

Abstract
Deamination at C6 of adenosine in RNA catalyzed by the ADAR enzymes generates inosine at the corresponding position. Because inosine is decoded as guanosine during translation, this modification can lead to codon changes in messenger RNA. Hydration of 8-azanebularine across the C6–N1 double bond generates an excellent mimic of the transition state proposed for the hydrolytic deamination reaction catalyzed by ADARs. Here, we report the synthesis of a phosphoramidite of 8-azanebularine and its use in the preparation of RNAs mimicking the secondary structure found at a known editing site in the glutamate receptor B subunit pre-mRNA. The binding properties of analogue-containing RNAs indicate that a tight binding ligand for an ADAR can be generated by incorporation of 8-azanebularine. The observed high-affinity binding is dependent on a functional active site, the presence of one, but not the other, of ADAR2’s two double-stranded RNA-binding motifs (dsRBMs), and the correct placement of the nucleoside analogue into the sequence/structural context of a known editing site. These results advance our understanding of substrate recognition during ADAR-catalyzed RNA editing and are important for structural studies of ADAR· RNA complexes.
Introduction
RNA editing refers to a wide variety of modification reactions that change the sequence of an RNA molecule from that encoded by the gene sequence.1 Deamination at C6 of adenosine (A) in RNA catalyzed by the ADAR family of enzymes generates inosine (I) at the corresponding nucleotide position.2 Because inosine is decoded as guanosine during translation, this RNA modification can lead to codon changes and the introduction of amino acids into a gene product not encoded in the gene.3,4 Inosines also appear in the untranslated regions of numerous mRNAs,5,6 and these may regulate whether a double-stranded RNA enters the RNA interference pathway.7 Interestingly, several targets of the A to I modification reaction are mRNAs encoding receptors for neurotransmitters, and editing appears to be necessary for a properly functioning central nervous system in metazoa.3,4,8–12 For instance, deletion of the gene encoding an ADAR homologue in Drosophila melanogaster leads to a morphologically normal fly with dramatic behavioral defects, such as tremors, uncoordinated locomotion, and an inability to fly or jump.11 Likewise, C. elegans containing deletion mutations in each ADAR gene show defects in behaviors such as chemotaxis.12 Thus, it appears the protein structural diversity that arises through editing of mRNA by adenosine deamination is used in the nervous system to produce complex behavior. The study of the effect changes in levels of RNA editing have on human behavior has only recently been initiated and appears to link abnormal editing to psychiatric disorders.13–15 During our ongoing studies to define the mechanism of the ADAR reaction and discover inhibitors of this process, we have endeavored to develop a mimic of the reaction transition state.16–20 Such a mimic would aid in the structural characterization of the protein–RNA complex and could lead to new approaches for inhibiting editing activity in vivo.
The development of an effective transition state analogue that binds an ADAR with high affinity requires knowledge of the RNA substrate recognition properties of the enzyme and the deamination reaction mechanism. ADARs recognize their substrates, at least in part, via an RNA-binding domain containing multiple copies of the double-stranded RNA binding motif (dsRBM).21 Consistent with the involvement of dsRBMs, duplex RNA secondary structure in the substrate is a requirement for the ADAR-catalyzed reaction.22 ADARs also have conserved sequences similar to the consensus sequence that makes up the active site of cytidine deaminases (CDAs).23,24 CDA uses a zinc-activated water molecule to carry out deamination of its nucleoside substrate via attack at C4 of the pyrimidine and loss of ammonia.25 Using the comparison to CDAs, amino acids identified as possible active site residues for the ADARs, including putative zinc-binding ligands and a residue involved in proton transfers (E396 in ADAR2), have been altered by site-directed mutagenesis with a corresponding loss of editing activity.26,27 It is believed that once an ADAR recognizes its duplex RNA substrate, it flips the reactive nucleotide into an active site similar to that found in CDAs.17,28 In analogy to the mechanism of CDA, a metal-bound hydroxide ion then attacks the purine ring to form a tetrahedral intermediate (Figure 1). This intermediate collapses, ejecting ammonia and the inosine-containing product. Nucleoside analogues that mimic the transition state leading to the tetrahedral intermediate, including compounds that are susceptible to covalent hydration of the pyrimidine ring, are excellent inhibitors of CDAs.29,30 This includes 5-fluorozebularine, which has been shown by X-ray crystallography to bind CDA as the covalent hydrate.31
Figure 1.
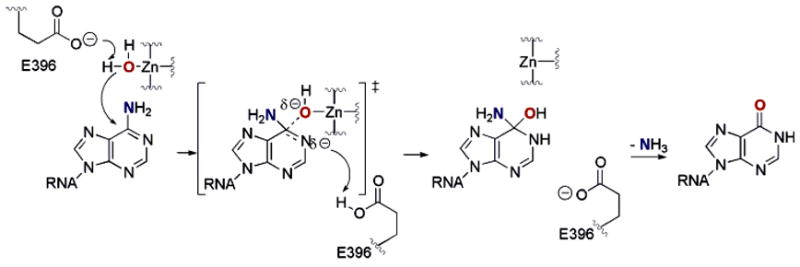
Proposed mechanism for the ADAR-catalyzed RNA-editing reaction. Hydration of adenosine forms a tetrahedral intermediate, and loss of ammonia from this structure generates the inosine-containing product.
8-Azaadenosine substitution in various RNA substrates accelerates the rate of deamination at these sites by ADAR2.20 Aza substitution at the 8-position of the purine ring system increases its susceptibility to covalent hydration.32 Thus, the increased rate of deamination for 8-azaadenosine-containing RNAs suggests that hydration is rate limiting for the ADAR2 reaction with these substrates. Consistent with the large 8-aza effect observed for the ADAR2 reaction, 8-azanebularine, an analogue with a hydrogen in place of the C6 amino group, inhibits the reaction at high concentrations (IC50 = 15 mM).20 8-Azanebularine inhibition can be explained if one considers that, once bound in the ADAR2 active site, the nucleoside can be hydrated to form a good mimic of the transition state for the hydrolytic deamination of adenosine (Figure 2). However, because this nucleoside lacks the duplex RNA structure found in RNA-editing substrates, the interaction with the enzyme is weak. For the generation of a high-affinity ligand for the enzyme, it would likely be necessary to incorporate 8-azanebularine into an RNA structure recognized by ADAR2.
Figure 2.
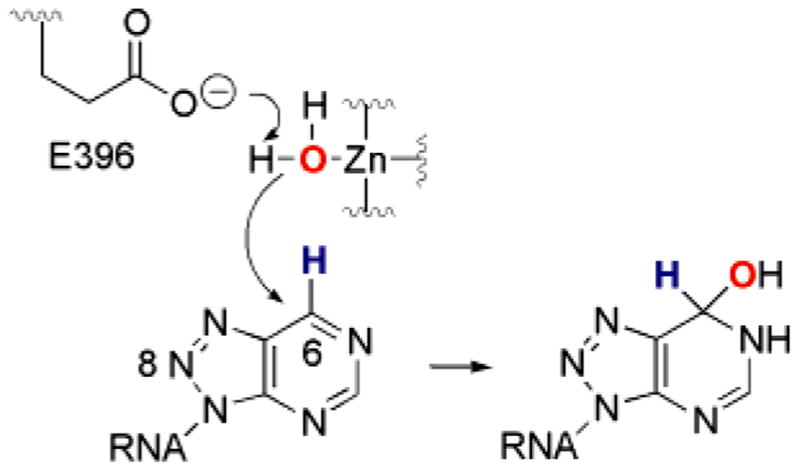
Hydration of 8-azanebularine in RNA generates a mimic of the proposed transition state for the ADAR RNA-editing reaction.
Here, we report the synthesis of a phosphoramidite of 8-azanebularine and its use in the preparation of an intermolecular duplex RNA similar to a hairpin structure found at a known editing site for ADAR2. The binding properties of the modified RNAs were analyzed in gel mobility shift experiments using deletion and point mutants of ADAR2. These studies indicate that a tight binding ligand for an ADAR can be generated by incorporation of 8-azanebularine into a duplex RNA. The observed high-affinity binding is dependent on a functional ADAR2 active site, the presence of dsRBM II, and correct placement of the nucleoside analogue into the sequence/structural context of a known editing site. These results have implications for future structural studies of ADAR·RNA complexes and in the development of inhibitors of adenosine deamination RNA editing.
Experimental Section
General Synthetic Procedures
All reagents were purchased from Sigma/Aldrich or Fischer Scientific and were used without further purification unless noted otherwise. Glassware for all reactions was oven dried at 125 °C overnight and cooled in a desiccator prior to use. Reactions were carried out under an atmosphere of dry nitrogen when anhydrous conditions were necessary. Liquid reagents were introduced by oven-dried microsyringes. Tetrahydrofuran was distilled from sodium metal and benzophenone; acetonitrile was distilled from CaH2. Thin-layer chromatography (TLC) was performed with Merck silica gel 60 F254 precoated TLC plates, eluting with the solvents indicated. Short-and long-wave visualization was performed with a Mineralight multi-band ultraviolet lamp at 254 and 365 nm, respectively. Flash column chromatography was performed on Mallinckrodt Baker silica gel 150 (60–200 mesh). 1H, 13C, and 31P nuclear magnetic resonance spectra of pure compounds were performed at 300, 75, and 121 MHz, respectively. Chemical shifts are reported in parts per million in reference to the solvent peak. Chemical shifts for phosphorus NMR are reported in parts per million using 85% phosphoric acid as an external standard. The abbreviations s, d, dd, t, and m represent singlet, doublet, doublet of doublets, triplet, and multiplet, in that order. Reversed-phase HPLC using a C-18 Vydac column, 218TP510, 5 μm particle size, 1 × 2.5 cm was performed with a Waters 600E system controller. UV peaks were monitored on a Waters 490 programmable multiwavelength detector at 260 nm. All HRFABMS spectra were obtained on a Finnigan MAT 95. All MALDI analyses were performed at the University of Utah Mass Spectrometry and Proteomics Core Facility on a Voyager-DE STR MALDI mass spectrometer.
2′,3′,5′-Tri-O-acetyl-8-azaadenosine (1) was synthesized according to a literature procedure. Spectroscopic data agreed with reported values.33
2′,3′,5′-Tri-O-acetyl-6-bromo-8-azanebularine (2)
2′,3′,5′-Tri-O-acetyl-8-azaadenosine (470 mg, 1.19 mmol) was dissolved in freshly distilled acetonitrile (40 mL). To this solution were added tBuONO (1.43 mL, 11.9 mmol) and TMS–Br (315 μL, 2.38 mmol). The reaction mixture was allowed to stir at 0 °C for 1 h. The mixture was diluted with EtOAc (15 mL) and washed with water (1 × 15 mL) and brine (1 × 15 mL). The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (CHCl3 followed by CHCl3/CH3OH 99:1) to give a white foam (328 mg, 60%). 1H NMR (CDCl3, 300 MHz): δ (ppm) 8.88 (s, 1H), 6.62 (d, J = 3 Hz, 1H), 6.17–6.14 (m, 1H), 5.80 (t, J = 6 Hz, 1H), 4.53–4.41 (m, 1H), 4.23–4.18 (m, 2H), 2.13 (s, 3H), 2.07 (s, 3H), 2.04 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ (ppm) 170.3, 169.4, 169.1, 155.7, 148.6, 146.3, 136.7, 87.8, 81.0, 73.0, 70.6, 62.5, 20.5, 20.3, 20.2. HRFABMS: calcd for C15H17BrN5O7 (M + H)+ 458.0312, obsd 458.0306.
2′,3′,5′-Tri-O-acetyl-8-azanebularine (3)
2′,3′,5′-Tri-O-acetyl-6-bromo-8-azanebularine (200 mg, 0.436 mmol) was dissolved in methanol (7 mL). 10% Pd/C (12 mg) and anhydrous NaOAc (72 mg, 0.873 mmol) were added to the solution. The flask was evacuated and refilled with hydrogen gas. The mixture was shaken in a Parr apparatus for 8 h under 2.5 atm of pressure. The mixture was filtered through Celite, and the residue was washed with methanol. The filtrate was concentrated under reduced pressure. The residue was redissolved in EtOAc (4 mL) and washed with brine (1 × 3 mL). The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (CH2-Cl2) to give a light-yellow syrup (133 mg, 80%). Spectroscopic data agreed with reported values.34
8-Azanebularine Ribonucleoside (4)
2′,3′,5′-Tri-O-acetyl-8-aza-nebularine (100 mg, 0.264 mmol) was dissolved in methanol saturated with NH3 (1.16 mL), and the mixture was allowed to stand at 4 °C overnight. The mixture was concentrated under reduced pressure and purified by flash column chromatography (preadsorbed on silica gel, 10% CH3OH/CHCl3) to give a light-yellow soft solid (54 mg, 81%). Spectroscopic data agreed with reported values.34
5′-O-(4,4′-Dimethoxytrityl)-8-azanebularine (5)
8-Azanebularine ribonucleoside (360 mg, 1.42 mmol) was dissolved in freshly distilled THF (15 mL). Anhydrous pyridine (690 μL, 8.53 mmol), 4,4′-dimethoxytrityl chloride (530 mg, 1.56 mmol), and AgNO3 (265.7 mg, 1.56 mmol) were added sequentially to this solution. The reaction mixture was stirred at room temperature overnight. The mixture was diluted with EtOAc (25 mL), filtered, and washed with saturated aqueous NaHCO3 (1 × 40 mL). The organic layer was dried (Na2-SO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography (DCM/MeOH/TEA 98:1:1) to give a light-orange foam (511 mg, 65%). 1H NMR (CD2Cl2, 300 MHz): δ (ppm) 9.54 (s, 1H), 9.13 (s, 1H), 7.37–7.34 (m, 2H), 7.26–7.23 (m, 4H), 7.19–7.16 (m, 3H), 6.73 (dd, J = 9, 3 Hz, 4H), 6.59 (d, J = 6 Hz, 1H), 5.18–5.16 (m, 1H), 4.82–4.78 (m, 1H), 4.35–4.30 (m, 1H), 3.74 (s, 6H), 3.40 (dd, J = 9, 3 Hz, 1H), 3.31–3.26 (m, 2H). 13C NMR (CD2Cl2, 75 MHz): δ (ppm) 159.0, 156.9, 152.3, 149.6, 145.3, 136.5, 136.3, 136.2, 130.5, 128.5, 128.2, 127.2, 113.5, 90.3, 86.7, 84.7, 74.6, 72.1, 64.1, 55.7.
5′-O-(4,4′-Dimethoxytrityl)-2′-O-(tert-butyldimethylsilyl)-8-azanebularine (6)
5′-O-(4,4′-Dimethoxytrityl)-8-azanebularine (460 mg, 0.828 mmol) was dissolved in freshly distilled THF (10 mL). Triethylamine (219 μL, 1.57 mmol) was added to the solution followed by the addition of TBDMSCl (137 mg, 0.911 mmol). After 5 min, AgNO3 (155 mg, 0.911 mmol) was added to the mixture. After being stirred for 8 h at room temperature, the reaction mixture was diluted with EtOAc (30 mL), filtered, and washed with saturated aqueous NaHCO3 (1 × 35 mL). The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel (EtOAc/hexanes 1:8) to give a white foam (234 mg, 42%). 1H NMR (CD2Cl2, 300 MHz): δ (ppm) 9.61 (s, 1H), 9.17 (s, 1H), 7.49–7.46 (m, 2H), 7.36 (dd, J = 9, 3 Hz, 4H), 7.27–7.20 (m, 3H), 6.82–6.77 (m, 4H), 6.57 (d, J = 6 Hz, 1H), 5.41–5.38 (m, 1H), 4.56–4.51 (m, 1H), 4.37–4.30 (m, 1H), 3.77 (s, 6H), 3.51–3.46 (m, 1H), 3.31–3.28 (m, 2H), 2.79 (d, J = 6 Hz, 1H), 0.86 (s, 9H), 0.05 (s, 3H), −0.13 (s, 3H). 13C NMR (CD2Cl2, 75 MHz): δ (ppm) 159.1, 157.1, 152.4, 150.0, 145.5, 136.7, 136.5, 136.3, 130.6, 128.6, 128.3, 127.2, 113.5, 90.2, 86.8, 85.4, 75.4, 72.4, 64.2, 55.7, 25.9, 18.3, −4.71, −4.76. HRFABMS: calcd for C36H44N5O6Si (M + H)+ 670.3062, obsd 670.3024.
5′-O-(4,4′-Dimethoxytrityl)-3′-O-[(2-cyanoethoxy)(N,N-diiso-propylamino)phosphino]-2′-O-(tert-butyldimethylsilyl)-8-azanebularine (7)
5′-O-(4,4′-Dimethoxytrityl)-2′-O-(tert-butyldimethylsilyl)-8-azanebularine (230 mg, 0.343 mmol) was dissolved in freshly distilled THF (2.0 mL). N,N-Diisopropylethylamine (359 μL, 2.06 mmol) followed by 2-cyanoethyl-(N,N-diisopropylamino)chlorophosphite (153 μL, 0.687 mmol) was added to the solution. The solution was allowed to stir at room temperature. After 8 h, the reaction mixture was diluted with EtOAc (45 mL), filtered, and washed with 5% (w/v) aqueous NaHCO3 (2 × 25 mL). The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel (EtOAc/hexanes/TEA 14:85:1) to give a light-straw foam (232 mg, 78%). 31P NMR (121 MHz, CH2Cl2, 85% H3PO4 as external standard): δ (ppm) 152.9, 151.0. HRFABMS: calcd for C45H70N7O7PSi (M + H)+ 870.4140, obsd 870.4136.
Deprotection and Purification of 8-Azanebularine-Xontaining RNA Oligonucleotides
The solid-phase synthesis of RNA oligonucleotides using 2′-O-TBDMS-protected β-cyanoethyl phosphoramidites was carried out as previously described.20 For deprotection, the controlled pore glass containing fully protected oligonucleotide was transferred to 1.5 mL of a saturated solution of NH3 in methanol and incubated at room temperature for 24 h. Solution was removed by pipet and lyophilized to give a slightly yellow solid. The TBDMS deprotection was allowed to proceed at room temperature for 24 h by incubation of the RNA oligonucleotide with 1 mL of Et3N·3HF. For trinucleotides prepared to test the sensitivity to solid-phase synthesis and deprotection conditions, the mixture was subjected to Waters reversed-phased C-18 cartridge and the oligonucleotide was eluted with a mixture of acetonitrile/water. After lyophilization of the oligonucleotide-containing fraction, the resulting solid was purified by reversed-phase HPLC using mobile phase A (0.1 M triethylammonium acetate (TEAA) buffer, pH = 7) and mobile phase B (a ratio of 80:20 acetonitrile:0.1 M TEAA buffer) over 48 min with a flow rate 1.5 mL/min and a solvent gradient 0–75% mobile phase B. Solvent was removed by lyophilization. MALDI analysis: calcd for trimer 5′-A8aza-NG-3′ (M + H)+, 928.19; obsd 928.30. For oligonucleotides longer than trimers, triethylamine trihydrofluoride was removed by dialysis, and the oligonucleotides were purified by gel electrophoreses as previously described.20 Deprotection conditions leading to modification/fragmentation of 8-azanebularine were as follows. Deprotection and purification of the trinucleotide was carried out as described above except the TBDMS deprotection was accomplished in the presence of 600 μL of 1.0 M TBAF/THF(wet) at room temperature for 48 h. MALDI analysis: calcd for 5′-AXG-3′, where X is a 5-amino-1,2,3-triazole-4-carbaldehyde ribosyl residue (see Figure 3) (M − H)−, 917.17; obsd 917.14.
Figure 3.
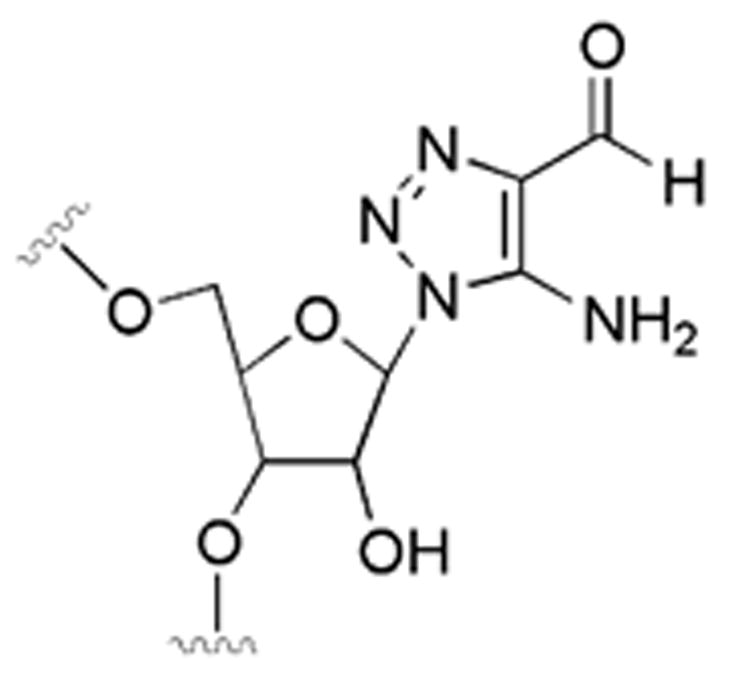
Putative hydrolysis product of 8-azanebularine in RNA deprotected with TBAF/THF.
General Biochemical Procedures
Distilled, deionized water was used for all aqueous reactions and dilutions. Biochemical reagents were obtained from Sigma/Aldrich unless otherwise noted. Common enzymes were purchased from Roche, Promega, or New England Biolabs. [γ-32P]-ATP (6000 Ci/mmol) was obtained from Perkin-Elmer Life Sciences. Storage phosphor autoradiography was performed utilizing imaging plates from Eastman Kodak Co. and a Molecular Dynamics Typhoon 9400. Full-length human ADAR2 (hADAR2a-LV(H)6), R-D (MS-(H)10ENLYFQG-hADAR2a216–711), and ADAR2300–711 (MS(H)10-ENLYFQG-hADAR2a300–711) were overexpressed in Saccharomyces cerevisiae and purified as previously described.35 After purification, R-D was treated with TEV protease to remove the (H)10 tag, which had a minimal effect on the measured dissociation constant.
Preparation of Duplex Structures
Oligonucleotides were 5′-end-labeled using [γ-32P]ATP (6000 Ci/mmol) and T4 polynucleotide kinase, purified on a 19% denaturing polyacrylamide gel, visualized by storage phosphor autoradiography, excised, and extracted by the crush and soak method. The labeled oligonucleotide was hybridized to its unlabeled complement strand by heating to 95 °C for 5 min in TE buffer (10 mM Tris-HCl, pH 7.5, 0.1 mM EDTA) with 200 mM NaCl followed by slow cooling.
Gel Mobility Shift Assay
5′-32P end-labeled RNA duplex (~60 pM) was added to 300 nM R-D, R-D (E396A), or ADAR2300–711 in 15 mM Tris-HCl, pH 7.5, 3% glycerol, 0.5 mM DTT, 60 mM KCl, 1.5 mM EDTA, 0.003% NP-40, 160 units/mL RNasin, 0.1 mg/mL BSA, and 1.0 μg/mL yeast tRNAPhe, and the reactions were incubated at 30 °C for 30 min. Samples were loaded onto a running 6% nondenaturing polyacrylamide gel (79:1 acrylamide:bisacrylamide) and electrophoresed in 0.5 X TBE buffer at 4 °C for 45 min. Storage phosphorimaging plates (Kodak) were pressed flat against the dried electrophoresis gels and exposed in the dark. For the determination of dissociation constants, varying concentrations of ADAR proteins were added to ~30 pM (8-azaN at position 24) or ~10 pM (8-azaN at R/G site) 5′-32P end-labeled RNA duplex, incubated at 30 °C for 10 min and treated as above. Variation in the incubation period from 10 to 30 min did not effect the measured dissociation constant. The data were analyzed by performing volume integrations of the regions corresponding to free RNA, protein–RNA complex, and background sites using ImageQuant software. The data were fit to the equation: fraction bound = A*[protein]/([protein] + KD), where KD is the fitted dissociation constant and A is the fitted maximum fraction RNA bound at that dissociation constant, using the least-squares method of KaleidaGraph.
Results
To accomplish the goal of incorporating 8-azanebularine into RNA, we synthesized the corresponding phoshoramidite (Scheme 1). Our synthesis begins with the known 2′,3′,5′-tri-O-acetyl-8-azaadenosine (1), which underwent diazotization/bromo-dediazoniation using a variant of the procedure recently reported by Francom and Robins to give the 6-bromonucleoside 2 (Scheme 1) in good yield.36 This compound was reductively dehalogenated using H2 and Pd/C to give acetyl protected 8-azanebularine 3. Although other synthetic routes to 8-azanebularine have been previously reported with similar overall yields,20,34 the generation of 6-bromonucleoside 2 in our scheme offers the advantage of ready access to other C6 derivatives of the 8-azapurine ring system by simple substitution reactions.37 Elaboration of 3 to the 5′-O-DMT, 3′-O-TBDMS β-cyanoethyl-di-iso-propyl phosphoramidite 7 was carried out using standard reaction conditions (see Experimental Section).
Scheme 1a.
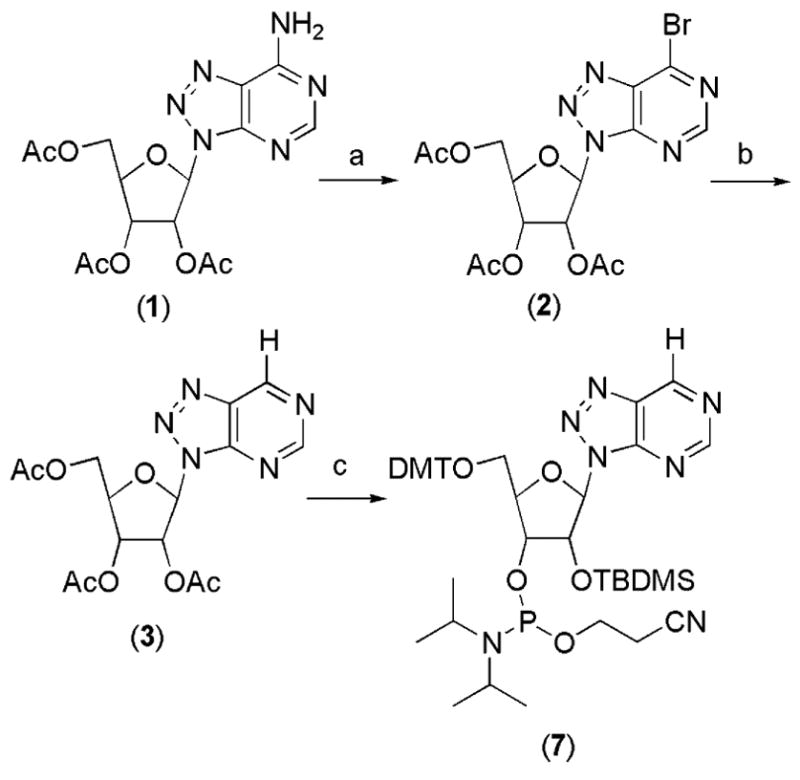
8-Azanebularine survives conditions of automated RNA synthesis and deprotection using NH3/MeOH followed by Et3N·3HF as indicated by mass spectrometric analysis of trinucleotides containing the analogue (see Experimental Section). However, when TBAF/THF was used for removal of silyl protecting groups, apparent modification at the nucleoside analogue occurred. The mass of the modified oligonucleotide product is consistent with a degradation pathway previously observed for 8-azapurines treated for extended periods with dilute aqueous acid or base involving covalent hydration followed by ring opening and fragmentation to give a 5-amino-1,2,3-triazole-4-carbaldehyde residue (Figure 3).38 Replacement of TBAF/THF with Et3N·3HF eliminated this unwanted side reaction. We used phosphoramidite 7 and the appropriate deprotection conditions to prepare mimics of the glutamate receptor B-subunit (GluR-B) pre-mRNA near the R/G editing site (Figure 4A).17
Figure 4.
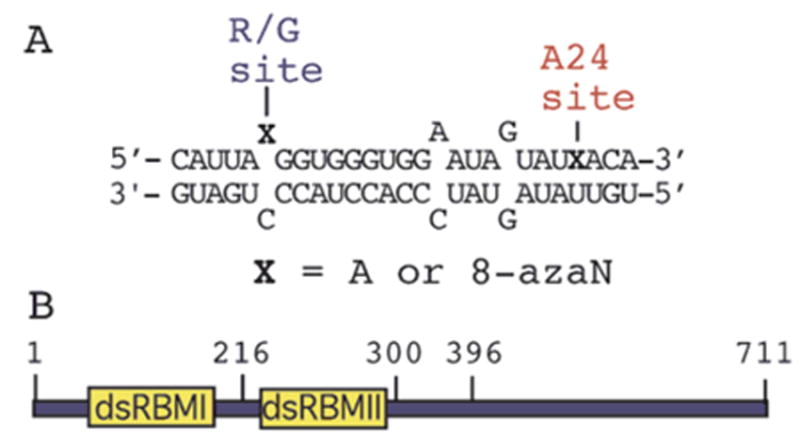
(A) Sequence of an RNA-editing substrate from the R/G site in GluR-B pre-mRNA, where X = adenosine (A) or 8-azanebularine (8-azaN) (R/G 27 mer).17 (B) Domain map for ADAR2 indicating approximate locations of dsRBM I, dsRBM II, and various amino acid positions.
We chose initially to analyze the effect 8-azanebularine has on the ADAR2 RNA binding affinity using a deletion mutant of the enzyme lacking dsRBM I (ADAR2216–711) (Figure 4B). We refer to this protein as R-D because it contains one dsRBM and the Deaminase domain. R-D has been shown to retain efficient RNA-editing activity at the GluR-B R/G site.35 By deleting dsRBM I from ADAR2, we anticipated the effects on RNA binding arising from interactions with the deaminase domain would be maximized. Importantly, a stable, slow moving complex is observed in gel mobility shift experiments upon addition of 300 nM R-D to the RNA duplex containing 8-azanebularine at the R/G editing site. Little binding is observed under these conditions when adenosine is at the editing site (Figure 5). Under the conditions of these experiments, adenosine at the R/G site is efficiently converted to inosine, so these observations indicate the protein–RNA complex is significantly less stable with inosine at the editing site.35 However, a more stable complex is formed with the 8-azanebularine at the reactive site in the duplex (Figure 5, lane 4).
Figure 5.
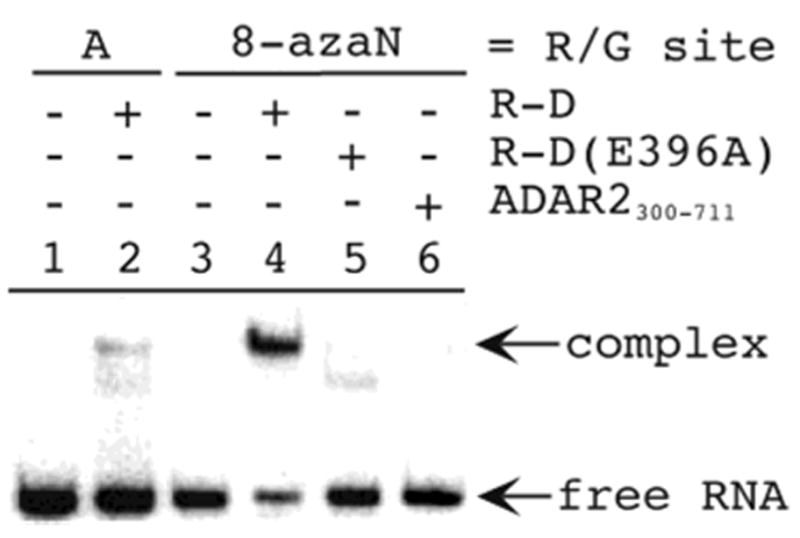
Gel mobility shift analysis of the binding of 8-azanebularine-containing RNA to ADAR2 mutants. Lane 1, unmodified GluR-B R/G duplex; lane 2, unmodified GluR-B R/G duplex + 300 nM R-D; lane 3, GluR-B R/G duplex with 8-azanebularine at the R/G site; lane 4, GluR-B R/G duplex with 8-azanebularine at the R/G site + 300 nM R-D; lane 5, GluR-B R/G duplex with 8-azanebularine at the R/G site + 300 nM R-D (E396A); lane 6, GluR-B R/G duplex with 8-azanebularine at the R/G site + 300 nM ADAR2300–711.
The calculated equilibrium constant for hydration of the 8-azapurine ring favors the dehydrated, fully aromatic system, and the hydrate has not been detected in solution.32 Thus, if hydration of the 8-azapurine ring system is required for stable complex formation as we have suggested, it is most likely facilitated by the enzyme. Therefore, one would predict that stable complex formation with 8-azanebularine-containing RNA would require a functional ADAR2 active site. We tested this hypothesis by analyzing the binding of the catalytically inactive E396A mutant of R-D. The analogous residue in E. coli CDA (E104) is critically important in forming the key tetrahedral intermediate for the CDA reaction.25 It is believed to both deprotonate the zinc-bound water and protonate the pyrimidine ring during its formation. Significantly, the production of a stable, high-affinity complex with 8-azanebularine-containing RNA is inhibited by the E396A mutation in R-D (Figure 5, lane 5). Indeed, the extent of binding observed is similar to the small amount seen with R-D and the RNA duplex with adenosine at the editing site (compare Figure 5, lanes 2 and 5) and likely arises from duplex RNA binding by dsRBM II. We also assessed the ability of a mutant of ADAR2 lacking both dsRBMs (ADAR2300–711) to bind the duplex with 8-azanebularine at the R/G editing site. Although this protein bears all of the residues conserved with CDAs and a demonstrated ability to catalyze deamination of adenosine in RNA, micromolar concentrations are required to bind the R/G 27 mer containing the aza analogue at the editing site with no binding observed at 300 nM (Figure 5, lane 6 and data not shown). Together these results indicate that both a functional active site and dsRBM II are required for the high-affinity binding to the nucleoside analogue-containing RNA. Interestingly, the dissociation constant for the analogue-containing RNA binding to full length ADAR2 is similar to that observed with R-D (see below), suggesting that dsRBM I of ADAR2 contributes little to specific binding to analogue-containing RNA.
To determine to what extent the tight binding was dependent on the sequence context of the natural editing site, we used a quantitative gel mobility shift assay to measure dissociation constants for binding of R-D to the RNA duplex with 8-azanebularine at the R/G editing site and to the duplex modified at a site that is not normally edited by ADAR2 (adenosine at position 24) (Figure 6).18 The RNA with the analogue at the editing site was bound with a KD = 2.0 ± 1.6 nM, whereas modification with the analogue at position 24 caused the RNA to be bound with a KD = 271 ± 101 nM (Table 1). The latter dissociation constant is similar in magnitude to that observed for unmodified RNA (KD ≈ 1 μM) (data not shown) and for R-D (E396A) binding to the RNA duplex with 8-azanebularine at the R/G editing site (KD = 308 ± 39 nM). Thus, over a 100-fold affinity difference is realized by positioning the analogue in the sequence/structural context of the GluR-B R/G editing site as compared to a site not edited in the same RNA structure. Furthermore, this increase in affinity requires the functional active site. Analysis of the binding of full length ADAR2 to the two RNAs again shows higher affinity binding to editing site-modified RNA, albeit with a smaller difference in the measured dissociation constants (~10-fold) (KD = 3.2 ± 1.6 nM for the R/G site; KD = 30 ± 6.7 nM for position 24) (Table 1). The difference is due to increased binding to position 24-modified RNA as R/G site-modified RNA binds the two proteins with the same affinity. The higher affinity measured for full length ADAR2 binding to position 24-modified RNA is a result of increased nonspecific binding arising from the presence of dsRBM I, because we have measured a similar affinity (KD = 22 ± 4 nM) for the unmodified duplex binding to full length ADAR2.17
Figure 6.
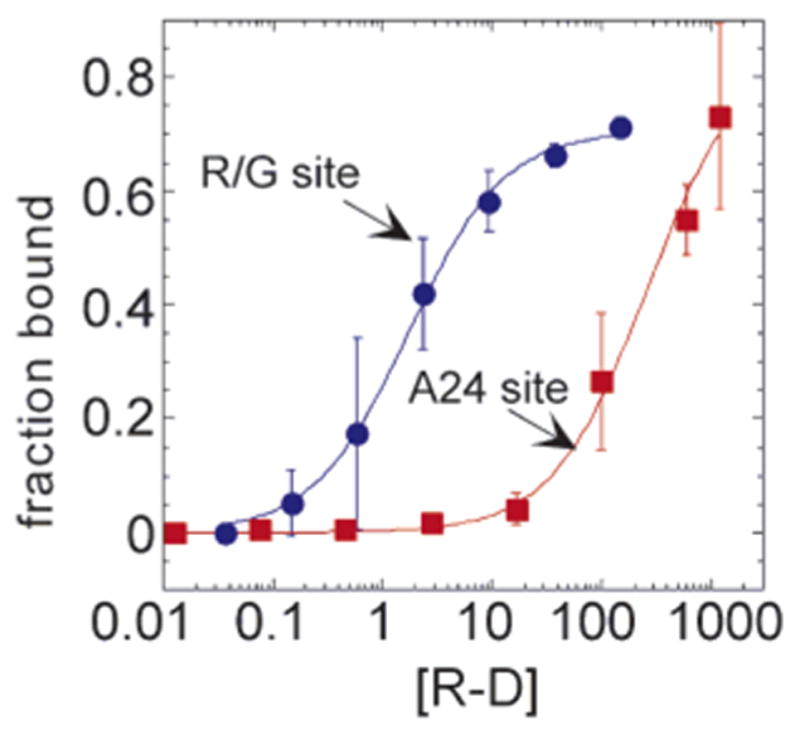
Quantitative gel mobility shift data for the binding of R-D to R/G 27 mer RNA shown in Figure 4 with 8-azanebularine at either the R/G site (blue) or the A24 site (red).
Table 1.
Dissociation Constants (KD) for Complexes with ADAR2 Mutants and 8-Azanebularine-Modified RNA Duplexesa
| site of 8-azanebularine modification
|
||
|---|---|---|
| protein | R/G site (KD, nM) | A24 site (KD, nM) |
| ADAR2full | 3.2 ± 1.6 | 30 ± 6.7 |
| R-D | 2.0 ± 1.6 | 271 ± 101 |
| R-D (E396A) | 308 ± 39 | NDb |
Dissociation constants were measured using quantitative gel mobility shift assays and are reported as the average ± standard deviation (see Experimental Section).
Not determined.
Discussion
There are currently no reports of high-resolution structural data for ADAR proteins or ADAR–RNA complexes. Such structural information would be extremely valuable in understanding protein–RNA recognition for these enzymes and for further definition of the deamination reaction mechanism. Crystallography, NMR spectroscopy, and even simple footprinting studies of dsRBM-containing RNA-binding proteins, like the ADARs, are complicated by the lack of sequence specificity for the dsRBMs.39,40 This leads to multiple protein–RNA complexes of similar affinities in solution. One approach to address this problem for the ADARs is to form a high-affinity complex involving a tight-binding deamination substrate analogue or transition state analogue in the RNA. The results presented here indicate that 8-azanebularine-containing RNA will be useful in these efforts, and relevant studies are currently underway. Furthermore, 8-azanebularine-containing oligonucleotides are likely to be selective inhibitors of ADARs and could be used to control ADAR-catalyzed deamination reactions.
8-Azanebularine is a known inhibitor of the nucleoside-modifying enzyme adenosine deaminase (ADA).41 ADA is structurally unrelated to ADARs, but does share some mechanistic similarities, such as the ability to displace alternate leaving groups from C6 and the 8-aza effect on the deamination rate.20 A crystal structure of ADA bound to nebularine (purine ribonucleoside) has been solved.42 Importantly, nebularine binds ADA as the covalent hydrate, a structure that is remarkably similar to the proposed hydrolytic deamination transition state. These studies suggested to us that 8-azanebularine, which is predicted to undergo covalent hydration more readily than does nebularine,32 might bind tightly in the ADAR active site as a covalent hydrate. Indeed, high concentrations of 8-azanebularine inhibit ADAR2, whereas similar concentrations of nebularine do not.20 Here, we show that 8-azanebularine can be incorporated into a known RNA ligand of ADAR2 and this leads to tighter binding to that RNA. This is consistent with the inhibition results with the free nucleoside and the observation that 8-azaadenosine in RNA is deaminated at a higher rate than is adenosine; that is, the 8-azapurine ring system is well-suited for interaction with the ADAR2 active site. However, the affinity achieved by the 8-azanebularine-containing RNA for R-D (KD = 2 nM) is still several orders of magnitude lower than that observed for the transition state analogue coformycin binding to ADA (KI ≈ 10 pM).43 This may be due to the requirement for formation of the unfavored hydrate of 8-azanebularine for ADAR binding, whereas coformycin, with its stable tetrahedral center at the reactive carbon, does not require this step for ADA binding. Furthermore, these observations suggest room for improvement in the development of new high-affinity ligands for the ADARs.
The data reported here support the hypothesis that the tight binding observed is in the form of a complex with 8-azanebularine at the R/G site bound into the ADAR2 active site. First, this binding requires substitution at a site on the duplex that is deaminated by ADAR2 (the R/G editing site). The presence of an adenosine within duplex RNA does not, by itself, indicate whether it will be deaminated by ADAR2. Its position on the duplex and the local sequence context play important roles in determining an adenosine’s susceptibility to ADAR-catalyzed deamination.44,45 The R/G site adenosine is deaminated by ADAR2 to ~80% yield with a kobs ≈ 1 min−1 in the duplex substrate shown in Figure 4, whereas no ADAR2-catalyzed deamination has been observed at the adenosine 24 position.17,18 Also, we have shown previously that the fluorescence of 2-aminopurine positioned at the R/G editing site in this duplex is enhanced by ADAR2, consistent with enzyme-induced base flipping at that site.18 No such change in fluorescence is observed when 2-aminopurine is placed at position 24.18 Here, we demonstrate that the increase in binding affinity arising from incorporation of 8-azanebularine into the RNA ligand is also dependent on its positioning on the duplex. Incorrect positioning prevents ADAR2 from productive interaction with the analogue. This may be because the A24 site is not in the right position relative to preferred binding registers of ADAR2’s dsRBMs or the flanking sequence may prevent the catalytic domain from maximum productive binding with adjacent nucleotides. Both of these factors could be contributing, and additional studies will be necessary to determine their relative importance.
The high-affinity binding to 8-azanebularine-containing RNA also requires a functional ADAR2 active site. Mutation of the critical conserved glutamate residue in the ADAR active site has been shown to inhibit enzyme activity, but has a minimal effect on RNA binding to unmodified RNA.26 However, we show here that this mutation dramatically reduces the affinity with which R-D binds the RNA duplex modified at the R/G site with 8-azanebularine (Figure 5, Table 1). This is most likely because tight binding requires formation of the covalent hydrate, as observed in the nebularine-ADA crystal structure, and hydration requires the catalytic residues of the enzyme.42 Importantly, formation of the covalent hydrate of 5-fluorozebularine in the active site of E. coli CDA is prevented by the E104A mutation in that enzyme as well.46
High-affinity binding to RNA modified at the R/G site also requires ADAR2’s dsRBM II. Comparing the affinities of R-D, which lacks dsRBM I, and ADAR2300–711, which lacks both dsRBMs, for R/G site-modified RNA clearly indicates a significant role for dsRBM II (at least a 1000-fold difference in affinity). Thus, in the complex leading to deamination at the R/G site, dsRBM II contacts the duplex and contributes to overall affinity. The presence of extensive duplex structure 3′ to the editing site undoubtedly positions the enzyme on the substrate with dsRBM II bound in this location. Furthermore, preferred binding registers on the RNA for dsRBM II may fine-tune the binding and positioning of the catalytic domain.
Interestingly, tight binding to 8-azanebularine-containing RNA does not require dsRBM I. Full length ADAR2 and R-D bind RNA modified at the R/G site with nearly identical dissociation constants (Table 1). The presence of dsRBM I does contribute to nonspecific binding considering the 10-fold difference in binding of these two proteins to RNA modified at position 24 (Table 1). How could dsRBM I contribute to nonspecific binding but not to specific binding to RNA containing the aza-analogue at the R/G site? It may be that dsRBM I and the catalytic domain of ADAR2 do not bind the RNA simultaneously. For the short RNA duplexes studied here, there simply may not be enough recognition surface for dsRBM I, dsRBM II, and the catalytic domain to bind this RNA at the same time. Alternatively, for dsRBM I and the catalytic domain to bind concurrently, a conformational change in the protein that disrupts favorable protein–protein interactions may be necessary.35 This could be compensated for by binding interactions between dsRBM I and RNA. For binding by the protein without dsRBM I (R-D), no conformational change would be necessary, but no additional affinity from dsRBM I·RNA binding would be possible either. The two processes would be energetically equivalent. The difference in affinity observed for the two proteins for the A24 site-modified RNA can simply be attributed to nonspecific duplex RNA binding arising from the dsRBMs, where full length ADAR2 binds more tightly because it carries two dsRBMs.
In summary, we have shown that 8-azanebularine can be incorporated into an RNA duplex substrate of ADAR2 via the phosphoramidite, creating a tight binding ligand for the enzyme. The observed high-affinity binding is dependent on a functional ADAR2 active site, the presence of dsRBM II, and correct placement of the nucleoside analogue into the sequence/structural context of a known editing site. ADAR2’s dsRBM I contributes little to the observed affinity for analogue-containing RNA. These results have implications in our understanding of substrate recognition during ADAR-catalyzed RNA editing, for future structural studies of ADAR·RNA complexes, and in the development of inhibitors of adenosine deamination RNA editing.
Supplementary Material
Acknowledgments
P.A.B. acknowledges support from the NIH (GM61115) and a Camille Dreyfus Teacher Scholar Award. We would also like to acknowledge the National Science Foundation (CHE-9002690) and the University of Utah for funds necessary to purchase the Finnigan MAT 95 mass spectrometer.
Footnotes
Supporting Information Available: NMR spectra for compounds 2, 5–7. MALDI mass spectra for trinucleotides containing 8-azanebularine after different deprotections conditions. Autoradiograms and plots of fraction RNA bound versus protein concentration for quantitative gel mobility shift assays. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Grosjean H, Benne R. Modification and Editing of RNA. ASM Press: Washington, DC; 1998. [Google Scholar]
- 2.Bass BL, Nishikura K, Keller W, Seeburg PH, Emeson RB, O’Connell MA, Samuel CE, Herbert A. RNA. 1997;3:947–949. [PMC free article] [PubMed] [Google Scholar]
- 3.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 4.Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, Seeburg PH. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- 5.Morse DP, Bass BL. Proc Natl Acad Sci USA. 1999;96:6048–6053. doi: 10.1073/pnas.96.11.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morse DP, Aruscavage PJ, Bass BL. Proc Natl Acad Sci USA. 2002;99:7906–7911. doi: 10.1073/pnas.112704299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tonkin LA, Bass BL. Science. 2003;302:1725. doi: 10.1126/science.1091340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoopengardener B, Bhalla T, Staber C, Reenan R. Science. 2003;301:832–836. doi: 10.1126/science.1086763. [DOI] [PubMed] [Google Scholar]
- 9.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Spengle R, Seeburg PH. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 10.Wang Q, Khillan J, Gadue P, Nishikura K. Science. 2000;290:1765–1768. doi: 10.1126/science.290.5497.1765. [DOI] [PubMed] [Google Scholar]
- 11.Palladino MJ, Keegan LP, O’Connell MA, Reenan RA. Cell. 2000;102:437–449. doi: 10.1016/s0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- 12.Tonkin LA, Saccomanno L, Morse DP, Brodigan T, Krause M, Bass BL. EMBO J. 2002;21:6025–6035. doi: 10.1093/emboj/cdf607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niswender CM, Herrick-Davis K, Dilley GE, Meltzer HY, Overholser JC, Stockmeier CA, Emeson RB, Sanders-Bush E. Neuropsychopharmacology. 2001;24:478–491. doi: 10.1016/S0893-133X(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 14.Iwamoto K, Kato T. Neurosci Lett. 2003;346:169–172. doi: 10.1016/s0304-3940(03)00608-6. [DOI] [PubMed] [Google Scholar]
- 15.Gurevich I, Tamir H, Arango V, Dwork AJ, Mann JJ, Schmauss C. Neuron. 2002;34:349–356. doi: 10.1016/s0896-6273(02)00660-8. [DOI] [PubMed] [Google Scholar]
- 16.Yi-Brunozzi HY, Easterwood LM, Kamilar GM, Beal PA. Nucleic Acids Res. 1999;27:2912–2917. doi: 10.1093/nar/27.14.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephens OM, Yi-Brunozzi HY, Beal PA. Biochemistry. 2000;39:12243–12251. doi: 10.1021/bi0011577. [DOI] [PubMed] [Google Scholar]
- 18.Yi-Brunozzi HY, Stephens OM, Beal PA. J Biol Chem. 2001;276:37827–37833. doi: 10.1074/jbc.M106299200. [DOI] [PubMed] [Google Scholar]
- 19.Easterwood LM, Veliz EA, Beal PA. J Am Chem Soc. 2000;122:11537–11538. [Google Scholar]
- 20.Veliz EA, Easterwood LM, Beal PA. J Am Chem Soc. 2003;125:10867–10876. doi: 10.1021/ja029742d. [DOI] [PubMed] [Google Scholar]
- 21.Doyle M, Jantsch MF. J Struct Biol. 2003;140:147–153. doi: 10.1016/s1047-8477(02)00544-0. [DOI] [PubMed] [Google Scholar]
- 22.Bass BL, Weintraub H. Cell. 1987;48:607–613. doi: 10.1016/0092-8674(87)90239-x. [DOI] [PubMed] [Google Scholar]
- 23.Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. Proc Natl Acad Sci USA. 1994;91:11457–11461. doi: 10.1073/pnas.91.24.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. Nature. 1996;379:460–464. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- 25.Carter CW. Biochemie. 1995;77:92–98. doi: 10.1016/0300-9084(96)88110-7. [DOI] [PubMed] [Google Scholar]
- 26.Lai F, Drakas R, Nishikura K. J Biol Chem. 1995;270:17098–17105. doi: 10.1074/jbc.270.29.17098. [DOI] [PubMed] [Google Scholar]
- 27.Cho DS, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. J Biol Chem. 2003;278:17093–17102. doi: 10.1074/jbc.M213127200. [DOI] [PubMed] [Google Scholar]
- 28.Hough RF, Bass BL. RNA. 1997;3:356–370. [PMC free article] [PubMed] [Google Scholar]
- 29.Liu PS, Marquez VE, Kelley J, Driscoll JS. J Org Chem. 1980;45:5225–5227. [Google Scholar]
- 30.Jeong LS, McCormack JJ, Cooney DA, Hao Z, Marquez VE. J Med Chem. 1998;41:2572–2578. doi: 10.1021/jm980111x. [DOI] [PubMed] [Google Scholar]
- 31.Betts L, Xiang S, Short SA, Wolfenden R, Carter CW. J Mol Biol. 1994;235:635–656. doi: 10.1006/jmbi.1994.1018. [DOI] [PubMed] [Google Scholar]
- 32.Erion MD, Reddy MR. J Am Chem Soc. 1998;120:3295–3304. [Google Scholar]
- 33.Seela F, Munster I, Lochner U, Rosemeyer H. Helv Chim Acta. 1998;81:1139–1155. [Google Scholar]
- 34.Nair V, Chamberlain SD. Synthesis. 1984:401–403. [Google Scholar]
- 35.Macbeth MR, Lingam AT, Bass BL. RNA. 2004 doi: 10.1261/rna.7920904. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Francom P, Robins MJ. J Org Chem. 2003;68:666–669. doi: 10.1021/jo020625a. [DOI] [PubMed] [Google Scholar]
- 37.Veliz EA, Beal PA. J Org Chem. 2001;66:8592–8598. doi: 10.1021/jo016078v. [DOI] [PubMed] [Google Scholar]
- 38.Albert A, Lin CJ. J Chem Soc, Perkins Trans 1. 1977:1819–1822. [PubMed] [Google Scholar]
- 39.Ryter JM, Schultz SC. EMBO J. 1998;17:7505–7513. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramos A, Grunert S, Adams J, Micklem DR, Proctor MR, Freund S, Bycroft M, St Johnston D, Varani G. EMBO J. 2000;19:997–1009. doi: 10.1093/emboj/19.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shewach DS, Krawczyk SH, Acevedo OL, Townsend LB. Biochem Pharm. 1992;44:1697–1700. doi: 10.1016/0006-2952(92)90061-m. [DOI] [PubMed] [Google Scholar]
- 42.Wilson DK, Rudolph FB, Quiocho FA. Science. 1991;252:1278–1284. doi: 10.1126/science.1925539. [DOI] [PubMed] [Google Scholar]
- 43.Agarwal RP, Spector T, Parks RE. Biochem Pharmacol. 1977;26:359–367. doi: 10.1016/0006-2952(77)90192-7. [DOI] [PubMed] [Google Scholar]
- 44.Polson AG, Bass BL. EMBO J. 1994;13:5701–5711. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dawson TR, Sansam CL, Emeson RB. J Biol Chem. 2004;279:4941–4951. doi: 10.1074/jbc.M310068200. [DOI] [PubMed] [Google Scholar]
- 46.Carlow DC, Short SA, Wolfenden R. Biochemistry. 1996;35:948–954. doi: 10.1021/bi951498y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.