

Abstract
Control over cell cycle exit is fundamental to the normal generation of the wide array of distinct cell types that comprise the mature vertebrate CNS. Here, we demonstrate a critical role for Cip/Kip class cyclin-kinase inhibitory (CKI) proteins in regulating this process during neurogenesis in the embryonic spinal cord. Using immunohistochemistry, we show that all three identified Cip/Kip CKI proteins are expressed in both distinct and overlapping populations of nascent and post-mitotic neurons during early neurogenesis, with p27Kip1 having the broadest expression, and both p57 kip2 and p21Cip1 showing transient expression in restricted populations. Loss- and gain-of-function approaches were used to establish the unique and redundant functions of these proteins in spinal cord neurogenesis. Using genetic lineage tracing, we provide evidence that, in the absence of p57, nascent neurons re-enter the cell cycle inappropriately but later exit to begin differentiation. Analysis of p57kip2; p27kip1double mutants, where p21 expression is confined to only a small population of interneurons, demonstrates that Cip/Kip CKI-independent factors initiate progenitor cell cycle exit for the majority of interneurons generated in the developing spinal cord. Our studies indicate that p57 plays a critical cell-autonomous role in timing cell cycle exit at G1/S by opposing the activity CyclinD1, which promotes cell cycle progression. These studies support a multi-step model for neuronal progenitor cell cycle withdrawal that involves p57kip2 in a central role opposing latent Cyclin D1 and other residual cell cycle promoting activities in progenitors targeted for differentiation.
Keywords: Neurogenesis, stem cells, proliferation, neural tube, differentiation, progenitor cyclin, p57kip2, p27kip1, p21Cip1
INTRODUCTION
The vertebrate central nervous system (CNS) contains thousands of functionally distinct neuronal cell types, the vast majority of which are produced within a brief period during embryogenesis. Normal CNS function depends critically on the generation of both the correct types and numbers of neurons in their proper positions and at the proper time. Post-mitotic neurons arise from multi-potent progenitors that can give rise to multiple distinct progeny types, making it critical to balance proliferation with cell cycle withdrawal so that the appropriate number of cells are generated at any given interval while also leaving sufficient numbers of progenitors available to generate subsequent neuronal, and glial, types. Although the core cellular mechanisms regulating progenitor cell cycle exit and differentiation in the CNS are similar to other developing organ systems, it is unclear whether similar factors play equivalent roles in all tissues or whether context-dependent differences exist in their function.
The current study addresses these issues using the vertebrate spinal cord as a model system. In this structure, distinct classes of neurons are generated during neurogenesis in discrete domains along the dorsoventral (DV) axis, and derive from similarly organized progenitor domains (Jessell, 2000). In the spinal cord, the production of post-mitotic neurons from neuronal progenitors (neurogenesis) begins shortly after the formation of the neural tube, and is largely complete by mid-gestation. As in most other regions of the developing CNS, proliferating cells in the spinal cord reside in the ventricular zone (VZ) and undergo interkinetic nuclear migration during progression through the cell cycle. During this process, cell nuclei translocate between the lumenal and pial sides of the VZ, with the position of the nucleus correlating with phases of the cell cycle: nuclei close to the lumen medially are in M-phase, those located laterally towards the pial surface are in S-phase, and those that are in transit are in G1 or G2.
Studies in cell culture have shown that the decision to continue or withdraw at each cycle is controlled by mitogen availability and is mediated by D-type cyclins at a point late in G1 phase, termed R (for restriction point; Sherr and Roberts, 1999). Many signals are thought to exert their control over the cell cycle at R. For example, various mitogens and integrin-mediated ECM signals stimulate cell cycle progression, while factors such as TGF-β can induce cell cycle withdrawal (Assoian and Schwartz, 2001). Completion of a cell cycle (passage through M-phase) after crossing R is independent of further inputs (Sherr and Roberts, 1999). In the spinal cord, nascent neurons that withdraw from the cell cycle enter a G0 state and migrate out of the VZ into the mantle zone (MZ), an area of accumulating neurons that becomes the adult gray matter. This transition takes place when their nuclei are positioned laterally in the VZ during late G1 phase.
A number of factors have been identified whose expression is either initiated in or confined to cells near the lateral margins of the VZ during neurogenesis, making them good candidates for regulating the transition of progenitor cells into neurons. Among these are p27Kip1 and 57Kip2, two of the three members of the vertebrate Cip/Kip family of cyclin- and cyclin-dependent-kinase inhibitor (CKI) proteins (Sherr and Roberts, 1999; Zhang, 1999). The expression and function of the third member, p21Cip1, has not been characterized extensively in the developing CNS.
Several lines of evidence indicate that Cip/Kip factors play an important role in many tissues to prevent cell cycle progression at the G1/S restriction point (R) via control of Rb phosphorylation, a factor that is a critical regulator of the E2F factors that control the transcription of S-phase DNA synthesis genes (Macleod, 1999; Hamel et al., 1992). First, the expression of these factors is largely confined to post-mitotic cells (Nagahama et al., 2001; Westbury et al., 2001). Second, loss-of-function studies in mice have demonstrated that p27Kip1 and p57Kip2 mutants exhibit various proliferative defects consistent with their requirement in arresting mitosis in various organs of the body (Fero et al., 1996; Kiyokawa et al., 1996; Nakayama et al., 1996; Yan et al., 1997; Zhang et al., 1998). Third, gain-of-function experiments have shown that these proteins are capable of driving proliferating cells out of the cell cycle (Sherr and Roberts, 1999; Dyer and Cepko, 2000; Dyer and Cepko, 2001).
In the mammalian CNS, the role of p27 and p57 has been studied in retinal histogenesis. In the absence of each of these CKIs, additional rounds of cell division are seen, along with changes in the proportions of retinal sub-types differing depending on which Cip/Kip CKI is lost (Dyer and Cepko, 2000; Dyer and Cepko, 2001). These studies are consistent with a model where individual Cip/Kip CKIs are required for cell-cycle exit in unique sub-populations of cells, and suggest that different populations employ distinct mechanisms to control cell cycle exit. However, a number of questions remain unanswered. First, it is unclear whether differences in the requirement for these factors represent functional differences between the proteins, or are related to specific patterns of expression in different tissues. Although previous studies have shown that there is some functional redundancy among and between the two major families of CKI proteins (Cip/Kip and INK4), it has not yet been determined whether all CKI activity is required for cell cycle exit. Second, prior studies have not established whether the requirement for Cip/Kip CKIs is cell-autonomous. Because loss of cell-cycle control in vivo often leads to apoptosis and non-cell-autonomous tissue defects, this question bears not only on understanding Cip/Kip CKI function but also on the broader mechanisms that balance neuronal proliferation with differentiation. Finally, although transfection studies in cell culture have demonstrated the ability of Cip/Kip CKI factors to block cell cycle progression via inhibition of G1 cyclin (cyclin D) activities, it is unclear whether similar mechanisms are responsible for this effect in vivo (Dyer and Cepko, 2000).
In this study, we set out to address these issues using a combination of in vivo approaches in the embryonic mouse and chick spinal cord. We find that all three identified Cip/Kip proteins are expressed in subsets of neurons in the spinal cord during neurogenesis, with both p21 and p57 being expressed transiently in the nuclei of distinct sub-classes of nascent interneurons exiting the ventricular zone, while p27 expression is initiated and maintained in the nuclei of post-mitotic neurons in the mantle zone. Thus, most or all post-mitotic neurons within the mouse spinal cord express at least one, and sometimes two, Cip/Kip CKI proteins at early neurogenic stages. In contrast, some INK4-family CKIs are only expressed in neuronal progenitor cells at these stages and therefore are not likely to function in controlling cell cycle exit or differentiation decisions (Zindy et al., 1997a; Zindy et al., 1997b). To determine the requirement for Cip/Kip CKI proteins in spinal cord neurogenesis, we examined the development of single and compound mutant mice. We find that loss of p57 results in the excess production of most, but not all, classes of early-generated interneurons. In contrast, loss of p27 has no consequences on early neurogenesis. Utilizing a method to genetically mark cells that express p57, we find that in the absence of this factor, many interneurons re-enter the cell cycle for at least one additional round of cell division before ultimately differentiating. We also demonstrate the requirement of the N-terminal CKI domain of p57 in functioning downstream of CyclinD1 to arrest proliferation in progenitors. Finally, our data shows that neuronal differentiation can proceed in the spinal cord, albeit abnormally, in the absence of all Cip/Kip CKI proteins. Our studies support a model whereby cell cycle exit involves multiple steps, with exit being triggered upstream of Cip/Kip CKIs, which function primarily to control the specific timing and number of spinal interneurons by opposing latent Cyclin D1 and other residual cell cycle promoting activities in progenitors targeted for differentiation.
MATERIALS & METHODS
Animals
p27kip1 (Kiyokawa et al., 1996) and p57kip2 knockout mice (Zhang et al., 1997) were maintained on C57BL/6 and CD1 backgrounds. Double mutant mice were generated by crossing heterozygous adult mice. Genotypes were determined by PCR as described (Zhang et al., 1998). The p57 allele is imprinted in mammals (Zhang et al., 1998), such that interbreeding of targeted heterozygous parents will give rise to both genetic (2 targeted alleles) and imprinted (1 targeted and 1 paternally imprinted allele) mutant offspring. However, to avoid potential variability in the phenotypes of these two populations, only genetically identified homozygote knockout embryos were analyzed in this study. To generate genetic p57−/− mutants expressing a LacZ reporter in p57+ cells, heterozygous male p57+/− mice were first crossed with transgenic mice generated from a modified 85kd BAC clone engineered to express LacZ in place of p57 (John et al., 2001), which were then intercrossed to generate litters containing both p57−/− null homozygotes and LacZ transgene allele/s. Transgenics were identified by PCR as described (Brinkmeier et al., 1998).
BrdU labelling
To specifically label neural progenitors in S-phase, a single injection of BrdU (5mg/ml in 0.9% NaCl) was given to pregnant female mice i.p. (50ug/g) at various time-points (2, 4, 6, 8, 10 and 12 hours) prior to sacrificing. Because BrdU is rapidly cleared from the maternal circulation, this protocol provides a means to “pulse-label” cells that are in S-phase only around the time of injection. During early neurogenic stages, cell-cycle length for proliferating neural tube progenitors in rodent embryos is about 8–10 hours, with S-phase comprising approximately 2–4 hours of this (Altman and Bayer, 1984). Thus, short intervals between BrdU injection and sacrifice (≤4 hours) will predominantly label cells that are actively dividing, while longer pulses (≥8–10 hours) are required to label cells that have withdrawn from the cell cycle. For chick embryos, a one-time application of BrdU was made by pipetting 0.2ml of a 5mg/ml solution onto embryos after windowing. Eggs were then sealed and returned to the incubator for 30 minutes before collecting. Because BrdU remains present throughout this period in eggs, this protocol only labels cells in S-phase.
Constructs
Full-length cDNAs and deletion constructs were sub-cloned into the pCIG vector that also encodes nuclear GFP under the control of an IRES element (Megason and McMahon, 2002). The p57 CKI domain (aa 1–92 of the 335 aa full-length protein) and CKI deleted (ΔCKI) form (aa 93–335) were generated by PCR and cloned in-frame into pCS2+MT. For co-electroporations, plasmid concentrations were adjusted to equal single-construct transfections.
In ovo electroporation
Constructs were injected into the lumen of the neural tube of stage 12–14 chick embryos (Hamburger and Hamilton, 1951) and electroporated as described (Lei et al., 2004). Embryos were collected 24–48 hour after transfection at stages 19–24.
In situ hybridization, immunohistochemistry, and cell counts
In situ hybridization and immunohistochemistry were performed on 12–14μm cryosections as described (Matise et al., 1998). Antibodies used were mouse anti-p21 & p27 (BD Pharmingen), BrdU (Sigma), cyclin D1 (Upstate Biotechnology), Isl1, Lhx1/2, Lhx3, Pax6, Pax7, c-myc (Developmental Studies Hybridoma Bank), Ngn2 (gift of L. Lo), NeuN (Chemicon), TuJ1 (Covance), Neurofilament (Sigma); rabbit anti-p57 (Santa Cruz), Pax2 (Zymed), En-1 (gift of A. Joyner), phosphorylated Histone-H3 (Upstate Biotechnology), caspase-3 (Idun Pharmaceuticals), Chx10 (gift of K. Sharma); goat anti-mouse p57 (Santa Cruz); guinea-pig anti-Prox-1 (K. Misra & M. Matise, unpublished), Lmx1b and Evx1 (gift of T. Jessell). RNA in situ probes were mouse p57 (gift of M. Lee), and chick p57 ESTs (accession numbers BM489375, BM491273). For quantification, more than 5 embryos for each group were examined and at least five sections were counted per marker. In Figures 6 and 7, GFP+ transfected cells expressing BrdU were compared between control and experimental vectors, and a ratio was derived for each experimental group as indicated. Statistical significance was determined by paired t-test. In bar charts, error bar=sem for all figures.
Fig. 6. Overexpression of CIP/KI family CKIs arrests cell proliferation in spinal cord progenitor cells.
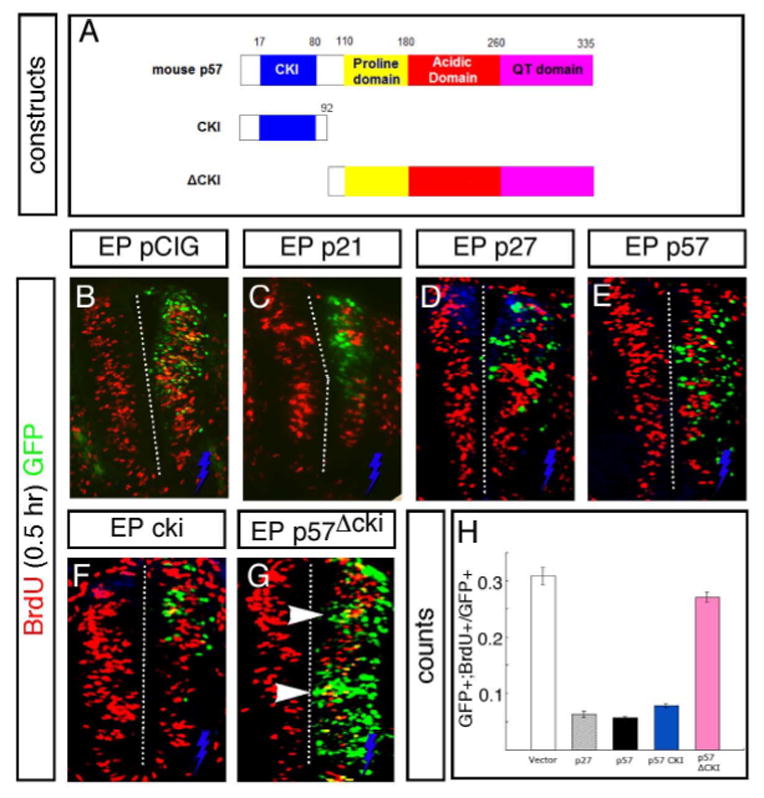
(A) Schematic showing the conserved domains of the p57 protein. The CKI cdk- and cyclin-dependent kinase binding domain is located at the N-terminus, the intermediate region contains a proline and acidic rich domain, and a unique “QT” domain of unknown function is found at the C-terminus. Below the full-length protein is shown deletion constructs used to assay the function of the cki domain in transfection assays. (B–E) Cells transfected with full length mouse cDNAs encoding p21 (C), p27 (D) and p57 (E) did not incorporate BrdU after a 0.5-hour pulse (tranfected cells are marked by GFP expression from a bi-cistronic vector) when analyzed 24 hours after transfection, compared to control vector transfections (B; yellow cells indicated by arrowhead). (F) Transfection of the p57 cki domain alone was sufficient to block BrdU incorporation, similar to the full-length protein. (G) In contrast, cells transfected with a construct lacking the N-terminal CKI domain incorporated BrdU (yellow cells, arrowhead) like control transfections. Electroporated side is on the right (indicated by blue bolt) in all figures. (H) Quantification of BrdU incorporation in GFP+ transfected cells comparing control and experimental vectors.
Fig. 7. The p57 CKI domain can antagonize cyclin-D1 activity in transfection assays and functions downstream of neurogenic bHLH factors.
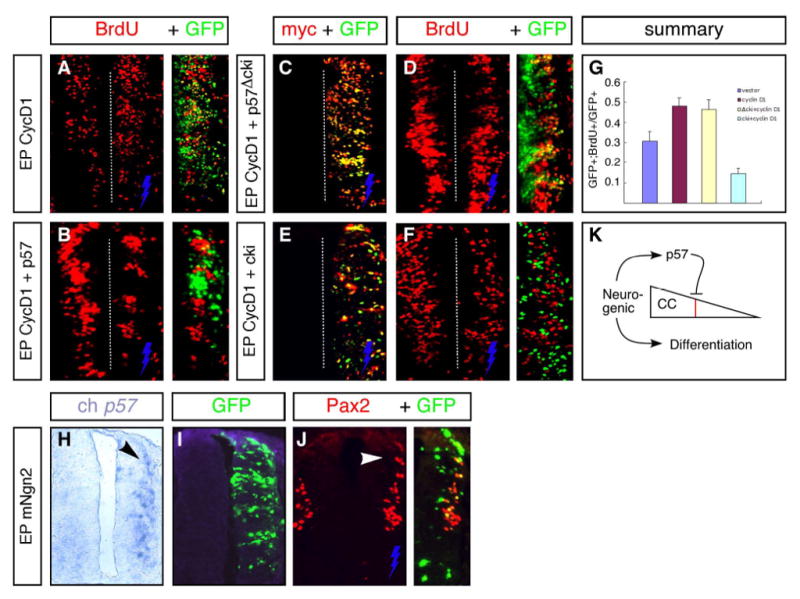
(A) Transfection of a full-length Cyclin-D1 cDNA (CycD1) promotes increased BrdU incorporation in progenitors. (B) Co-transfection of full-length p57 with cycD1 blocks this effect and prevents BrdU incorporation in transfected cells. Co-expression was determined by using a mouse-specific p57 antisera that does not cross-react with endogenous chick p57 (not shown); GFP expression marks CycD1 tranfected cells in these panels. (C–F) The p57 CKI domain is sufficient and required to block cycD1-stimulated increase in BrdU incorporation. For these experiments, myc-tagged p57 truncation/deletion constructs were used in co-transfection experiments, and staining with anti-Myc antibodies shows a high degree of co-expression of these with CycD1 (GFP) (C,E). The p57 cki domain alone, but not a construct encoding the remainder of the p57 protein absent the CKI domain, could block the activity of CycD1. (G) Quantification of BrdU incorporation in GFP+ transfected cells comparing control and experimental vectors. (H–J) Embryos electroporated with Ngn2 up-regulate p57 expression precociously (H) while inducing cell cycle withdrawal and neurogenesis (indicated by Pax2 expression in J) in transfected progenitors (arrowheads). At this stage (st. 19), endogenous p57 expression is not yet activated in normal embryos at lower thoracic levels (note lack of staining on contralateral un-transfected side). Electroporated side is on the right (indicated by blue bolt) in all figures. (K) Model for the role of p57 in arresting residual cell-cycle (CC) activity sharply (red line) after withdrawal is initiated upstream by neurogenic factors that simultaneously stimulate the onset of neuronal differentiation.
RESULTS
Cip/Kip family CKIs display distinct spatial and temporal patterns of expression in differentiating neurons in the embryonic mouse spinal cord
To address the role of Cip/Kip class cyclin kinase inhibitors (CKIs) in controlling neuronal differentiation in the developing spinal cord, we used immunohistochemistry to localize p21cip1, p27kip1 and p57kip2 proteins during neurogenesis between embryonic day (E) 9.5–13.5 (Fig. 1 and Supplemental Fig. S1). In this structure, progenitor cells residing in the ventricular zone (VZ) exit the cell cycle and migrate laterally to settle in the mantle zone (MZ) to differentiate. During early neurogenesis, functionally distinct neuronal types are generated in specific and discrete domains along the dorsoventral (DV) axis, and can be identified by a unique homeodomain (HD) protein expression profile (Jessell, 2000). All three CKI factors were expressed in overlapping but distinct groups of cells located at the lateral border of the VZ. p21cip1 expression was detected medial to cells expressing Chx10, which marks ventral interneurons within the V2 domain dorsal to motoneurons (MNs) (Fig. 1A). In contrast, p57 was expressed more broadly along the DV axis in two stripes of cells at the lateral margin of the VZ/proximal part of the MZ, but was excluded specifically from the ventral V2 and MN domains (Fig. 1B). A similar pattern of expression was seen for p57 mRNA in both mouse and chicken spinal cords at equivalent stages (Fig. 1D,E). p27kip1 had the broadest pattern of the three Cip/Kip CKIs, being expressed in nearly every cell within the MZ but not in progenitors (Fig. 1C; see also Novitch et al., 2001 for chick expression). As a result, p27 is co-expressed with both p21 and p57 in the lateral aspects of their expression domains (Fig. 1F; 2E; data not shown). Thus, most or all post-mitotic neurons within the mouse spinal cord express at least one, and sometimes two, Cip/Kip CKI proteins at early neurogenic stages (Fig. 1F).
Fig. 1. Cip/Kip family CKIs display distinct spatial expression patterns in the developing vertebrate spinal cord.

(A) p21cip1 protein is expressed in cells located medial to Chx10+ V2 interneurons in the ventral spinal cord. (B) p57kip2 is expressed in two groups of cells at the lateral margins of the ventricular zone everywhere along the DV axis except in V2 and motor neuron (MN) domains (bracketed). (C) p27kip1 is widely expressed in cells located within the mantle zone (MZ). (D,E) p57 mRNA expression in E11.5 mouse and E4 chick embryos show a similar pattern of expression. (F) Summary of overlapping and unique expression patterns of CKIs during neurogenesis. (A–D) Show E11–11.5 mouse embryo, (E) is from a stage 22 chick embryo. All sections are through the thoracic region.
Fig. 2. p57kip2 expression is initiated in nascent neurons at G1/G0 phase.
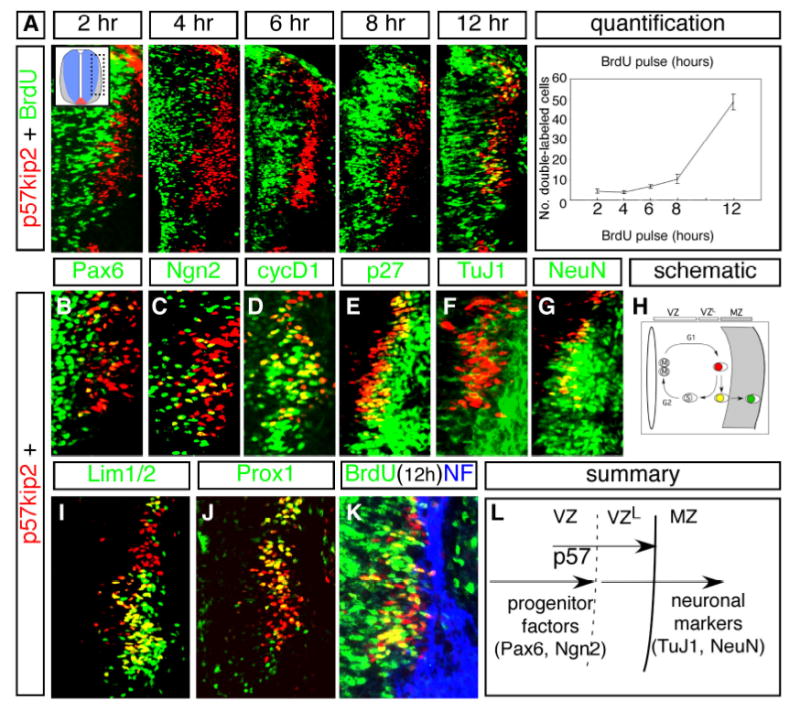
(A) Incorporation of BrdU by E11.5 embryos at various times prior to sacrifice, as indicated. Dotted box in inset shows region of spinal cord enlarged in each panel. When pulsed at 2, 4, 6 and 8hrs prior to sacrifice, p57+ cells were rarely labeled with BrdU. Significant numbers of double stained cells were observed only in embryos pulsed at 12 hours prior to sacrifice (yellow cells). Chart at right shows quantification of BrdU incorporation results. Bottom axis indicates interval between BrdU injection (pulse) and sacrifice (see Materials & Methods for details). (B–J) Expression of p57 protein in comparison to markers of progenitors and post-mitotic neurons. (B) Medially positioned p57+ cells express low levels of Pax6 protein at the lateral edge of its expression domain in neural progenitors. (C) Many Ngn2+ cells in the lateral VZ co-express p57. (D) Many p57+ cells co-express the G1-phase marker cyclin D1 (cycD1). (E–G,K) Lateral p57+ cells co-express p27 protein (E), TuJ1 (F), NeuN (G), and Neurofilament (NF) (K) in post-mitotic MZ cells. (H) Schematic showing the timing of p57 expression in relation to progenitor nuclear position as cell progress through different phases of the cell cycle. The decision to exit the cell cycle occurs late in G1 when nuclei are positioned in the lateral part of the VZ, where p57 expression is seen. (I) p57 is co-expressed with the homeodomain (HD) factor Lim1/2 that marks a subset of newly differentiating neurons. (J) p57 expression overlaps with Prox1, which also marks nascent neurons. (K) 12 hour BrdU pulse/sacrifice interval labels p57+ cells but not mature neuronal markers that come on in the MZ such as neurofilament (NF). (L) Schematic diagram of p57 expression showing overlap with progenitor markers (Pax6) and mature neuronal markers (TuJ1, NeuN) in the developing vertebrate spinal cord. VZL=lateral aspect of the ventricular zone.
p57 is transiently expressed in nascent interneurons at G1/G0
To begin to address the role of Cip/Kip CKIs in neural differentiation, we focused on p57, which shows widespread but transient expression in most early primary interneuron classes. BrdU labeling was used to determine the timing of p57 expression relative to S-phase of the cell cycle (see Materials & Methods for details of the labeling protocol). Single injections of BrdU given to pregnant females 2, 4 and 6 hours prior to sacrifice labeled only a small number of p57+ cells in embryos, while at 8 hours the number increased slightly above this (Fig. 2A). Co-expression increased sharply (5-fold) 12-hours after BrdU administration (Fig. 2A). This finding suggests that strong p57 expression occurs at the terminal G1 phase after the final mitosis.
To further examine this, we compared p57 expression to other factors that mark specific progenitor and neural populations. p57+ cells are largely located lateral to Pax6+ and Ngn2+ progenitor cells in the VZ, while a small number co-express both factors in the region of overlap between the two expression domains (Fig. 2B, C). The majority of medially positioned p57+ cells co-expressed the G1 phase marker Cyclin D1 (Fig. 2D), while laterally positioned p57 cells co-expressed mature neuronal markers p27, TuJ1, NeuN, and Neurofilament (NF) (Fig. 2E–G,K), as well as the Lim1/2 and Pax2 HD protein, whose expression is initiated in post-mitotic cells (Fig. 2I; Matise and Joyner, 1997; data not shown). p57 was also co-expressed with Prox1 (Fig. 2J), a factor whose expression is similarly confined to newly-born neurons exiting the VZ (K.Misra & M.P.M., unpublished). Together, these data indicate that p57 expression marks nascent interneurons as they undergo a transition from late G1 to early G0 phase after their terminal cell cycle in the lateral aspect of the VZ (Fig. 2H,L).
Loss of p57 results in ectopic cell division
To address the role of p57 in nascent interneurons, we examined neurogenesis in targeted p57−/− mutant mouse embryos (Zhang et al., 1997). Consistent with a role in inhibiting cell cycle progression, p57−/− homozygotes showed an increase in the number of proliferating cells within the VZ, indicated by increased BrdU (Fig. 3A–E) and phosphorylated Histone-H3 expression (pHH3) (Fig. 3F–I). Notably, pHH3, which is expressed during G2/M phases of the cell cycle and is normally detected only in cells adjacent to the central canal where mitosis occurs in wild-type (WT) embryos (Fig. 3F,H), was also seen in lateral cells adjacent to and within the proximal MZ in p57−/− mutants (Fig. 3G,I). Similarly, BrdU was detected in Prox1+ cells at the lateral aspect of the VZ in p57−/− mutants only 4 hours after administration, an interval which does not normally result in significant Brdu/Prox1 co-localization in WT embryos (Fig. 3J,K; data not shown). Together, these results indicate that loss of p57 leads to sites of ectopic cell division within the developing spinal cord and an increase in the total number of S- and M-phase cells in p57−/− spinal cords (Fig. 3E).
Fig. 3. Loss of p57 results in an increase in dividing progenitors and ectopic cell division.
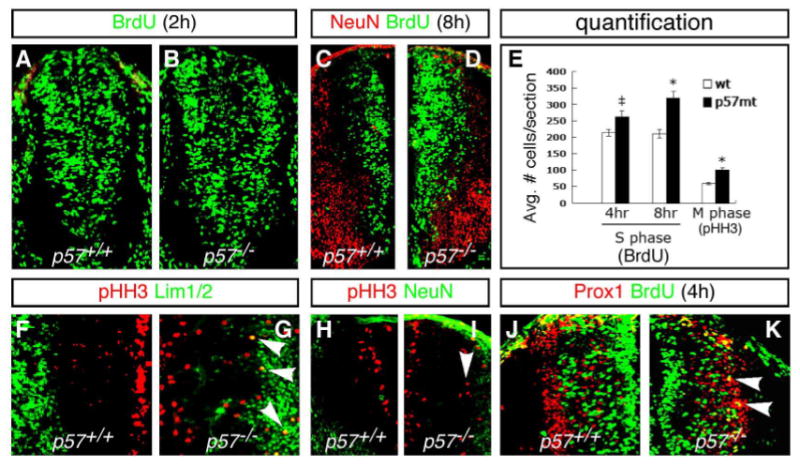
(A–D) Increased BrdU incorporation is seen in p57−/− mutants at E10.5d (B) and E11.5d (D) compared to the wild type embryos (A, C). (E) Quantification of BrdU incorporation results. There is a significant increase in BrdU incorporation in p57−/− mutants, compared to WT, after 4 and 8 hour pulses prior to sacrifice. (F–I) Expression of the G2-M marker phosphorylated Histone H-3 (pHH3) in WT and p57−/− mutants. Ectopic pHH3 expressing cells are detected laterally among Lim1/2+ (G) and NeuN+ (I) cells in the marginal zone (arrowheads). As a consequence, the total number of pHH3+ cells is significantly increased in p57−/− mutants (E). (J) In WT embryos, Prox1+ cells are not labeled after a 4-hour BrdU pulse. (K) In p57−/− null mice, a 4-hour interval following BrdU pulse labelled numerous Prox1+ cells in the lateral VZ (yellow cells marked by arrowheads). Panels (C–K) show left-right hemi-sections through p57+/+ (left) and p57−/− (right) mutant embryos. Significance: ‡p<0.05, *p<0.01.
Nascent neurons targeted for differentiation re-enter the cell cycle in p57−/− mutants
Our results above suggest that nascent neurons re-enter the cell cycle inappropriately in the absence of p57. To address this possibility, we used two approaches. First, we compared BrdU incorporation in cells expressing differentiated markers in WT and p57−/− mutants. In WT embryos, short (2–4) hour intervals between BrdU injection and sacrifice did not label cells expressing markers that identify early differentiating post-mitotic cells, such as Pax2, Lim1/2, and NeuN. (Fig. 4A,C; data not shown). In contrast, in p57−/− mutants, many Pax2+/Lim1/2+ cells co-expressed BrdU after a 2-hour interval (Fig. 4B,D).
Fig. 4. Nascent neurons re-enter S-phase inappropriately in p57−/− embryos.

(A–D) Incorporation of BrdU after short pulses in HD-expressing, newly differentiating neurons. In WT embryos, neither Pax2 (A) nor Lim1/2 (B) co-localize with BrdU after a 2-hour pulse (prior to sacrifice) that only labels actively dividing cells. In p57−/− mutants, BrdU is detected in both Pax2+ (B) and Lim1/2+ (C) cells at this interval. (E-E”) Expression of βgal in sections taken from a transgenic reporter mouse line generated from an 85kd BAC clone containing the p57 gene that has been engineered to express lacZ in place of p57 (John et al., 2001). Fewer cells express βgal compared to endogenous p57 protein, but the overall patterns are similar (E”). (F, G) BrdU is detected in βgal+ cells after a 2-hour pulse prior to sacrifice in p57−/− mutant but not WT embryos (arrowheads). (H, I) Many pHH3+ cells are co-labelled with βgal in mutant, but not WT, embryos. (J) Quantification of BrdU expression in transgenic βgal mice pulsed 2-hours prior to sacrifice on a WT or p57−/− mutant background. (K,L) Schematic summarizing the requirement of p57 at G1/G0 transition to prevent differentiating neurons from re-entering the cell cycle. In the absence of p57, nascent neurons re-enter at S-phase and incorporate BrdU after only a short, 2-hour pulse. E10.5 (A–D) and E11.5 (E–I) embryos shown. Left/right panels show p57+/+ and p57−/− hemi-sections, respectively, as in Fig. 3. Significance in J:*p<0.01.
Next, we examined BrdU incorporation in a transgenic mouse reporter line generated from a modified 85kD mouse C57/B6 BAC clone containing the p57 gene and 85kD of upstream sequence, which has been engineered using homologous recombination to express lacZ/β-gal in place of p57 (John et al., 2001). It has been previously shown that lacZ expression from this transgenic reporter line is similar to endogenous p57 in the embryonic CNS (John et al., 2001). Closer examination of reporter expression in the spinal cord showed that β-gal staining was detected in most, but not all, cells expressing endogenous p57 protein, while some cells expressed β-gal but not p57, likely due to the greater perdurance of beta;-gal protein compared to p57, which is down-regulated rapidly as cells migrate into the MZ (Fig. 4E). As with endogenous p57 protein, co-expression of BrdU and beta;-gal was detected only after BrdU injection/collection intervals exceeding 8–12 hours, but not at 2 hours (Fig. 4F; data not shown). Thus, the modified 85kD p57 BAC transgenic reporter mouse line faithfully recapitulates the timing and overall pattern of endogenous p57 expression in nascent spinal cord interneurons, although not all p57+ cells are marked.
The stability and perdurance of β-gal protein provides a means to track the fate of cells that normally express p57 in a p57−/− mutant background. To do this, we crossed the transgenic p57 BAC reporter line onto targeted p57−/− mutants to generate p57−/−;p57BAC/+ transgenic reporter/mutant embryos. LacZ reporter expression does not require p57 function in these embryos since β-gal expression is detected in a similar pattern in p57+/−, p57+/+ and p57−/− backgrounds (Fig. 4G). Consistent with our results above, we found a significant increase in the number of β-gal+ (p57+) cells that incorporated BrdU after a short (2-hour) interval between injection and sacrifice in p57−/−;p57BAC/+ mutants, compared to p57+/−;p57BAC/+ or p57+/+;p57BAC/+ embryos which show very little co-expression at this interval (39±6 vs. 7±2; p<0.0002) (Fig. 4F,G, J). In addition, many β-gal+ cells also co-expressed the M-phase marker pHH3 in p57−/−;p57BAC/+ transgenic embryos (Fig. 4H,I). Taken together, these results indicate that cells targeted for differentiation re-enter the cell cycle abnormally in the absence of p57 function in the spinal cord, and demonstrate a cell-autonomous requirement for this factor for neurogenesis (Fig. 4K, L).
Increased production of postmitotic neurons in p57−/− mutants
While our results above demonstrate that p57 is required for normal cell cycle exit in spinal cord interneurons, it is apparent from our analysis that significant numbers of differentiated neurons are present in p57−/− homozygotes (cf. Fig. 3G,I), suggesting that mutant cells are ultimately able to cease proliferation and differentiate even in the absence of p57. To address this issue, we counted the number of neurons within the MZ in mutant embryos using markers that identify specific neuronal classes to allow comparison of cells that normally express p57 (e.g., En1+ V1 cells) with those that do not (Chx10+ V2 cells, MN) (Fig. 1B,F). Loss of p57 resulted in a significant increase in the number of post-mitotic V1 interneurons, while the number of V2 interneurons was not different from WT, consistent with the idea that loss of p57 results in cell-autonomous changes in neuronal differentiation (Fig. 5A–D; data not shown). Notably, the time course for the generation of excess neurons paralleled neurogenesis, with the peak number of additional neurons being attained at E11.5 and then declining thereafter but still remaining above WT levels (Fig. 5D). At this stage, more than 2-fold more post-mitotic neurons were found in mutant embryos compared to WT (Fig. 5D). In addition, a greater number of cells expressing activated caspase-3 were detected in p57−/− mutants compared to WT, indicating that some nascent neurons may undergo apoptosis in the absence of p57 (data not shown). Taken with our results above showing re-entry into the cell cycle for p57−/− mutant cells, our findings are consistent with the idea that, in the absence of p57 function, neuronal progenitors targeted for differentiation undergo at least 1 additional round of cell division before ultimately leaving the cell cycle to differentiate or, less frequently, undergo apoptosis.
Fig. 5. Increased numbers of post-mitotic neurons in p57−/− mutant embryos and similarity to p27;p57 double-mutants.
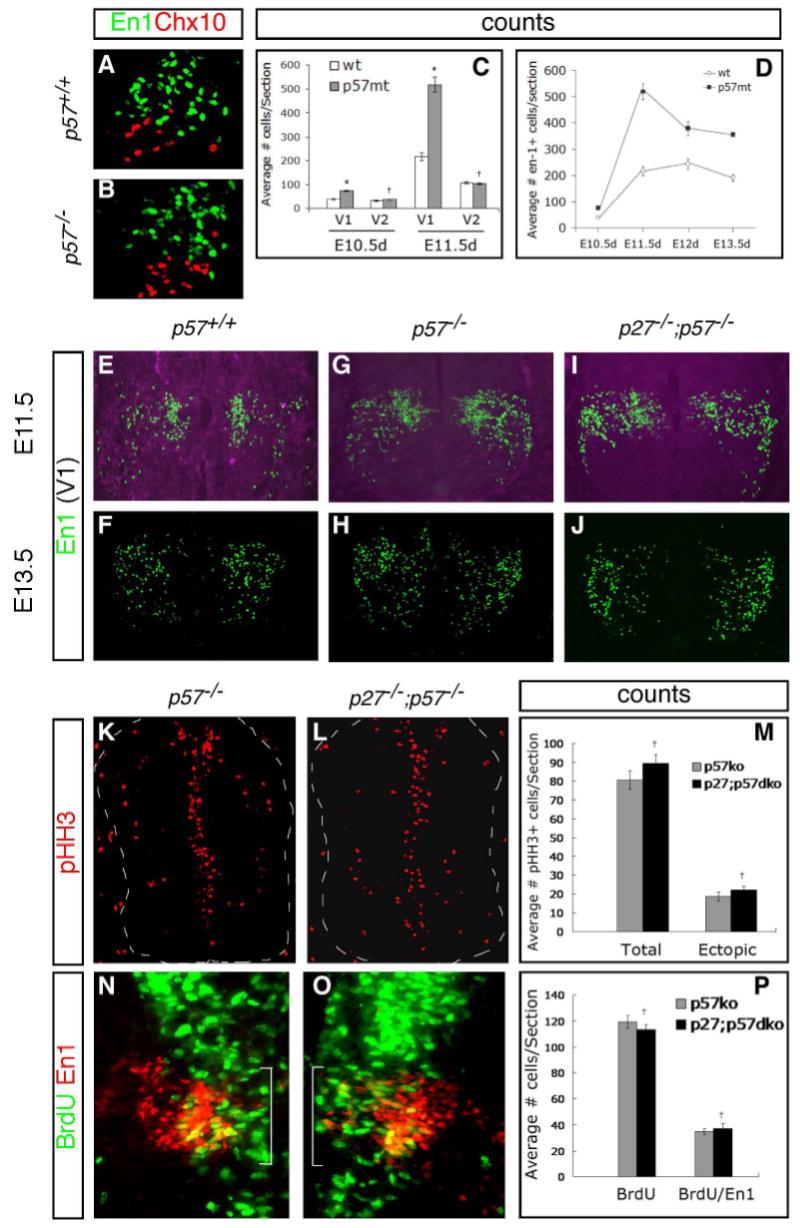
(A–D) En1+ V1 interneurons that co-express p57 are generated in excess numbers in p57−/− mutants, while Chx10+ V2 interneurons that do not normally express p57 are not affected (A,B). (C) Quantification of changes in V1 and V2 interneuron numbers at E10.5 and E11.5 shows a significant (*=p<0.001; +p>0.05) increase over WT for V1 but not V2 subtypes. Counts were made at mid-thoracic levels. (D) Quantitative depiction of changes in V1 (En1+) cell numbers during the course of neurogenesis between E10.5–E13.5. The greatest difference was seen at the peak of neurogenesis at E11.5, with a 2-fold increase in p57−/− mutants compared to WT at this stage (p<0.001). (E–J) Analysis of neuronal subtype marker expression in p27−/−; p57−/− double mutant embryos. No significant difference in the number of En1+ V1 cells was detected in double mutant embryos compared to single p57−/− mutants at E11.5 or E13.5. (K–M) Comparison of ectopic pHH3 expression in single and double mutant embryos shows no significant differences. (N–P) Comparison of BrdU incorporation in V1 interneurons after a 2-hour injection/collection interval shows a similar increase in single and double mutants.
Cip/Kip class CKIs are dispensable for neuronal cell cycle exit
Our finding that spinal cord neuronal progenitors can exit the cell cycle even in the absence of p57 raises the possibility that other CKI factors might compensate for the loss of p57 in mutant embryos. To address this, we examined neurogenesis in double p27−/−;p57−/− mutants, since we found that p21 remains restricted to cells within the V2 domain in these embryos (Supplemental Fig. S1). Thus, most cells in p27−/−;p57−/− embryos (with the exception of those in the V2 domain that continue to express p21) lack expression of all Cip/Kip family CKI proteins in the spinal cord. We studied neuronal proliferation and differentiation in these mutants spanning the neurogenic period, between E10.5 and E13.5. Notably, neurogenesis in p27−/−;p57−/− double mutants was similar to either single mutant alone at all stages examined, with both an increase in the number of proliferating cells as well as excess generation of post-mitotic differentiated neurons (Fig. 5E–P, Supplemental Fig. S2). In contrast, single p27−/− embryos had no detectable neurogenic defect (data not shown). These results demonstrate that Cip/Kip CKI proteins are not required for cell cycle exit during neurogenesis in the spinal cord.
Overexpression of Cip/Kip CKI proteins forces cell cycle arrest in neural progenitor cells
To address the function of p57 and other Cip/Kip proteins in controlling the timing of cell cycle exit in nascent interneuron progenitors, we used a gain-of-function approach in chick embryos. Using in ovo electroporation, we transfected each factor into neural progenitor cells using a bi-cistronic pCIG vector (Megason and McMahon, 2002) that also encodes GFP to allow the fate of transfected cells to be monitored. All three Cip/Kip family proteins were capable of arresting cell cycle progression in progenitors at 24 and 48 hours post-transfection, as indicated by the total absence of BrdU incorporation in transfected cells and decreased incorporation of BrdU on the transfected control side (Fig. 6B–E,H, and data not shown). For p57, the N-terminal CKI domain (Fig. 6A) was both sufficient and required for this activity (Fig. 6F–H). These results are consistent with p57 functioning in a cell-autonomous manner to inhibit cyclin-dependent kinase activity in neural cells via the N-terminal cki domain.
To examine this further, we assayed whether p57 could interfere with the ability of cyclin D1 to promote cell cycle progression. In agreement with similar results in other systems (Sherr and Roberts, 1999; Bartkova et al., 1997), forced overexpression of a cyclin D1 cDNA elicited an increase in BrdU uptake in tranfected cells compared to control (GFP-only) transfections (Fig. 7A,G). Co-transfection of full-length p57 with cyclin D1 blocked BrdU incorporation in co-transfected cells, indicating that it is capable of antagonizing cyclin D1 cell-division-promoting activity (Fig.7B). To further assay the involvement of the p57 cki domain, we co-transfected either the p57 cki domain alone with cyclin D1, or a construct containing a deletion of the cki domain (see Fig. 6A,F,G). Only the p57 cki domain, but not a C-terminal construct that lacked the cki domain, was capable of blocking cyclin D1-stimulated increase in BrdU incorporation (Fig. 7C–G). These findings are consistent with the idea that p57 functions to arrest cell cycle progression at G1 by antagonizing cyclin D1 function in neural progenitor cells via the N-terminal cki domain.
Taken together, our data suggests that the p57 Cip/Kip CKI is dispensable for neuronal cell cycle exit but is required for timely withdrawal, suggesting that the initial trigger for termination of the cell cycle occurs in progenitors upstream of p57. To test this idea, we mis-expressed Ngn2, a neurogenic factor that has been previously shown to drive neuronal progenitors toward differentiation (Lo et al., 2002) and is co-expressed with p57 in many nascent interneurons (Fig. 2C). Indeed, over-expression of Ngn2 in chick neural progenitor cells resulted in the up-regulation of neuronal markers as well as precocious activation of p57 expression (Fig. 7H–J). In contrast, Ngn2 expression was not activated by mis-expression of p57 (data not shown), consistent with a genetic heirarchy placing bHLH neurogenic factors upstream of the p57 Cip/Kip protein in enacting cell cycle withdrawal in neural progenitors in the spinal cord (Fig. 7K).
DISCUSSION
Control over cell cycle exit is fundamental to the normal generation of the wide array of distinct cell types that comprise the mature vertebrate CNS. Here, we demonstrate a central role for the p57kip2 cyclin-kinase inhibitory (CKI) protein in regulating this process in a subset of interneurons during neurogenesis in the embryonic spinal cord. We provide direct genetic evidence supporting a cell-autonomous requirement for p57 in controlling cell cycle exit for spinal cord interneurons. In homozygous p57−/− mouse mutants, many interneurons targeted for cell cycle withdrawal and differentiation undergo additional mitotic cycles outside of the VZ. Because Chx10+ V2 interneurons, which do not normally express p57, are generated in normal numbers, these results demonstrate that p57 activity is required to establish the appropriate proportions of interneuron sub-classes in the developing spinal cord. Gain-of-function studies in chick embryos show that forced expression of p57 in proliferating progenitor cells blocks cell cycle progression by interfering with the cell-cycle promoting activities of Cyclin D1. Our analysis of compound p27;p57 homozygous mutant mice which lack Cip/Kip protein expression in most spinal interneurons provides evidence that Cip/Kip family CKIs are not required for neuronal cell cycle exit during neurogenesis, but rather function to control the precise timing, and hence overall number, of the majority of interneurons generated during spinal cord development. Our studies support a multi-step model for neuronal progenitor cell cycle withdrawal that involves p57kip2 in an important role opposing latent Cyclin D1 and other residual cell cycle promoting activities in progenitors targeted for differentiation. Taken with previous studies, our results demonstrate that the functional requirement for Cip/Kip CKI proteins during development varies in different tissue contexts.
p57 controls interneuron cell cycle exit via the N-terminal CKI domain
The p57 protein contains several distinct conserved domains (see Fig. 6A). One of these, the N-terminal CKI domain, is highly conserved among all three Cip/Kip family CKI proteins (40–50% identity within 65 amino acids comprising this domain; Hashimoto et al., 1998). Indeed, all three vertebrate Cip/Kip family CKI proteins share the property of being capable of arresting or delaying cell cycle progression when mis-expressed in progenitors in various tissues (Zhang et al., 1998; Cunningham et al., 2002; Dyer and Cepko, 2000; Tarui et al., 2005), as well as being required to prevent cell cycle re-entry (Zindy et al., 1999; Dyer and Cepko, 2000; Dyer and Cepko, 2001).
Our analysis of p57−/− and compound p27−/−;p57−/− mutants indicates that p57 is uniquely required to prevent cell cycle re-entry in differentiating spinal interneurons. In addition, in p57−/− mutant mice, we observed hyper-phosphorylated Rb (Fig. S2), indicative of increased Cdk activity, and consistent with the idea that reduced CKI function is responsible for this phenotype. To test the function of the CKI domain further, we used gain-of-function studies in chick embryos. All three Cip/Kip CKI proteins were capable of preventing cell cycle progression in transfected spinal neuronal progenitors. Furthermore, we found that the N-terminal CKI domain of p57 was both sufficient and required for this activity. Consistent with the notion that cyclin D promotes cell cycle progression (Sherr, 1995), overexpression of cyclin D1 increased cell proliferation, and this activity that can be completely abrogated by co-transfection with p57 or its CKI domain. These results indicate that p57 functions downstream of cyclin D, and are in agreement with results from earlier studies showing that p57 inhibits the activity of Cdk2 at the G1/S checkpoint (Zhang et al, 1997; Li et al., 2004).
Notably, we did not uncover evidence in our studies that the role of the Cip/Kip CKI proteins is critical for establishing distinct neuronal fates in spinal cord interneurons, unlike their role in the mammalian retina (Dyer and Cepko, 2000; Dyer and Cepko, 2001). Together, these results suggest that the process of retinal histogenesis is more critically linked to the precise timing of progenitor cell cycle exit than in the spinal cord, where neuronal classes are distinguished primarily by spatial, rather than temporal, parameters.
Cip/Kip class CKI proteins are dispensable for cell cycle exit during spinal cord neurogenesis
p57 is one of three identified vertebrate members of the Cip/Kip CKI class, the other two members being p21 and p27. Because these proteins have both common (cell cycle inhibitory) and unique functions, the observation that cell differentiation is not entirely inhibited in various tissues in single and double mutants for these factors has been reasonably ascribed to functional redundancy, but this possibility has been difficult to test genetically. To understand their unique and redundant functions in the developing neural tube, we began by examining their expression during neurogenesis. Virtually all differentiating neurons in the spinal cord express at least one Cip/Kip CKI during neurogenesis, and although there is some overlap, each has a unique spatial and temporal pattern (Fig 1). Notably, our observation that p21, which shows the most restricted expression of the three Cip/Kip members in the spinal cord (to a single ventral interneuron domain), is not significantly altered in p27;p57 double mutants has afforded us an ideal opportunity to address this issue in the developing spinal cord.
We found that p27−/−;p57−/− double mutants had a similar phenotype as single p57−/− mutants. Nascent interneurons re-entered cell cycle, but ultimately exited to differentiate in excess numbers. Furthermore, V2 interneurons, which express only p21 and p27, did not show any abnormalities in cell cycle or differentiation (Fig. S2). These results indicate that Cip/Kip class CKIs are dispensable for cell cycle exit during neurogenesis, and that p21 and p27 do not compensate for the loss of p57 to block cell cycle re-entry in nascent interneurons. Interestingly, in lower vertebrates and invertebrates, Cip/Kip CKIs also appear to be dispensable for cell cycle exit (de Nooij et al., 1996; Lane et al., 1996; Hong et al., 1998). Therefore, while the number of Cip/Kip CKI homologs has expanded over the course of evolution, it appears that, remarkably, their basic functional requirement has been conserved.
Cip/Kip proteins regulate the timing of cell cycle exit in the spinal cord
Although neuronal differentiation can proceed in the spinal cord in the absence of p57 or all three Cip/Kip CKIs, excess numbers of neurons are generated. Using genetic lineage tracing, we provide direct evidence that interneurons targeted for differentiation re-enter the cell-cycle abnormally in p57 mutants, but ultimately exit to differentiate in the mantle zone. That the loss of a single factor, p57, can result in cell-cycle re-entry indicates that a pool of nascent neurons still possess the inherent capacity to enter S- and M-phases even though they are in the process of exiting the cell cycle to begin differentiation, and thus should formally be regarded as “progenitors”. The observation that the G1 cyclin D1, as well as some well-characterized progenitor factors such as Pax6, are co-expressed with p57 in cells at the lateral margins of the VZ is consistent with this idea. The increase in the number of post-mitotic neurons at early neurogenic stages is therefore likely to result from a transient expansion of this neuronally-restricted “progenitor” pool in p57−/− and p27−/−;p57−/− mutants. Consistent with this, loss of p57 does not affect the generation of oligdendrocyte glial progenitors at later stages (P. Casaccia-Bonnefil, personal communication), indicating that it is specifically required in this restricted pool and not the common neuronal/glial pool that populates the VZ at these stages. p57 expression thus appears to define the transition between a distinct neural “progenitor” state, where G1 cyclin activity predominates, and a post-proliferative G0 state, where cell-cycle promoting activities are down-regulated and neuronal differentiation has begun. Interestingly, a similar function for regulating cell cycle exit for a sub-population of neuronal stem cells residing in the sub-ventricular zone of the adult rodent brain has been assigned to p27 (Doetsch et al., 2002). These observations suggest that Cip/Kip CKIs may play a similar regulatory role in controlling G1-G0 transitions in both embryonic and adult neurogenesis in mammals.
Taken together with previously published studies on the molecular control of neurogenesis, our studies support a model whereby cell cycle withdrawal is a multistep process. In this view, the initial signal/s that stimulate cell-cycle exit likely occur prior to the final mitosis or early in G1 phase, and the expression and function of several bHLH neurogenic factors make them likely candidates for playing a central role in this process. For example, Ngn2 can induce full-blown neuronal differentiation as well as precocious p57 expression when mis-expressed in chick neural progenitors (Fig. 7), while over-expression of p57 does not induce this factor (data not shown) or neuronal differentiation. However, the direct activities of bHLH factors alone are not likely to be sufficient to provide the sharp termination of cyclin D or E activity necessary for precise cell cycle exit in proliferating neural progenitors at early neurogenic stages, requiring a second inhibitory mechanism that links directly to the cell cycle machinery and which functions to precisely control exit in sub-populations of nascent neurons—a role played by p57.
Supplementary Material
Acknowledgments
We thank Rosalind John for providing transgenic p57BAC reporter mice and Steven Elledge for the targeted p57 mutants. We also thank J. Harper, A. Koff, M. Lee, and F. Guillemot for providing reagents, and K. Misra for data in Fig. 1A. This work is dedicated to the memory of Dr. Ira Black.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altman J, Bayer SA. Advances in Anatomy, Embryology and Cell Biology. Vol. 85. Springer-Verlag; Berlin: 1984. The development of the rat spinal cord. [DOI] [PubMed] [Google Scholar]
- Assoian R, Schwartz M. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Current Opinions in Genetics & Development. 2001;11:48–53. doi: 10.1016/s0959-437x(00)00155-6. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Lukas J, Bartek J. Aberrations of the G1- and G1/S-regulating genes in human cancer. Prog Cell Cycle Res. 1997;3:211–20. doi: 10.1007/978-1-4615-5371-7_16. [DOI] [PubMed] [Google Scholar]
- Brinkmeier ML, Gordon DF, Dowding JM, Saunders TL, Kendall SK, Sarapura VD, Wood WM, Ridgway EC, Camper SA. Cell-specific expression of the mouse glycoprotein hormone alpha-subunit gene requires multiple interacting DNA elements in transgenic mice and cultured cells. Mol Endocrinol. 1998;12:622–33. doi: 10.1210/mend.12.5.0103. [DOI] [PubMed] [Google Scholar]
- Cunningham JJ, Levine EM, Zindy F, Goloubeva O, Roussel MF, Smeyne RJ. The cyclin-dependent kinase inhibitors p19(Ink4d) and p27(Kip1) are coexpressed in select retinal cells and act cooperatively to control cell cycle exit. Mol Cell Neurosci. 2002;19:359–74. doi: 10.1006/mcne.2001.1090. [DOI] [PubMed] [Google Scholar]
- de Nooij JC, Letendre MA, Hariharan IK. A cyclin-dependent kinase inhibitor, Dacapo, is necessary for timely exit from the cell cycle during Drosophila embryogenesis. Cell. 1996;87:1237–47. doi: 10.1016/s0092-8674(00)81819-x. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Verdugo JM, Caille I, Alvarez-Buylla A, Chao MV, Casaccia-Bonnefil P. Lack of the cell-cycle inhibitor p27Kip1 results in selective increase of transit-amplifying cells for adult neurogenesis. J Neurosci. 2002;22:2255–64. doi: 10.1523/JNEUROSCI.22-06-02255.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer MA, Cepko CL. p57(Kip2) regulates progenitor cell proliferation and amacrine interneuron development in the mouse retina. Development. 2000;127:3593–605. doi: 10.1242/dev.127.16.3593. [DOI] [PubMed] [Google Scholar]
- Dyer MA, Cepko CL. p27Kip1 and p57Kip2 regulate proliferation in distinct retinal progenitor cell populations. J Neurosci. 2001;21:4259–71. doi: 10.1523/JNEUROSCI.21-12-04259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996;85:733–44. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HC. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Hamel P, Gallie B, Phillips R. The retinoblastoma protein and cell cycle regulation. Trends in Genetics. 1992;8:180. doi: 10.1016/0168-9525(92)90221-o. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Kohri K, Kaneko Y, Morisaki H, Kato T, Ikeda K, Nakanishi M. Critical role for the 310 helix region of p57(Kip2) in cyclin-dependent kinase 2 inhibition and growth suppression. J Biol Chem. 1998;273:16544–50. doi: 10.1074/jbc.273.26.16544. [DOI] [PubMed] [Google Scholar]
- Hong Y, Roy R, Ambros V. Developmental regulation of a cyclin-dependent kinase inhibitor controls postembryonic cell cycle progression in Caenorhabditis elegans. Development. 1998;125:3585–97. doi: 10.1242/dev.125.18.3585. [DOI] [PubMed] [Google Scholar]
- Jessell TM. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nature Reviews Genetics. 2000;1:20–29. doi: 10.1038/35049541. [DOI] [PubMed] [Google Scholar]
- John RM, Ainscough JF, Barton SC, Surani MA. Distant cis-elements regulate imprinted expression of the mouse p57(Kip2) (Cdkn1c) gene: implications for the human disorder, Beckwith--Wiedemann syndrome. Hum Mol Genet. 2001;10:1601–9. doi: 10.1093/hmg/10.15.1601. [DOI] [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1) Cell. 1996;85:721–32. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- Lane ME, Sauer K, Wallace K, Jan YN, Lehner CF, Vaessin H. Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell. 1996;87:1225–35. doi: 10.1016/s0092-8674(00)81818-8. [DOI] [PubMed] [Google Scholar]
- Lei Q, Zelman AK, Kuang E, Li S, Matise MP. Transduction of graded Hedgehog signaling by a combination of Gli2 and Gli3 activator functions in the developing spinal cord. Development. 2004;131:3593–604. doi: 10.1242/dev.01230. [DOI] [PubMed] [Google Scholar]
- Li G, Domenico J, Lucas JJ, Gelfand EW. Identification of multiple cell cycle regulatory functions of p57Kip2 in human T lymphocytes. J Immunol. 2004;173:2383–91. doi: 10.4049/jimmunol.173.4.2383. [DOI] [PubMed] [Google Scholar]
- Lo L, Dormand E, Greenwood A, Anderson DJ. Comparison of the generic neuronal differentiation and neuron subtype specification functions of mammalian achaete-scute and atonal homologs in cultured neural progenitor cells. Development. 2002;129:1553–67. doi: 10.1242/dev.129.7.1553. [DOI] [PubMed] [Google Scholar]
- Macleod K. pRb and E2f-1 in mouse development and tumorigenesis. Curr Opin Genet Dev. 1999;9:31–9. doi: 10.1016/s0959-437x(99)80005-7. [DOI] [PubMed] [Google Scholar]
- Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development. 1998;125:2759–70. doi: 10.1242/dev.125.15.2759. [DOI] [PubMed] [Google Scholar]
- Matise MP, Joyner AL. Expression patterns of developmental control genes in normal and Engrailed-1 mutant mouse spinal cord reveal early diversity in developing interneurons. The Journal of Neuroscience. 1997;17:7805–7816. doi: 10.1523/JNEUROSCI.17-20-07805.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megason SG, McMahon AP. A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development. 2002;129:2087–98. doi: 10.1242/dev.129.9.2087. [DOI] [PubMed] [Google Scholar]
- Nagahama H, Hatakeyama S, Nakayama K, Nagata M, Tomita K. Spatial and temporal expression patterns of the cyclin-dependent kinase (CDK) inhibitors p27Kip1 and p57Kip2 during mouse development. Anat Embryol (Berl) 2001;203:77–87. doi: 10.1007/s004290000146. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–20. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- Novitch BG, Chen AI, Jessell TM. Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron. 2001;31:773–89. doi: 10.1016/s0896-6273(01)00407-x. [DOI] [PubMed] [Google Scholar]
- Sapir T, Geiman EJ, Wang Z, Velasquez T, Mitsui S, Yoshihara Y, Frank E, Alvarez FJ, Goulding M. Pax6 and engrailed 1 regulate two distinct aspects of renshaw cell development. J Neurosci. 2004;24:1255–64. doi: 10.1523/JNEUROSCI.3187-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. D-type cyclins. Trends Biochem Sci. 1995;20:187–90. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Tarui T, Takahashi T, Nowakowski RS, Hayes NL, Bhide PG, Caviness VS. Overexpression of p27 Kip 1, probability of cell cycle exit, and laminar destination of neocortical neurons. Cereb Cortex. 2005;15:1343–55. doi: 10.1093/cercor/bhi017. [DOI] [PubMed] [Google Scholar]
- Westbury J, Watkins M, Ferguson-Smith AC, Smith J. Dynamic temporal and spatial regulation of the cdk inhibitor p57(kip2) during embryo morphogenesis. Mech Dev. 2001;109:83–9. doi: 10.1016/s0925-4773(01)00512-3. [DOI] [PubMed] [Google Scholar]
- Yan Y, Frisen J, Lee MH, Massague J, Barbacid M. Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 1997;11:973–83. doi: 10.1101/gad.11.8.973. [DOI] [PubMed] [Google Scholar]
- Zhang P. The cell cycle and development: redundant roles of cell cycle regulators. Curr Opin Cell Biol. 1999;11:655–62. doi: 10.1016/s0955-0674(99)00032-0. [DOI] [PubMed] [Google Scholar]
- Zhang P, Liegeois NJ, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePinho RA, Elledge SJ. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387:151–8. doi: 10.1038/387151a0. [DOI] [PubMed] [Google Scholar]
- Zhang P, Wong C, DePinho RA, Harper JW, Elledge SJ. Cooperation between the Cdk inhibitors p27(KIP1) and p57(KIP2) in the control of tissue growth and development. Genes Dev. 1998;12:3162–7. doi: 10.1101/gad.12.20.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Cunningham JJ, Sherr CJ, Jogal S, Smeyne RJ, Roussel MF. Postnatal neuronal proliferation in mice lacking Ink4d and Kip1 inhibitors of cyclin-dependent kinases. Proc Natl Acad Sci U S A. 1999;96:13462–7. [Google Scholar]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997a;15:203–11. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- Zindy F, Soares H, Herzog KH, Morgan J, Sherr CJ, Roussel MF. Expression of INK4 inhibitors of cyclin D-dependent kinases during mouse brain development. Cell Growth Differ. 1997b;8:1139–50. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.