

Abstract
ADARs are RNA editing enzymes that target double-stranded regions of nuclear-encoded RNA and viral RNA. These enzymes are particularly abundant in the nervous system, where they diversify the information encoded in the genome, for example, by altering codons in mRNAs. The functions of ADARs in known substrates suggest that the enzymes serve to fine-tune and optimize many biological pathways, in ways that we are only starting to imagine. ADARs are also interesting in regard to the remarkable double-stranded structures of their substrates and how enzyme specificity is achieved with little regard to sequence. This review summarizes ongoing investigations of the enzyme family and their substrates, focusing on biological function as well as biochemical mechanism.
Keywords: double-stranded RNA, inosine, deaminase, neurotransmission
INTRODUCTION
What is RNA Editing?
Like RNA splicing, RNA editing alters the sequence of an RNA from that encoded in the DNA. Typically, a single RNA splicing reaction removes a large block of contiguous sequence, whereas each RNA editing reaction changes only one or two nucleotides. In this regard the nomenclature is apt: in general, splicing is a cut-and-paste mechanism and editing is one of fine-tuning. There are many mechanistically diverse types of RNA editing, which together have a correspondingly diverse list of functions. RNA editing can create, delete, or alter the meaning of a codon, create a splice site, or alter RNA structure [see (1, 2)]. Some types of RNA editing repair or correct the information encoded by the genome, whereas others act to diversify this information, offering an organism the potential for greater complexity.
RNA editing was discovered in an mRNA encoded by the kinetoplastid mitochondria of trypanosomes (3), and at first most scientists believed the phenomenon would be limited to these unusual protozoa. However, only a year later the first example of RNA editing in a mammal, in a nuclear-encoded mRNA, was reported (4). Two types of RNA editing have now been found in nuclear-encoded mRNAs, and both involve deamination of encoded nucleotides [reviewed in (5)]. One type involves the deamination of cytidine (C) to create uridine (U), and the other, deamination of adenosine (A) to create inosine (I). This review focuses on the type of editing that occurs by adenosine deamination to change A to I in the nuclear-encoded RNAs of metazoa.
An Overview of RNA Editing by Adenosine Deamination
RNA editing by adenosine deamination is catalyzed by members of an enzyme family known as adenosine deaminases that act on RNA (ADARs) (6). ADARs were first discovered in Xenopus laevis (7–9) and have now been cloned and characterized in many metazoa, including mammals [ADAR1 (10, 11), ADAR2 (12–14)], birds (15), frogs (16), fish (17, 18), flies (19), and worms (Tonkin et al., unpublished data). New sequences are constantly being added to various databases, and it seems likely that ADARs will be found in all metazoa, where they are usually found in the nucleus.
ADARs act on RNA that is completely, or largely, double-stranded and catalyze the deamination of adenosine to produce inosine (Figure 1). Inosine is translated as a guanosine (G) (21), and most enzymes recognize inosine as a guanosine. Thus, ADARs change the primary sequence information in an RNA. In addition, because inosine base-pairs with cytidine, ADARs can change the structure of an RNA by changing an AU base-pair to an IU mismatch. Conceivably, ADARs could affect any biological process that involves sequence- or structure-specific interactions with RNA, and ultimately, numerous roles of ADARs may be discovered. So far, ADARs have been definitively shown to alter the meaning of codons, create splice sites, and sequester an RNA to the nucleus.
Figure 1.
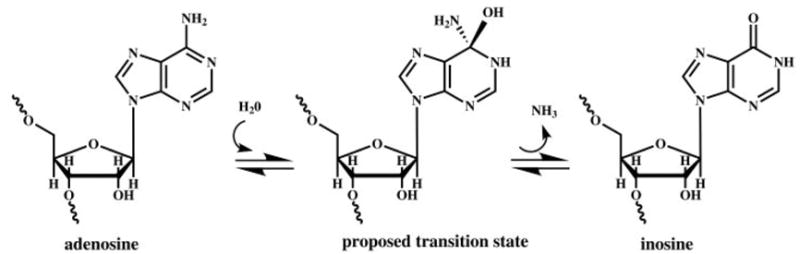
ADARs convert adenosines to inosines by hydrolytic deamination. The stereochemistry of the proposed tetrahedral intermediate has not been investigated. However, given the similarities of ADARs and ADATs (adenosine deaminases that act on tRNAs) to cytidine deaminases (CDAs), the intermediate is drawn as if water attacks from the same side of the base as observed with the CDA enzymes.
THE ENZYME FAMILY
The ADARs
ADARs from all organisms have a common domain structure that includes variable numbers of double-stranded RNA (dsRNA) binding motifs (dsRBMs) (Figure 2) [reviewed in (22)] followed by a highly conserved C-terminal catalytic domain. Different organisms differ in the number of ADAR genes they express, as illustrated in Figure 2, which shows ADARs expressed by humans, flies, and worms. As depicted, ADARs differ not only in the number of dsRBMs they contain, but also in regard to the precise distance between domains. Further diversity is gained through the use of multiple promoters (19, 23), splice sites (12, 14, 19, 24; Tonkin et al, unpublished data), and translation initiation sites [reviewed in (25) and see (19; Tonkin et al, unpublished data)].
Figure 2.

Human, fly, and worm ADAR open reading frames (ORFs) are shown with Z-DNA binding motifs (oval with a Z), dsRBMs (gray ovals), and catalytic domains (blue). Splice forms are designated by a letter of the alphabet following the ADAR name (6), and ORF lengths correlate with the relative number of amino acids for the particular splice-form. Insertions (triangle, apex up) and deletions (triangle, apex down) relative to other splice forms are indicated. An ADAR3 has been detected in mammals, but so far deaminase activity cannot be detected with this protein (26, 27). For comparison an ADAT is also shown.
Despite the diversity in primary structure, with only a few exceptions (26, 27), all ADARs deaminate adenosines within completely base-paired dsRNA, regardless of the identity of their natural, endogenous substrates. Typically, this is the assay used to detect ADAR activity of cell extracts or recombinant proteins [e.g., see (9, 10, 28, 29)]. Vertebrate ADAR1 and ADAR2 have slight differences with regard to which adenosines they target within dsRNA (see Preferences, below), and although not yet investigated, all ADARs may exhibit slightly different specificities. Some studies suggest that splice variants may further fine-tune specificity (30) or catalytic activity (12, 14).
Several observations suggest that ADARs of different vertebrates are likely to be functional homologs. For example, glutamate receptor (gluR) mRNAs are natural targets of ADARs in many different vertebrates [e.g., see (31)]. ADAR1 enzymes from X. laevis and H. sapiens, in addition to having an almost identical domain structure, have identical sequence specificities, which has been demonstrated in vitro (32) and in vivo (33). So far, none of the RNAs identified as substrates in vertebrates have been shown to be substrates in nonvertebrates, such as flies or worms. So outside of the vertebrates, it is not clear which ADARs are homologs, and in regard to nomenclature, the numbering of a particular ADAR does not necessarily imply homology to an ADAR from another organism.
Vertebrate ADAR1 has several intriguing characteristics that suggest its expression is highly regulated [reviewed in (25)]. For example, in contrast to other ADARs, vertebrate ADAR1 has a long amino-terminal extension that contains two Z-DNA binding domains (34). Although not yet proven, these domains may help to localize the enzyme close to DNA that is actively transcribing ADAR substrates (35). In addition, vertebrate ADAR1 is expressed as a long and short form, although the mechanisms that lead to the two forms may differ between organisms. In humans the use of two promoters leads to alternate exons (1A and 1B) at the 5′ end of the ADAR1 mRNA (23), which leads to the synthesis of the long and short forms of the protein. The long form is produced from an interferon-inducible promoter, and in contrast to the nuclear localization typical of most ADARs, this protein can also be found in the cytoplasm. The constitutive form of the protein is shorter, because its exon 1B excludes the methionine at position 1, leading to initiation at a downstream methionine. Genes induced by interferons are important for establishing an antiviral state, and by analogy, the long form of ADAR1 may function in viral defense. Because the long form of ADAR1 can be found in the cytoplasm, possibly it functions to target viruses that replicate in the cytoplasm. Recent studies show that human ADAR1 is a nucleo-cytoplasmic shuttling protein, with nuclear localization signals as well as nuclear export signals (36, 37), revealing yet another unique characteristic of ADAR1.
Extended Family Members: The ADATs
Although ADARs are only found in metazoa, many organisms, if not all, express members of a related gene family known as adenosine deaminases that act on tRNAs (ADATs) (Figure 2) [reviewed in (5, 38)]. These genes encode proteins that have an obvious sequence similarity to the C-terminal catalytic domain of the ADARs and essentially look like an ADAR without the dsRBMs. The current view is that ADARs evolved from ADATs after they acquired dsRBMs (see Figure 6).
Figure 6.
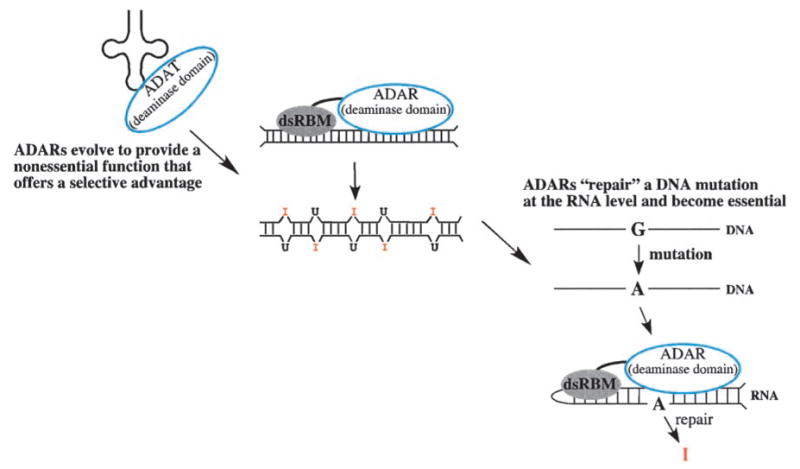
A model of how ADARs evolved.
As the name implies, ADATs convert adenosine to inosine in tRNAs. Two ADAT activities have been identified, one that deaminates A34 in the tRNA wobble position (39) and one that deaminates A37, immediately adjacent to the anticodon (40). Adenosine deamination at A34, but not A37, is essential for viability in yeast, and presumably this will prove true for other organisms as well. In prokaryotes A34 of tRNA2Arg is deaminated by a single polypeptide (Tad2p), whereas in eukaryotes two proteins are required (Tad2p and Tad3p), and multiple tRNAs are targeted. The A37 deamination only occurs in eukaryotes, and only on A37 of tRNAala. A single polypeptide, Tad1p, catalyzes this reaction, and this enzyme has been characterized in yeast (40), humans (41), and flies (42). The Tad2p/Tad3p proteins show sequence similarities to the ADARs, as well as the cytidine deaminases, and as described later, provide strong support for the idea that ADARs evolved from the cytidine deaminase superfamily.
WHAT DO ADARs DO IN VIVO?
Overview
As illustrated in Figure 3, ADARs target double-stranded structures that encompass coding sequences, introns, and 5′ and 3′ untranslated regions (UTRs). Many of the editing sites within coding sequences alter codon meaning so that more than one protein isoform can be synthesized from a single gene (see Table 1). In this capacity ADARs enhance the complexity offered by a genome, and consistent with this, the levels of ADARs and their substrates are very high in tissues of the nervous system [(43) and references therein]. Some evidence suggests that the most common site of editing is within noncoding sequences (45; D. P. Morse et al., unpublished data), but in most cases the function of this editing is unknown. However, as described below, in one case editing in an intron is known to create a splice-site. Recent data show that RNAs that contain many inosines are retained in the nucleus (46); this mechanism is used by polyoma virus to reduce expression of its early transcripts. Cellular RNAs that use this mechanism have not been identified, but the data suggest yet another way ADARs can regulate gene expression.
Figure 3.

(a) The cartoon illustrates that ADARs can target double-stranded regions in 5′ and 3′ untranslated regions (UTRs), exons, and introns. An exon complementary sequence (ECS) is shown in green. (b) The hairpin targeted by ADARs at the R/G site of gluR-B, -C, and -D is shown as an example of a structure required for deamination in a codon. The identities of sequences in blue are strictly conserved, and those indicated with a red dot covary so as to maintain an unpaired state (31). (c) The structure targeted by ADARs in the 3′ UTR of syntaxin (unc-64) is shown as an example of a structure that mediates deamination in a UTR (D. P. Morse et al., unpublished data).
TABLE 1.
ADAR substrates with editing sites in coding sequences
| Cell type | RNA | Base-pairing | Codon changesa | References |
|---|---|---|---|---|
| Mammalian | gluR-B | Exon/intron | Q/R, R/G | (56, 53) |
| gluR-C | Exon/intron | R/G | (53) | |
| gluR-D | Exon/intron | R/G | (53) | |
| gluR-5 | Exon/intron | Q/R | (56) | |
| gluR-6 | Exon/intron | Q/R, I/V, Y/C | (56, 59) | |
| Serotonin receptor | Exon/intron | I/V, I/M, N/D, N/S, N/G | (65) | |
| HDV antigenomeb | Antigenome | Amber/W | (33) | |
| Measles virus | Unknown | Hypermutation | (85, 121) | |
| Parainfluenza virus 3 | Unknown | Hypermutation | (77) | |
| Respiratory syncytial virus | Unknown | Hypermutation | (122) | |
| VSV DI particlec,d | Hairpin | Hypermutation | (78) | |
| Polyoma virus | Antisense | Hypermutation | (80) | |
| Avian | ALV (retrovirus)e | Hairpinf | Hypermutation | (79) |
| RAV-1 (retrovirus)g | Unknown | Hypermutation | (82) | |
| Xenopus | bFGF | Antisense | Hypermutation | (94) |
| Drosophila | 4F rnp | Unknown | Hypermutation | (123) |
| Calcium channel (cac) | Exon/intron | S/G, M/I, N/S, M/V, N/G, N/D, R/G | (50, 72) | |
| Sodium channel (para) | Exon/intron | Q/R, Y/C, M/V, N/D, K/R, N/S | (50, 69, 70) | |
| Chloride channel | Unknown | I/V, K/R, N/S | (71) | |
| Squid | Potassium channel | Unknown | I/V, Y/C, K/D, M/V, I/M, D/G, S/G | (73) |
Editing sites are named according to the amino acid change they produce, unedited/edited.
HDV, hepatitis delta virus
VSV, vesicular stomatitis virus
DI, defective-interfering
ALV, avian leukosis virus
This hairpin is not a natural sequence, but engineered into the retroviral construct.
RAV-1, Rous-associated virus type 1
Tables 1 and 2 list most of the RNAs that have been identified as ADAR substrates. The majority of these substrates were identified by chance when a sequence discrepancy between a gene and its cDNA was noticed. (A hallmark of an ADAR editing site is a genomically encoded A that appears as a G in a cDNA). An exception is the substrates with inosines in noncoding regions, shown in Table 2. These substrates were identified using a method specifically developed to identify inosine-containing RNAs in cellular RNA fractions (45; D. P. Morse et al., unpublished data).
TABLE 2.
ADAR substrates with editing sites in noncoding sequencesa
| Cell type | RNAb (Wormbase ID or Accession #) | Base-pairing |
|---|---|---|
| C. elegansc | M05B5.3 | 5′ UTRd |
| 52G (F55A4.9) | Noncoding | |
| pop-1 (W10C8.2) | 3′ UTR | |
| 9A (ZC239.6) | 3′ UTR | |
| 16G (Y6D11A.1) | 3′ UTR | |
| Syntaxin (F56A8.7a) | 3′ UTR | |
| Laminin-γ (C54D1.5) | 3′ UTR | |
| 36A (C35E7.6) | 3′ UTR | |
| SSADH (F45H10.1) | 3′ UTR | |
| 12A (D2024.3) | 3′ UTR | |
| H. sapiense | CDK8-like (Z84480) | Intron |
| NrCAM (AF172277) | Intron | |
| PDE8A (AF056490) | Intron | |
| Ubiquitin hydrolase-like (AK001647) | Intron | |
| HsC7-I, proteasome subunit (D26599) | 3′ UTR | |
| NADH-dehydrogenase (BC007323) | 3′ UTR | |
| Tankyrase (AF082557) | 3′ UTR | |
| PP2C-β (AF294792) | Intron | |
| PAR-SN (U55937) | Noncoding |
Data derived from (45; L.A. Tonkin et al. and D.P. Morse et al., unpublished data).
For mRNAs of unknown function, substrates are designated as described (45; D. P. Morse et al., unpublished data).
Whole worms, all developmental stages.
UTR, untranslated region
Brain
Below I review the phenotypes of animals lacking ADAR activity and provide details about representative editing events. Editing is essential for only two of the editing sites characterized to date. Mice that cannot edit the Q/R site of mammalian gluR-B are prone to epilepsy and die shortly after birth (47). Without editing at the amber/W site of the hepatitis delta virus (HDV) antigenome, the virus cannot assemble viral particles and is not infectious (reviewed in 48). From what we know about other editing sites and the fact that ADARs are not essential in Drosophila and C. elegans, it seems likely that the primary role of ADARs is not to provide essential functions. Rather, ADARs modulate and optimize cellular functions and biological pathways, in sometimes subtle ways, to increase an organism’s chance of survival.
One way ADARs may exert these effects is by fine-tuning the myriad of protein-protein interactions that occur in a cell. For example, ADARs can create amino acid changes that alter the affinity of interacting proteins, as described below for editing of the G protein–coupled 5-HT2C serotonin receptor mRNA (49). Because ADARs also act in UTRs of mRNAs (45), editing may sometimes regulate the actual levels of an RNA or its translatability; in this way an ADAR could alter the amount of a complex by changing the concentration of one of its protein partners.
Phenotypes of Knock-Out Animals
Consistent with the idea that the primary biological role of ADARs is an important, but nonessential one, D. melanogaster and C. elegans strains that lack all ADAR activity are viable. However, these animals are not normal: They exhibit defects that are largely behavioral, as expected from the high expression of ADARs in the nervous system (19; Tonkin et al, unpublished data). Flies containing a homozygous deletion in their single ADAR gene have defects in locomotion, grooming, and mating, as well as tremors, and these defects all become more severe with age (50). In general, mutant animals exhibit normal morphology, but anatomical defects are observed in the retina, and as the animals age, lesions can be observed in all areas of the brain. C. elegans strains containing homozygous deletions in either or both of their ADAR genes have more subtle defects, but again related to behavior (Tonkin et al, unpublished data). Mutant worms have chemotaxis defects, and both ADARs contribute to these defects.
In stark contrast to the phenotypes of the ADAR knock-outs in worms and flies, ADARs are absolutely essential in mammals. In mice, knocking out a single allele of ADAR1 leads to lethality by embryonic day 14.5 and defects in the proliferation and differentiation of blood cells (51). Whereas ADAR2 heterozygote mice are viable, homozygotes die shortly after birth, between postnatal day 0 (P0) to P20, and become progressively seizure prone after P12 (47). α-Amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptor–mediated currents recorded in ADAR2−/− brain slices show pronounced rectification, increased desensitization rates, and higher calcium permeability compared with wild-type. Remarkably, the defects of the ADAR2−/− animals are completely rescued by replacing each wild-type gluR-B allele with a glurR-B allele encoding the edited R codon. This is interesting in light of the fact that editing of many other sites is significantly reduced in the ADAR2−/− animals, including the R/G site of gluR-B, -C and -D, the Q/R site of gluR-5 and −I/V and Y/C of gluR-6, and the B, C, and D editing sites of the 5-HT2C serotonin receptor. Presumably alterations in editing at these sites, although not lethal, produce subtle defects that are more difficult to discern.
RNA Editing in Mammalian Glutamate Receptor RNAs
Glutamate-gated ion channels assemble from multiple glutamate receptor (gluR) subunits, and most evidence indicates there are four subunits per channel. The channels mediate fast excitatory neurotransmission in the brain and, as might be expected given this important role, exhibit enormous diversity in their electro-physical properties. This diversity comes from the particular constellation of gluR subunits that comprise a given channel, as well as from the fact that RNA splicing and RNA editing lead to multiple isoforms of the individual gluR subunits.
ADARs target mRNAs of five gluR subunits [reviewed in (52)], and as shown in Table 1, each different receptor has a characteristic subset of editing sites. All editing occurs within dsRNA that forms between an exon sequence and an intron “exon complementary sequence” (ECS). As represented in Figure 3, in most cases the dsRNA forms between an exon sequence and a nearby intron ECS (53, 54), but the Q/R site of gluR-5 and −6 involves an intronic ECS that is >1700 nucleotides away from its complementary exon sequence (55).
The Q/R editing site occurs in the AMPA receptor subunit gluR-B, as well as in the kainate receptor subunits gluR-5 and −6, and was the first gluR editing site discovered (56). As the name implies, editing at this site changes a Q to R deep within the ion channel in the “pore loop” of membrane segment 2 [reviewed in (57)]. This position is critical for determining the ion permeability of the channel, and in most cases inclusion of subunits containing the edited R form lowers the calcium permeability of the channel (56, 58). However, for gluR-6, editing of the Q/R site may lead to higher calcium permeability if the I/V and Y/C sites of membrane segment 1 are also edited (59) (Table 1).
As mentioned above, editing at the Q/R site of gluR-B, but not gluR-5 or gluR-6, is essential. Mice with reduced levels (60) of or completely lacking all editing at this site (47) are born with epilepsy and die about 3 weeks after birth. However, almost all (99%) of the gluR-B subunit found in a cell is the edited R form, and further, mice that lack any of the unedited Q form appear completely healthy. This raises the question of why editing exists for the Q/R site. Why not just encode an R at this site in the genome?
It is easier to understand why editing exists at other sites within the gluR subunits because both unedited and edited isoforms coexist (allowing greater diversity), and further, the amount of editing is often regulated. For example, the Q/R site in gluR-5 and −6 is developmentally regulated, existing in the unedited form in the rat embryo and rising to 55% (gluR-5) and 85% (gluR-6) by birth (61). The R/G site in gluR-B, -C, and -D is also largely unedited in the embryonic brain and rises after birth (53). However, in contrast to the Q/R site of gluR-B, editing at these other sites is not essential and appears to play a more subtle role. For example, whereas editing of gluR-6 at the Q/R site also alters calcium permeability of the ion channel (58, 59), mice lacking editing at this site are viable and for the most part, appear normal. However, careful studies show that this editing event has subtle effects on synaptic plasticity as well as seizure vulnerability (62). In particular, in contrast to wild-type mice, the kainate receptors of mice that lack Q/R site editing in gluR-6 are capable of NMDA receptor–independent long-term potentiation and are more susceptible to kainate-induced seizures. The number of seizures correlates inversely with the amount of editing, which is interesting in light of the fact that gluR-6 editing increases during human seizures, possibly as an adaptive mechanism (63, 64). The role of editing at the R/G editing site found in gluR-B, -C, and -D, is also more subtle (53), and the unedited and edited forms of these AMPA receptor channels recover from desensitization at different rates. As shown in Table 1, two additional amino acid changes (I/V, Y/C) are introduced into gluR-6 in membrane segment 1. Although these sites are less well characterized, editing also appears to modulate calcium permeability (59).
RNA Editing in Mammalian Serotonin Receptor RNAs
The neurotransmitter serotonin binds to a large family of receptors to elicit signaling events that are also crucial for proper neurotransmission. ADARs target the mRNA encoding the 2C subtype of serotonin receptor (5-HT2CR), which is a member of the G protein– coupled receptor superfamily that stimulates phospholipase C to produce phosphates and diacylglycerol. Five editing sites (A–E) have been identified within 5-HT2CR mRNA (65, 66), and these allow five distinct amino acid changes (see Table 1) in the putative second transmembrane loop, a region known to be important for G protein coupling. cDNA analyses of RNA isolated from brain show that the editing events occur in various combinations that are predicted to produce at least 7 protein isoforms in rat (65) and at least 12 protein isoforms in human (49). The different isoforms exist at different levels depending on the part of the brain analyzed (65), and interestingly, the relative abundance of the different isoforms differs between rat and human brain (49). Like other examples of ADAR editing, the editing sites occur within dsRNA that involves base-pairing between an exon and intron; the structure is conserved among mammals (66).
As a first step in understanding the function of editing within the 5-HT2CR mRNA, various isoforms were expressed in mammalian cell lines and their function evaluated by measurements of inositol monophosphates. (This is an indicator of the efficiency with which the serotonin receptor couples to the G protein). These studies revealed that isoforms corresponding to the most heavily edited mRNAs require a higher level of serotonin to stimulate phospholipase C, possibly because the edited receptors couple less efficiently to the G protein. More recent studies suggest that unedited isoforms have a higher level of constitutive activity because of a greater tendency to isomerize to an active conformation that can couple to the G protein (49). These authors hypothesize that RNA editing may silence constitutive activity, thus increasing the signal: noise ratio at sites where editing efficiency is high.
The different coupling efficiencies lead to different affinities of ligands that have a preference for binding to the coupled or uncoupled receptor (49). For example, fully edited human receptor is less sensitive to lysergic acid diethyl-amide (LSD) (67). Intriguingly, a recent study comparing schizophrenic and control brains reports a significant difference in the abundance of certain isoforms found in tissue isolated from the frontal cortex (68).
RNA Editing in Drosophila Sodium Channel RNAs
In Drosophila the primary sodium channel of the nervous system is encoded by the para gene, and ADARs target para transcripts at a number of sites (69). Editing sites are found throughout the mRNA and include codon changes in transmembrane domains as well as intracellular domains (50). As observed for the R/G hairpin of gluRs (31) (Figure 3b), the sequences that base-pair to form the dsRNA substrate are highly conserved between D. melanogaster and D. virilis (70). Editing at some sites is developmentally regulated (70). It is not clear if, or how, editing of para transcripts correlates with the behavioral defects in flies lacking a functional ADAR gene. However, some of the editing sites are observed in D. melanogaster as well as D. virilis, consistent with the idea that they are functionally important (70).
Other Neuronal Substrates
A to G changes indicative of editing by ADARs have also been observed in transcripts encoding a subunit of a Drosophila neurotransmitter-gated chloride channel (50, 71) and a subunit of a Drosophila calcium channel (50, 72). Although editing alters coding sequences in these cases, the consequences of editing are not known, and other aspects of the editing have not been characterized.
In squid the voltage-gated Kv2 potassium channel (sqKv2) (73) mRNA is targeted by ADARs at multiple sites (see Table 1). The function of editing at two of the most highly edited sites, a Y/C change in the pore region between S5 and S6 and an I/V change within the S6 segment, has been evaluated by monitoring ion currents of channels expressed in Xenopus oocytes. Whereas the conductance-voltage relationships are not altered by editing, the rate of channel closure upon repolarization is affected. Squid Kv2 channels exhibit a slow inactivation process characterized by a long-lived nonconducting state, and both editing events also show altered kinetics in this process. Although it has not been determined whether ADARs are essential in squid, these effects of editing are subtle and would not be expected to be essential for viability.
Creation of a Splice Site by RNA Editing
Mammalian ADAR2 acts on its own pre-mRNA to create a new splice site (74). Editing alters an AA dinucleotide to generate an AI dinucleotide, which is recognized as the canonical AG typically found at 3′ splice sites. The alternative splicing results in a 47-nucleotide insertion that is predicted to cause a frameshift and the production of an 88–amino acid polypeptide, lacking both dsRBMs and the catalytic domain. An obvious interpretation of these data is that the alternative splicing is part of an auto-feedback loop designed to tightly regulate the endogenous levels of ADAR2. However, it is not clear that editing of the ADAR2 mRNA can actually change the overall enzyme activity in a cell (74). Interestingly, D. melanogaster also targets its own mRNA, in this case altering a codon in the variable region of the catalytic domain (19).
So far, the creation of a splice site in mammalian ADAR2 pre-mRNA is the only example where an ADAR alters a splice site. In theory, ADARs could create both 5′ and 3′ splice sites as well as destroy a 3′ splice site, and it seems likely that other examples will be discovered in the future.
Does RNA Editing Affect RNA splicing?
As illustrated in Figure 3, the dsRNA required for editing often encompasses sequences that are also important for splicing. Thus, it seems likely that RNA editing and splicing are somehow coordinated. Although this has not yet been rigorously investigated, several observations suggest it is true. For example, elimination of editing at the Q/R site of gluR-B leads to an accumulation of unspliced precursor RNA (47). In Drosophila the maleless (mle) gene encodes an RNA helicase required for dosage compensation, and interestingly, the napts allele of mle confers a temperature-sensitive paralytic phenotype that is nearly indistinguishable from mutations in the para gene itself. A recent study helps explain this phenotype by demonstrating that many para transcripts of mlenapts flies contain exon skipping events in the region of RNA editing (69). The dsRNA required for editing in this region contains a splice donor, and the current view is that the RNA helicase encoded by mle is required to resolve the structure required for editing so splicing can occur.
Editing in Noncoding RNAs, 5′ UTRs, and 3′ UTRs
Analyses of the amounts of inosine in various rat tissues suggest there are many more ADAR substrates to be discovered (43). In hopes of identifying some of these unknown substrates, a systematic way of identifying ADAR substrates was developed. This method has been applied to polyA+ RNA isolated from mixed populations of C. elegans (45), as well as human brain tissue (D. P. Morse et al., unpublished data). Surprisingly, in both cases inosines were identified in non-coding sequences, rather than in coding sequences. As shown in Table 2, inosines were found in sequences that were completely noncoding, as well as 5′ and 3′ UTRs and introns. For both C. elegans and H. sapiens samples, the inosines were found in remarkably stable structures that in many cases contain hundreds of nearly contiguous base-pairs. The function of these structures, or the inosines within them, is unknown. Possibly the structures have roles in RNA translation, localization, or stability, and ADARs serve to modulate these functions (45).
The method for identifying ADAR substrates was developed and optimized for its ability to identify inosines in the coding regions of known mammalian substrates, namely, gluR and serotonin receptor mRNAs. Whereas it can readily identify these substrates under controlled conditions, it did not lead to the discovery of new substrates with inosines in coding regions. Although RNAs with inosines in coding regions have not yet been identified in C. elegans, they clearly exist among human RNAs and by inference, probably in C. elegans as well. The most likely explanation for why the method did not identify inosines in coding sequences is that these sequences are much more rare than those in noncoding sequences.
RNA Editing by ADARs in Viruses
A number of reports describe viruses, or their transcripts, that show sequence changes consistent with modification by an ADAR (Table 1) [reviewed in (75, 76)]. In some cases the sequence discrepancies are the typical A to G changes, but for RNA viruses that replicate through a complementary intermediate, U to C changes are sometimes observed. This suggests that after replication (or transcription), an A to I change in one strand becomes a U to C change in the complementary strand. Of course, if both plus and minus strands are deaminated, multiple rounds of replication will yield molecules with both A to G and U to C transitions (e.g., see 77).
With only one exception [hepatitis delta virus (HDV)], the sequence changes observed in viral sequences are the nonselective or “hypermutation” type of deamination that occurs in ADAR substrates that are completely double-stranded (see ADAR Specificity). In some cases it is clear that the virus, or viral transcripts, form intramolecular hairpins (78, 79) or interact with an antisense molecule (80). In other cases how the dsRNA forms is unclear. For RNA viruses that replicate through an RNA intermediate, possibly, as proposed (81), aberrant double-stranded structures form between the plus and minus strands or the minus strands and an mRNA.
Among the viral substrates listed in Table 1, editing of measles virus, polyoma virus transcripts, and HDV, have been most well characterized, and as described below, all data indicate these viral sequences are true ADAR targets. In most other cases the question of whether ADARs are responsible for the observed sequence transitions has not been directly addressed, but indirect evidence makes it likely. For example, when the sequence contexts of the observed A to G or U to C changes have been published (77–79, 82), it is clear that the changes occur at sites with nearest neighbors preferred by ADARs (83) (see Preferences, below). Except for the examples discussed below, the viral hypermutations have not been correlated with a functional consequence. Possibly, some viruses just get unavoidably caught up with ADARs, and the editing has no biological role. Alternatively, although hypermutations are unlikely to lead to functional protein isoforms, a more global effect seems plausible, such as the triggering of degradation (84) or a change in nuclear export (46, 80) (see Polyoma Virus, below).
MEASLES VIRUS
Shortly after ADARs were discovered a viral RNA isolated from the brain of a patient who died of a persistent measles virus infection (85) was found to contain sequence changes indicative of the action of an ADAR. In cDNAs synthesized from the viral matrix (M) mRNA, ~50% of the uridines appeared as cytidines, and these hypermutations were proposed to result from adenosine to inosine conversions in the negative strand RNA genome (81). Both U to C and A to G transitions have now been observed in cDNAs derived from other cases of persistent measles virus infection, in the M gene and others (75, 86). Deamination sites all show the nearest neighbor preferences characterized for ADARs (see Preferences, below). The function of the measles virus hyper-mutations is still unclear, but it has been speculated they somehow lead to viral persistence. When the measles virus hypermutation events were first discovered, it was a mystery as to how the virus, which replicates in the cytoplasm, ever encountered ADAR in the nucleus (87). As mentioned above, an interferon-inducible form of human ADAR1 has been identified (23), and a significant fraction of this ADAR localizes to the cytoplasm. Although not yet explored, possibly a measles virus infection triggers the cytoplasmic form of ADAR1.
HEPATITIS DELTA VIRUS
The HDV genome is a negative strand RNA of ~1700 nucleotides that replicates through a positive strand RNA intermediate called the antigenome [reviewed in (48)]. Both the genome and antigenome exist as covalently closed circles that are predicted to base-pair along their entire length (Figure 4a). Consistent with this, the HDV genome appears rod-shaped by electron microscopy (88). RNA editing by ADARs targets a single adenosine in the only expressed HDV open-reading frame, to convert an amber stop codon to a tryptophan (33, 89, 90). RNA editing at the amber/W site allows the virus to make a short (HD-Ag-S) and a long (HD-Ag-L) form of the viral protein, delta antigen. Both proteins are essential for the viral life cycle: HD-Ag-S, is required for viral replication (91) and HD-Ag-L for assembly of new viral particles (92).
Figure 4.

ADARs are capable of a wide range of specificities depending on the structure of the RNA substrate. (a) RNAs that are predicted to form rod-shaped molecules of similar lengths. While an 800– base-pair dsRNA is deaminated promiscuously by an ADAR, the ~1700 nucleotide HDV antigenome is specifically targeted at the amber/W site (red). (b) ADARs (green) reacting with four dsRNAs of differing stabilities. Because ADARs change AU base-pairs to IU mismatches, ADAR substrates become increasingly single-stranded as the reaction proceeds. The model proposes that an ADAR reaction stops when the RNA is too single-stranded to be bound by an ADAR. Substrates that are shorter, or contain mismatches, are more selectively deaminated because it takes fewer deaminations to reach the critical “thermodynamic” threshold. In the far left and far right panels blue lines represent a specific sequence. The sequence is modified more selectively when placed between internal loops, as in the barbell molecule.
As depicted in Figure 4a, although the HDV antigenome is highly base-paired, the double-helical structure contains many mismatches, bulges and loops. These disruptions to the helix preclude the nonselective hypermutation type of editing (see Selectivity, below) and promote a very specific deamination event, which in this case occurs at a single position (1012) within the antigenomic RNA. As evidence that the specificity is intrinsic to the ADAR enzyme and does not require accessory factors, the same specificity is observed with purified Xenopus ADAR1, and point mutations near the amber/W site have identical effects on in vivo and in vitro editing (33). The latter result, as well as the fact that mammalian liver contains high levels of ADAR1 but not ADAR2, suggests that ADAR1 deaminates the amber/W site in vivo. However, both enzymes are capable of editing the amber/W site (93).
POLYOMA VIRUS AND RETENTION OF INOSINE-CONTAINING RNAS IN THE NUCLEUS
Polyoma virus has a circular, double-stranded DNA genome with early and late transcription units that are expressed from opposite strands. Late transcripts accumulate only after DNA replication begins, and while early transcripts continue to be synthesized, they are downregulated by a posttranscriptional process. During the late stage of infection RNA polymerase II encircles the genome multiple times to create giant, multimer transcripts that have regions of complementarity to early transcripts. Downregulation of the early transcripts occurs because ADARs target duplex structures that form between the early and late transcripts, and these hypermutated RNAs are retained in the nucleus (80). Recent evidence indicates that nuclear retention of hypermutated RNAs, i.e., those containing many inosines, is a general phenomenon (46) and involves an inosine-specific RNA binding protein p54nrb, the splicing factor PSF, and the inner nuclear matrix structural protein, matrin 3. It has been proposed that this complex keeps hyperedited RNAs in the nucleus, away from the translational machinery, while allowing more selectively modified RNAs to be exported.
STRUCTURES OF ADAR SUBSTRATES
ADARs contain dsRBMs so it is not surprising that their substrates are highly base-paired molecules. What is surprising, however, is that the sequences targeted by ADARs are almost entirely double-stranded. Figures 3 and 4 show secondary structures for representative ADAR substrates. In contrast to the familiar secondary structures of tRNA and rRNA, which consist of short double helices branching off from nonhelical sequences, secondary structures of ADAR substrates usually consist of one long, unbranched double helix. Whereas the R/G hairpin is one of the shortest helices known to be targeted by an ADAR in vivo (Figure 3b) (31, 53), others consist of hundreds of nearly contiguous base-pairs (45) or, like HDV, a long helix interrupted by mismatches, bulges, and loops (Figure 4a).
Most of the known ADAR substrates form their structures when a polynucleotide “folds back” to create an intramolecular hairpin, as shown in Figure 3. However, there are also examples of substrates that form through intermolecular interactions, such as the bFGF mRNA pairing with a naturally occurring antisense RNA (94) and the early and late transcripts of polyoma virus (80).
RNA structures are commonly derived through a rigorous battery of techniques, including biochemical, phylogenetic, and biophysical methods. By these criteria, very few of the structures of ADAR substrates are rigorously proven. However, in many cases compensatory mutations have been analyzed, and these studies verify the interaction of the complementary sequences, albeit not their details (as cited in 76). The structures of the long hairpins found in noncoding sequences have not been proven (45) (Figure 3c). However, pairing seems almost certain in these molecules, and the presence of inosines is proof of their double-stranded character because ADARs will not target single-stranded RNA (8, 95).
The R/G site hairpin is an exception to the above because it has been proven with mutagenesis, biochemistry, and phylogenetics (31, 53). Interestingly, the pattern of covariation observed in the phylogenetic analysis of the R/G hairpin was quite different than expected (31). Phylogenetic analyses of tRNA and rRNA show that, in general, the identity of bases within helical regions are less conserved than those in nonhelical regions. In contrast, for the R/G site hairpin, bases in helices are most conserved, and bases in nonhelical regions covary so as to maintain their nonhelical state (Figure 3b). This pattern is not so surprising because, as described below, the exact position of loops and other unpaired sequences found in ADAR substrates is critical in maintaining enzyme specificity. The conservation of sequences predicted to be in helical regions has also been noted for the structure important for editing serotonin receptor mRNA (66).
ADAR SPECIFICITY
Overview
As illustrated in Figure 4, ADARs are capable of a remarkably wide range of specificities. Depending on the substrate, the enzyme can deaminate half of the adenosines before the reaction stops, precisely target one adenosine in the midst of hundreds, or do anything in between these two extremes. These various specificities are intrinsic to ADAR enzymes and do not require accessory factors [e.g., see (33)]. Although ADARs do have slight preferences for adenosines within certain sequence contexts, specificity is dictated in large part by the structure of the RNA rather than its sequence [reviewed in (76)]. Substrates that are completely base-paired, and longer than ~50 base-pairs, are deaminated nonselectively (83, 96). In these molecules ~50% of the adenosines are deaminated at complete reaction. Shorter molecules, or molecules that are less stable because they contain mismatches, bulges, or loops, are deaminated at many fewer sites at reaction completion. The latter, more selective deamination is very important when ADARs alter codons, where they must act selectively so that deamination occurs at only one, or a few, sites.
ADARs are dsRNA binding proteins (dsRBPs), and any discussion of how ADARs achieve specificity must start with a consideration of what it means to be a dsRBP. RNA forms a double-helical structure known as A-form, which has a more deep and narrow major groove than B-form DNA. Most of the functional groups that allow a protein to distinguish one base from another are within the major groove, and it is difficult for a protein to access this information within an A-form helix. In fact, X-ray crystallographic and nuclear magnetic resonance studies of dsRBMs in complex with dsRNA show that most interactions are sequence-independent and occur with the minor groove and the phosphodiester backbone (97–100).
Consistent with the above discussion, ADARs bind tightly to dsRNA of any sequence, with dissociation constants in the nM range (29, 101–103). The ways that ADARs achieve specificity, even though they bind indiscriminately of sequence, are quite intriguing because the principles are so different from those that dictate specificity of other RNA binding proteins. The terms preferences and selectivity have been coined to describe the specificity that allows an ADAR to target a specific adenosine over others (83), and these terms are described below.
Preferences
Although ADARs will bind to any dsRNA, they exhibit slight sequence preferences for deaminating certain adenosines over others. Evidence of these preferences derives from surveys of where inosines are found within endogenous ADAR substrates, as well as from in vitro studies (32, 83). In vitro studies with Xenopus ADAR1 and synthetic substrates show that this enzyme targets adenosines with a 5′ neighbor preference of U = A > C > G and rarely targets adenosines less than three nucleotides from the 5′ terminus, or eight nucleotides from the 3′ terminus (83). Similar studies show that human ADAR1 exhibits identical preferences, whereas human ADAR2 has a similar but distinct 5′ neighbor preference (U≈A > C = G) (32). ADAR2 can deaminate adenosines as close as three nucleotides from either termini, if not closer (32), since other studies show deamination directly at the 5′ terminus (104). Unlike human ADAR1, human ADAR2 also has a 3′ neighbor preference (U = G > C = A). Comparisons of the ability of mammalian ADAR1 and ADAR2 to deaminate editing sites in a natural substrate, gluR-B RNA, emphasize that the enzymes have overlapping but distinct preferences (12–14, 105).
Although ADARs from nonvertebrates have not been characterized with regard to their sequence specificity, the location of inosines in RNAs isolated from these organisms suggests the enzymes will have similar nearest neighbor preferences. For example, one of the easiest ways to visualize the nearest neighbor preferences is to look for adenosines that have a 5′ guanosine. These sites mark adenosines in very poor context for ADARs and, consistent with this, are infrequently targeted in substrates of Drosophila (69), squid (73) and C. elegans (45).
Whereas many aspects of ADAR specificity are probably dictated by the dsRBMs, several considerations suggest that preferences reflect the architecture of the ADAR active site. For example, the 5′ and 3′ neighbors are obviously close to the targeted adenosine, and the latter must occupy the active site during deamination. Another nucleotide that may be near the target adenosine when it occupies the active site is its pairing partner in the complementary strand. Consistent with the idea that preferences are dictated by the active site, a recent study shows that discrimination between various pairing partners is determined by the catalytic domain rather than the dsRBMs (106). This study showed that for both mammalian ADAR1 and ADAR2, an AC mismatch allows more efficient editing than an AA or AG mismatch. An AC mismatch was observed to be optimal compared with an AU base-pair, in most cases.
Selectivity
Clearly the slight preferences for the identity of neighboring nucleotides cannot explain the exquisite specificity observed in some ADAR substrates. For example, whereas the HDV antigenome has more than 300 adenosines, ADARs precisely deaminate the amber/W site (Figure 4a) (33, 107). As mentioned, additional specificity derives from the structure and thermodynamic stability of the substrate. These features dictate what fraction of the preferred adenosines will be deaminated before the reaction stops, or the selectivity of the reaction. Although there are very few experimental studies of selectivity, in theory it can be understood by considering the fact that ADARs have a structural specificity: They bind dsRNA but not single-stranded RNA (8, 95). In the following discussion it is important to remember that a particular sequence of RNA is never completely paired or unpaired but at equilibrium favors one state over the other. Anything that shifts the equilibrium of an ADAR substrate toward the single-stranded state will decrease the enzyme’s affinity for the substrate.
WHY DOES THE REACTION STOP?
The concept of selectivity rests on the fact that an ADAR reaction stops before all of the adenosines in good context are deaminated. Although long, completely base-paired molecules are deaminated at many sites; when about 50–60% of their adenosines are deaminated the reaction stops (83, 96). In substrates that are very specifically deaminated, the reaction stops after only one or two deamination events. One reason the reaction stops is that deamination of an adenosine in an AU base-pair creates the less stable IU mismatch. As an ADAR reaction proceeds, the substrate becomes increasingly single-stranded. Because ADARs are dsRBPs, at a certain point in the reaction a substrate becomes too single-stranded to be bound by the enzyme.
This simple idea is illustrated in Figure 4b and helps explain why shorter substrates, or substrates that are less stable because they contain mismatches, are deaminated at fewer sites than long, completely base-paired molecules. In less stable substrates fewer deamination events are required before the “thermodynamic threshold” is reached, at which point the enzyme can no longer bind its substrate. This idea does not explain everything, and there is certainly more to the story. For example, deamination of the adenosine at the amber/W site of HDV, and that at the R/G site of the gluR-B hairpin, converts an AC mismatch into an IC base-pair, thus increasing rather than decreasing stability.
INTERNAL LOOPS CAN INCREASE SELECTIVITY
The above discussion leads to the conclusion that selectivity is increased by structural disruptions in a helix, such as mismatches, or by decreasing the length of a helix. Although HDV has many disruptions to its overall base-paired structure, it is certainly not a short helix. Considerations of the exquisite specificity observed in HDV raised the possibility that ADARs do not recognize the antigenome as a single helix but as a series of shorter ones. This led to the hypothesis that internal loops within ADAR substrates define the ends of individual helical substrates. Indeed a recent study provides strong evidence that this is true, at least for some loops (108). As shown in Figure 4b, internal loops can divide a long double-stranded substrate into a series of shorter substrates. Because ADARs deaminate fewer adenosines in short RNA helices than in longer ones, the insertion of loops decreases the number of deaminations and increases selectivity.
By comparing the far left and far right panels of Figure 4b, it is apparent that decreasing the size of the loops in a molecule, or base-pairing them entirely as in the far left panel, can dramatically decrease selectivity. This helps explain why the unpaired sequences of the R/G hairpin covary between organisms to precisely maintain a specific helical length (Figure 3b) (31).
DO ADARS EVER BIND IN REGISTER?
When an ADAR is mixed with a completely base-paired dsRNA, because binding is not sequence-specific, the enzyme binds at a different site on each molecule. After the first round of deamination the products are heterogeneous. Although not well studied, most evidence indicates ADARs are not processive (83), and after one or two deamination events, the enzyme dissociates and then rebinds to deaminate a different site. Presumably this process continues until all molecules are too unstable for further modification (see Figure 4b); the final reaction products are each deaminated at a slightly different subset of preferred sites, which depends on the location of the initial binding event.
Whereas there are many studies documenting ADAR products that are heterogeneous, it is also clear that ADARs can efficiently deaminate a single adenosine to generate molecules that are all deaminated at the same site, as occurs for the R/G hairpin. Recent studies provide a way out of this conundrum and suggest that the structural features of certain substrates may restrict the initial binding event to a single site (32, 102). This could allow binding in register and result in uniformly deaminated products. Possibly, in some cases this is achieved by making a duplex very short, so that there is only one way to bind (32). In other substrates a structural feature of the enzyme, for example a mismatch, may serve the purpose (102). Interestingly, although all known dsRBPs bind tightly to any sequence of dsRNA, two recent studies of other dsRBPs indicate there are sequences in the dsRBM that can bind to unpaired, nonhelical sequences that flank a dsRNA helix, thus providing a means for binding in register (109, 110, 110a).
PUTTING IT ALL TOGETHER: HOW TO GET OPTIMAL SPECIFICITY
In summary, optimal specificity is achieved by:
Locating as many adenosines as possible in poor sequence context, as specified by preferences.
Flanking the helix with unpaired sequences, such as loops. This will uncouple the helix from adjacent double-stranded regions and allow its effective length to be very short.
Including mismatches and other structural disruptions so the stability of the helix will be close to the “thermodynamic threshold” (see Figure 4b). This will cause the reaction to stop after a minimal number of deaminations.
Including structural features that allow ADARs to bind in register.
ADAR CATALYSIS
Overview
When ADAR reactions are performed in 18O-labeled water, the heavy oxygen is incorporated at C6 of the inosine product (111). This indicates that water is the nucleophile in the reaction and that the reaction proceeds through a tetravalent intermediate, as shown in Figure 1. This mechanism is used by deaminases that act on nucleosides and nucleotides, such as the adenosine deaminases (ADAs) (111a) and cytidine deaminases (CDAs) (112), and it is likely the mechanism used by ADARs and ADATs as well. Beyond this, the details of ADAR catalysis are almost entirely unproven, although as described below, they have been inferred from what is known about ADAs and CDAs.
Although the ADA and CDA enzymes use similar mechanisms, involving activation of the attacking water by a catalytic zinc, the details of catalysis are quite different [see discussions in (113, 114)]. ADA is monomeric with an α-β triose phosphate isomerase (TIM) barrel architecture, whereas CDA family members are dimeric or tetrameric, with a unique and different fold. Both enzymes have a catalytic glutamate near in sequence to one of the zinc ligands, but CDA uses a histidine and two cysteines to coordinate zinc (see Figure 5), and ADA uses three histidines and a single aspartate. Crystallographic studies suggest that the critical active site residues curl around opposite sides of the zinc cofactor. This difference indicates that the attacking water (hydroxyl) approaches the electrophilic carbon (C4 or C6) from opposite sides of the base for the two enzymes (see Figure 1). As discussed below, most evidence suggests ADARs and ADATs are more similar to the CDA than the ADA enzymes, but the structure of an ADAR or ADAT has not yet been solved. However, all evidence suggests the ADA and CDA families exemplify a case of convergent evolution. In an ironic twist to this story, a recent crystallographic study of cytosine deaminase shows that this enzyme has a fold like ADAs, not CDAs (115).
Figure 5.
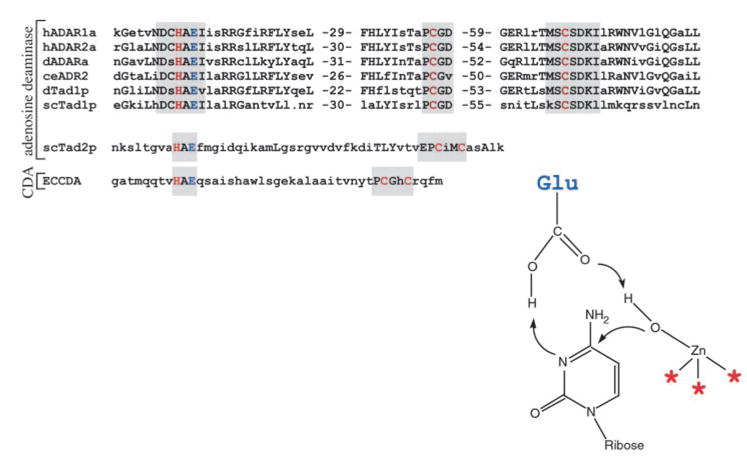
ADARs and ADATs share sequence similarities with the cytidine deaminase (CDA) family. At the top an alignment of six sequences includes representative members of the ADARs, and the ADATs that act at A37 (Tad1p). Highly conserved sequences are shown in capital letters (identities four of six shown), and regions proposed to function in binding zinc are shaded in gray. The S. cerevisiae enzyme Tad2p represents the ADATs that target A34 of tRNA. This group of enzymes shows similarities to the ADARs and Tad1p ADAT, as well as the cytidine deaminase sequence shown below, here represented by the E. coli enzyme. To the right, amino acids that coordinate zinc (red) and the glutamate involved in proton shuttling (blue) are shown interacting with cytidine, according to the crystal structure of E. coli cytidine deaminase (ECCDA) (112). The same color scheme is used to highlight residues proposed to serve a similar function in the ADARs and ADATs. Nomenclature and accession numbers for the sequences are as follows: hADAR1a, human ADAR1a, NM-001111; hADAR2a, human ADAR2a, NM-001112; dADARa, D. melanogaster ADARa, AF208535; ceADR2, C. elegans ADR2, AF051275; dTad1p, D. melanogaster Tad1p, AF192530; scTad1p, S. cerevisiae Tad1p, AJ007297; scTad2p, S. cerevisiae Tad2p, AJ242667; ECCDA, E. coli cytidine deaminase, M60916.
Do ADARs and ADATs Require Zinc?
Because ADAs and CDAs utilize a zinc to activate water for nucleophilic attack (see Figure 5), it has long been assumed that ADARs also have a catalytic zinc. The metal is presumably tightly bound because in vitro ADAR reactions proceed without adding any cofactors and are typically performed in high concentrations of ethylene diamine tetracetic acid (EDTA). This is not surprising in light of the fact that the zinc cofactors of CDA and ADA are also difficult to chelate [e.g., see (116)]. Although o-phenanthroline, a zinc-chelator, inhibits ADAR activity (10), as pointed out in these studies, the inhibition may be due to interference with other aspects of ADAR catalysis.
The first available cDNA sequences for the ADARs showed conserved sequences that more closely matched the zinc coordinating residues of the CDAs than the ADAs (Figure 5), and it was proposed that ADARs evolved from CDAs rather than ADAs (10). The recent discovery of the ADATs Tad2p/Tad3p provides strong support for this idea (39). As mentioned above, these enzymes act as a heterodimer to deaminate A34 at the wobble position of certain tRNAs. These enzymes are adenosine deaminases and produce inosine, and they do not deaminate cytidine or cytosine. However, they show clear sequence similarity to the CDA family, particularly in the regions containing amino acids known to be important for CDA catalysis. Figure 5 illustrates these sequence relationships and, by analogy to E. coli cytidine deaminase, shows how the critical residues of ADARs and ADATs would function in catalysis. Although the roles of these amino acids in catalysis have not been proven, mutations at the highlighted amino acids inhibit ADAR activity (101, 105).
Do ADARs and ADATs Act as a Dimer?
E. coli cytidine deaminase acts as a dimer, and its crystal structure in complex with a transition state inhibitor shows that both monomers make contributions to the active site (112). The fact that Tad2p and Tad3p act as a heterodimer to deaminate tRNA, again suggests that the ADAR/ADAT family may be more closely related to the CDA family than the ADA family.
A recent pre–steady state kinetic analysis of ADAR2a reacting at the R/G site of gluR-B suggests that the active form of this enzyme is an RNA-dependent dimer (D. C. J. Jaikaran, C. H. Collins, A. M. MacMillan, unpublished data). Kinetic, gel shift, and cross-linking experiments are consistent with a reaction scheme in which the rate determining step is binding of a second enzyme (E) to an already assembled ADAR · RNA complex (ES) to produce the ESE complex. The scheme is attractive in that it accounts for the substrate inhibition observed with ADARs (117). That is, an increase in substrate over enzyme would lead to an accumulation of the ES complex and preclude formation of the ESE complex. As mentioned, a previous foot-printing analysis using the same enzyme and substrate indicates that the initial binding event covers the target adenosine at the R/G editing site (102). As the authors of the kinetic analysis suggest, it seems likely that the ES complex correlates with this initial binding event, and that the subsequent binding event serves to activate the complex by promoting a conformational change, or possibly by providing a portion of the active site.
Are ADARs Base-Flipping Enzymes?
As pointed out above, dsRNA exists in an A-form helix, which has a very deep and narrow major groove. The amino group of adenine that must be removed during the ADAR reaction lies deep within the major groove, and it is hard to imagine how an enzyme active site could access this functional group. In analogy to some DNA modifying enzymes [reviewed in (118)], it has been proposed that ADARs use a base-flipping mechanism in which the target nucleotide base is extracted from the helix into the enzyme active site (83). Many of the DNA modifying enzymes that utilize this mechanism are methyltransferases, and intriguingly, the ADAR catalytic domain shows distant sequence similarities to conserved motifs in the DNA-(adenine-N6Aα) aminomethyltransferases [(16); reviewed in (25)]. Although not yet proven, a recent study provides experimental support for the idea that ADARs use a base-flipping mechanism (119).
PERSPECTIVES AND QUESTIONS FOR THE FUTURE
The Relationship of ADARs to Other Processes that Involve dsRNA
Very little is known about how ADARs interface with other biological pathways. Amazingly, not a single two-hybrid analysis has been reported for an ADAR, and the only proteins so far known to interact with ADARs are those that mediate nuclear export of ADAR1 (37). Certainly this area will be a focus of future studies. Because ADARs and the RNA splicing machinery sometimes act at the same sequences of pre-mRNAs, some interacting partners may turn out to be splicing factors.
As discussed above, dsRBPs, including ADARs, bind to dsRNA without regard to its specific sequence. This means that the dsRNA substrates of one dsRBP will also be substrates for other dsRBPs. Hopefully future studies will reveal how RNA editing by ADARs intersects with other processes that involve dsRNA, such as RNA interference (120).
What is the Primary Role of ADARs In Vivo?
Gaining insight into how a biological macromolecule has evolved often leads to an enhanced understanding of its function in modern cells. The amino acid sequence similarity between the ADAR catalytic domain and the ADATs suggests ADARs evolved from the tRNA-modifying enzymes. As shown in Figure 6, in this scenario ADARs would arise when an ADAT acquired a dsRBM. Of course, this raises the question of why dsRBMs existed. If their function at the time they fused with the deaminase domain was to bind dsRNA, what was the primordial role of dsRNA?
By necessity, essential functions evolve from nonessential ones. Although I envision the primordial function of ADARs as a nonessential one, it is easy to imagine how ADARs might become essential, as they are in current day mammals. As shown in the far right panel of Figure 6, if a mutation that normally is lethal is “repaired” by an ADAR, the mutation will become locked into the genome, and ADARs will thereafter be essential. This scenario fits the essential Q/R site of mammalian gluR-B quite well. In addition, because mice that lack the unedited sequence appear completely normal, gluR-B Q/R site editing may have arisen quite recently. Further evolution would be predicted to generate regulatory pathways that make use of both unedited and edited sequences, as occurs for most other known editing sites.
Although certainly the role of ADARs in diversifying codons is immensely important, particularly in neurotransmission, this may not be the primary role of ADARs. No doubt, if we knew the primordial role of ADARs, and its purpose in the middle panel of Figure 6, we would have insight into its other roles in modern organisms. We would probably also have insight into the role of the remarkable structures found in UTRs and other noncoding sequences (45; D. P. Morse et al., unpublished data), as well as the roles of the inosines in these structures. As illustrated in Figure 6, possibly the primordial function of ADARs was to modulate the function of dsRNA by modulating its structure. Obviously in this capacity ADARs could function in many different pathways. Understanding these other roles is what I await in future studies.
LITERATURE CITED
- 1.Bass BL, editor. RNA Editing. Oxford: Oxford Univ. Press; 2001. p. 210. [Google Scholar]
- 2.Gott JM, Emeson RB. Annu Rev Genet. 2000;34:499–531. doi: 10.1146/annurev.genet.34.1.499. [DOI] [PubMed] [Google Scholar]
- 3.Benne R, Van den Burg J, Brakenhoff JP, Sloof P, Van Boom JH, et al. Cell. 1986;46:819–26. doi: 10.1016/0092-8674(86)90063-2. [DOI] [PubMed] [Google Scholar]
- 4.Powell LM, Wallis SC, Pease RJ, Edwards YH, Knott TJ, et al. Cell. 1987;50:831–40. doi: 10.1016/0092-8674(87)90510-1. [DOI] [PubMed] [Google Scholar]
- 5.Gerber AP, Keller W. Trends Biochem Sci. 2001;26:376–84. doi: 10.1016/s0968-0004(01)01827-8. [DOI] [PubMed] [Google Scholar]
- 6.Bass BL, Nishikura K, Keller W, Seeburg PH, Emeson RB, et al. RNA. 1997;3:947–49. [PMC free article] [PubMed] [Google Scholar]
- 7.Rebagliati MR, Melton DA. Cell. 1987;48:599–605. doi: 10.1016/0092-8674(87)90238-8. [DOI] [PubMed] [Google Scholar]
- 8.Bass BL, Weintraub H. Cell. 1987;48:607–13. doi: 10.1016/0092-8674(87)90239-x. [DOI] [PubMed] [Google Scholar]
- 9.Bass BL, Weintraub H. Cell. 1988;55:1089–98. doi: 10.1016/0092-8674(88)90253-x. [DOI] [PubMed] [Google Scholar]
- 10.Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. Proc Natl Acad Sci USA. 1994;91:11457–61. doi: 10.1073/pnas.91.24.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Connell MA, Krause S, Higuchi M, Hsuan JJ, Totty NF, et al. Mol Cell Biol. 1995;15:1389–97. doi: 10.1128/mcb.15.3.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai F, Chen CX, Carter KC, Nishikura K. Mol Cell Biol. 1997;17:2413–24. doi: 10.1128/mcb.17.5.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, et al. Nature. 1996;379:460–64. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- 14.Gerber A, O’Connell MA, Keller W. RNA. 1997;3:453–63. [PMC free article] [PubMed] [Google Scholar]
- 15.Herbert A, Lowenhaupt K, Spitzner J, Rich A. Proc Natl Acad Sci USA. 1995;92:7550–54. doi: 10.1073/pnas.92.16.7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hough RF, Bass BL. RNA. 1997;3:356–70. [PMC free article] [PubMed] [Google Scholar]
- 17.Slavov D, Crnogorac-Jurcevic T, Clark M, Gardiner K. Gene. 2000;250:53–60. doi: 10.1016/s0378-1119(00)00175-x. [DOI] [PubMed] [Google Scholar]
- 18.Slavov D, Clark M, Gardiner K. Gene. 2000;250:41–51. doi: 10.1016/s0378-1119(00)00174-8. [DOI] [PubMed] [Google Scholar]
- 19.Palladino MJ, Keegan LP, O’Connell MA, Reenan RA. RNA. 2000;6:1004–18. doi: 10.1017/s1355838200000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deleted in proof
- 21.Basilio C, Wahba AJ, Lengyel P, Speyer JF, Ochoa S. Proc Natl Acad Sci USA. 1962;48:613–16. doi: 10.1073/pnas.48.4.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fierro-Monti I, Mathews MB. Trends Biochem Sci. 2000;25:241–46. doi: 10.1016/s0968-0004(00)01580-2. [DOI] [PubMed] [Google Scholar]
- 23.George CX, Samuel CE. Proc Natl Acad Sci USA. 1999;96:4621–26. doi: 10.1073/pnas.96.8.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, George CX, Patterson JB, Samuel CE. J Biol Chem. 1997;272:4419–28. doi: 10.1074/jbc.272.7.4419. [DOI] [PubMed] [Google Scholar]
- 25.Hough RF, Bass BL. 2001. pp. 77–108. See Ref. 1. [Google Scholar]
- 26.Chen CX, Cho DS, Wang Q, Lai F, Carter KC, et al. RNA. 2000;6:755–67. doi: 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, et al. J Biol Chem. 1996;271:31795–98. doi: 10.1074/jbc.271.50.31795. [DOI] [PubMed] [Google Scholar]
- 28.O’Connell MA, Keller W. Proc Natl Acad Sci USA. 1994;91:10596–600. doi: 10.1073/pnas.91.22.10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim U, Garner TL, Sanford T, Speicher D, Murray JM, et al. J Biol Chem. 1994;269:13480–89. [PubMed] [Google Scholar]
- 30.Liu Y, Samuel CE. J Biol Chem. 1999;274:5070–77. doi: 10.1074/jbc.274.8.5070. [DOI] [PubMed] [Google Scholar]
- 31.Aruscavage PJ, Bass BL. RNA. 2000;6:257–69. doi: 10.1017/s1355838200991921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lehmann KA, Bass BL. Biochemistry. 2000;39:12875–84. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 33.Polson AG, Bass BL, Casey JL. Nature. 1996;380:454–56. doi: 10.1038/380454a0. [DOI] [PubMed] [Google Scholar]
- 34.Herbert A, Alfken J, Kim YG, Mian IS, Nishikura K, et al. Proc Natl Acad Sci USA. 1997;94:8421–26. doi: 10.1073/pnas.94.16.8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herbert A, Rich A. Proc Natl Acad Sci USA. 2001;98:12132–37. doi: 10.1073/pnas.211419898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eckmann CR, Neunteufl A, Pfaffstetter L, Jantsch MF. Mol Biol Cell. 2001;12:1911–24. doi: 10.1091/mbc.12.7.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poulsen H, Nilsson J, Damgaard CK, Egebjerg J, Kjems J. Mol Cell Biol. 2001;21:7862–71. doi: 10.1128/MCB.21.22.7862-7871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keller W, Wolf J, Gerber A. FEBS Lett. 1999;452:71–76. doi: 10.1016/s0014-5793(99)00590-6. [DOI] [PubMed] [Google Scholar]
- 39.Gerber AP, Keller W. Science. 1999;286:1146–49. doi: 10.1126/science.286.5442.1146. [DOI] [PubMed] [Google Scholar]
- 40.Gerber A, Grosjean H, Melcher T, Keller W. EMBO J. 1998;17:4780–89. doi: 10.1093/emboj/17.16.4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maas S, Gerber AP, Rich A. Proc Natl Acad Sci USA. 1999;96:8895–900. doi: 10.1073/pnas.96.16.8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keegan LP, Gerber AP, Brindle J, Leemans R, Gallo A, et al. Mol Cell Biol. 2000;20:825–33. doi: 10.1128/mcb.20.3.825-833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paul MS, Bass BL. EMBO J. 1998;17:1120–27. doi: 10.1093/emboj/17.4.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deleted in proof
- 45.Morse DP, Bass BL. Proc Natl Acad Sci USA. 1999;96:6048–53. doi: 10.1073/pnas.96.11.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Z, Carmichael GG. Cell. 2001;106:465–75. doi: 10.1016/s0092-8674(01)00466-4. [DOI] [PubMed] [Google Scholar]
- 47.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, et al. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 48.Taylor JM. Adv Virus Res. 1999;54:45–60. doi: 10.1016/s0065-3527(08)60365-6. [DOI] [PubMed] [Google Scholar]
- 49.Niswender CM, Copeland SC, Herrick-Davis K, Emeson RB, Sanders-Bush E. J Biol Chem. 1999;274:9472–78. doi: 10.1074/jbc.274.14.9472. [DOI] [PubMed] [Google Scholar]
- 50.Palladino MJ, Keegan LP, O’Connell MA, Reenan RA. Cell. 2000;102:437–49. doi: 10.1016/s0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- 51.Wang Q, Khillan J, Gadue P, Nishikura K. Science. 2000;290:1765–68. doi: 10.1126/science.290.5497.1765. [DOI] [PubMed] [Google Scholar]
- 52.Seeburg PH, Higuchi M, Sprengel R. Brain Res Brain Res Rev. 1998;26:217–29. doi: 10.1016/s0165-0173(97)00062-3. [DOI] [PubMed] [Google Scholar]
- 53.Lomeli H, Mosbacher J, Melcher T, Hoger T, Geiger JR, et al. Science. 1994;266:1709–13. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- 54.Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, et al. Cell. 1993;75:1361–70. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- 55.Herb A, Higuchi M, Sprengel R, Seeburg PH. Proc Natl Acad Sci USA. 1996;93:1875–80. doi: 10.1073/pnas.93.5.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sommer B, Kohler M, Sprengel R, Seeburg PH. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-j. [DOI] [PubMed] [Google Scholar]
- 57.Seeburg PH, Single F, Kuner T, Higuchi M, Sprengel R. Brain Res. 2001;907:233–43. doi: 10.1016/s0006-8993(01)02445-3. [DOI] [PubMed] [Google Scholar]
- 58.Egebjerg J, Heinemann SF. Proc Natl Acad Sci USA. 1993;90:755–59. doi: 10.1073/pnas.90.2.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kohler M, Burnashev N, Sakmann B, Seeburg PH. Neuron. 1993;10:491–500. doi: 10.1016/0896-6273(93)90336-p. [DOI] [PubMed] [Google Scholar]
- 60.Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, et al. Science. 1995;270:1677–80. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 61.Bernard A, Khrestchatisky M. J Neurochem. 1994;62:2057–60. doi: 10.1046/j.1471-4159.1994.62052057.x. [DOI] [PubMed] [Google Scholar]
- 62.Vissel B, Royle GA, Christie BR, Schiffer HH, Ghetti A, et al. Neuron. 2001;29:217–27. doi: 10.1016/s0896-6273(01)00192-1. [DOI] [PubMed] [Google Scholar]
- 63.Bernard A, Ferhat L, Dessi F, Charton G, Represa A, et al. Eur J Neurosci. 1999;11:604–16. doi: 10.1046/j.1460-9568.1999.00479.x. [DOI] [PubMed] [Google Scholar]
- 64.Grigorenko EV, Bell WL, Glazier S, Pons T, Deadwyler S. NeuroReport. 1998;9:2219–24. doi: 10.1097/00001756-199807130-00013. [DOI] [PubMed] [Google Scholar]
- 65.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, et al. Nature. 1997;387:303–8. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 66.Niswender CM, Sanders-Bush E, Emeson RB. Ann NY Acad Sci. 1998;861:38–48. doi: 10.1111/j.1749-6632.1998.tb10171.x. [DOI] [PubMed] [Google Scholar]
- 67.Niswender CM, Herrick-Davis K, Dilley GE, Meltzer HY, Overholser JC, et al. Neuropsychopharmacology. 2001;24:478–91. doi: 10.1016/S0893-133X(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 68.Sodhi MS, Burnet PW, Makoff AJ, Kerwin RW, Harrison PJ. Mol Psychiatry. 2001;6:373–79. doi: 10.1038/sj.mp.4000920. [DOI] [PubMed] [Google Scholar]
- 69.Reenan RA, Hanrahan CJ, Barry G. Neuron. 2000;25:139–49. doi: 10.1016/s0896-6273(00)80878-8. [DOI] [PubMed] [Google Scholar]
- 70.Hanrahan CJ, Palladino MJ, Ganetzky B, Reenan RA. Genetics. 2000;155:1149–60. doi: 10.1093/genetics/155.3.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Semenov EP, Pak WL. J Neurochem. 1999;72:66–72. doi: 10.1046/j.1471-4159.1999.0720066.x. [DOI] [PubMed] [Google Scholar]
- 72.Smith LA, Peixoto AA, Hall JC. J Neurogenet. 1998;12:227–40. doi: 10.3109/01677069809108560. [DOI] [PubMed] [Google Scholar]
- 73.Patton DE, Silva T, Bezanilla F. Neuron. 1997;19:711–22. doi: 10.1016/s0896-6273(00)80383-9. [DOI] [PubMed] [Google Scholar]
- 74.Rueter SM, Dawson TR, Emeson RB. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- 75.Cattaneo R. Curr Opin Genet Dev. 1994;4:895–900. doi: 10.1016/0959-437x(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 76.Bass BL. Trends Biochem Sci. 1997;22:157–62. doi: 10.1016/s0968-0004(97)01035-9. [DOI] [PubMed] [Google Scholar]
- 77.Murphy DG, Dimock K, Kang CY. Virology. 1991;181:760–63. doi: 10.1016/0042-6822(91)90913-v. [DOI] [PubMed] [Google Scholar]
- 78.O’Hara PJ, Horodyski FM, Nichol ST, Holland JJ. J Virol. 1984;49:793–98. doi: 10.1128/jvi.49.3.793-798.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hajjar AM, Linial ML. J Virol. 1995;69:5878–82. doi: 10.1128/jvi.69.9.5878-5882.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kumar M, Carmichael GG. Proc Natl Acad Sci USA. 1997;94:3542–47. doi: 10.1073/pnas.94.8.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bass BL, Weintraub H, Cattaneo R, Billeter MA. Cell. 1989;56:331. doi: 10.1016/0092-8674(89)90234-1. [DOI] [PubMed] [Google Scholar]
- 82.Felder MP, Laugier D, Yatsula B, Dezelee P, Calothy G, et al. J Virol. 1994;68:4759–67. doi: 10.1128/jvi.68.8.4759-4767.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Polson AG, Bass BL. EMBO J. 1994;13:5701–11. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scadden AD, Smith CW. EMBO J. 2001;20:4243–52. doi: 10.1093/emboj/20.15.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cattaneo R, Schmid A, Eschle D, Baczko K, ter Meulen V, et al. Cell. 1988;55:255–65. doi: 10.1016/0092-8674(88)90048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Billeter MA, Cattaneo R, Spielhofer P, Kaelin K, Huber M, et al. Ann NY Acad Sci. 1994;724:367–77. doi: 10.1111/j.1749-6632.1994.tb38934.x. [DOI] [PubMed] [Google Scholar]
- 87.Horikami SM, Moyer SA. Virus Res. 1995;36:87–96. doi: 10.1016/0168-1702(94)00103-j. [DOI] [PubMed] [Google Scholar]
- 88.Kos A, Dijkema R, Arnberg AC, van der Meide PH, Schellekens H. Nature. 1986;323:558–60. doi: 10.1038/323558a0. [DOI] [PubMed] [Google Scholar]
- 89.Casey JL, Gerin JL. J Virol. 1995;69:7593–600. doi: 10.1128/jvi.69.12.7593-7600.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Luo GX, Chao M, Hsieh SY, Sureau C, Nishikura K, et al. J Virol. 1990;64:1021–27. doi: 10.1128/jvi.64.3.1021-1027.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kuo MYP, Chao M, Taylor J. J Virol. 1989;63:1945–50. doi: 10.1128/jvi.63.5.1945-1950.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chang FL, Chen PJ, Tu SJ, Wang CJ, Chen DS. Proc Natl Acad Sci USA. 1991;88:8490–94. doi: 10.1073/pnas.88.19.8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sato S, Wong SK, Lazinski DW. J Virol. 2001;75:8547–55. doi: 10.1128/JVI.75.18.8547-8555.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saccomanno L, Bass BL. RNA. 1999;5:39–48. doi: 10.1017/s1355838299981335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wagner RW, Smith JE, Cooperman BS, Nishikura K. Proc Natl Acad Sci USA. 1989;86:2647–51. doi: 10.1073/pnas.86.8.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nishikura K, Yoo C, Kim U, Murray JM, Estes PA, et al. EMBO J. 1991;10:3523–32. doi: 10.1002/j.1460-2075.1991.tb04916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bycroft M, Grunert S, Murzin AG, Proctor M, St Johnston D. EMBO J. 1995;14:3563–71. doi: 10.1002/j.1460-2075.1995.tb07362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kharrat A, Macias MJ, Gibson TJ, Nilges M, Pastore A. EMBO J. 1995;14:3572–84. doi: 10.1002/j.1460-2075.1995.tb07363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. EMBO J. 1998;17:5458–65. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ryter JM, Schultz SC. EMBO J. 1998;17:7505–13. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lai F, Drakas R, Nishikura K. J Biol Chem. 1995;270:17098–105. doi: 10.1074/jbc.270.29.17098. [DOI] [PubMed] [Google Scholar]
- 102.Öhman M, Källman AM, Bass BL. RNA. 2000;6:687–97. doi: 10.1017/s1355838200000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stephens OM, Yi-Brunozzi HY, Beal PA. Biochemistry. 2000;39:12243–51. doi: 10.1021/bi0011577. [DOI] [PubMed] [Google Scholar]
- 104.Yang JH, Sklar P, Axel R, Maniatis T. Nature. 1995;374:77–81. doi: 10.1038/374077a0. [DOI] [PubMed] [Google Scholar]
- 105.Maas S, Melcher T, Herb A, Seeburg PH, Keller W, et al. J Biol Chem. 1996;271:12221–26. doi: 10.1074/jbc.271.21.12221. [DOI] [PubMed] [Google Scholar]
- 106.Wong SK, Sato S, Lazinski DW. RNA. 2001;7:846–58. doi: 10.1017/s135583820101007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Polson AG, Ley HL, 3rd, Bass BL, Casey JL. Mol Cell Biol. 1998;18:1919–26. doi: 10.1128/mcb.18.4.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lehmann KA, Bass BL. J Mol Biol. 1999;291:1–13. doi: 10.1006/jmbi.1999.2914. [DOI] [PubMed] [Google Scholar]
- 109.Chanfreau G, Buckle M, Jacquier A. Proc Natl Acad Sci USA. 2000;97:3142–47. doi: 10.1073/pnas.070043997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ramos A, Grunert S, Adams J, Micklem DR, Proctor MR, et al. EMBO J. 2000;19:997–1009. doi: 10.1093/emboj/19.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110a.Nagel R, Ares M., Jr RNA. 2000;6:1142–56. doi: 10.1017/s1355838200000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Polson AG, Crain PF, Pomerantz SC, McCloskey JA, Bass BL. Biochemistry. 1991;30:11507–14. doi: 10.1021/bi00113a004. [DOI] [PubMed] [Google Scholar]
- 111a.Wilson DK, Rudolph FB, Quiocho FA. Science. 1991;252:1278–84. doi: 10.1126/science.1925539. [DOI] [PubMed] [Google Scholar]
- 112.Betts L, Xiang S, Short SA, Wolfenden R, Carter CW., Jr J Mol Biol. 1994;235:635–56. doi: 10.1006/jmbi.1994.1018. [DOI] [PubMed] [Google Scholar]
- 113.Carter CW., Jr . In: Modification and Editing of RNA. Grosjean H, Benne R, editors. Washington, DC: ASM Press; 1998. pp. 363–75. [Google Scholar]
- 114.Carter CW., Jr Biochimie. 1995;77:92–98. doi: 10.1016/0300-9084(96)88110-7. [DOI] [PubMed] [Google Scholar]
- 115.Ireton GC, McDermott G, Black ME, Stoddard BL. J Mol Biol. 2002;315:687–97. doi: 10.1006/jmbi.2001.5277. [DOI] [PubMed] [Google Scholar]
- 116.Cooper BF, Sideraki V, Wilson DK, Dominguez DY, Clark SW, et al. Protein Sci. 1997;6:1031–37. doi: 10.1002/pro.5560060509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hough RF, Bass BL. J Biol Chem. 1994;269:9933–39. [PubMed] [Google Scholar]
- 118.Roberts RJ, Cheng X. Annu Rev Biochem. 1998;67:181–98. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- 119.Yi-Brunozzi HY, Stephens OM, Beal PA. J Biol Chem. 2001;276:37827–33. doi: 10.1074/jbc.M106299200. [DOI] [PubMed] [Google Scholar]
- 120.Bass BL. Cell. 2000;101:235–38. doi: 10.1016/s0092-8674(02)71133-1. [DOI] [PubMed] [Google Scholar]
- 121.Wong TC, Ayata M, Hirano A, Yoshikawa Y, Tsuruoka H, et al. J Virol. 1989;63:5464–68. doi: 10.1128/jvi.63.12.5464-5468.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rueda P, Garcia-Barreno B, Melero JA. Virology. 1994;198:653–62. doi: 10.1006/viro.1994.1077. [DOI] [PubMed] [Google Scholar]
- 123.Petschek JP, Mermer MJ, Scheckelhoff MR, Simone AA, Vaughn JC. J Mol Biol. 1996;259:885–90. doi: 10.1006/jmbi.1996.0365. [DOI] [PubMed] [Google Scholar]