the hepatocytes, the same volume of buffer used for each point was administered to cultures in the absence of growth factors. The results showed that addition of TBS had no effect on the hepa-tocytes, and the results were comparable with cultures in which either nothing was added or factor Xa alone was added (data not shown). As shown in Fig. 1, HGF/NK1 elicited a mitogenic response in primary cultured hepatocytes. Furthermore, the level of maximal stimulation by HGF/NK1 was comparable with that of scHGF/SF. Whereas the scHGF/SF is a pro-form of HGF/SF, previous work (42) has shown that cultured hepatocytes produce uPA and are fully capable of activating scHGF/SF. The half-maximal level of stimulation by scHGF/SF was estimated to be 51 pm (5 ng), whereas the half-maximal stimulation of HGF/NK1 was estimated to be 714 pm (15 ng) (Fig. 1).
Fig. 1. Effect of HGF/NK1 and scHGF/SF on DNA synthesis by primary rat hepatocytes.
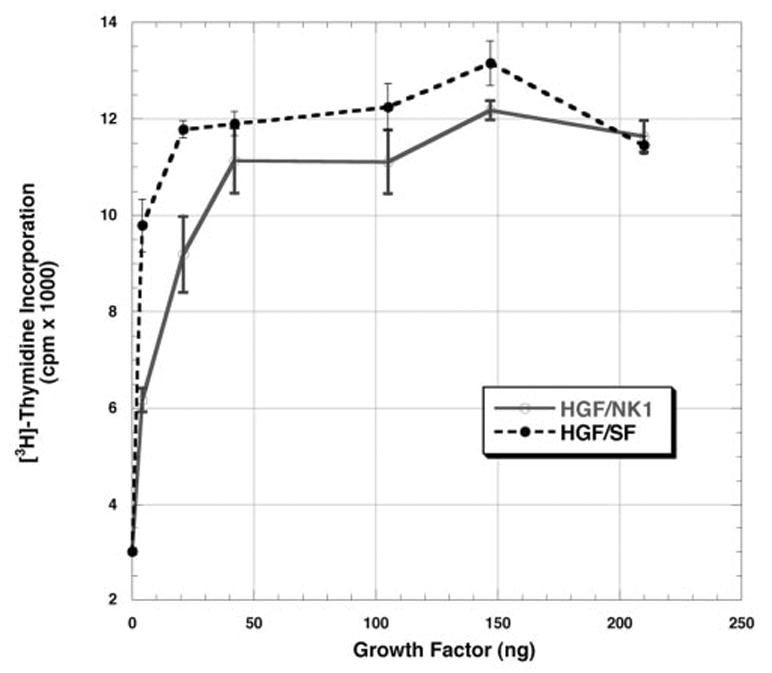
Stimulation of [3H]thymidine incorporation by HGF/NK1 (open circles) and scHGF/SF (closed circles) in 1-ml cultures of rat hepatocytes, which were cultured for 48 h as described under “Materials and Methods.” The values represent the mean values ± S.E. from one representative experiment expressed as cpm × 10−3. Similar results were seen in the other two experiments.
Processing of scHGF/SF and HGF/NK1 by Coagulation Factor Xa
Fig. 2 shows the autoradiograms from time course digestion of scHGF/SF (1 μg, 10.2 pmol) or HGF/NK1 (0.5 μg, 23.8 pmol) by coagulation factor Xa (fXa). The reactions were sampled at indicated times and analyzed either by non-reducing PAGE (Fig. 2, A and C) or by reducing PAGE (Fig. 2, B and D). The autoradiograms run under reducing PAGE clearly demonstrate that fXa is capable of cleaving scHGF/SF (Fig. 2B) and HGF/NK1 (Fig. 2D). The rate of disappearance by 2 h of HGF/NK1 is greater than the rate of disappearance by 2 h of scHGF/SF. Because the reaction volumes were identical, HGF/NK1 appears to be a better substrate for fXa than scHGF/SF (Fig. 2, B and D). This is likely due to the accessibility of the fXa to the proteolytic site. The proteolytic site in the scHGF/SF may be further shielded by the three additional kringles as well as the light chain than is the proteolytic site in HGF/NK1.
Fig. 2. Autoradiograms of time course digestions of scHGF/SF and HGF/NK1 by factor Xa.
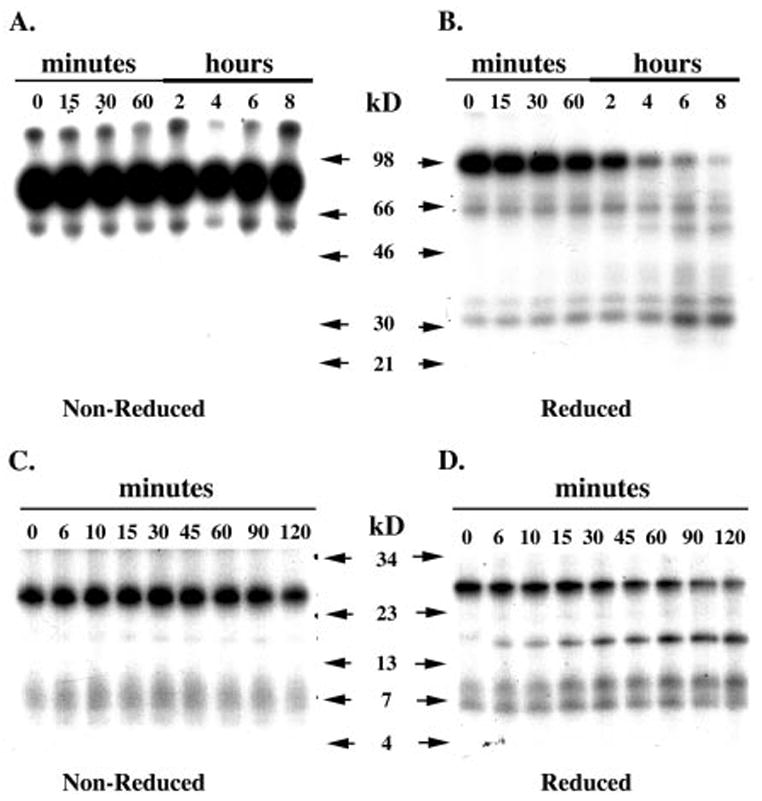
One μg of scHGF/SF (A and B) and 0.5 μg of HGF/NK1 (C and D) were digested with factor Xa as described under “Materials and Methods.” At indicated times, the reactions were sampled and analyzed by PAGE. A, scHGF/SF under non-reducing conditions. B, scHGF/SF under reducing conditions. C, HGF/NK1 under non-reducing conditions. D, HGF/NK1 under reducing conditions.
Examination of the scHGF/SF digestion autoradiogram under non-reducing PAGE shows no apparent fragments; therefore, the primary cleavage site must be located between the disulfide bonds at Cys70 and Cys604 (Fig. 2A). If cleavage occurred in the light chain region of HGF/SF, one would expect diminished intensity of the band corresponding to the light chain (32-kDa doublet, Fig. 2B). This did not occur; rather, at the last time points (6 and 8 h, Fig. 2B), the amount of light chain appeared to increase indicating that activation of scHGF/SF at the Arg494–Val495 occurred.
To confirm that the primary cleavage site of fXa is located within the heavy chain, the splice variant isoform, HGF/NK1, was cleaved with fXa. Under reducing conditions, the cleavage reaction produced two fragments with apparent molecular masses of 15 and 10 kDa (Fig. 2D). PAGE analysis of HGF/NK1 by fXa under non-reducing conditions showed no significant bands other than the band corresponding to a single HGF/NK1 molecule (Fig. 2C). Although a very faint band is seen measuring about 15 kDa in the non-reduced HGF/NK1 autoradiogram, this band is most likely due to inadvertent reduction of a disulfide bond within the HGF/NK1 molecule during sample preparation.
Identification of the fXa Cleavage Site by N-terminal Sequencing
In order to obtain the N-terminal sequence of the fragments and to identify the cleavage site, HGF/NK1 (8 μg) was digested with fXa. Fig. 3 shows the Coomassie Blue-stained polyvinylidene difluoride membrane that was used for sequencing of the HGF/NK1 fragments. The N-terminal sequence of the whole molecule of scHGF/SF is GQRKR (Gly31–Arg35) and was confirmed by N-terminal sequencing of the lone band in the lane HGF/NK1 (Fig. 3). The band that appears in the HGF/NK1 + fXa lane with an apparent molecular mass of 17 kDa corresponds to the light chain of fXa (Fig. 3). The band with an apparent molecular mass of 15 kDa had a sequence that was identical to the N-terminal sequence of the intact molecule (GQRKR) (Fig. 3). The band with an apparent molecular mass of 11 kDa had the sequence SYKGT (Fig. 3). This sequence corresponded to Ser135–Thr139 of HGF/NK1. To confirm this site, HGF/NK2 was subjected to fXa digestion, and the results identified the same cleavage site (data not shown). Attempts were made to sequence directly scHGF/SF following fXa digestion, but we lacked sufficient protein to generate enough products for sequencing. In the literature, the preferred cleavage site of fXa is defined by the linear sequence Ile-(Glu or Asp)-Gly-Arg, but fXa can also cleave at secondary sites defined by the sequence Gly-Arg (43).
Fig. 3. Factor Xa digestion HGF/NK1 used for protein sequencing.
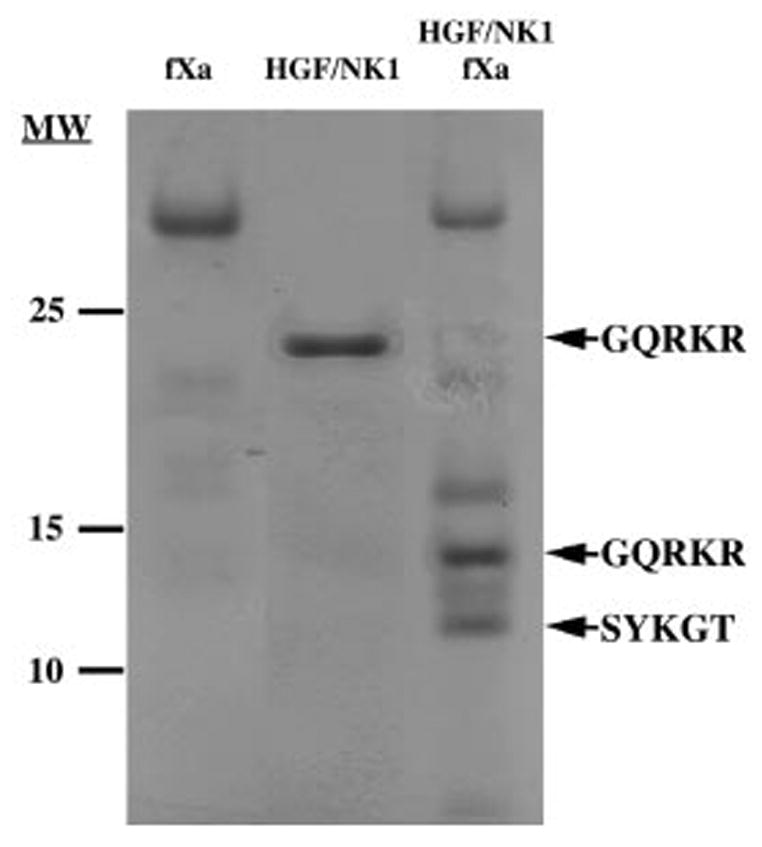
HGF/NK1 was digested with factor Xa overnight as described under “Materials and Methods.” The lane FXa contains only fXa with no HGF/NK1. The lane HGF/NK1 was 5 μg of HGF/NK1 incubated without factor Xa. The lane HGF/NK1 FXa was 8 μg of HGF/NK1 digested with factor Xa. Molecular weights are marked on the left side of the blot, and five amino acid sequences obtained from N-terminal sequencing are labeled on the right side of the blot.
Biological Activity of the Factor Xa-processed scHGF/SF and HGF/NK1
Fig. 4 is the dose-response curve of the growth factors (HGF/NK1 and scHGF/SF) incubated overnight either with or without fXa. Following digestion with fXa, the mitogenic activity of HGF/NK1 is significantly diminished, whereas the activity of scHGF/SF remains unaffected (Fig. 4). In earlier work, Zhou et al. (34) showed that heparin stabilizes HGF/NK1 and protects its structure from reduction by dithiothreitol. To examine if heparin stabilizes HGF/NK1 and prevents the loss of activity following fXa digestion, reactions (final volume 115 μl) of scHGF/SF or HGF/NK1 were carried out in the presence or absence of 1 μg of heparin (Fig. 5A). The reaction mixtures were then added to cultures of primary hepatocytes. The final concentration of heparin in the cultures was kept at 0.125 μg/ml. The results show that even in the presence of heparin, the mitogenic activity of HGF/NK1 is largely abolished. As expected, addition of heparin to undigested HGF/NK1 prior to the addition to the hepatocyte culture enhances the biological potency of HGF/NK1. This is especially apparent below the 42-ng data point (Fig. 5A). There was no effect of heparin on the scHGF/SF mitogenic response whether the scHGF/SF was digested with or without fXa, and scHGF/SF potency at very low doses was unchanged by addition of heparin to the fXa cleavage reaction (Fig. 5B). Interestingly, heparin in the reaction accelerated the cleavage of scHGF/SF (Fig. 6A) and HGF/NK1 (Fig. 6B) by fXa. This may be due either to the ability of heparin to induce a conformational change in the HGF/SF isoforms, thus further exposing Arg134, or to the possibility that heparin acts as a nucleation center bringing both substrate and enzyme together. To verify that the affinity of the HGF/SF isoforms for heparin was unaltered following their cleavage by the fXa, high pressure liquid chromatography analysis was used. Following overnight digestion of scHGF/SF and HGF/NK1 with fXa, the products of the reaction were loaded onto a heparin affinity column and eluted as described under “Materials and Methods.” The retention time of scHGF/SF and HGF/NK1 either digested or undigested with fXa was 27.04 and 28.18 min, respectively.
Fig. 4. Effect of HGF/NK1 and scHGF/SF digested with or without factor Xa on DNA synthesis by primary rat hepatocytes.
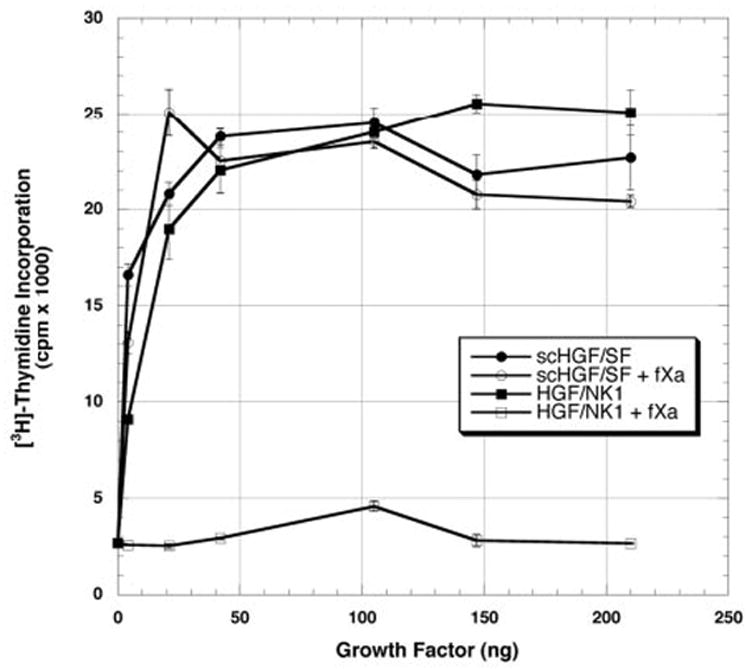
Stimulation of [3H]thymidine incorporation by HGF/NK1 (closed squares), scHGF/SF (closed circles), HGF/NK1 digested with fXa (open squares), and scHGF/SF digested with fXa (open circles) in 1-ml cultures of rat hepatocytes cultured for 48 h as described under “Materials and Methods.” The values represent the mean values ± S.E. from one representative experiment expressed as cpm × 10−3. Similar results were seen in the other two experiments.
Fig. 5. Effect of HGF/NK1 and scHGF/SF digested with factor Xa in the presence or absence of heparin on DNA synthesis by primary rat hepatocytes.
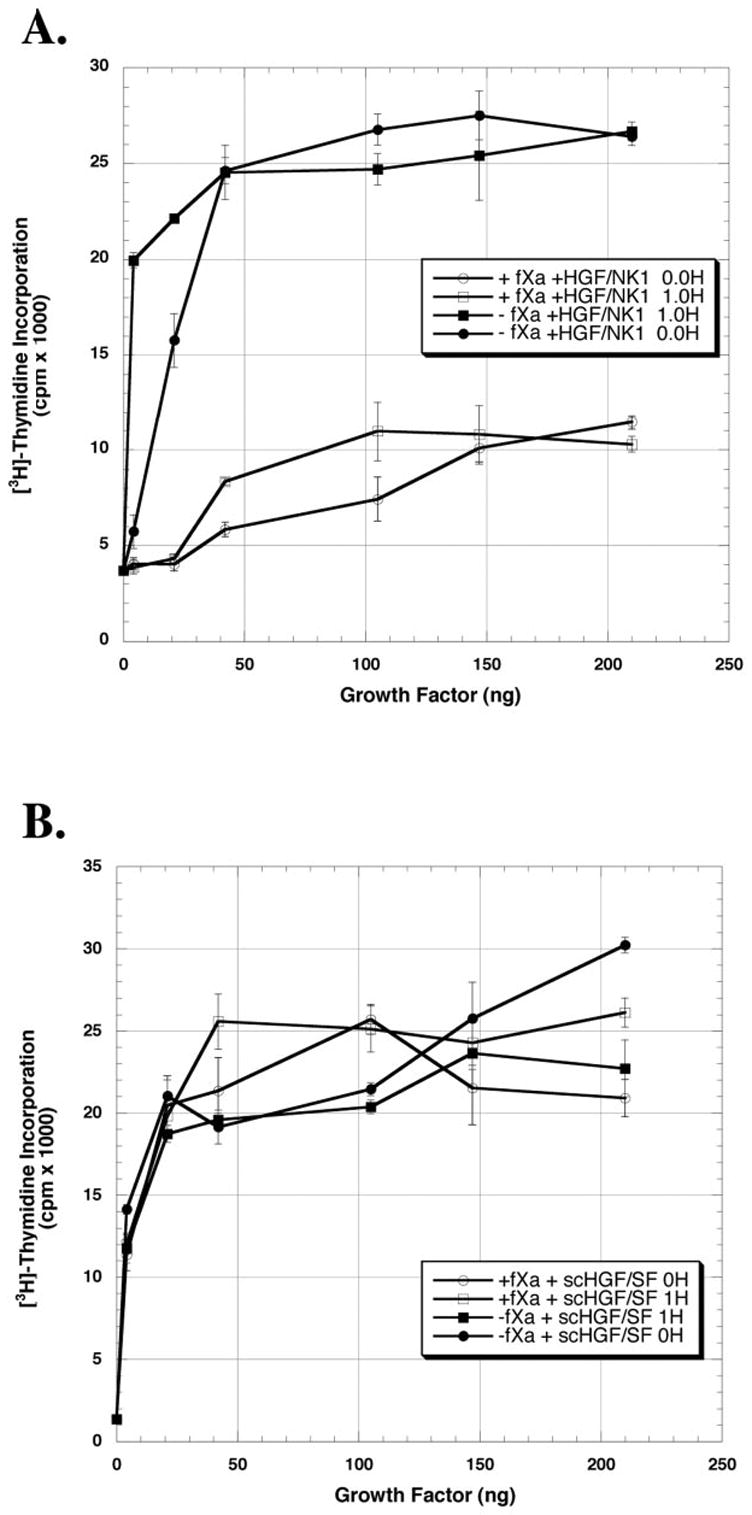
HGF/NK1 (A) or scHGF/SF (B) was digested with or without fXa in the presence or absence of 1 μg of heparin in a final reaction volume of 115 μl. The values represent the mean values ± S.E. from one representative experiment expressed as cpm × 10−3. Similar results were seen in the other two experiments.
Fig. 6. Autoradiograms of time course digestions of scHGF/SF and HGF/NK1 by factor Xa in the presence of heparin.
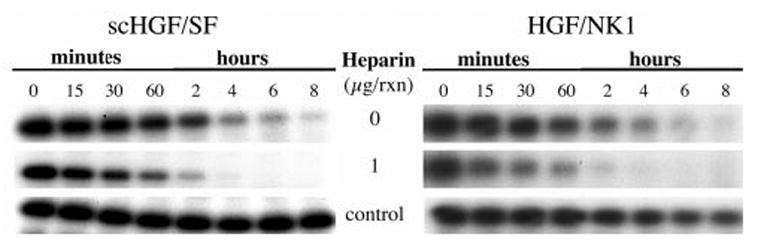
A, scHGF/SF (1 μg) was digested in the presence or absence of 1 μg of heparin. B, HGF/NK1 (0.5 μg) was digested in the presence or absence of 1 μg of heparin. A control reaction was identical in composition to the cleavage reactions except fXa was replaced with TBS.
DISCUSSION
In previous studies, the mitogenic response of this HGF/NK1 was examined with B5/589 cells, a mammary epithelial cell line (41). The investigators showed that the maximum level of stimulation by HGF/NK1 was only 60% of the level that was achieved with activated HGF/SF and that the half-maximal response to HGF/NK1 corresponded to a concentration of about 5 nm (41). In our work with primary cultures of rat hepatocytes, we show that HGF/NK1 can stimulate a mitogenic response that is ~90% of the maximal level of stimulation achieved with scHGF/SF. The concentration for the half-maximal response, which we observed with hepatocytes, was 0.714 nm (15 ng/ml). Several reasons can account for this discrepancy in mitogenic responses of cells to HGF/NK1. First and foremost is that different cell types were used. This is supported by the fact that different cell types exhibit varying degrees of mitogenic stimulation in response to HGF/SF. The cell surface heparin sulfate proteoglycan content could also affect the mitogenic response and may also influence the sensitivity of the cells to HGF/NK1 stimulation. Additionally, culture size and population density of cells used in the respective assays may affect the mitogenic response.
In the course of looking for proteases that are capable of activating or cleaving scHGF/SF other than the previously described proteases uPA and HGF activator, we found that fXa was capable of specifically cleaving scHGF/SF at a site other than the normal activation site (Arg494). Furthermore, this processing greatly reduced the mitogenic activity of cultured hepatocytes in response to HGF/NK1, while not affecting the mitogenic response of the cultured hepatocytes to scHGF/SF.
Factor Xa is a trypsin-like serine protease. The normally described cleavage site of fXa is at the C-terminal bond of Arg in the linear sequence, Ile-(Asp/Glu)-Gly-Arg. A secondary cleavage site of fXa has also been described which consists of only two amino acids Gly-Arg (43). Within the scHGF/SF molecule, none of the primary cleavage sites exist, but three of the secondary sites do exist: Arg134, Arg556, and Arg695. To identify which of the possible fXa cleavage sites was actually utilized, the reaction products were examined by PAGE under reducing and non-reducing conditions (Fig. 2, A and B). Because only a single band was seen in the scHGF/SF digestion products under non-reducing conditions, the site Arg695 can be ruled out (Fig. 2A) because Arg695 lies C-terminal to the last cysteine involved in the disulfide bridge which holds the light chain of tcHGF/SF to the heavy chain of tcHGF/SF. We also ruled out the fXa site on the light chain at Arg556 as the cleavage site. If Arg556 were the cleavage site, then we should see a disappearance of light chain in the fXa-digested scHGF/SF autoradiogram, which did have a small amount of activated tcHGF/SF in it (Fig. 2B). Instead we saw an increase in light chain indicating that fXa may also be capable of cleaving scHGF/SF at Arg494 (Fig. 2B). Only Arg134 remained as a likely cleavage site for fXa and would yield fragments similar in size to those we observed.
To confirm that Arg134 was indeed the cleavage site, we utilized the HGF/NK1 molecule and examined its products by PAGE under reducing as well as non-reducing conditions (Fig. 2, C and D). The non-reduced autoradiogram showed only a single band; this suggested to us that the cleavage site must be located between Cys70 and Cys206 (Fig. 2C). Under reducing conditions, the fragment sizes were ~15 and 10 kDa. Although this evidence strongly supports the notion that Arg134 is the cleavage site, we digested 8 μg of HGF/NK1 with fXa and sequenced the reaction products (Fig. 3). To verify this site, an fXa cleavage reaction was also performed on HGF/NK2 (6 μg). It yielded identical results (data not shown).
To examine whether cleavage of scHGF/SF or HGF/NK1 by fXa altered the associated mitogenic activity, we evaluated the mitogenic activity of the proteins following fXa digestion. We found that scHGF/SF mitogenic activity was unaffected by fXa cleavage at Arg134, whereas the HGF/NK1 activity was severely blunted (>90%) (Fig. 4). Because fXa-cleaved scHGF/SF is still fully capable of eliciting a potent mitogenic response, it is reasonable to assume that the receptor binding domain of scHGF/SF via the first kringle domain and as well as the ability of c-Met to transduce the mitogenic signal were unaffected by fXa processing. Therefore, the reason for the significant reduction in HGF/NK1 activity following digestion with fXa is not likely due to the capacity of HGF/NK1 to bind c-Met; rather, it must be due to the ability for HGF/NK1 to induce dimerization of the c-Met receptor.
Although a complete understanding of how tcHGF/SF dimerizes the c-Met receptor is not yet known, the manner in which HGF/NK1 dimerizes the receptor is more clearly understood. The N domain has been shown to possess most of the heparin binding activity associated with the whole HGF/SF molecule, whereas the first kringle domain retains all of the receptor binding activity of tcHGF/SF. In solution or on the cell surface, heparin acts as a bridge to join two distinct HGF/NK1 molecules leaving the exposed kringle 1 to bind to the receptor (34). This explains why HGF/NK1 has been described as an antagonist. If heparin is not present at a sufficient level, then HGF/NK1 is not capable of dimerizing c-Met. Thus HGF/NK1 acts as an HGF/SF antagonist (37). The first kringle, whether heparin is present or not, is fully capable of binding to the c-Met receptor with equal avidity as full-length HGF/SF (35).
Our data suggest that fXa-cleaved HGF/NK1 fails to induce dimerization of c-Met receptors. A possible explanation why this occurs may be explained by the spatial orientation of the HGF/NK1 molecule and the manner by which HGF/NK1 acts to dimerize the c-Met receptor. The crystal-derived structure for HGF/NK1 shows that the primary heparin-binding site in the N domain is located on the opposite side of the molecule as the residues Glu159, Ser161, Glu195, and Arg197 of kringle 1, which are known to be involved in receptor binding (44, 45) (Fig. 7). Because the cleavage site at Arg134 is located six amino acids into the first kringle domain, this may allow for relaxation of the N domain of the HGF/NK1 molecule in such a way as to perturb its orientation with regard to the first kringle domain. Cleavage at Arg134 would allow for the entire N domain to have complete freedom of rotation around the disulfide bond of Cys128–Cys206, which may prevent efficient dimerization of c-Met by heparin and HGF/NK1. Another possibility is that the cleavage of HGF/NK1 by fXa significantly alters the affinity of HGF/NK1 for heparin thus preventing effective HGF/NK1 dimerization. This is unlikely given the fact that the retention times of cleaved and uncleaved HGF/NK1 on a heparin column were identical.
Fig. 7. A three-dimensional representation of HGF/NK1.
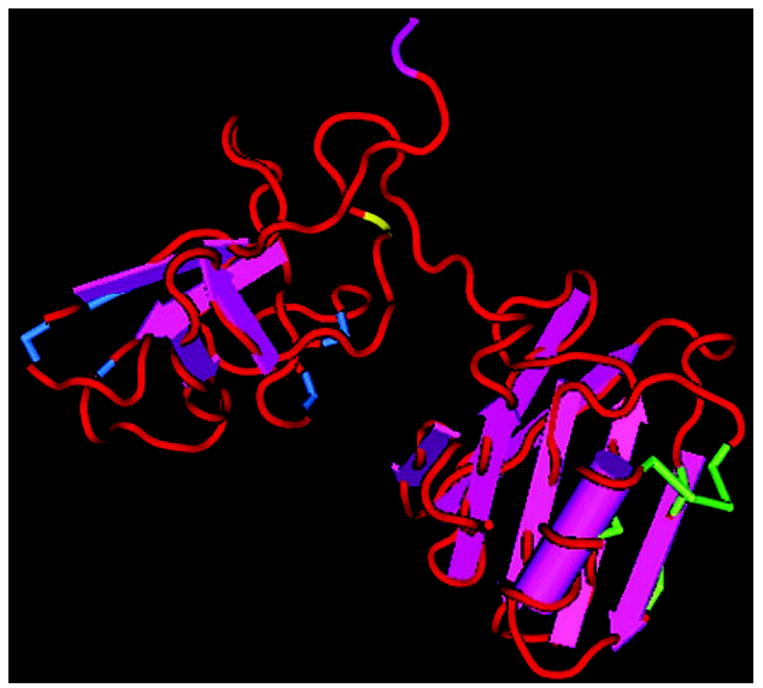
The three-dimensional representation of HGF/NK1 was rendered with CN3D and is based on the solved structure (Protein Data Bank code 1NK1) by Chirgadze et al. (48). The amino acids marked in green represent the residues involved in heparin binding and lie in the N domain. The amino acids marked in blue represent residues that are involved in receptor binding and lie in the first kringle domain. The yellow residue represents the cleavage site of HGF/NK1 by factor Xa, arginine 135.
Our results show that the scHGF/SF dose-response curve was unaffected by fXa cleavage (Fig. 4). This implies that the receptor-binding domain in the first kringle is still capable of binding to c-Met with equal avidity as uncleaved scHGF/SF. Furthermore, addition of heparin to the reaction did not alter the results; the scHGF/SF dose response with or without heparin demonstrated that heparin is not a requirement for full-length HGF/SF-induced dimerization of the c-Met receptor. This leads us to conclude that dimerization of c-Met by full-length HGF/SF utilizes a mechanism other than heparin. This is supported by other studies, which showed that HGF/SF activity is not affected by the presence or absence of HSPGs on the cell surface, whereas for HGF/NK1 the presence of heparin is a requirement for mitogenic activity (37, 46).
The fact that the fXa cleavage of scHGF/SF did not alter the mitogenic response leads to some interesting possibilities. One would expect that cleavage at Arg134, which is located at the bottom of the kringle domain, would disrupt the kringle domain and significantly alter its ability to bind ligand. If this happened, then one would predict that the mitogenic response of hepatocytes to scHGF/SF would be affected. Our experiments clearly show that digestion of scHGF/SF by fXa does not alter the associated mitogenic activity. It is possible that subdomains within the first kringle exist which are sufficient to allow high affinity binding of HGF/SF to c-Met, such as fibronectin type II sub-domains contained with in the kringle (47).
The cleavage of scHGF/SF and HGF/NK1 by fXa or other serine proteases may be a likely occurrence in vivo in a rich proteolytic environment, where many trypsin-like serine pro-teases are active, such as at sites of inflammation or injury in which scHGF/SF is commonly present. Cleavage by fXa or other similar serine protease may be used as a mechanism by which the less active and potentially antagonistic activity of HGF/NK1 can be eliminated, whereas the main agonistic activity of HGF/SF is employed.
The abbreviations used are
- HGF/SF
hepatocyte growth factor/scatter factor
- tcHGF/SF
activated or two-chain hepatocyte growth factor/scatter factor
- HSPG
heparan sulfate proteoglycan
- fXa
factor Xa
- uPA
urokinase plasminogen activator
- CAPS
3-(cyclohexylamino)propanesulfonic acid
- Tricine
N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
Footnotes
This work was supported by National Institutes of Health Grants CA30241 and CA35373 (to G. K. M.).
References
- 1.Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, Sugimura A, Tashiro K, Shimizu S. Nature. 1989;342:440–443. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 2.Stoker M, Perryman M. J Cell Sci. 1985;77:209–223. doi: 10.1242/jcs.77.1.209. [DOI] [PubMed] [Google Scholar]
- 3.Zarnegar R, Michalopoulos G. Cancer Res. 1989;49:3314–3320. [PubMed] [Google Scholar]
- 4.Galimi F, Brizzi MF, Comoglio PM. Stem Cells. 1993;11(Suppl 2):22–30. doi: 10.1002/stem.5530110805. [DOI] [PubMed] [Google Scholar]
- 5.Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, Kitamura N. Nature. 1995;373:702–705. doi: 10.1038/373702a0. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, Gherardi E, Birchmeier C. Nature. 1995;373:699–702. doi: 10.1038/373699a0. [DOI] [PubMed] [Google Scholar]
- 7.Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Nature. 1995;376:768–771. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- 8.Mizuno S, Kurosawa T, Matsumoto K, Mizuno-Horikawa Y, Okamoto M, Nakamura T. J Clin Invest. 1998;101:1827–1834. doi: 10.1172/JCI1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, Dai C, Liu Y. Gene Ther. 2001;8:1470–1479. doi: 10.1038/sj.gt.3301545. [DOI] [PubMed] [Google Scholar]
- 10.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 11.Sonnenberg E, Weidner KM, Birchmeier C. Exs (Basel) 1993;65:381–394. [PubMed] [Google Scholar]
- 12.Nakamura T. Princess Takamatsu Symp. 1994;24:195–213. [PubMed] [Google Scholar]
- 13.Birchmeier C, Gherardi E. Trends Cell Biol. 1998;8:404–410. doi: 10.1016/s0962-8924(98)01359-2. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto K, Date K, Ohmichi H, Nakamura T. Cancer Chemother Pharmacol. 1996;38:S42–S47. doi: 10.1007/s002800051037. [DOI] [PubMed] [Google Scholar]
- 15.Stoker M, Gherardi E, Perryman M, Gray J. Nature. 1987;327:239–242. doi: 10.1038/327239a0. [DOI] [PubMed] [Google Scholar]
- 16.Tuck AB, Park M, Sterns EE, Boag A, Elliott BE. Am J Pathol. 1996;148:225–232. [PMC free article] [PubMed] [Google Scholar]
- 17.Rong S, Jeffers M, Resau JH, Tsarfaty I, Oskarsson M, Vande Woude GF. Cancer Res. 1993;53:5355–5360. [PubMed] [Google Scholar]
- 18.Prat M, Narsimhan RP, Crepaldi T, Nicotra MR, Natali PG, Comoglio PM. Int J Cancer. 1991;49:323–328. doi: 10.1002/ijc.2910490302. [DOI] [PubMed] [Google Scholar]
- 19.Natali PG, Nicotra MR, Di Renzo MF, Prat M, Bigotti A, Cavaliere R, Comoglio PM. Br J Cancer. 1993;68:746–750. doi: 10.1038/bjc.1993.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Natali PG, Prat M, Nicotra MR, Bigotti A, Olivero M, Comoglio PM, Di Renzo MF. Int J Cancer. 1996;69:212–217. doi: 10.1002/(SICI)1097-0215(19960621)69:3<212::AID-IJC11>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 21.Olivero M, Rizzo M, Madeddu R, Casadio C, Pennacchietti S, Nicotra MR, Prat M, Maggi G, Arena N, Natali PG, Comoglio PM, Di Renzo MF. Br J Cancer. 1996;74:1862–1868. doi: 10.1038/bjc.1996.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gherardi E, Sharpe M, Lane K. Exs (Basel) 1993;65:31–48. [PubMed] [Google Scholar]
- 23.Walz DA, Hewett-Emmett D, Seegers WH. Proc Natl Acad Sci U S A. 1977;74:1969–1972. doi: 10.1073/pnas.74.5.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lokker NA, Mark MR, Luis EA, Bennett GL, Robbins KA, Baker JB, Godowski PJ. EMBO J. 1992;11:2503–2510. doi: 10.1002/j.1460-2075.1992.tb05315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mars WM, Zarnegar R, Michalopoulos GK. Am J Pathol. 1993;143:949–958. [PMC free article] [PubMed] [Google Scholar]
- 26.Shimomura T, Miyazawa K, Komiyama Y, Hiraoka H, Naka D, Morimoto Y, Kitamura N. Eur J Biochem. 1995;229:257–261. doi: 10.1111/j.1432-1033.1995.tb20463.x. [DOI] [PubMed] [Google Scholar]
- 27.Shima N, Nagao M, Ogaki F, Tsuda E, Murakami A, Higashio K. Biochem Biophys Res Commun. 1991;180:1151–1158. doi: 10.1016/s0006-291x(05)81187-8. [DOI] [PubMed] [Google Scholar]
- 28.Hartmann G, Naldini L, Weidner KM, Sachs M, Vigna E, Comoglio PM, Birchmeier W. Proc Natl Acad Sci U S A. 1992;89:11574–11578. doi: 10.1073/pnas.89.23.11574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cioce V, Csaky KG, Chan AM, Bottaro DP, Taylor WG, Jensen R, Aaronson SA, Rubin JS. J Biol Chem. 1996;271:13110–13115. doi: 10.1074/jbc.271.22.13110. [DOI] [PubMed] [Google Scholar]
- 30.Chan AM, Rubin JS, Bottaro DP, Hirschfield DW, Chedid M, Aaronson SA. Science. 1991;254:1382–1385. doi: 10.1126/science.1720571. [DOI] [PubMed] [Google Scholar]
- 31.Jakubczak JL, LaRochelle WJ, Merlino G. Mol Cell Biol. 1998;18:1275–1283. doi: 10.1128/mcb.18.3.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizuno K, Inoue H, Hagiya M, Shimizu S, Nose T, Shimohigashi Y, Nakamura T. J Biol Chem. 1994;269:1131–1136. [PubMed] [Google Scholar]
- 33.Kinosaki M, Yamaguchi K, Murakami A, Ueda M, Morinaga T, Higashio K. Biochim Biophys Acta. 1998;1384:93–102. doi: 10.1016/s0167-4838(98)00002-8. [DOI] [PubMed] [Google Scholar]
- 34.Zhou H, Casas-Finet JR, Heath Coats R, Kaufman JD, Stahl SJ, Wingfield PT, Rubin JS, Bottaro DP, Byrd RA. Biochemistry. 1999;38:14793–14802. doi: 10.1021/bi9908641. [DOI] [PubMed] [Google Scholar]
- 35.Rubin JS, Day RM, Breckenridge D, Atabey N, Taylor WG, Stahl SJ, Wingfield PT, Kaufman JD, Schwall R, Bottaro DP. J Biol Chem. 2001;276:32977–32983. doi: 10.1074/jbc.M105486200. [DOI] [PubMed] [Google Scholar]
- 36.Lokker NA, Presta LG, Godowski PJ. Protein Eng. 1994;7:895–903. doi: 10.1093/protein/7.7.895. [DOI] [PubMed] [Google Scholar]
- 37.Schwall RH, Chang LY, Godowski PJ, Kahn DW, Hillan KJ, Bauer KD, Zioncheck TF. J Cell Biol. 1996;133:709–718. doi: 10.1083/jcb.133.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Date K, Matsumoto K, Shimura H, Tanaka M, Nakamura T. FEBS Lett. 1997;420:1–6. doi: 10.1016/s0014-5793(97)01475-0. [DOI] [PubMed] [Google Scholar]
- 39.Jirtle RL, Michalopoulos G, McLain JR, Crowley J. Cancer Res. 1981;41:3512–3518. [PubMed] [Google Scholar]
- 40.Schagger H, von Jagow G. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 41.Stahl SJ, Wingfield PT, Kaufman JD, Pannell LK, Cioce V, Sakata H, Taylor WG, Rubin JS, Bottaro DP. Biochem J. 1997;326:763–772. doi: 10.1042/bj3260763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mars WM, Kim TH, Stolz DB, Liu ML, Michalopoulos GK. Cancer Res. 1996;56:2837–2843. [PubMed] [Google Scholar]
- 43.Stevens RC. Structure Fold Des. 2000;8:R177–R185. doi: 10.1016/s0969-2126(00)00193-3. [DOI] [PubMed] [Google Scholar]
- 44.Lietha D, Chirgadze DY, Mulloy B, Blundell TL, Gherardi E. EMBO J. 2001;20:5543–5555. doi: 10.1093/emboj/20.20.5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ultsch M, Lokker NA, Godowski PJ, de Vos AM. Structure. 1998;6:1383–1393. doi: 10.1016/s0969-2126(98)00138-5. [DOI] [PubMed] [Google Scholar]
- 46.Sakata H, Stahl SJ, Taylor WG, Rosenberg JM, Sakaguchi K, Wingfield PT, Rubin JS. J Biol Chem. 1997;272:9457–9463. doi: 10.1074/jbc.272.14.9457. [DOI] [PubMed] [Google Scholar]
- 47.Ozhogina OA, Trexler M, Banyai L, Llinas M, Patthy L. Protein Sci. 2001;10:2114–2122. doi: 10.1110/ps.15801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chirgadze DY, Hepple JP, Zhou H, Byrd RA, Blundell TL, Gherardi E. Nat Struct Biol. 1999;6:72–79. doi: 10.1038/4947. [DOI] [PubMed] [Google Scholar]