

Abstract
The neuroprotective effects of Δ9-tetrahydrocannabinol (THC) were examined using an in vitro model in which the AF5 CNS cell line was exposed to toxic levels of N-methyl-D-aspartate (NMDA), an agonist of the NMDA glutamate receptor. NMDA toxicity was reduced by THC, but not by the more specific cannabinoid receptor agonist, WIN55,212-2. Addition of dibutyryl cAMP (dbcAMP) to the culture medium did not alter the neuroprotective effect of THC and did not unmask a neuroprotective effect of WIN55,212-2. The cannabinoid antagonist SR141716A did not inhibit the neuroprotection induced by THC or alter the response to WIN55,212-2, even in the presence of dbcAMP, indicating that the neuroprotective effect of THC was cannabinoid receptor-independent. On the other hand, both THC and WIN55,212-2 produced cellular toxicology at higher dosages, an effect which was blocked in part by SR141716A. Capsaicin, an antioxidant and vanilloid receptor agonist, also produced a protective effect against NMDA toxicology. The protective effect of capsaicin was blocked by co-application of ruthenium red, but was not blocked by the specific vanilloid receptor antagonist capsazepine, and the transient receptor potential vanilloid type 1 (TRPV1) and ANKTM1 transcripts were not detected in AF5 cells. Thus, the neuroprotective effects of THC and capsaicin did not appear to be mediated by TRP ion channel family receptors. The antioxidant α-tocopherol prevented neurotoxicity in dose-dependent manner. Therefore, THC may function as an antioxidant to increase cell survival in NMDA-induced neurotoxicity in the AF5 cell model, while higher dosages produce toxicity mediated by CB1 receptor stimulation.
Keywords: Cannabinoids, NMDA, Excitotoxicity, Neuroprotection, Antioxidant
1. Introduction
Cannabinoids, such as the pharmacologically active component of marijuana (-)Δ9-tetrahydrocannabinol (THC), and the endogenous cannabinoid anandamide, exert a variety of biological actions which are mediated by activation of cannabinoid (CB) receptors [1,2], receptors of the transient receptor potential (TRP) family of ion channels [3,4], and antioxidant effects which are not mediated by receptors [5–7].
One of the most promising potential medical applications of cannabinoids involves their ability to protect cells from a variety of toxic events [8]. Cannabinoids have been reported to protect neurons from death caused by glutamatergic overstimulation, ischemia and oxidative damage [9]. The mechanisms of these cellular protectant effects are not, however, entirely clear. On one hand, there are several studies which suggest that for cell death induced by glutamate, hydrogen peroxide, retinoids, and serum deprivation, the protectant actions of THC are mediated primarily or exclusively by chemical antioxidant effects [5,7,10]. For example, Hampson et al. [5] reported that cell death in neuronal cultures induced by stimulation of the AMPA/kainate glutamate receptor was similarly inhibited by THC and by cannabidiol, which does not stimulate CB1 receptors, and that the CB1 antagonist SR141716A did not inhibit the protective effect of either THC or cannabidiol. Marsicano et al. [10] found that alteration of CB1 receptor expression using knockout animals, or gene transfer in the HT22 cell line, did not influence the protectant effect of THC against hydrogen peroxide toxicity in vitro.
On the other hand, THC and the synthetic cannabinoid WIN55,212-2, which does not have an antioxidant effect [5,7,10] prevent glutamate- and NMDA-induced neurotoxicity in isolated neurons [11] as well as in the brain [12] via activation of the cannabinoid CB1 receptor [13,14]. Moreover, El-Remessy et al. [15] investigated the neuroprotective effect of THC in a model of NMDA-induced retinal toxicity. In this model, the effect of THC was partially, although not completely, blocked by the CB1 antagonist SR141716A. Therefore, it appears that in some circumstances THC exerts neuroprotective effects exclusively through non-receptor mediated antioxidant properties, while in other models or other cell types, stimulation of CB1 receptors by THC or WIN55212-2 can produce neuroprotection. Differences between effects observed in different models may be related to the cell type or model system, and to differences in the toxic events which have been employed.
The purpose of the present study was to further explore the mechanisms of neuroprotection induced by THC, using a neural progenitor cell line model [16,17]. The AF5 cell line retains its plasticity in culture and possesses some of the characteristics of primary mesencephalic neural progenitors [18]. In the present study, the AF5 cell line was examined for expression of NMDA and CB1 receptors and susceptibility to toxicity mediated by NMDA. NMDA was employed as a toxic agent since most of the studies which have found CB1 receptor-mediated neuroprotection, mentioned above, have employed NMDA-induced toxicity. This model was then employed to assess the neuroprotectant properties of THC and the antioxidants α-tocopherol and capsaicin.
2. Materials and Methods
2.1. Chemicals and Materials
N-methyl-D-aspartate (NMDA), N-methyl-L-aspartate (NMLA), Δ9-THC, Hoechst 33342, Ruthenium red, and α-tocopherol were obtained from Sigma (St. Louis, MO). (R)-(+) WIN55,212-2 mesylate salt, (+)-MK-801, Capsazepine, and (E)-Capsaicin were from Tocris Cookson (Ellisville, MO). SR141716A was obtained from the National Institute on Drug Abuse drug supply system. Dibutyryl cAMP (dbcAMP) was from Calbiochem (San Diego, CA). The MTT cell proliferation assay kit was purchased from ATCC (Manassas, VA). RNA STAT-60 was purchased from TEL-TEST (Friendswood, TX). The reverse transcription reagents were purchased from Promega (Madison, Wisconsin). The ECL chemiluminescence kit was purchased from Amersham Biosciences (Piscataway, NJ). The NMDAR1 antibody was obtained from BD Pharmingen (San Diego, CA), ssDNA antibody (MAb F7-26) from Chemicon (Temecula, CA), and the CB1 receptor antibody was obtained from Calbiochem (San Diego, CA). The fluorescein-conjugated anti-mouse IgG antibody (Alexa Fluor 594) was from Molecular Probes (Eugene, OR). The fluorescein-conjugated anti-mouse IgM was from Jackson Immunoresearch (West Grove, PA). TUNEL staining kits were from Roche Applied Science (Indianapolis, IN).
2.2. Cell cultures
The AF5 rat mesencephalic cell line, which was immortalized using a fragment of the SV40 large T antigen gene known as T155g, has been described previously [16,18]. Cultures were maintained in a 5% CO2 incubator, generally in medium consisting of Dulbecco’s Modified Eagle’s Medium/Ham’s F12 (DMEM/F12, 1:1, Gibco Life Technologies, Gaithersburg, MD), 10% fetal calf serum (FSC), 2 mM L-glutamine, 100U/ml penicillin G, and 100μg/ml streptomycin. Cells were counted and 5x104 cells per well were seeded into the wells of 96 multiwell plates for the cell proliferation assay, and 4x104 cells per well were seeded into 24 well plates for other experiments. The medium was changed to DMEM/F12 without serum on the next day.
2.3. NMDA-mediated toxicity and drug treatment procedures
Cell cultures (2 days in vitro) were washed with Mg2+, Ca2+-free Dulbecco’s phosphate buffered saline (PBS) solution once and maintained in varying concentrations (5 mM, 7.5 mM, 8.5 mM and 10 mM) of NMDA in vehicle solution, Hanks' balanced salt solution (HBSS) without calcium and magnesium, containing 5 μM glycine at 37°C for 30 min. The medium was then replaced by DMEM/F12 medium without serum and cultures were returned to the incubator and maintained at 37°C for an additional 16–18 h.
For cannabinoid treatment, the cells were first incubated with cannabinoids: WIN55,212-2, or THC in serum-free DMEM/F12 for 6 h, then the media were replaced with media containing varying concentrations of NMDA in HBSS for 30 min. After exposure, cells were cultured in fresh serum-free DMEM/F12 for 16–18 h. For some experiments, 1 mM dbcAMP was added to the culture medium after NMDA exposure and then the cells were incubated for 16–18h. The CB1 antagonist SR141716A, and the TRPV1 antagonists capsazepine and ruthenium red, were first added to the cultures for 20 min. The medium was then replaced with medium containing both the appropriate antagonist and THC or WIN55,212-2 for 6 h prior to NMDA exposure. For capsaicin and α-tocopherol treatment, the cells were first incubated with capsaicin or α-tocopherol alone for 20 min, and the media were then changed to media containing NMDA.
2.4. Quantitation of toxicity
Cell toxicity was determined by a colorimetric assay using 3-[4, 5-dimethylthiazol-2-yl]-2–5 diphenyltetrazolium bromide (MTT). Briefly, 5x104 AF5 cells were plated in 96-well plates in final volume of 100 μl per well. After stimulation for the period indicated, 10 μl of MTT reagent was added to each well. The plates were returned to the incubator for 2.5 h. Then, 100 μl of detergent reagent was added to each well, and the plates were covered and kept in the dark for 2 h at room temperature (rt). Finally, optical density (OD) was measured at 570 nm. The OD value of the untreated sample was regarded as 100%. Mitochondrial activity results in conversion of MTT to purple formazan crystals which absorb light at 570 nm; thus, loss of cell viability results in decreased OD values.
2.5. ssDNA immunostaining
ssDNA immunostaining was performed according to manufacturer’s directions (ssDNA MAb F7-26, Chemicon) with minor modifications. Adherent AF5 cells were fixed in methanol-phosphate buffered saline (PBS) (6:1) for 24 h at −20°C. After fixation, cells were incubated in 0.25 ml of formamide at 70°C for 5 min. Two ml of 1% non-fat dry milk in PBS was added and the cells were incubated at rt for 15 min. After washing, cells were incubated with 1:10 dilution of mouse anti-ssDNA antibody for 15 min. Single-stranded DNA, which is a specific indicator for apoptotic cell death [19], was visualized by using fluorescein-conjugated anti-mouse IgM (1:200, Jackson Immunoresearch). The plates were imaged and analyzed under a Zeiss Axiovert inverted fluorescence microscope (magnification x 200). Numbers of cells positive for ssDNA and total cells (a minimum of 250 cells/group) were counted in a randomly chosen area of each well. Data shown are the means ± S.E.M. of three independent experiments.
2.6. Hoechst staining
AF5 cells were fixed with 4% paraformaldehyde for 10 min at rt and rinsed with PBS. The fixed cells were incubated for 10 min in the dark with 100 ug/ml Hoechst 33342. Cells were imaged and analyzed under a Zeiss Axiovert inverted fluorescence microscope (magnification x 200) with a UV excitation filter. Between 250 and 800 cells were counted in each group from six areas chosen randomly, and the experiment was repeated three times.
2.7. Terminal deoxynueotidyl transferase (TdT)-mediated dUTP nick-ended labeling (TUNEL)
TUNEL histochemical staining was performed according to manufacturer's directions with minor modifications. Briefly, cultured cells were fixed in 4% paraformaldehyde for 30 min, followed by 0.3% Triton-X 100 permeabilization solution treatment for 2 min. About 150 μl TUNEL reaction mixture was subsequently added to each chamber well to cover the entire bottom. After incubation for 60 min at 37°C, the chambers were removed, and the slides were cover-slipped. For statistical analysis, numbers of positive signals and total cells were counted in a randomly chosen area of each well under a 20X objective lens. About eight areas were counted for each group, and the experiment was repeated three times. Data from the three experiments were collected for statistical analysis.
2.8. Immunocytochemistry
Cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% triton-X100. After blocking with 5% goat serum, the cells were incubated with mouse anti-NMDAR1 antibody (1:200, BD Pharmingen) overnight at 4°C. Localization of antigens was visualized with an Alexa Fluor 594 goat anti-mouse secondary antibody (1:500, Molecular Probes). PBS was used for washing, dilution of antibodies and blocking buffer. Images were collected by using a Zeiss Axiovert inverted fluorescence microscope (magnification x 200) and photographed with a Cooke SensiCam digital camera. No specific immunoreactivity was present in negative controls incubated with mouse IgG instead of the specific primary antibody.
2.9. Western blotting
Cells were harvested and pellets were washed once with ice-cold PBS and resuspended in 0.25 ml of homogenization buffer (0.1% SDS, 1X protease inhibitor in PBS). Suspensions were sonicated for 10 sec. The cell membrane lysates were centrifuged at 10,000g for 15 min at 4°C and the supernatant was collected as the membrane fraction. Membrane fractions containing equal amounts of protein were loaded in each lane and separated on 8% SDS-polyacrylamide gels, and proteins were transferred to nitrocellulose membranes by electroblotting. After blocking with 3.5% non-fat milk and 0.5% BSA in 1x phosphate buffer, the membranes were incubated with primary antibodies against the CB1 receptor (1:500) overnight at 4°C. Subsequently, the membranes were treated with secondary IgG conjugated to horseradish peroxidase (1: 2000) for 1 h at rt. Secondary antibody bound to proteins was detected using the Amersham ECL chemiluminescence kit.
2.10. Reverse transcriptase-polymerase chain reaction (PCR) to detect CB1 receptor and TRP receptor mRNA
Total RNA was extracted from AF5 cells using the RNA STAT-60 kit. cDNA was synthesized by using reverse transcription reagents with 1μg total RNA in a 25 μl reaction according to the manufacturer’s instructions. For each PCR reaction, 1μl of cDNA template was used in 25 μl of reaction volume with the Taq DNA polymerase. The cycling parameters were 95°C 30 sec, 60°C 30 sec, 72°C 30 sec for 35 cycles. The PCR cycle was preceded by initial denaturation of 2 min at 95°C and followed by a final extension of 10 min at 72°C. The PCR primers were as follows: rat CB1 receptor forward 5′- CAT CAT CAT CCA CAC GTC AG-3′; and reverse 5′-ATG CTG TTG TCT AGA GGC TG-3′. Primers for rat TRPV1 receptor were forward 5′-TGA CCC TCT TGG TGG AGA ATG G-3′ and reverse 5′-TAA GCG ATC ACC TCC AGA ACC G-3′. ANKTM1 primers were selected from ANKTM1 sequences in the mouse gene bank. The primers were forward 5′-AGG ATG TGG TAT ATG AGC CTC TTA-3′ and reverse 5′-TTG AGG AAC AAG GGC AAC ACG-3′. The β-actin “housekeeper gene” was used as an internal control. Primer sequences were forward 5′-TTG TAA CCA ACT GGG ACG ATA TGG -3′; and reverse 5′- GAT CTT GAT CTT CAT GGT GCT AGG-3′. Amplification products were electrophoresed on 2% of agarose gel and stained with ethidium bromide. Photographed images of gels were digitized with a scanner.
2.11. Statistical analysis
All cell culture experiments were conducted independently three to six times for each treatment group. Statistical analyses were performed using GraphPad InStat (V 3.05). Data were analyzed by one-way ANOVA and post-hoc Tukey multiple comparison tests. In all cases, differences were considered to be significant when ρ ≤0.05. Data are presented as means ± S.E.M.
3. Results
3.1. NMDA receptor expression and NMDA-induced cell death
AF5 cells were found to express the NMDA receptor using immunocytochemistry, which showed that the NMDA1 receptor protein was present in AF5 cell bodies (Fig. 1A).
Fig. 1.
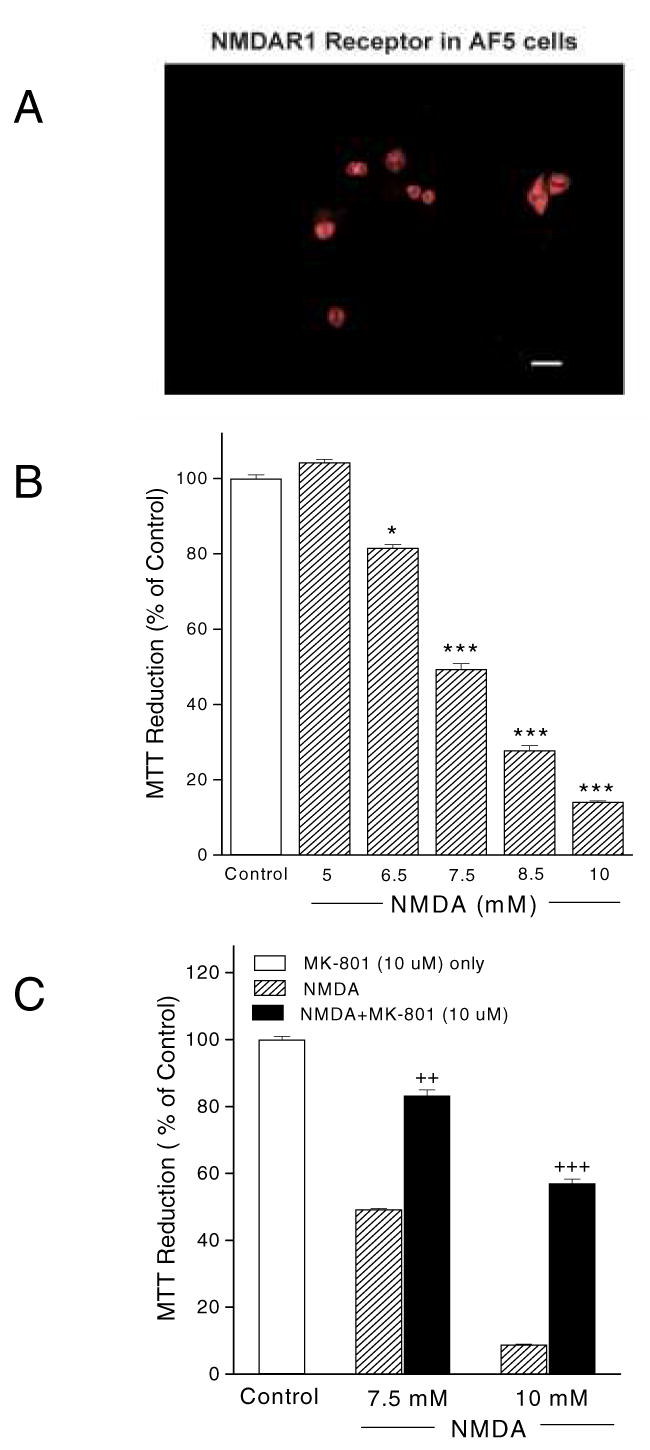
NMDA-mediated AF5 cell death. (A) NMDAR1 receptor staining was present in AF5 cell bodies. Scale bar = 20 μm; (B) Dose-dependent induction of loss of cell viability by NMDA. (C) MK-801 (10 μM) blocked the NMDA-induced loss of cell viability at 7.5 and 10 mM. The total OD value of control group was normalized to 100%. p< 0.05, *** p< 0.001 vs. control. ## p< 0.01 vs. 7.5 mM NMDA. ### p<0.001 vs. 10 mM NMDA.
Exposure of AF5 cells to 5 to 10 mM NMDA reduced cell viability in a dose-dependent manner, as measured by the MTT assay (Fig. 1B). At 7.5 mM NMDA, cell survival was reduced to approximately 40–50% of vehicle-treated cells. Only about 20% of cells survived following exposure to 10 mM NMDA. Co-treatment with 10 μM MK-801, a selective non-competitive NMDA receptor antagonist, substantially blocked the NMDA-induced loss of cell viability (Fig. 1C).
N-methyl-L-aspartate, the inactive stereoisomer of N-methyl-aspartic acid, in 5 to 10 mM concentrations, had no effect on cell viability (data not shown).
3.2. CB1 receptor expression and toxicity of THC and WIN55,212-2
Although the CB1 receptor transcript was previously detected in AF5 cells by microarray analysis [18], expression of the CB1 receptor in this cell line has not been confirmed. We also tested whether exposure to THC or WIN55,212-2 influenced CB1 receptor expression in AF5 cells. CB1 receptor mRNA was present in AF5 cells and cannabinoid exposure at the maximum non-toxic dosages (see below) did not cause apparent changes in expression of the CB1 receptor or β-actin mRNA (Fig. 2A). Western blot analysis revealed that the CB1 receptor protein was detectable in its glycosylated form (60KDa), and there were no apparent changes after exposure to NMDA (not shown), or to THC and WIN55212-2 (Fig. 2B).
Fig. 2.
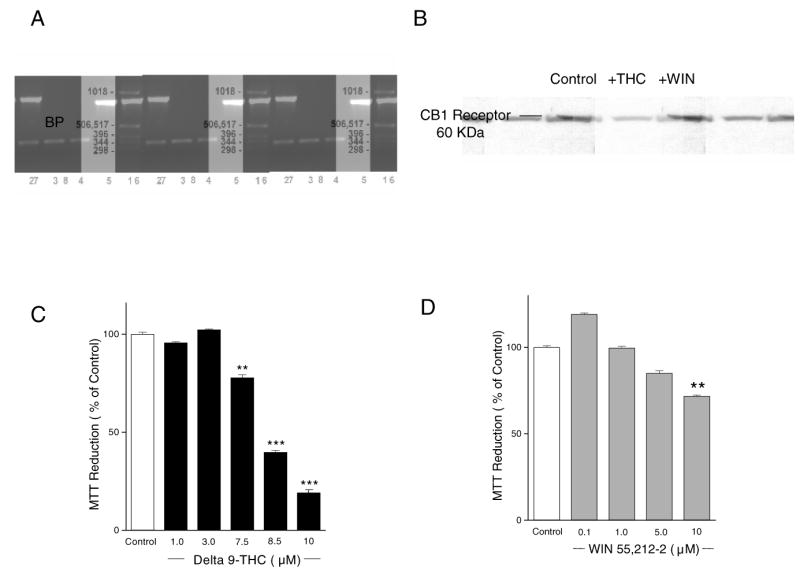
CB1 receptor expression in AF5 cells and toxicity of THC and WIN55,212-2. (A) CB1 receptor gene expression in AF5 cells. Lane 1 is base pair (BP) size, Lanes 2, 3, 4 represent the CB1 receptor transcript in untreated cells, after THC exposure, and after WIN55,212-2 exposure respectively; lanes 5, 6, 7 respectively represent the β-actin housekeeper gene in untreated cells, and after THC exposure and WIN55,212-2 exposure respectively. Lane 8 is a control, containing no cDNA. (B) Western blotting detected a 60-kDa band corresponding to the molecular mass of the CB1 receptor in the untreated cells, and after exposure to THC or WIN55,212-2. (C) Cell viability in AF5 cells treated with varying concentrations of THC. (D) Cell viability after treatment with different concentrations of WIN55,212-2. The total OD value of the control group was normalized to 100%. ** and *** indicate p< 0.01 and p< 0.001, respectively, vs. control.
Cells were incubated with varying concentrations of THC or WIN55,212-2 according to the cannabinoid treatment procedure. Significant toxicity as measured by MTT was produced by THC at doses of 7.5 μM and higher (Fig. 2C) and by WIN55,212-2 at 10 μM (Fig. 2D). THC at 3 μM and WIN55,212-2 at 5 μM were not toxic (Fig. 2C and D) and were therefore used for subsequent experiments on neuroprotective effects.
To determine whether the toxic effects of THC and WIN55,212-2 were CB1 receptor-mediated, we tested whether the specific CB1 receptor antagonist SR141716A could block the toxic effects of THC and WIN55,212-2. SR141716A partially blocked the toxic effect of 8.5 μM THC, but was not effective for the higher concentration of THC (10 μM). When cells were treated with SR141716A prior to incubation with 10 μM WIN55,212-2, the toxic effect of WIN55212-2 was almost completely abolished (Fig. 3).
Fig. 3.
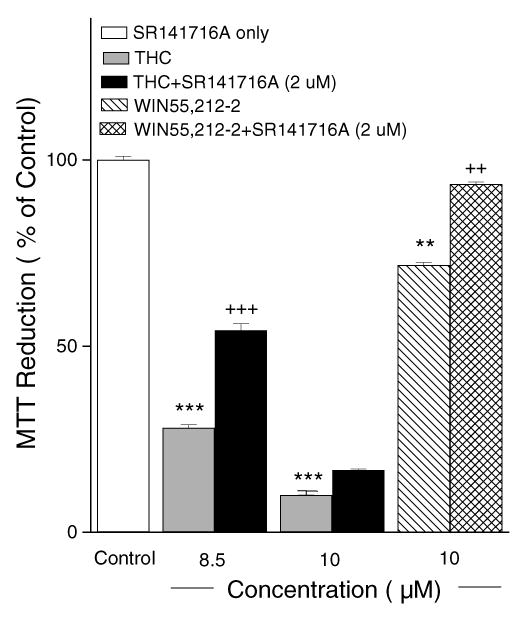
Toxic effects of THC and WIN are mediated by CB1 receptor stimulation. The CB1 receptor antagonist SR 141716A (2μM) partially blocked the toxic effects induced by 8.5μM THC and almost completely abolished the toxic effects induced by 10μM WIN55,212-2.
3.3. TRP receptor expression
Because the transient receptor potential (TRP) ion channel family proteins TRPV1 and ANKTM1 have been reported to mediate some cannabinoid effects, we also tested the AF5 cell line for expression of these receptors. RT-PCR was employed using rat TRPV1 primers and primers for mouse ANKTM1. Expression of the TRPV1 receptor and the ANKTM1 receptor was not detected (not shown).
3.4. NMDA-induced toxicity is prevented by THC but not WIN55,212-2
Figures 4 and 5 represent the overall means (± S.E.M.) of MTT assay data. Figure 4A shows a dose-response curve for neuroprotection induced by THC or WIN55,212-2 against 7.5 mM NMDA measured by MTT assay. THC prevented NMDA toxicity in a dose-dependent manner. WIN55,212-2 had no protective effect, either at lower or higher concentrations. Both cannabinoids produced toxic effects at 10 μM, consistent with the results shown in Fig 2A and B for cells treated with the cannabinoids alone. Neuroprotective effects of the maximum non-toxic doses of THC and WIN55,212-2 were compared for two concentrations of NMDA. As shown in Fig. 4B, the cell loss induced by 7.5 mM NMDA was almost completely prevented, and cell loss caused by 10 mM NMDA was greatly reduced by 3 μM THC. In contrast, WIN55,212-2 did not protect against NMDA-induced toxicity for either dose of NMDA. Addition of dbcAMP after NMDA removal did not increase the neuroprotection induced by THC and not unmask a neuroprotective effect of WIN55,212-2 (data not shown). Figure 5 reveals that THC showed a neuroprotective effect in the presence of the specific cannabinoid receptor antagonist, SR141716A (2μM); however, SR141716A itself did not affect cell survival. Also, SR141716A did not inhibit the neuroprotective effect of THC even in the presence of 1 mM dbcAMP after NMDA exposure (data not shown).
Fig. 4.
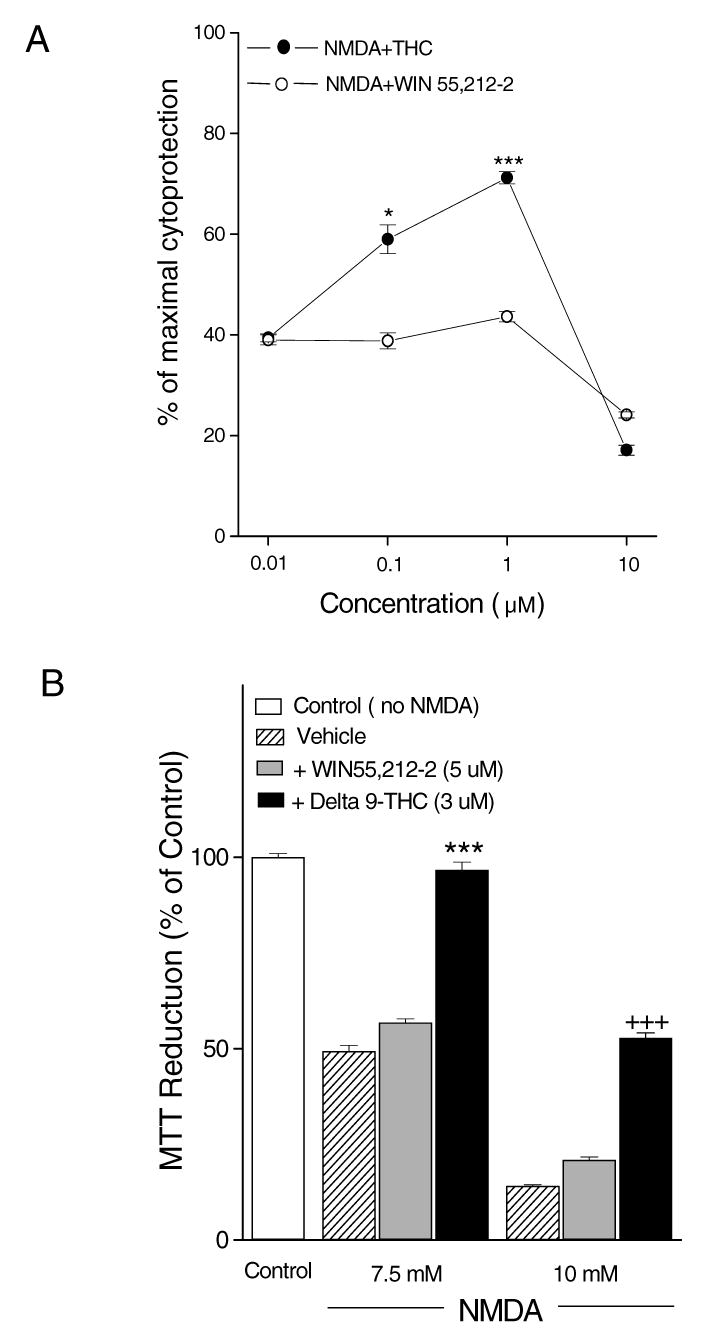
THC protected AF5 cells from NMDA neurotoxcity. (A) Dose-response curve for THC or WIN55,21202 against 7.5 mM NMDA. * indicates p< 0.05 and *** indicates p< 0.001, vs. 7.5 mM NMDA treatment. (B) Cell survival was increased by THC at both 7.5 mM and 10 mM NMDA doses, but not WIN55,212-2. *** indicates p< 0.001 vs. 7.5 mM NMDA. +++ indicates p< 0.001 vs. 10 mM NMDA. Data are presented as means ± S.E.M.
Fig. 5.
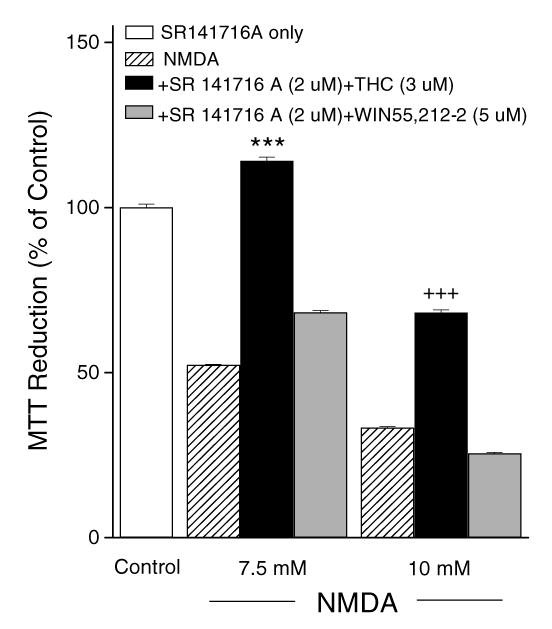
Neuroprotection induced by THC was CB1 receptor-independent. The CB1 receptor antagonist SR 141716A (2 μM) did not abolish neuroprotection by THC from NMDA-induced toxicity. *** and +++ indicate p< 0.001, respectively, as compared to 7.5 mM NMDA or 10 mM NMDA alone. Data are means ± S.E.M.
The efficacy of THC or WIN55,212-2 in preventing apoptosis induced by NMDA was tested using a ssDNA assay and TUNEL. As shown in Fig. 6A, THC (3 μM) attenuated the NMDA-induced increase in cells positive for ssDNA at 7.5 mM and 10 mM concentrations. WIN55,212-2 did not decrease ssDNA staining after exposure to NMDA. Statistical analysis (Fig. 6B) showed that 3 μM THC improved cell survival to about 40% for the 7.5 mM NMDA-treated group. This dose of THC also caused a smaller but significant protection against ssDNA staining induced by 10 mM NMDA. WIN55,212-2 did not have a protective effect.
Fig. 6.
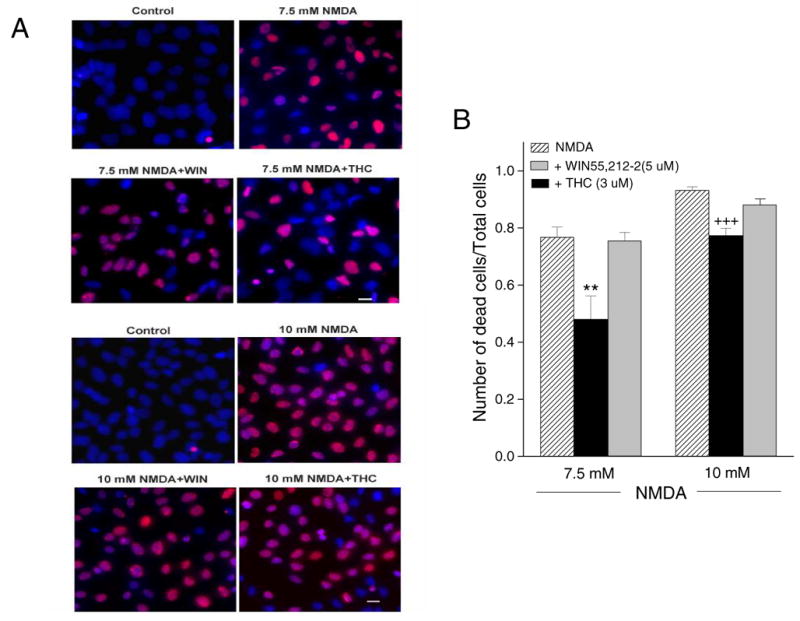
THC attenuated NMDA-induced ssDNA-positive staining. (A) Cells were immunostained for ssDNA (red) and counterstained with Hoechst 33342 (blue). Almost no ssDNA-positive cells were found in untreated cells. NMDA treatment resulted in numerous cells positive for ssDNA. Pretreatment with THC decreased the number of ssDNA positive cells. Scale bar = 20 μM. (B) Statistical analysis was based on at least 250 cells for each group, ** indicates p< 0.01 vs. 7.5 mM NMDA, +++ indicates p< 0.001 vs. 10 mM NMDA.
A typical result from one of three independent TUNEL assays is shown in Fig. 7A. Very few TUNEL-positive cells were observed in control wells while NMDA caused a marked increase in the number of TUNEL-positive cells. As shown, about 61% and 97% of cells were positive for TUNEL after 7.5 mM and 10 mM NMDA treatment, respectively. Protection was seen in the THC-treated groups, particularly for 7.5 mM NMDA. There was no protective effect of WIN55,212-2. The quantitative results are shown in Fig. 7B.
Fig. 7.
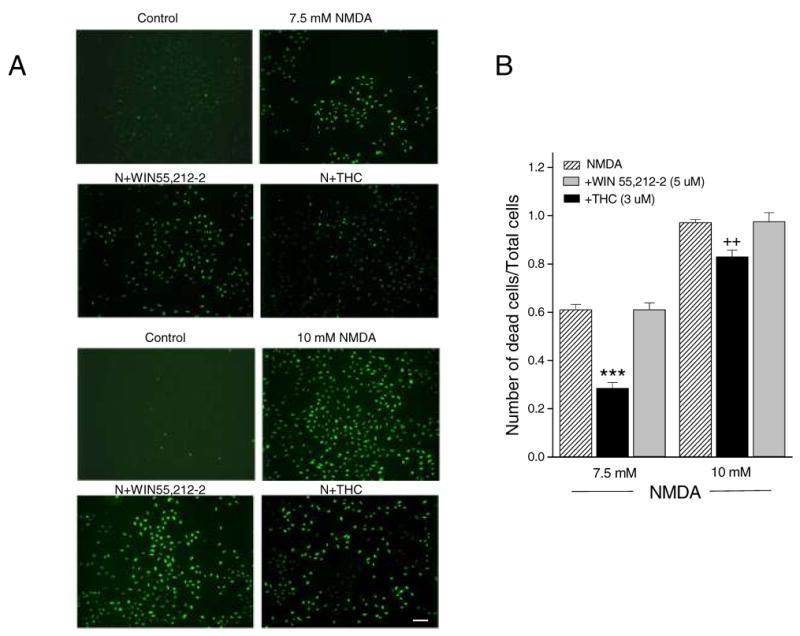
THC attenuated TUNEL staining induced by NMDA. (A) Few TUNEL-positive cells were found in untreated cells. NMDA treatment at both 7.5 and 10 mM concentration caused increases in TUNEL-positive cells. Significant protection from NMDA toxicity was found only for THC-treated cells, particularly for the 7.5 mM NMDA treated group. Scale bar = 100um. (B) Quantitative analysis of cannabinoid-induced protection as measured by TUNEL staining. THC produced a significant neuroprotective effect. *** p< 0.001 and ++ p< 0.01 for THC compared to the respective controls.
3.5. Effects of capsaicin, capsazepine, ruthenium red, and α-tocopherol
The neuroprotective effect of THC was mimicked by capsaicin (1μM – 10 μM) in a dose-dependent manner (Fig. 8A). The competitive TRPV1 antagonist capsazepine (10 μM) did not, however, block the neuroprotective effect of THC for either the 7.5 mM or 10 mM doses of NMDA (Fig. 8B) and it also did not block the protective effect of capsaicin (data not shown). Ruthenium red (1 μM), a channel blocker and inhibitor of vanilloid responses, blocked the protective effect of THC on NMDA-induced excitotoxicity for both the 7.5 mM and 10 mM concentrations (Fig. 8C). Neither capsazepine or ruthenium red altered the response to WIN55,212-2.
Fig. 8.
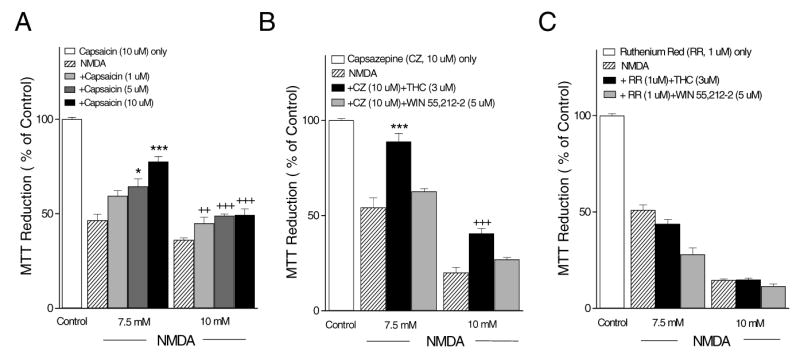
Effects of capsaicin, capsazepine, and ruthenium red. (A) The TRPV1 agonist capsaicin protected against NMDA toxicity in a dose-dependent manner. (B) The TRPV1 antagonist capsazepine failed to block the neuroprotective effect of THC against NMDA toxicity. * indicates p< 0.05 and *** p< 0.001 vs. 7.5 mM NMDA, +++ p< 0.01, +++ 0.001 vs. 10 mM NMDA respectively. (C) Brief incubation of cells with ruthenium red (1uM) prior to NMDA stimulation blocked the protective effect of THC.
Finally, we investigated the effects of the antioxidant α-tocopherol. α-tocopherol alone did not affect cell viability in concentrations of 30–300μM (not shown). α-tocopherol attenuated NMDA toxicity in a concentration-dependent manner, but also produced increased toxicity at high concentrations (1000 μM) (Fig. 9A). Figure 9B shows a comparison of THC, capsaicin and α-tocopherol for prevention of NMDA toxicity.
Fig. 9.
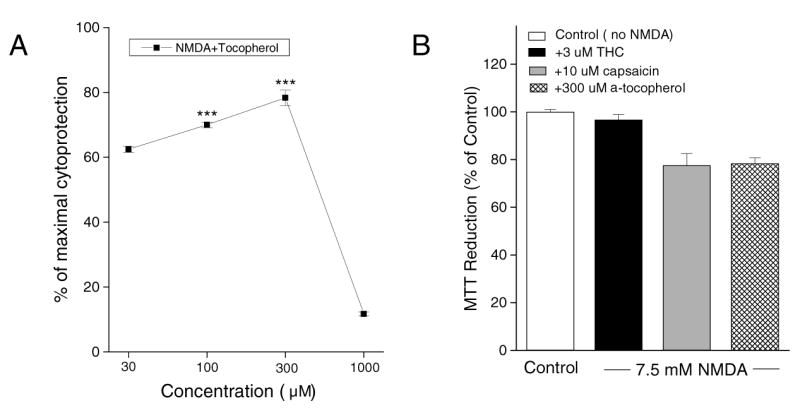
Effects of α-tocopherol and comparison to THC and capsaicin. (A) Protection induced by the antioxidant α-tocopherol against NMDA-induced cell death. Results are expressed as % cell survival. *** indicates p< 0.001 vs. 7.5 mM NMDA. (B) Comparison of THC (Fig. 4), capsaicin (Fig. 8A) and α-tocopherol for their ability to prevent glutamate toxicity in neurons. The effects of THC (3 μM), capsaicin (10 μM) and α-tocopherol (300uM) were examined. The total OD value of control group was normalized to 100%.
4. Discussion
The present study demonstrates that the AF5 cell line can be employed to investigate the effects of neuroprotectants against NMDA-induced neurotoxicity. The AF5 cell line expresses functional NMDA1 receptors and is vulnerable to toxicity induced by exposure to NMDA in high concentrations. NMDA-induced toxicity was blocked by the NMDA antagonist MK-801, and the inactive stereoisomer of NMDA, N-methyl-L-aspartate, was inactive. These results suggest that NMDA-induced cell death in AF5 cells is mediated by stimulation of NMDA receptors.
The AF5 cell line was found to express both the CB1 message and the CB1 receptor protein. Nevertheless, THC produced a neuroprotective effect, as measured by both cell viability and apoptosis assays, whereas WIN55,212-2 was ineffective. The mechanisms through which THC and WIN55,212-2 exert neuroprotective effects are apparently complex [13]. Several studies have shown that WIN55,212-2 and THC, both of which are CB1 receptor agonists, exert similar neuroprotective effects against glutamate- or NMDA-induced neurotoxicity in isolated neurons [11,14] or in the brain [12,20]. These effects are mediated by activation of the cannabinoid receptor subtype CB1, as evidenced by blockade with SR141716A. A recent study by Hampson and Grimaldi [21] showed that cannabinoids were neuroprotective in vitro only when dbcAMP or forskolin was added to the culture medium. In the present model, however, dbcAMP did not alter the response to THC or not unmask a neuroprotective effect of WIN55,212-2. Also, the neuroprotection produced by THC was not blocked by the CB1 receptor antagonist SR141716A, even in the presence of dbcAMP after NMDA exposure. SR141716A also did not alter the response to WIN55,212-2. Therefore, in the present model the neuroprotective effect of THC against NMDA-induced cell death did not appear to be mediated by CB1 receptors.
In the present study, both THC and WIN55,212-2 produced toxic effects in AF5 cells at higher concentrations; especially, THC produced a toxic effect at doses which were about 2-3 fold higher than those which were neuroprotective. The toxic effects of THC at 8.5 μM and WIN55,212-2 at 10 μM were partially blocked by the CB1 receptor antagonist, SR141716A. Therefore, the toxic effects of both THC and WIN55,212-2 were at least in part related to CB1 receptor stimulation. Several prior studies directly or indirectly suggest that THC can be neurotoxic. Toxic effects of THC as measured by brain morphology or function have been reported [22]. Toxic effects on neurons which are mediated by CB1 receptor activation have also previously been identified [22,23]. Notably, Chan et al. [23] found that THC was toxic for cultured hippocampal neurons in concentrations similar to those which we found to be toxic for AF5 cells in the present study. Non-toxic doses of THC and WIN55,212-2 were, however, used for neuroprotection experiments in our study.
Some endogenous cannabinoids, such as arvanil, afford protection against oubain-induced in vivo excitotoxicity through activity at both the CB1 receptor and vanilloid receptor 1 (VR1), also termed TRPV1 [24], which is a member of the transient receptor potential (TRP) ion channel family. The ANKTM1 receptor was recently identified as a new member of the TRP channel family [25], and a receptor for THC [4]. The ANKTM1 receptor is likely to be responsible for non-CB1, non-VR1 mediated effects of THC on calcitonin gene-related peptide release in sensory neurons reported earlier by Zygmunt et al. [26]. VR1 (or TRPV1) is the only receptor which is known to be activated by capsaicin [3]. We did not detect either the ANKTM1 or TRPV1 receptor in AF5 cells. Capsaicin produced a neuroprotective effect in AF5 cells, which was not blocked by the VR1 antagonist capsazepine. Therefore, it is not likely that neuroprotective effects produced by THC or capsaicin in the AF5 cell line involves receptors of the TRP ion channel family.
On the other hand, in certain model systems THC can produce receptor-independent neuroprotective or cellular protective effects at micromolar concentrations as a result of its antioxidant properties [5,7,10]. Interestingly, Marsicano et al. [10] directly measured the antioxidant properties of WIN55,212-2 using protein and membrane preparations, and showed that it does not have an antioxidant effect. This finding is consistent with the lack of neuroprotective effect of WIN55,212-2 in the present study. Nevertheless, Nagayama et al. [12] found that WIN55,212-2, which had a receptor-mediated stereoselective neuroprotective effect in vivo, had a non-stereoselective neuroprotective effect against hypoxia in vitro, and this protective effect was not shared by THC. This in vitro neuroprotective effect of WIN55212-2 [12] would not seem to be consistent with either an antioxidant effect, ANKTM1 receptor effects, or CB1 receptor stimulation.
Capsaicin has also been reported to have antioxidant effects at micromolar concentrations [27–29]. In comparison to α-tocopherol, capsaicin is approximately 15–25 times more potent as an antioxidant [27,28]. We also examined α-tocopherol in the AF5 cell model, and found that it was neuroprotective in concentrations that were approximately consistent with the relative antioxidant potency of capsaicin. THC was, however, more effective than either compound, producing a more complete protective effect in lower concentrations than either capsaicin or α-tocopherol.
The response to THC was blocked by ruthenium red. Ruthenium red is a functional and non-selective antagonist of VR1 receptors, in addition to blocking ANKTM1 receptor function [4]. Ruthenium red has a number of effects on ion transport, including inhibition of ion channels other than members of the TRPV family [30], and inhibition of mitochondrial calcium transport [31,32]. Thus, the combination of ruthenium red and THC may produce complex changes in intracellular calcium levels that negate the beneficial effect of THC alone [33,34]. It does not appear that the effect of ruthenium red in the present study was likely to have been related to antagonism of TRPV1 or ANKTM1 receptors.
In conclusion, THC produces a potent neuroprotective effect in AF5 cells which appears to be mediated by antioxidant properties and is independent of the cannabinoid receptor CB1. The neuroprotective effect of THC is shared by capsaicin and α-tocopherol, but not WIN55,212-2. Because of the toxic effect of THC at higher doses which is mediated by CB1 receptor activation, development of the neuroprotective antioxidant cannabinoids which lack CB1 receptor efficacy may be the most useful route for the application of cannabinoids in preventing neurodegenerative disorders related to oxidative stress or excitotoxicity. The AF5 cell line may be a useful in vitro model to investigate neuroprotective and toxic mechanisms of cannabinoids, antioxidants, and drugs to block glutamatergic neurotoxicity through CB receptor-dependent or non-CB1 receptor pathways.
References
- 1.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 2.Pertwee R. The evidence for the existence of cannabinoid receptors. Gen Pharmacol. 1993;24:811–824. doi: 10.1016/0306-3623(93)90154-p. [DOI] [PubMed] [Google Scholar]
- 3.Gunthorpe MJ, Benham CD, Randall A, Davis JB. The diversity in the vanilloid (TRPV) receptor family of ion channels. Trends Pharmacol Sci. 2002;23:183–191. doi: 10.1016/s0165-6147(02)01999-5. [DOI] [PubMed] [Google Scholar]
- 4.Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- 5.Hampson AJ, Grimaldi M, Axelrod J, Wink DK. Cannabidiol and (-)Δ9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA. 1998;95:8268–8273. doi: 10.1073/pnas.95.14.8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hampson AJ, Grimaldi M, Lolic M, Wink D, Rosenthal R, Axelrod J. Neuroprotective antioxidants from marijuana. Ann N Y Acad Sci. 2000;899:282. [PubMed] [Google Scholar]
- 7.Chen Y, Buck J. Cannabinoids protect cells from oxidative cell death: A receptor-independent mechanism. J Pharmacol Exp Ther. 2000;293:807–812. [PubMed] [Google Scholar]
- 8.Guzman M, Sanchez C, Galve-Roperh I. Control of the cell survival/death decision by cannabinoids. J Mol Med. 11;2001;78:613–625. doi: 10.1007/s001090000177. [DOI] [PubMed] [Google Scholar]
- 9.Hansen HH, Azcoitia I, Pons S, Romero J, Garcia-Segura LM, Ramos JA, Hansen HS, Fernandez-Ruiz J. Blockade of cannabinoid CB(1) receptor function protects against in vivo disseminating brain damage following NMDA-induced excitotoxicity. J Neurochem. 2002;82:154–158. doi: 10.1046/j.1471-4159.2002.00961.x. [DOI] [PubMed] [Google Scholar]
- 10.Marsicano G, Moosmann B, Hermann H, Lutz B, Behl C. Neuroprotective properties of cannabinoids against oxidative stress: Role of the cannabinoid receptor CB1. J Neurochem. 2002;80:448–456. doi: 10.1046/j.0022-3042.2001.00716.x. [DOI] [PubMed] [Google Scholar]
- 11.Shen M, Thayer SA. Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol. 1998;54:459–462. doi: 10.1124/mol.54.3.459. [DOI] [PubMed] [Google Scholar]
- 12.Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci. 1999;19:2987–2995. doi: 10.1523/JNEUROSCI.19-08-02987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Stelt M, Veldhuis WB, Maccarrone M, Bar PR, Nicolay K, Veldink GA, Di Marzo V, Vliegenthart JF. Acute neuronal injury, excitotoxicity, and the endocannabinoid system. Mol Neurobiol. 2002;26:317–346. doi: 10.1385/MN:26:2-3:317. [DOI] [PubMed] [Google Scholar]
- 14.Grigorenko E, Kittler J, Clayton C, Wallace D, Zhuang S, Bridges D, Bundey S, Boon A, Pagget C, Hayashizaki S, Lowe G, Hampson R, Deadwyler S. Assessment of cannabinoid induced gene changes: Tolerance and neuroprotection. Chem Phys Lipids. 2002;121:257–266. doi: 10.1016/s0009-3084(02)00161-5. [DOI] [PubMed] [Google Scholar]
- 15.El-Remessy AB, Khalil IE, Matragoon S, Abou-Mohamed G, Tsai NJ, Roon P, Caldwell RB, Caldwell RW, Green K, Liou GI. Neuroprotective effect of (-)Δ9-tetrahydrocannabinol and cannabidiol in N-methyl-D-aspartate-induced retinal neurotoxicity: Involvement of peroxynitrite. Am J Pathol. 2003;163:1997–2008. doi: 10.1016/s0002-9440(10)63558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Truckenmiller ME, Tornatore C, Wright RD, Dillon-Carter O, Meiners S, Geller HM, Freed WJ. A truncated SV40 large T antigen lacking the p53 binding domain overcomes p53-induced growth arrest and immortalizes primary mesencephalic cells. Cell Tissue Res. 1998;291:175–189. doi: 10.1007/s004410050989. [DOI] [PubMed] [Google Scholar]
- 17.Pleasure SJ, Lee VM. NTera 2 cells: A human cell line which displays characteristics expected of a human committed neuronal progenitor cell. J Neurosci Res. 1993;35:585–602. doi: 10.1002/jnr.490350603. [DOI] [PubMed] [Google Scholar]
- 18.Truckenmiller ME, Vawter MP, Zhang P, Conejero-Goldberg C, Dillon-Carter O, Morales N, Cheadle C, Becker KG, Freed WJ. AF5, a CNS cell line immortalized with an n-terminal fragment of SV40 large T: Growth, differentiation, genetic stability and gene expression. Exp Neurol. 2002;175:318–337. doi: 10.1006/exnr.2002.7898. [DOI] [PubMed] [Google Scholar]
- 19.Frankfurt OS, Robb JA, Sugarbaker EV, Villa L. Monoclonal antibody to single-stranded DNA is a specific and sensitive cellular marker of apoptosis. Exp Cell Res. 1996;226:387–397. doi: 10.1006/excr.1996.0240. [DOI] [PubMed] [Google Scholar]
- 20.van der Stelt M, Veldhuis WB, Bar PR, Veldink GA, Vliegenthart JF, Nicolay K. Neuroprotection by Δ9-tetrahydrocannabinol, the main active compound in marijuana, against ouabain-induced in vivo excitotoxicity. J Neurosci. 2001;21:6475–6479. doi: 10.1523/JNEUROSCI.21-17-06475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hampson AJ, Grimaldi M. Cannabinoid receptor activation and elevated cyclic AMP reduce glutamate neurotoxicity. Eur J Neurosci. 2001;13:1529–1536. doi: 10.1046/j.0953-816x.2001.01536.x. [DOI] [PubMed] [Google Scholar]
- 22.Sarne Y, Keren O. Are cannabinoid drugs neurotoxic or neuroprotective? Med Hypotheses. 2004;63:187–192. doi: 10.1016/j.mehy.2004.02.043. [DOI] [PubMed] [Google Scholar]
- 23.Chan GC, Hinds TR, Impey S, Storm DR. Hippocampal neurotoxicity of Delta9-tetrahydrocannabinol. J Neurosci. 1998;18:5322–5332. doi: 10.1523/JNEUROSCI.18-14-05322.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veldhuis WB, van der Stelt M, Wadman MW, van Zadelhoff G, Maccarrone M, Fezza F, Veldink GA, Vliegenthart JF, Bar PR, Nicolay K, Di Marzo V. Neuroprotection by the endogenous cannabinoid anandamide and arvanil against in vivo excitotoxicity in the rat: role of vanilloid receptors and lipoxygenases. J Neurosci. 2003;23:4127–4133. doi: 10.1523/JNEUROSCI.23-10-04127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112:819–829. doi: 10.1016/s0092-8674(03)00158-2. [DOI] [PubMed] [Google Scholar]
- 26.Zygmunt PM, Andersson DA, Hogestatt ED. Δ9-tetrahydrocannabinol and cannabinol activate capsaicin-sensitive sensory nerves via a CB1 and CB2 cannabinoid receptor-independent mechanism. J Neurosci. 2002;22(11):4720–4727. doi: 10.1523/JNEUROSCI.22-11-04720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ochi T, Takaishi Y, Kogure K, Yamauti I. Antioxidant activity of a new capsaicin derivative from capsicum annuum. J Nat Prod. 2003;66:1094–1096. doi: 10.1021/np020465y. [DOI] [PubMed] [Google Scholar]
- 28.Kogure K, Goto S, Nishimura M, Yasumoto M, Abe K, Ohiwa C, Sassa H, Kusumi T, Terada H. Mechanism of potent antiperoxidative effect of capsaicin. Biochim Biophys Acta. 2002;1573:84–92. doi: 10.1016/s0304-4165(02)00335-5. [DOI] [PubMed] [Google Scholar]
- 29.Henderson DE, Slickman AM, Henderson SK. Quantitative HPLC determination of the antioxidant activity of capsaicin on the formation of lipid hydroperoxides of linoleic acid: a comparative study against BHT and melatonin. J Agric Food Chem. 1999;47:2563–2570. doi: 10.1021/jf980949t. [DOI] [PubMed] [Google Scholar]
- 30.Hamilton MG, Lundy PM. Effect of ruthenium red on voltage-sensitive Ca++ channels. J Pharmacol Exp Ther. 1995;273:940–947. [PubMed] [Google Scholar]
- 31.Watson EL, Vincenzi FF, Davis PW. Ca 2+-activated membrane ATPase: selective inhibition by ruthenium red. Biochim Biophys Acta. 1971;249:606–610. doi: 10.1016/0005-2736(71)90140-4. [DOI] [PubMed] [Google Scholar]
- 32.Moore CL. Specific inhibition of mitochondrial Ca++ transport by ruthenium red. Biochem Biophys Res Commun. 1971;42:298–305. doi: 10.1016/0006-291x(71)90102-1. [DOI] [PubMed] [Google Scholar]
- 33.Dubinsky JM, Rothman SM. Intracellular calcium concentrations during "chemical hypoxia" and excitotoxic neuronal injury. J Neurosci. 1991;11:2545–2551. doi: 10.1523/JNEUROSCI.11-08-02545.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee WT, Yin HS, Shen YZ. The mechanisms of neuronal death produced by mitochondrial toxin 3-nitropropionic acid: the roles of N-methyl-D-aspartate glutamate receptors and mitochondrial calcium overload. J Neurosci. 2002;112:707–716. doi: 10.1016/s0306-4522(02)00097-0. [DOI] [PubMed] [Google Scholar]