

Abstract
Recently, the human ATP-binding cassette transporter-1 (ABC1) gene has been demonstrated to be mutated in patients with Tangier disease. To investigate the role of the ABC1 protein in an experimental in vivo model, we used gene targeting in DBA-1J embryonic stem cells to produce an ABC1-deficient mouse. Expression of the murine Abc1 gene was ablated by using a nonisogenic targeting construct that deletes six exons coding for the first nucleotide-binding fold. Lipid profiles from Abc1 knockout (−/−) mice revealed an ≈70% reduction in cholesterol, markedly reduced plasma phospholipids, and an almost complete lack of high density lipoproteins (HDL) when compared with wild-type littermates (+/+). Fractionation of lipoproteins by FPLC demonstrated dramatic alterations in HDL cholesterol (HDL-C), including the near absence of apolipoprotein AI. Low density lipoprotein (LDL) cholesterol (LDL-C) and apolipoprotein B were also significantly reduced in +/− and −/− compared with their littermate controls. The inactivation of the Abc1 gene led to an increase in the absorption of cholesterol in mice fed a chow or a high-fat and -cholesterol diet. Histopathologic examination of Abc1−/− mice at ages 7, 12, and 18 mo demonstrated a striking accumulation of lipid-laden macrophages and type II pneumocytes in the lungs. Taken together, these findings demonstrate that Abc1−/− mice display pathophysiologic hallmarks similar to human Tangier disease and highlight the capacity of ABC1 transporters to participate in the regulation of dietary cholesterol absorption.
Low serum high density lipoprotein cholesterol (HDL-C) concentrations have been identified as a good predictor for coronary artery disease (1, 2). A variety of factors contribute to low HDL-C levels, including genes harboring a basic defect, modifying genes, and environmental factors (3). Low HDL syndromes are genotypically heterogeneous, and understanding their molecular basis could explain the essential role of HDL in plasma cholesterol homeostasis and atherosclerosis.
By using elegant linkage analysis and positional cloning, three separate groups identified mutations in the human ATP-binding cassette transporter 1 (ABC1) gene that are linked to Tangier disease (4–6). ABC1 is part of a large family of evolutionarily conserved transmembrane proteins that includes the cystic fibrosis transregulator protein and the multidrug-resistance proteins. Proteins of this gene family are involved in energy-dependent transport of a wide variety of substrates across the membrane (7). Although the physiological functions and substrates of ABC transporters are largely unknown, defects in most of the mammalian ABC transporters have been associated with clinically relevant phenotypes.
ABC1 possesses all of the distinguishing features of other ATP-binding proteins, including two ATP-binding segments and two transmembrane domains (8). ABC1 is activated by protein kinases (9) and is modulated at the transcriptional level by increased cellular cholesterol stores (10). Recently, ABC1 has been associated with the initial steps of reverse cholesterol transport from cells (4). In addition, ABC1 may have critically important functions in the body by virtue of its ability to function as a cholesterol gate keeper.
Gaining insight into the role of ABC1 in vivo represents a fundamental step in understanding HDL metabolism, cholesterol trafficking, and disease. To address these questions, we used gene targeting in embryonic stem cells to create mice that lack ABC1. In this study, we report the generation of Abc1 knockout mice and provide a detailed description of the murine ABC1 mutation with respect to plasma lipids, cholesterol absorption, expression profiling, and physiopathology.
Materials and Methods
Creation of Abc1-Deficient Mice.
All of the experimentation, except construction of the Abc1 targeting vector, was conducted at Pfizer Central Research and was performed under approved protocols by the Pfizer Institutional Animal Care and Use Committee. Mouse genomic DNA for the targeting vector was cloned from commercially available 129/svJ genomic library (Stratagene). The targeting construct (Fig. 1A) contains 1.8 kb of 5′ homology and ≈12.0 kb of 3′ homology cloned adjacent to pMCNeo polyA. The construct also contains a PGK-tk cassette 3′ to the 12.0-kb homology arm, allowing positive/negative selection (11). The targeting vector was electroporated into both E14 (129/Ola) and DBA/1lacJ embryonic stem (ES) cells (12). ES cells surviving the +/− selection were analyzed by Southern blot after BamHI restriction enzyme digestion and hybridization to a 0.5-kb probe from a region 5′ of the 1.2-kb homology arm. Targeted clones were identified in both ES cell types by the presence of a 4.4-kb BamHI RFLP introduced by the deletion of 5.7 kb, including a BamHI site, compared with a 6.8-kb wild-type fragment (Fig. 1A). Targeted ES clones were confirmed by Southern analysis of 3′ end. Two independently targeted cell lines from DBA/1 ES cells were microinjected into C57BL/6 blastocysts, and chimeric mice were backcrossed to DBA/1J mates to identify germline transmission. Abc1−/− mice were derived from crosses of Abc+/− males and females. Expression of the Abc1 gene was evaluated by using reverse-transcription–PCR (RT-PCR) on total RNA isolated from livers with a forward primer at nucleotide no. 3110 (5′-AGCAGAGATGGAGCAGATGG-3′) and a reverse primer at nucleotide no. 3627 (5′-CAGAGACATCGATGGTCAGA-3′).
Figure 1.

Gene targeting of the murine Abc1 gene. (A) Strategy to disrupt Abc1 expression. The targeting vector construct was designed to replace exons 17–22 that encode the first nucleotide-binding fold. The resulting targeted allele has pMC-neo replacing the six exons deleting an endogenous BamHI (B) site resulting in a restriction fragment length polymorphism, from 6.8 kb to 4.4 kb, with a downstream BamHI (B′) site by using an external 5′ probe. (B) Southern analysis of offspring from Abc1+/− matings. All three genotypes are observed by using the genomic probe strategy described above. Lanes A, C, and E, samples from +/+ mice; lanes B and D, samples from +/− mice; lanes F and G, samples from −/− mice. (C) RT-PCR results from Abc1+/+ and −/− mice. Lanes B and C are DNA and no DNA controls, respectively. Lane D is RT-PCR of hepatic RNA for Abc1 from wild-type mouse. Lane D is RT-PCR from Abc1−/− mice. Lane A is a 100-bp molecular weight marker.
Plasma Lipids and Lipoprotein Analysis.
All phenotypic characterizations were performed in +/+, +/−, and −/− littermates of the F2 generation fed either a chow diet (Purine Prolab RMH 3000 rodent diet) or a Western-type diet containing 0.15% cholesterol and 20% saturated fat (Harlan Teklad, Madison, WI, TD 88137) for 4 wk. Mouse plasma was isolated from blood collected retroorbitally. Cholesterol, triglycerides, and phospholipid levels were determined by using enzymatic colorimetric assays (Wako Biochemicals, Osaka). HDL was separated from non-HDL lipoproteins by dextran sulfate precipitation. Apolipoprotein (apo)AI and apoB were determined by ELISA as described previously (13). Lipoproteins were isolated from 200 μl of pooled plasma from six +/+ and six −/− mice fed a chow or Western-type diet and fractionated by FPLC as previously described (14).
Intestinal Absorption of Cholesterol.
Cholesterol absorption was determined in sex- and age-matched Abc1+/+ and −/−, apoAI −/−, and C57BL6 mice by using a modified form of the dual isotope single-meal feeding method (15). Mice were placed in metabolic cages for 24 h, fasted for 4–6 h, and then given an intragastric bolus of 150 μl liquid diet (Universal Test Diets, Frenchtown, NJ) containing 5 μCi of [14C]cholesterol (Amersham Pharmacia) and 5 μCi of [5–6 3H]β-sitostanol (American Radiolabeled Chemicals, St. Louis). Mice were returned to metabolic cages, food intake was monitored, and feces were collected for 24 h. Fecal samples were frozen (−20°C), lyophilized, and combusted in a Model 306 Tissue Oxidizer (Packard). The efficiency of cholesterol absorption was expressed as percentage of administered dose absorbed by using the formula:
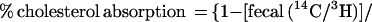 |
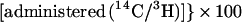 |
RNA Isolation and Expression Profiling.
Total liver RNA was isolated from age-matched Abc1+/+ and −/− mice by using TRISOLV reagent, purified by using RNA Easy reagent (Qiagen), and treated with DNase to remove residual DNA contamination. Eight micrograms of total RNA was reverse transcribed, amplified, and labeled as described (16). Labeled cRNA was hybridized to a pair of oligonucleotide arrays containing probes for 11,000 murine genes (Affymetrix, Santa Clara, CA, Mouse 11K SubA and SubB). Arrays were washed, stained with antibiotin streptavidin–phycoerythrin-labeled antibody, and scanned by using the GeneChip system (Hewlett–Packard) and genechip 3.0 software to determine the expression of each gene. Intensity values were scaled by calculating the overall signal for each array type. Differential expression was calculated by averaging intensity values of +/+ (n = 3) and −/− (n = 3) mice and calculating the ratio. The significance of differences in gene expression levels between +/+ and −/− mice was determined by using the S-Plus (mathsoft) implementation of Student's two-sample t test. Calculations were performed on the scaled intensity values, and variances in the two samples were allowed to differ. The resulting probabilities (P values) were used to rank genes by the significance of the difference in the measured gene expression level between the two groups of mice.
Morphologic Evaluation.
Six Abc1−/− mice (7, 12, and 18 mo old), three +/+ controls (7 and 12 mo old), and three +/− mice (18 mo old) fed a chow diet were examined for morphologic changes related to the ablation of the Abc1 gene by using standard histologic techniques (17). To determine the contents of foamy cells observed within the lung, frozen sections of formalin-fixed lung were stained with Oil red O or Nile blue or were immersed in 2% glutaraldehyde in 0.1 M sodium phosphate buffer, processed, embedded in Spurr's resin, and examined by using a Hitachi 7100 (Tokyo) electron microscope (17).
Results
Creation of an Abc1-Deficient Mouse.
To ensure functional inactivation of the ABC1 protein, a gene-targeting vector was constructed such that homologous recombination would introduce a deletion of the entire N-terminal ATP-binding cassette in the endogenous mouse Abc1 gene. The introduced mutation is a 5.7-kb deletion that includes exons 17–22 (Fig. 1A), resulting in a murine Abc1 allele lacking the coding sequence for amino acids Q788 to K1093. The efficiency of gene targeting was 10% in E14 ES and 3% in DBA/1 ES cells, respectively suggesting a 3-fold increase in targeting efficiency with isogenic DNA. Three targeted E14 and two DBA/1 ES cell lines were injected into C57BL/6 blastocysts. A single male chimera transmitted the ABC1 mutation through the DBA/1 germline, whereas no germline transmission was established from the breeding of seven 129/C57 chimeras. Therefore, the induced murine ABC1 mutation was established on the DBA/1J inbred background only. Homozygous Abc1−/− mice were produced by crossing Abc1+/− males and females; however, at weaning, a distortion in the Mendelian inheritance of the −/− genotype was observed: of 84 mice born of +/− by +/− crosses, we observed 30 +/+:41 +/−:13 −/− genotypes (Fig. 1B), suggesting a perinatal lethality of Abc1−/− pups. Further studies are under way to characterize the developmental aspects of the murine Abc1 mutation (G.C. et al., unpublished observation). Loss of expression of the Abc1 gene was demonstrated by using RT-PCR of total liver RNA (Fig. 1C). This result indicates that the expression of the targeted allele is completely ablated; therefore, we consider the Abc1−/− a functional null.
Plasma Lipids and Lipoprotein Analysis.
Abc1-deficient mice were severely hypocholesterolemic and had a near complete absence of HDL. On a chow diet, plasma cholesterol was decreased by ≈70% (P < 0.0001) when compared with wild-type littermates (Fig. 2). This decrease could be accounted for largely by a 99.5% and 99.8% reduction in HDL-C and apoAI, respectively. Plasma phospholipid levels were decreased by 68% (P < 0.0001) when compared with control mice, whereas triglycerides were not different. Lipid profiles were similar in female and male −/− mice. Fig. 2 also shows that plasma lipids, apoAI and apoB levels for +/− male and female Abc1 mice are intermediate, suggesting a gene-related dose response.
Figure 2.

Lipid analysis of +/+, +/−, and −/− male (solid columns), and female (shadowed columns) mice fed a chow diet. Bar graph represents the means ± standard deviations from the number of animals in parentheses of each group. *, **, and ***: P < 0.05, P < 0.005, and P < 0.0001 compared with wild-type mice, respectively.
The distribution of cholesterol, apoAI, and apoB among plasma lipoproteins was determined by FPLC (Fig. 3). This confirmed that HDL-C and apoAI were dramatically decreased in Abc1−/− mice and revealed a 70% and 20% decrease in LDL-C and apoB in −/− and +/− mice, respectively, compared with their wild-type littermates. To determine whether the decrease in HDL-C and apoAI is accompanied by a parallel decrease in phospholipid, FPLC fractions corresponding to HDL were pooled and concentrated to determine phospholipid and apoAI levels. HDL from Abc1−/− mice, which represent less than 1% of wild-type levels, are enriched in phospholipids relative to apoAI (12.9-fold more than HDL from controls), suggesting the presence of small phospholipid-rich HDL particles. HDL subclass distribution, as determined by two-dimensional nondenaturing gradient gel electrophoresis (18), revealed the complete absence of α-migrating HDL particles in Abc1−/− mice (data not shown).
Figure 3.

Plasma lipoprotein cholesterol, apoB and apoAI distribution in mice of different genotypes fed a chow diet, determined by FPLC. Two hundred microliters of pooled mouse plasma from the different genotypes was fractionated by FPLC as described under Methods. Cholesterol, apoAI and apoB content was determined and plotted as a function of FPLC fractions. The positions at which known lipoproteins eluted from the column are indicated.
To evaluate further the changes in plasma lipoprotein levels, mice from all three genotypes were fed a Western-type diet for 4 wk. In response to this diet, plasma cholesterol and phospholipids were increased in each of the three genotypes studied. As observed in the chow diet, Abc1−/− and +/− mice had significantly lower cholesterol and phospholipid levels than control mice, whereas plasma triglyceride and apoB levels were essentially unchanged (Table 1). HDL-C and apoAI were decreased in Abc1+/− mice and undetectable in −/− mice, despite the significant increases in non-HDL-C.
Table 1.
Plasma lipids, HDL–cholesterol, apoAI and apoB levels in +/+, +/−, and −/− Abc1 mice fed a Western-type diet
| Mice | TC | PL | TG | HDL-C | ApoAI | ApoB |
|---|---|---|---|---|---|---|
| +/+ | 159 ± 39 | 338 ± 61 | 310 ± 121 | 93 ± 22 | 1,587 ± 284 | 93 ± 36 |
| +/− | 107 ± 16* | 234 ± 52* | 270 ± 132 | 57 ± 13** | 1,032 ± 215** | 105 ± 14 |
| −/− | 72 ± 31** | 168 ± 60** | 212 ± 112 | 0.0 ± 0.0*** | 6.5 ± 1.1*** | 80 ± 43 |
TC, total cholesterol; TG, triglycerides; PL, phospholipids; HDL-C, HDL–cholesterol, apoAI, and apoB. Six mice per genotypes were fed to a Western-type diet for 4 wks. TC, TG, PL, and HDL-C values are mg/dl, whereas apoAI and apoB values are μg/ml. Littermates were matched for sex and were 6–8 mo old.
*, **, ***, P < 0.05, P < 0.005, and P < 0.0001 compared to wild-type mice, respectively.
Cholesterol Absorption.
The absorption of cholesterol from the intestine plays a major role in determining plasma lipoprotein levels, yet the regulation of this process is only partially understood. The role of ABC1 in the absorption of dietary cholesterol from the intestine was assessed by comparing the amount of radiolabeled sterol excreted in the feces after its infusion into the stomach of −/− and +/+ mice fed either a chow or a Western-type diet. Consistent with previous studies (19), the percentage of cholesterol absorbed is decreased in wild-type mice fed a Western-type diet (chow diet 69% ± 6% vs. Western diet 49% ± 11%, P < 0.005). The Abc1−/− mice fed a chow diet experienced a moderate but statistically significant increase (P < 0.05) in the absorption of cholesterol compared with their wild-type littermates (Table 2). When fed a high-fat and -cholesterol diet, the extent of cholesterol absorption was increased by 62% (P < 0.005) in Abc1−/− mice compared with control littermates. To rule out the possibility that the Abc1-induced increase in cholesterol absorption results from reduced plasma HDL, cholesterol absorption was determined in apoAI knockout and their control wild-type littermates. The percentage of cholesterol absorbed was not different between the two groups of mice (73% ± 6% (n = 6) vs. 70% ± 8% (n = 6) in apoAI knockout and control mice, respectively). These results suggest that Abc1 plays a critical role in the absorption of dietary cholesterol. Although the exact location of ABC1 in the enterocyte is unknown, it is likely that ABC1 acting as a unidirectional transporter efflux cholesterol to the intestinal lumen and determines the net gradient of cholesterol taken up by the enterocyte. Interestingly, this increase in cholesterol absorption in Abc1−/− mice is accompanied by an accumulation of cholesterol in lipoproteins eluting in fractions corresponding to VLDL in Abc1−/− mice fed ad libitum a Western-type diet for 4 wks (Fig. 4). Relative to apoB, these lipoproteins are enriched in cholesterol (cholesterol to apoB ratio, 41 and 11 for Abc1−/− and +/+, respectively), suggesting an accumulation of intestinally derived lipoproteins enriched in cholesterol.
Table 2.
Cholesterol absorption by +/+ and Abc1−/− mice
| Diet | Abc1 +/+ | Abc1 −/− |
|---|---|---|
| Chow | 69% ± 6% (n = 6) | 81% ± 9%* (n = 3) |
| Western | 49% ± 11% (n = 6) | 79% ± 8%** (n = 5) |
Cholesterol absorption was determined in sex- and age-matched mice fed either a chow or a Western-type diet for 4 wk as described in Methods. Mice received a gastric bolus of liquid diet containing [14C]cholesterol and [5-63H]β-sitostanol placed in metabolic cages, and feces were collected for 24 h. Percentage of cholesterol absorbed was calculated as described in Methods. *, P < 0.05; **, P < 0.005 compared to wild-type littermates on the same diet, respectively.
Figure 4.

Plasma lipoprotein cholesterol distribution in Abc1−/− and control mice fed a Western-type diet ad libitum for 4 wk. Two hundred microliters of pooled plasma from six mice was analyzed as described in Methods.
Gene Expression Analysis of Abc1−/− Mice.
To examine more broadly the effects of the ablation of Abc1, we analyzed the expression of ≈11,000 murine genes by using oligonucleotide microarrays. A total of 131 genes exhibited greater than 1.8-fold differential regulation in the −/− mice compared with +/+ littermates. These were grouped into the following categories: sterol and lipid metabolism (n = 21), protein synthesis and degradation (n = 16), transport proteins (n = 9), energy and mitochondrial function (n = 11), intracellular signaling (n = 8), cell growth (n = 7), cytoskeletal proteins (n = 4), and expressed sequence tags and miscellaneous function (n = 55) (see the supplemental data, www.pnas.org). Shown in Table 3 are examples of sterol- and lipid metabolism-related genes exhibiting differential regulation in Abc1 null mice.
Table 3.
Summary of sterol and lipid related genes in liver exhibiting differential regulation in Abc1−/− mice
| Accession no. | Description | Fold change* |
|---|---|---|
| U37226 | Plasma phospholipid transfer protein | 2.6 |
| AA065811 | ApoE | 2.3† |
| W35864 | ApoA-II | 2.0† |
| M21285 | Stearoyl-CoA desaturase | −1.9 |
| AA014996 | ApoB-100 | −2.1 |
| L23754 | Cholesterol α-7-hydroxylase | −2.3 |
| M62766 | HMG-CoA reductase | −2.6 |
| AA036251 | Farnesyl pyrophosphate synthetase | −3.1† |
| D42048 | Squalene epoxidase | −3.2 |
Calculated from the average signal intensity from three animals of each genotype.
† Average fold change of multiple probe sets for these genes.
Morphologic Evaluation of Abc1−/− Mice.
A complete necropsy was performed to determine morphologic changes related to ablation of the Abc1 gene in Abc1−/−, +/−, and +/+ mice fed a chow diet. Multiple pale foci were visible in the lungs of Abc1−/− mice. These lesions increased in severity with age, affecting <5–10% of pulmonary parenchyma in 7-mo-old mice to as much as 30% of the pulmonary parenchyma in 18-mo-old mice. Although macroscopic lesions were not observed in +/+ or +/− mice, microscopic lesions were present in two of three heterozygous mice, consistent with an age-related and gene dose-response similar to that observed with plasma lipids. Microscopically, lesions consisted of foci of foamy type II pneumocytes, intraalveolar macrophages, and cholesterol clefts (Fig. 5B). Alveolar septae were thickened by lymphoid aggregates, scant increased stroma, and hypertrophy of type II pneumocytes. In more advanced lesions, alveolar architecture collapsed, and remnant alveoli were epithelialized because of marked type II pneumocyte hypertrophy and hyperplasia.
Figure 5.

Pulmonary lesions in Abc1−/− mice. A contains a section of normal lung from a 12-mo-old +/+ mouse with a normal terminal bronchiole and alveoli lined predominantly with flattened type I pneumocytes (hematoxylin/eosin stain, ×400). B demonstrates an early lesion from a 12-mo-old −/−mouse including numerous foamy cells (arrows) and cholesterol clefts (arrowheads). Alveolar septae are focally expanded by mild type II pneumocyte hypertrophy, macrophages, and aggregates of lymphocytes and plasma cells. An Oil red O-stained section of lung from a 7-mo-old −/− mouse demonstrates lipid accumulation within alveolar cells (arrow, C). By electron microscopy, these cells are identified primarily as type II pneumocytes (D, arrows) and intraalveolar macrophages (arrowhead, ×3,500).
To identify the contents of foamy cells, frozen sections of lung were stained with Oil red O and Nile blue stains. Red staining with Oil red O was consistent with lipid accumulation (Fig. 5C), which was characterized as neutral lipid by Nile blue staining (data not shown). Vacuolated cells were identified by electron microscopy as markedly distended type II pneumocytes and alveolar macrophages (Fig.5D). Interestingly, normal surfactant-containing lamellar bodies were not observed within the type II pneumocytes. Instead of phospholipid, the membrane-bound vacuoles contained amorphous lipid and scant lamellar material, consistent with aberrant unsecreted lamellar bodies. Foamy alveolar macrophages contained variable amounts of lipid and cell debris. Sporadic lesions observed in remaining tissues including spleen, liver, proximal aorta, peripheral nerve, and intestine were not attributed to ablation of the Abc1 gene.
Discussion
We used gene targeting in ES cells to replace the first nucleotide-binding cassette region of the mouse Abc1 gene with a neomycin resistance gene. We found that this strategy disrupted Abc1 expression and resulted in phenotypic consequences in both heterozygous and homozygous mice, revealing an essential role of Abc1 in cholesterol homeostasis. This is compatible with recent reports where homozygous mutations of the ABC1 gene in humans results in Tangier disease, with near absence of HDL, reduced levels of LDL and accumulation of foam cells. The phenotypic evaluation of our inbred Abc1-deficient mice suggests this induced mutation is a good model of human Tangier disease. The mice demonstrate near complete loss of HDL, decreased LDL, and pulmonary accumulation of lipid-laden type II pneumocytes and macrophages. Some Tangier disease patients also demonstrate peripheral neuropathy and hepatosplenomegaly; however, these observations are disparate, suggesting influence of other genetic loci. The inbred nature of this induced Abc1 mutation may allow the identification of other genetic loci involved in traits associated with Tangier disease by quantitative trait locus identification and positional cloning.
Previous studies demonstrated that in fibroblasts from patients with Tangier disease, both translocation of cholesterol and phospholipids to the cell membrane and efflux onto lipid-poor apoAI particles are defective (20), resulting in increased cholesteryl ester in macrophages (21). Although this biochemical defect has been known for many years, its molecular identification remained unknown. Recent reports (4–6) identifying mutations on the ABC1 gene in patients with Tangier disease raised the possibility that ABC1 is a functional cholesterol gatekeeper that participates in the synthesis of HDL. In the present study, we provide compelling evidence in support of this hypothesis. The ablation of the murine Abc1 gene causes the near complete lack of HDL despite normal levels of liver and intestinal apoAI mRNA (data not shown). This suggests, as demonstrated on patients with Tangier disease (22), that the extremely low levels of HDL cholesterol and apoAI are the consequence of an exceptionally rapid catabolism rather than defective synthesis. HDL-C and apoAI levels are strikingly similar between male and female knockout mice and remain essentially unchanged on cholesterol feeding, despite the accumulation of VLDL and LDL. Therefore, in the absence of Abc1, neither surface remnants (23) nor phospholipids and cholesterol transferred by phospholipid transfer protein (24) from triglyceride-rich lipoproteins onto apoAI are sufficient to generate HDL particles. This study implies that the apolipoprotein-mediated lipid removal pathway is an absolute requirement for generating HDL, whereas alternative efflux mechanisms (25) may occur subsequent to the initial cellular lipidation of apolipoproteins. Although markedly reduced compared with wild type, Abc1−/− HDL have pre-β mobility. This suggests that the ability of these lipid-poor apoAI particles to mature to spherical α-migrating HDL is impaired. This finding is consistent with the proposed role of Abc1 as a cholesterol and probably phospholipid transporter that provides the initial cell-derived lipidation of apoAI triggering the synthesis and maturation of HDL (26, 27).
Although cellular cholesterol efflux to apolipoproteins would be reduced markedly in a patient with Tangier disease, the cholesterol content of most cells would be unaffected, because cell cholesterol is regulated almost exclusively by the two major sterol delivery pathways, cholesterol biosynthesis and LDL receptor-mediated endocytosis (28). Consistent with this hypothesis, the transcript imaging of Abc1−/− livers demonstrates that cholesterol biosynthetic enzymes are decreased compared with wild-type littermates. Macrophages, however, internalize cholesterol-rich lipoproteins and cell debris by receptor and phagocytotic pathways that are not repressed when cells acquire excess cholesterol, making these cells dependent on apolipoprotein-mediated cholesterol-removal mechanisms to prevent excess deposition of sterols. In tissues such as lung, where alveolar macrophages are involved in the uptake and clearance of surfactant phospholipid and cholesterol (29), the absence of ABC1 leads to the accumulation of large amounts of lipids in the cytosol, giving the cells the “foamy” appearance. This accumulation most likely represents a protective phagocytic response but also causes chronic stress and impairs cellular functions, resulting in the production of cytokines and extracellular degrading enzymes that injure lung cells and matrix (30). Alternatively, ABC1 could be involved in the maturation, differentiation, and migration of circulating monocytes to different organs such as lungs.
Our findings also suggest that ABC1 is involved in the intracellular trafficking of phospholipids and may play a role in the production and release of pulmonary surfactant from type II pneumocytes (31). The accumulation of vacuoles within type II pneumocytes in Abc1−/− mice suggests defective secretion of surfactant. In addition, the presence of abnormal lamellar bodies suggests either abnormal incorporation of neutral lipids or altered composition of saturated phospholipids. Further experiments will be required to ascertain the answers to these questions.
In conclusion, our findings demonstrate that ABC1 has an essential role in lipoprotein metabolism and HDL biogenesis. Our results also highlight additional important functions of ABC1, such as the regulation of cholesterol absorption. Taken together, these findings provide the basis for additional experiments to elucidate the role of apolipoprotein-mediated efflux in foam cell formation and atherosclerosis and could lead to novel strategies for the treatment of atherosclerosis.
Supplementary Material
Acknowledgments
We thank Germaine G. Boucher, Kim P. Kowsz, David Krull, Karen L. Steever, Eliot Sugarman, Melanie Allen, Jane Bennett, Deb DiMattia, Petra Koza-Taylor, Nicole Segal, and Jeff Stock for excellent technical assistance. The member who communicated this paper is a Director of Pfizer, Inc. This work was supported by Pfizer Central Research, Groton, CT.
Abbreviations
- ABC1
ATP-binding cassette transporter 1
- apo
apolipoprotein
- HDL
high density lipoproteins
- LDL
low density lipoproteins
- ES
embryonic stem
- RT-PCR
reverse transcription–PCR
- apo
apolipoprotein
Note Added in Proof
Shortly after the acceptance of our manuscript for publication, a paper was published describing the phenotype of mice with targeted inactivation of Abc1 (32). With the exception of generation of the Abc1 targeting vector, the animals used in the studies reported by Orsó et al. were created at Pfizer Central Research under protocols approved by the Pfizer Institutional Animal Care and Use Committee. As reported in the current article, a complete necropsy and light microscopic evaluation of Abc1−/− mice was performed at Pfizer, Inc. In contrast to the report from Orsó et al., macroscopic and light microscopic changes in the spleen and adrenal gland could not be attributed to ablation of Abc1 but varied with age or gender. By light microscopy, no gene-related changes were evident in the liver, spleen, adrenal gland, or intestinal tract of Abc1−/− mice.
References
- 1.Miller N E, Forde O H, Thelle D S, Mjos O D. Lancet. 1977;1:965–968. doi: 10.1016/s0140-6736(77)92274-7. [DOI] [PubMed] [Google Scholar]
- 2.Keys A. Lancet. 1980;2:603–606. doi: 10.1016/s0140-6736(80)90280-9. [DOI] [PubMed] [Google Scholar]
- 3.Shaefer E J. Arteriosclerosis. 1984;4:303–322. doi: 10.1161/01.atv.4.4.303. [DOI] [PubMed] [Google Scholar]
- 4.Brooks-Wilson A, Marcil M, Clee S M, Zhang L-H, Roomp K, van Dam M, Yu L, Brewer C, Collins J A, Molhuizen H O F, et al. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 5.Bodzioch M, Orsó E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S, Buchler C, Porsch-Ozcurumez M, et al. Nat Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 6.Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette J-C, Deleuze J-F, Brewer H B, Duverger N, Denèfle P, et al. Nat Genet. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 7.Higgins C F. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 8.Luciani M F, Denizot F, Savary S, Mattei M G, Chimini G. Genomics. 1994;21:150–159. doi: 10.1006/geno.1994.1237. [DOI] [PubMed] [Google Scholar]
- 9.Becq F, Hamon Y, Bajetto A, Gola M, Verrier B, Chimini G. J Biol Chem. 1997;272:2695–2699. doi: 10.1074/jbc.272.5.2695. [DOI] [PubMed] [Google Scholar]
- 10.Langmann T, Kluchen J, Reil M, Liebisch G, Luciani M-F, Chimini G, Kaminski W E, Schmitz G. Biochem Biophys Res Commun. 1999;258:73–76. doi: 10.1006/bbrc.1999.0406. [DOI] [PubMed] [Google Scholar]
- 11.Mansour S L, Thomas K R, Capecchi M R. Nature (London) 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 12.Roach M L, Stock J L, Byrum R, Koller B H, McNeish J D. Exp Cell Res. 1995;221:520–525. doi: 10.1006/excr.1995.1403. [DOI] [PubMed] [Google Scholar]
- 13.Ueda Y, Royer L, Gong E, Zhang J, Cooper P N, Francone O, Rubin E M. J Biol Chem. 1999;274:7165–7171. doi: 10.1074/jbc.274.11.7165. [DOI] [PubMed] [Google Scholar]
- 14.Francone O L, Haghpassand M, Bennett J A, Royer L, McNeish J. J Lipid Res. 1997;38:813–822. [PubMed] [Google Scholar]
- 15.Zilversmit D B, Hughes L B. J Lipid Res. 1974;15:465–473. [PubMed] [Google Scholar]
- 16.Mahadevappa M, Warrington J A. Nat Biotechnol. 1999;17:1134–1136. doi: 10.1038/15124. [DOI] [PubMed] [Google Scholar]
- 17.Bancroft J D. Histochemical Techniques. 2nd Ed. Butterworths; 1975. pp. 141–154. [Google Scholar]
- 18.Francone O L, Royer L, Haghpassand M. J Lipid Res. 1996;37:1268–1277. [PubMed] [Google Scholar]
- 19.Kirk E A, Moe G L, Caldwell M T, Lernmark J A, Wilson D L, LeBoeuf R C. J Lipid Res. 1995;36:1522–1532. [PubMed] [Google Scholar]
- 20.Francis G A, Knopp R H, Oram J F. J Clin Invest. 1995;96:78–87. doi: 10.1172/JCI118082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrans V J, Fredrickson D S. Am J Pathol. 1975;78:101–158. [PMC free article] [PubMed] [Google Scholar]
- 22.Bojanovski D, Gregg R E, Zech L A, Meng M S, Bishop C, Ronan R, Brewer H B. J Clin Invest. 1987;80:1742–1747. doi: 10.1172/JCI113266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tall A R, Small D M. N Engl J Med. 1978;30:1232–1236. doi: 10.1056/NEJM197811302992207. [DOI] [PubMed] [Google Scholar]
- 24.Jiang X-C, Bruce C, Mar J, Lin M, Ji Y, Francone O L, Tall A R. J Clin Invest. 1999;103:907–914. doi: 10.1172/JCI5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothblat G H, de la Llera-Moya M, Atger V, Kellner-Weibel G, Williams D L, Phillips M C. J Lipid Res. 1999;40:781–796. [PubMed] [Google Scholar]
- 26.Castro G R, Fielding C J. Biochemistry. 1988;27:25–29. doi: 10.1021/bi00401a005. [DOI] [PubMed] [Google Scholar]
- 27.Francone O L, Gurakar A, Fielding C J. J Biol Chem. 1989;264:7066–7072. [PubMed] [Google Scholar]
- 28.Brown M S, Goldstein Science. 1983;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 29.Wright J R, Youmans D C. Am J Physiol. 1995;268:L772–L780. doi: 10.1152/ajplung.1995.268.5.L772. [DOI] [PubMed] [Google Scholar]
- 30.Wright J R. Physiol Rev. 1997;77:931–962. doi: 10.1152/physrev.1997.77.4.931. [DOI] [PubMed] [Google Scholar]
- 31.Dobbs L G. Annu Rev Med. 1989;40:431–446. doi: 10.1146/annurev.me.40.020189.002243. [DOI] [PubMed] [Google Scholar]
- 32.Orsó E, Broccardo C, Kaminski W E, Böttcher A, Liebish G, Drobnik W, Götz A, Chambenoit O, Diederich W, Langmann T, et al. Nat Genet. 2000;24:192–196. doi: 10.1038/72869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.