

Abstract
The fundamental principles that govern drug therapy are often overlooked by the busy clinician. This disregard frequently results in the use of particular drugs and regimens that may be less ideal for the clinical situation being managed. By convention, these principles are categorized as pharmacokinetic and pharmacodynamic. Pharmacokinetic processes include drug absorption, distribution, biotransformation (metabolism), and elimination—essentially reflecting the influence of the body on the drug administered. These principles were addressed in the preceding issue of this journal. Pharmacodynamics deals with the actual mechanisms of action and effects a drug produces on the patient and is the topic for this continuing education article.
Keywords: Drug therapy, Pharmacodynamics, Dental pharmacology
Pharmacodynamics concerns the actions and effects drugs produce on living tissues. Two basic correlates must be emphasized before this topic is addressed. First, drugs can be developed to modify virtually any physiologic function, but they cannot create a new function or effect. For example, salivary acini can be stimulated or inhibited from secreting saliva; they cannot be stimulated to secret insulin. Secondly, drugs must demonstrate some degree of specificity in action. Otherwise, they would produce a spectrum of undesirable effects that overshadow their usefulness.
DRUG ACTION
The mechanism by which a drug produces an effect is described as its action. In many cases, a drug's action involves interaction with specific macromolecular components of cells. These components are operationally defined as receptors. Most receptors are protein in structure and represent the cellular component with which endogenous molecules interact to produce normal physiologic responses.
Any substance that binds to a specific receptor is called a ligand. Although receptors are intended for the body's endogenous ligands, drugs have been synthesized to interact with them in 2 manners. Those that bind to the receptor and initiate a response are called agonists. Those that bind to receptors but are unable to initiate a response are called antagonists. These function clinically as blockers, denying agonists or endogenous ligands access to the receptor. Agonists and antagonists each demonstrate receptor affinity, but only agonists generate intrinsic activity (biochemical events) within the receptor and thereby alter cell function (Figure 1). Binding and unbinding of ligands is a dynamic process. Although some may have greater tenacity (binding affinity) than others, the net result from competition between ligands for a receptor is largely dependent on their relative concentration in the vicinity of the receptor.
Figure 1.
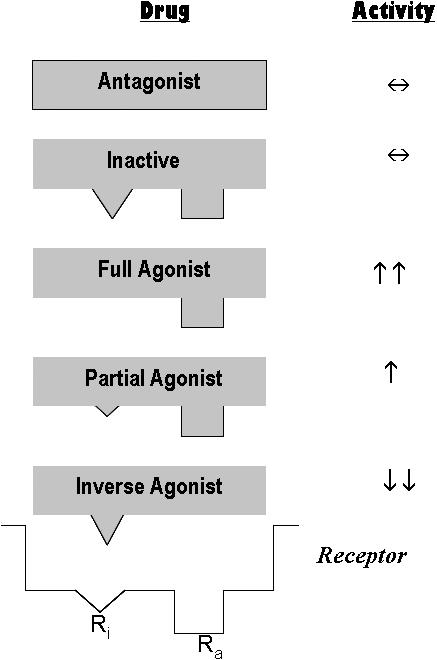
Receptors exist in both active (Ra) and inactive (Ri) states. Drugs may interact in a variety of manners, based on their ability to bind and activate these states. Antagonists bind to receptors but have no ability to activate either receptor state. Agonists bind and also activate the receptor. Agonists that activate both states equally are essentially inactive and behave similar to antagonists. Full agonists bind the active state selectively and produce a full response. Partial agonists also have some activity at the inactive state leading to a response that is less intense than that produced by a full agonist. Inverse agonists selectively activate the inactive state causing the cellular response to proceed in a manner opposite that generated by a natural agonist. Clinically, the effect may be indistinguishable from that produced by antagonists or inactive agonists.
Agonists can behave in several manners because receptors are known to exist in 2 conformational states: active and inactive.1 Full agonists selectively bind and trigger the active state while inverse agonists selectively bind and trigger the inactive state producing an opposite effect on the cell. Inverse agonists generally produce clinical effects resembling those produced by antagonists for the receptor. This would also be true for a drug that has equal activity for both active and inactive states. Other drugs can function as partial agonists by acting at both states but more so at the active one. These drugs cannot produce as great an effect as a full agonist. The beta blockers are a perfect example of a drug class that demonstrates these principles. They are known for their ability to reduce heart rate by interfering with sympathetic influence, but they accomplish this effect using several of the mechanisms just described. Propranolol (Inderal) acts as a pure antagonist while metoprolol (Toprol XL) acts as an inverse agonist. Pindolol (Visken) acts as a partial agonist; it can increase heart rate but much less than the body's endogenous agonists, nor-epinephrine and epinephrine.
Another property of receptors deserves consideration. Continued stimulation by agonists may cause receptors to become desensitized or “down-regulated.” This leads to a phenomenon called tachyphylaxis and presents as a diminished response to the drug. The repeated use of beta2 agonists as bronchodilators in patients with asthma is a prime example. Following excessive use of their inhalers, asthma patients experience less and less benefit. Conversely, receptors become supersensitive or “up-regulated” when exposed continuously to antagonists. Abrupt withdrawal of beta-blocker therapy may result in a dramatic rebound of sympathetic stimulation to the heart.1
Obviously, the structure and function of receptors is a complex science and is, understandably, a casual interest for most of us. Certain principles have significant clinical value, however. If the general effects of specific receptors are understood, one can predict the indications and effects of many drugs used in clinical practice. For example, beta receptors in the heart mediate excitatory events. Drugs acting as agonists at beta receptors increase heart rate and are used to manage bradycardia. Conversely, beta-receptor antagonists decrease heart rate and can be used to manage episodes of tachycardia. Several receptors of clinical relevance for dental practice are summarized in the Table.
Drugs may also produce effects by interacting with enzymes that regulate specific metabolic pathways. In the purest sense, enzymes are receptors, but it may be more clinically useful to regard drug actions on enzymes as a separate concept. Drugs that increase a particular enzyme's activity are regarded as enzyme inducers, while those that diminish enzyme activity are called enzyme inhibitors. Nonsteroidal anti-inflammatory drugs (NSAIDs) provide an excellent example of enzyme inhibition. Most of their effects can be explained by an inhibitory effect on cyclo-oxygenases that convert arachidonic acid to prostaglandins. Prostaglandins are involved in the production of pain, fever, and inflammation. By inhibiting those enzymes responsible for prostaglandin synthesis, NSAIDs produce analgesic, antipyretic, and anti-inflammatory effects.
DRUG EFFICACY AND POTENCY
Drug effects are the physiologic changes that result from a drug's action. Drugs are incapable of producing a single effect; all produce a variety of effects, some desirable and others that are not. Desired effects are generally described as primary and others are designated as side effects or adverse effects. Drug toxicity is a more emphatic term, conventionally reserved for the most serious side effects. It should be emphasized that adjectives used to describe drug effects are arbitrary. A particular effect may be regarded as primary in one patient, but adverse in another. Constipation is a bothersome side effect when using opioids (narcotics) to manage pain, but this same effect is primary if managing diarrhea.
Efficacy refers to the potential intensity of a particular drug effect, ie, its effectiveness. When choosing between drugs of comparable cost and safety, preference must be given to the one demonstrating greatest efficacy. Potency refers to the quantity of drug required to produce a designated intensity of effect. The potency of a drug does not correlate reliably with efficacy or toxicity.2 There is simply no basis for regarding a more potent drug as more effective, or more dangerous for that matter. It is unfortunate that potency is used carelessly as a synonym for efficacy, but this practice is not likely to change.
Drugs belonging to the same class generally produce comparable efficacy, provided one administers appropriate doses. Such doses are designated as equipotent. For example, fentanyl is more potent than morphine, which is more potent than meperidine as analgesics, but they are equivalent in efficacy. When administered intravenously, using equipotent doses, ie, 50 μg, 5 mg, and 50 mg, respectively, they exhibit comparable analgesic efficacy and a side effect profile.
Dose-response curves are used to compare the potency and efficacy of similar drugs. In Figure 2, Drug A is more potent, but also demonstrates greater efficacy than Drug B for increasing heart rate. However, 5 mg of Drug A and 20 mg of Drug B are equipotent for increasing heart rate by 20%. Also, notice that both drugs exhibit a ceiling to their response. This is to say that a point is reached whereby increasing the dose further will not produce a greater response. A 30-mg dose of Drug B will produce a 40% increase in heart rate. Increasing the dose to 40 mg or even 70 mg will not produce a greater response. This dose-response is typical of analgesia provided by acetaminophen and NSAIDs. Drug C is less potent but more effective than either Drug A or B. It produces no effect at 20 mg, but the response is much greater than either of the other drugs as its dose is increased. In fact, it appears that its response may be unlimited; there appears to be no ceiling. This is a typical dose-response for the analgesic effect of opioids.
Figure 2.
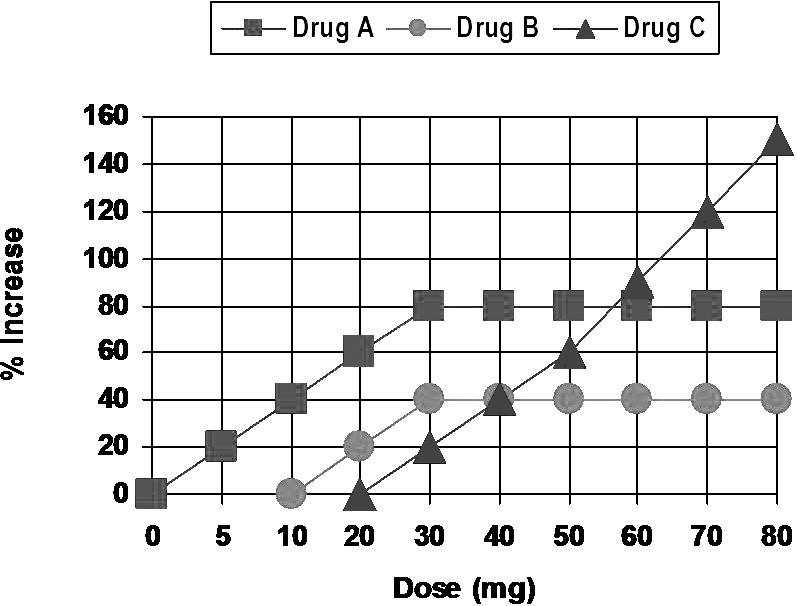
Dose-response curves. Drug A is more potent and has greater efficacy that Drug B. However, Drug A 5 mg and Drug B 20 mg are equipotent doses for increasing heart rate 20%. Drug C is less potent than Drugs A or B, but is more effective, having no apparent ceiling to its response.
The concepts of potency and efficacy are extremely important for clinical practice. Too often a particular agent is prescribed or administered based on misconceptions regarding these properties. In general, potency has little relevance; the drug is formulated in dosages that are equipotent with competitors. Furthermore, it is extremely rare for a drug within a given class to have efficacy greater than similar products. At equipotent doses, drugs within a particular class are equally effective. However, there may be differences in the actual frequency of side effects or a particular patient's ability to tolerate the medication.
DRUG SAFETY
During the research and development of a drug, scientists determine its relative safety by comparing the effective dose (ED) and lethal dose (LD) for 50% of the animals studied. This ratio is the drug's therapeutic index. For example:
For obvious reasons, these identical parameters cannot be used to establish the therapeutic index in humans. Therefore, it is recalculated, using more acceptable values such as toxic dose for 1% and effective dose for 90%. In some instances it is calculated using even more precise parameters such as serum concentrations producing therapeutic effect versus those producing toxicity. In any case, the therapeutic index represents the ratio between an effective and a toxic dose.
Generally, drugs prescribed with great frequency have a high therapeutic index. For example, the therapeutic index for diazepam (Valium) is < 100. Some agents, however, leave little margin for error. Digoxin, for example, has a therapeutic index of 2 or 3. Merely doubling or tripling its recommended dose introduces a significant risk for toxicity.
Any drug at any dosage has the potential to do harm. However, adverse effects are more likely if recommended dosages are exceeded. This can be attributed to an exaggerated primary effect such as hypoglycemia due to excess insulin, or an enhanced effect on a nontargeted tissue such as hepatotoxicity following excessive acetaminophen intake. Many adverse effects are not dose-related. Allergic reactions and so-called idiosyncratic reactions occur at any dose. A familiar example for the latter is the patient who becomes excited after receiving a sedative.
DRUG INTERACTIONS
Adverse effects may follow the interaction between 2 or more medications. It is an extremely important topic for clinical practice and a more detailed presentation will be offered as a future continuing education article in this journal. For now, an overview will suffice. At times an interaction is desirable, such as the enhanced sedation provided by sedatives and opioids during intravenous sedation, but our theme here is adverse effects. The staggering number of potential drug interactions defies memorization, but an understanding of their mechanisms provides a conceptual framework for prevention. For convenience, drug interactions may be categorized as pharmaceutical, pharmacodynamic, or pharmacokinetic.
Pharmaceutical interactions represent incompatibilities of solutions. These interactions occur before drugs are actually administered to the patient. If barbiturates are mixed in the same syringe or intravenous line with opioids, a precipitate forms.
Pharmacodynamic interactions occur when the action or effects of multiple drugs combine to produce an adverse effect. The interaction may involve primary or secondary effects of the drugs. For example, NSAIDs should not be taken concurrently with warfarin. Erosion and bleeding of gastrointestinal mucosa produced by NSAIDs may be enhanced in the presence of anticoagulant therapy.2
Pharmacokinetic interactions occur when one drug alters the delivery of another to its target. The interaction may involve absorption, metabolism, or elimination and is the most common category of drug interaction. Most frequently, pharmacokinetic interactions alter activity of hepatic microsomal enzymes. These enzymes are known as the cytochrome P450 enzymes (CYP) and are categorized using a system of numbers and letters representing genetic families, eg, 3A4 and 2D6. Erythromycin and codeine derivatives will serve as examples of this interaction.3,4
Erythromycin inhibits the activity of CYP2A4. This group of enzymes is responsible for the metabolism and clearance of carbamazepine (Tegretol), protease inhibitors for HIV infection, and the statins such as simvastatin (Zocor) used for cholesterol reduction. Prescribing erythromycin for patients taking these medications can allow them to accumulate and produce their toxic effects.
Codeine, hydrocodone, and oxycodone are prodrugs that are converted to their active morphine derivative by CYP2D6. The selective serotonin reuptake inhibitor (SSRI) antidepressants such as fluoxetine (Prozac) and paroxetine (Paxil) inhibit this family of enzymes and decrease the activation of codeine derivatives. Patients taking these medications may not experience pain relief from these particular opioids.
Receptors Accounting for Actions of Drugs Prescribed or Administered Therapeutically in Dental Practice

CONTINUING EDUCATION QUESTIONS
- Morphine produces its effects, including analgesia and respiratory depression, by acting as a full agonist at μ opioid receptors. Buprenorphine (Buprenex) is a partial agonist at these receptors. Conventional equipotent intramuscular doses are 10 mg and 0.4 mg, respectively. All of the following are correct regarding buprenorphine EXCEPT:
- buprenorphine has less analgesic efficacy than morphine
- buprenorphine is more potent than morphine
- naloxone, a μ receptor antagonist, will reverse morphine but is ineffective for reversing buprenorphine
- as doses are increased for both drugs, morphine will produce greater respiratory depression
- Beckerdal is a newly developed drug that acts as an antagonist at alpha and cholinergic (muscarinic) receptors. Primary and secondary effects produced by this drug may include any of the following EXCEPT:
- xerostomia
- hypertension
- bronchodilation
- constipation
- Morphine, 10 mg intramuscularly, and oxycodone, 20 mg orally, are considered equipotent analgesic doses. How many 5-mg oxycodone tablets will provide pain relief equivalent to that provided by a 15-mg intramuscular injection of morphine?
- 2
- 4
- 6
- 8
- Your patient has a negative medical history except for over-the-counter cimetidine (Tagamet) for gastritis. You administer diazepam, 20 mg orally, for sedation, and the patient calls the next day complaining of continued lethargy and sluggishness. You find that cimetidine inhibits the CYP450 enzymes responsible for metabolizing diazepam. This interaction would be classified as:
- idiosyncratic
- pharmaceutical
- pharmacodynamic
- pharmacokinetic
REFERENCES
- Buxton ILO. Pharmacokinetics and pharmacodynamics. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 11th ed. New York, NY: McGraw-Hill; 2006. pp. 1–39. [Google Scholar]
- Oates JA. The science of drug therapy. In: Brunton LL, Lazo JS, Parker KL, editors. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 11th ed. New York, NY: McGraw-Hill; 2006. pp. 117–136. [Google Scholar]
- Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352:2211–2221. doi: 10.1056/NEJMra032424. [DOI] [PubMed] [Google Scholar]
- Abramowicz M, editor. The Medical Letter on Drugs and Therapeutics. 2003. pp. 46–48. ed. [PubMed]