

Abstract
Fosfomycin is a cell wall inhibitor used mainly for the treatment of uncomplicated lower urinary tract infections. As shown here, resistance to fosfomycin develops rapidly in Escherichia coli under experimental conditions, but in spite of the relatively high mutation rate in vitro, resistance in clinical isolates is rare. To examine this apparent contradiction, we mathematically modeled the probability of resistance development in the bladder during treatment. The modeling showed that during a typical episode of urinary tract infection, the probability of resistance development was high (>10−2). However, if resistance was associated with a reduction in growth rate, the probability of resistance development rapidly decreased. To examine if fosfomycin resistance causes a reduced growth rate, we isolated in vitro and in vivo a set of resistant strains. We determined their resistance mechanisms and examined the effect of the different resistance mutations on bacterial growth in the absence and presence of fosfomycin. The types of mutations found in vitro and in vivo were partly different. Resistance in the mutants isolated in vitro was caused by ptsI, cyaA, glpT, uhpA/T, and unknown mutations, whereas no cyaA or ptsI mutants could be found in vivo. All mutations caused a decreased growth rate both in laboratory medium and in urine, irrespective of the absence or presence of fosfomycin. According to the mathematical model, the reduced growth rate of the resistant strains will prevent them from establishing in the bladder, which could explain why fosfomycin resistance remains rare in clinical isolates.
Fosfomycin is a bactericidal antibiotic that acts as a cell wall inhibitor by interfering with the first step in peptidoglycan biosynthesis. It functions as a phosphoenolpyruvate analogue and binds to UDP-N-acetylglucosamine-3-O-enolpyruvyl transferase. The binding inhibits the formation of UDP-N-acetylglucosamine-3-O-enolpyruvate from UDP-N-acetylglucosamine and phosphoenolpyruvate (18, 31). In Escherichia coli, fosfomycin is transported into the cell via the GlpT and UhpT transporters. Expression of these genes requires the presence of the cyclic AMP- cyclic AMP receptor protein complex (30, 35, 42), and for the uhpT gene, high-level expression also requires the regulatory genes uhpA, uhpB and uhpC (16, 35). Defects in one or both of the transport systems (caused by mutations in the uhpT and glpT structural genes or the regulators) can confer fosfomycin resistance (17, 18, 31). In addition, plasmid-encoded fosfomycin resistance conferred by an enzyme that inactivates the antibiotic has been described (3-5).
Clinically, fosfomycin in the form of its trometamol salt is mainly used in treatment of uncomplicated lower urinary tract infections (UTI). The antibiotic is active against both gram-positive and gram-negative bacteria (31). When given orally, it is rapidly absorbed and excreted at high concentrations in urine for several days, making use of a single dose clinically feasible (for a review, see reference 7). In 80 to 90% of uncomplicated UTIs, the causative agent is E. coli, and increasing resistance to other commonly used antibiotics like fluoroquinolones and trimethoprim is a growing problem with this bacterium. However, the frequency of fosfomycin resistance in clinical isolates of E. coli remains low. For example, in certain countries in Europe, fosfomycin has been used for many years in treatment of UTIs and still only about 1% of uropathogenic E. coli are fosfomycin-resistant (19).
Antibiotic resistances often confers a biological cost to the resistant bacteria that can be observed as a decreased growth rate in vitro and/or in vivo as well as decreased virulence (2, 8-11, 33, 37, 40, 41, 43). This cost can often be reduced by compensatory mutations, and such genetic compensation has been described for several antibiotics and bacterial species both in vitro and in vivo (2, 8-11, 29, 33, 37, 40, 41, 43). At a given antibiotic pressure, the biological cost of resistance is one of the main determinants of how rapidly and to what extent a resistant mutant will establish itself within an individual or a host population. Thus, it is of importance to examine the magnitude and mechanism of this cost to understand how rapidly and under which conditions resistance might appear (25, 26). The effect of resistance mutations on fitness is of particular importance for uropathogens because of the limits that the bladder dynamics pose on the minimal growth rate needed to establish an infection (14).
Here we identified several fosfomycin resistance mutations and examined their effect on biological fitness, i.e., growth in urine and laboratory medium. Our experimental results and the modeling suggest that most of the resistant mutants are unable to grow fast enough to become established in the bladder in the presence of fosfomycin.
MATERIALS AND METHODS
Isolation in vitro of fosfomycin-resistant strains.
Fosfomycin-resistant mutants were isolated by plating Escherichia coli strain NU14 on Luria agar (LA) plates with 200 or 500 mg of fosfomycin per liter (Sigma). Strain NU14 was originally isolated from a patient with a UTI and has been used for studies of bacterial interactions with epithelial cells (15, 23, 32). Resistant colonies (one per independent culture) were picked, restreaked on LA plates with fosfomycin, and subsequently frozen at −70°C.
Another set of fosfomycin-resistant strains were isolated on LA plates containing 50 mg of fosfomycin per liter and 50 mg of glucose 6-phosphate (G6P) per liter (Sigma), added in order to induce the UhpT transport system. The MIC for fosfomycin was determined with E-test (AB Biodisk, Uppsala, Sweden) according to the manufacturer's instructions. As the E-tests contain G6P, the MIC in the absence of G6P was determined by streaking bacteria on LA plates with different concentrations of fosfomycin. A strain was defined as fosfomycin resistant when its MIC clearly deviated from the MICs of the normal wild-type population (20, 22). This definition avoids the use of arbitrarily chosen MICs to define a strain as resistant.
Mutation frequency determination.
A Luria-Delbrück fluctuation test was performed to determine the mutation frequency to fosfomycin resistance. For each strain, 25 independent 3-ml Luria-Bertani (LB) broth cultures were inoculated with approximately 50 cells and grown to saturation (ca. 3 × 109 cells/ml) at 37°C. From each culture, 100 μl was spread on LB with 200 mg of fosfomycin per liter and LB with 50 mg of fosfomycin per liter supplemented with 50 mg of G6P per liter. The number of resistant colonies appearing after 24 h of incubation at 37°C was counted. A viable count was performed on four of the cultures to estimate the total number of cells. The mutation frequency was calculated from the median number of resistant cells divided by the total number of cells per milliliter of culture (24).
Clinical isolates.
In a recent study, one of the authors (G.K.) screened 2,478 isolates of E. coli from lower UTIs in women obtained between 1999 and 2000 in 16 European countries and Canada for resistance to 12 antibiotics, one of which was fosfomycin, commonly used in UTIs (19). The few resistant isolates recovered were investigated further in the present study. In addition, fosfomycin-resistant isolates collected from women with an uncomplicated UTI in Sweden during 1997 were studied.
Growth on carbohydrates.
Growth on different carbohydrates as the sole carbon source was determined by streaking bacteria on M9 minimal medium agar plates supplemented with nicotinic acid and a carbohydrate at 0.2%. The carbohydrates tested were lactose, mannitol, glycerol, G6P, and dl-α-glycerophosphate (α-GP). When included, the concentration of cyclic AMP (cAMP) was 1 mM. The plates were inspected for growth after 48 h of incubation at 37°C except for plates containing G6P or α-GP, which were incubated for 72 h.
Growth rate measurements.
Growth rates at 37°C were measured in Luria-Bertani broth (LB) and in sterile-filtered pooled human urine in both the presence and absence of fosfomycin. The urine used was pooled from one of the authors (A.N.). The urine pH was 6. The bacteria were grown in LB and urine overnight. Approximately 105 cells were then inoculated into 400 μl of growth medium on a bioscreen plate. The absorbance at 540 nm was read with a BioscreenC (Labsystems, Helsinki, Finland). For each strain and condition, growth rates were measured in quadruplicate in two separate experiments. Relative growth rates were calculated as the ratio of the growth rate of a reference strain divided by the growth rate of the test strain.
Sequencing.
The gene of interest was PCR amplified (primer sequences are available upon request), and the PCR product was purified from solution with GFX PCR DNA and gel band purification kit (Amersham Pharmacia Biotech Inc.). The purified PCR product was subsequently used as the template in a sequencing reaction with the BigDye terminator cycle sequencing ready reaction kit (Applied Biosystems, Warrington, United Kingdom), and the sequences were read with an ABI Prism 3100 genetic analyzer. Sequence changes in resistant mutants derived from E. coli NU14 were identified by pairwise alignment of the sequence from the strain analyzed to that of NU14. Each mutation was confirmed by one additional sequencing reaction. For the clinical fosfomycin-resistant isolates, only sequence changes causing frameshifts, deletions, insertions, or stop codons were scored as mutations that inactivate gene function. However, it should be noted that some missense mutations might also inactivate gene function.
Modeling the probability of resistance development during treatment.
We developed a mathematical model to allow stringent examination of the conditions under which resistance development would occur when an antibiotic selective pressure is applied to a population of bacteria growing in the bladder (the complete model is available upon request). A basic characteristic is the cyclic pattern of growth while the bladder is filling up with urine (rate λ, milliliters per hour), followed by purging of the bacteria when the bladder is emptied. The bacteria are assumed to grow with a constant growth rate (k, replications per hour). If the volume increases from Vmin to Vmax and is then reduced to Vmin again in a regular pattern, bacteria will first grow in number by a factor G = exp[k(Vmax − Vmin)/λ]. When the bladder is emptied, the bacteria will decrease in number by a factor β−1 = Vmin/Vmax. Thus, the net growth factor from cycle to cycle is G/β = (Vmin/Vmax)exp[k(Vmax − Vmin)/λ]. If this factor is larger than 1, the bacteria will become established in the bladder. This is the Gordon-Riley criterion for bladder establishment (14):
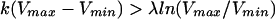 |
(1a) |
or, equivalently,
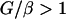 |
(1b) |
The model assumes that the susceptible bacteria have become established before the antibiotic is added and reached a steady state where their numbers at the beginning of each cycle are the same, N0. A resistance mutation appears with probability u in each replication, and the resistant bacteria are assumed to have a relative growth disadvantage of s (<0). At the steady state, resistance will be present on average in a fraction u/|s| of all bacteria (the mutation-selection balance). The model accounts for the probability distribution of having a certain number of resistant mutants present at the time when antibiotic is added and then calculating the probability that these will grow in number, rather than be washed out, during the subsequent cycles. The general model also accounts for the probability that resistance mutations appear after antibiotic is added and that these will grow in number. However, with the parameter values that are relevant for this problem, this last probability turns out to be negligible.
After antibiotic has been added, the susceptible bacteria grow with rate constant k0 and the resistant ones with rate constant k1. Basic parameters of the model are the growth factor of resistant bacteria, G = exp[k1(Vm − V0)/λ], bladder expansion β = Vmax/Vmin, mutation pressure uN0, the relative growth disadvantage s (<0) of the resistant bacteria before addition of antibiotic, and the growth ratio γ = k0/k1 between susceptible and resistant bacteria. The results are particularly simple in the case when the antibiotic is added at the moment when the bladder volume is minimal (or maximal). In this case, the probability for resistance fixation can be expressed as
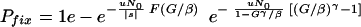 |
(2) |
The first exponential factor in equation 2 is the probability that all preexisting resistant bacteria are lost, and the second exponential factor is the probability that all resistance mutants that appear after antibiotic is added are lost. The function F(G/β) in the first exponential function is defined as
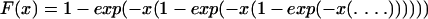 |
(3) |
The exponential functions continue ad infinitum. F(x) can also be defined implicitly from x = −(1/F)ln(1 − F). With x = G/β, F(G/β) corresponds to the ultimate survival probability through all subsequent growth cycles in the presence of the antibiotic for a single resistant mutant that exists at the beginning of a cycle. For G/β > 3, F(G/β) is very nearly equal to 1. F(G/β) → 0 when G/β → 1, i.e., close to the Gordon-Riley criterion. This smallness of F accounts for the probability that a small number of bacteria could be lost stochastically even if the Gordon-Riley criterion is formally satisfied. It can be noted that when fixation is caused primarily by preexisting resistant bacteria, the probability of fixation (Pfix) depends on only two parameter combinations, G/β and uN0/|s|.
RESULTS
Frequency of antibiotic-resistant E. coli isolated from UTIs in European countries.
We examined the frequency of resistance in 2,478 E. coli strains isolated during the Eco.Sens study (19). All strains were tested for resistance with the disk diffusion method standardized according to the Swedish Reference Group of Antibiotics (http://www.srga.org). Fosfomycin resistance was rare in all investigated European countries, the highest figure being from Greece, with 1.5% resistant isolates. There was no difference in resistance frequencies between countries with no fosfomycin consumption (Denmark, 1.2%; Norway, 1.2%) and countries with a long tradition of fosfomycin use (France, 1.0%; Finland, 1.1%; and Belgium, 0.7%). Furthermore, we examined the frequency of fosfomycin resistance among 1,381 E. coli strains isolated in Sweden during 1997. In this strain collection, the frequency of fosfomycin resistance was 1.0%.
Mutation frequency to resistance in vitro.
With strain NU14, we performed a Luria-Delbrück fluctuation test to determine the mutation frequency to fosfomycin resistance in both the presence and absence of G6P (28). The mutation frequency was calculated from the median value of resistant mutants divided by the total cell number (24). The mutation frequency to fosfomycin resistance in the absence of G6P (200 mg of fomycin per liter) was approximately 10−7 and in the presence of G6P (50 mg of fosfomycin per liter) 10−8.
Probability of resistance development during UTI treatment.
We developed a mathematical model to examine the conditions under which it is predicted that resistant mutants would become fixed when a population of bacteria growing in the bladder is exposed to antibiotic (see Materials and Methods). The model shows that the probability of resistant mutants appearing during treatment is dependent on several parameters, including the growth rates of the resistant mutants (k1), the ratio β = Vmax/Vmin (where Vmax is a full bladder and Vmin is an emptied bladder), and the flow rate of urine (λ). In Fig. 1 we plotted Pfix as a function of G/β = (Vmin/Vmax)exp[k1(Vmax − Vmin)/λ] at different values of uN0 (mutation rate to resistance times population size of bacteria) values.
FIG. 1.

Modeling of probability of fixation (Pfix from equation 2) of fosfomycin-resistant mutants as a function of G/β. s = −0.2 was assumed for the relative growth disadvantage of resistant bacteria to susceptible ones in the absence of the antibiotic. The solid curves show Pfix with uN0 values of 0.001, 0.01, 0.1, and 1 (bottom to top). The dotted curves show the corresponding results if treatment is initiated when the bladder is half full.
Here u was experimentally determined to be 10−7 in this study. N0 varied quite extensively between patients, but about 60 to 70% of the patients had bacterial titers of > 105/ml of urine (36, 38, 44). As can be seen, when N0 was > 105/ml, the probability of fixation was high (>10−2), whereas when the growth rate was reduced below a certain level, Pfix dropped to zero. In other words, a rather moderate decrease in the growth rate will prevent the resistant mutants from becoming established.
If we assume that Vmin and λ are essentially constant, 1 ml and 60 ml per h, respectively, a doubling time longer than approximately 36 min (with Vmax = 300 ml, typical for a healthy adult) will not allow establishment of the bacteria in the bladder (Fig. 2). As UTIs usually cause a reduction in Vmax (more frequent voiding of urine), this will decrease Pfix further. Thus, if Vmax is decreased to 150 ml, the bacterial doubling time has to be shorter than 21 min to allow establishment. Finally, we also examined if the time of addition of the antibiotic with respect to how filled the bladder is would have an effect on Pfix. It was found that initiating treatment at an intermediate time during bladder expansion had only a marginal effect on the calculated Pfix irrespective of the values chosen for uN0 and Vmax (Fig. 1).
FIG. 2.

Gordon-Riley criterion, equation 2. τmax is the maximum doubling time (= ln [2]/k), in minutes, that can allow establishment, plotted as a function of Vmax at a given value of Vmin. Curves are for λ = 60 h−1 and Vmin = 1, 5, 10, 20, and 50 ml (from bottom to top). Doubling times shown below each line allow establishment under the given conditions.
Identification of mechanisms of fosfomycin resistance in mutants isolated in vitro.
We isolated a set of fosfomycin-resistant mutants, examined their resistance mechanism, and determined the effect of these mutations on bacterial growth. We isolated 13 resistant mutants from strain NU14, 3 in the absence of G6P and 10 in the presence of G6P. Resistance levels ranged from 6 to > 1,024 mg per liter (Table 1). To determine the resistance mechanism, we used DNA sequencing, PCR analysis, and phenotypic tests (growth on various carbon sources). The main mechanism of fosfomycin resistance is inhibition of transport of the antibiotic into the cell due to defects in the Uhp and/or GlpT transport system (1, 17, 31, 45). Strains defective in fosfomycin uptake are identified by their inability to grow on the carbon sources preferentially taken up by GlpT and UhpT, α-GP and G6P, respectively.
TABLE 1.
Characteristics of fosfomycin-resistant E. coli mutants isolated in vitro
| Strain | Fosfomycin concn (mg/liter) used for isolationa | MIC (mg/liter)b
|
Growth on carbohydrates mM
|
Growthc
|
Resistance mutationd | Genes sequenced | |||
|---|---|---|---|---|---|---|---|---|---|
| With G6P | Without G6P | Without cAMP | With 1 mM cAMP | On α-GP | On G6P | ||||
| NU14 (parent) | 1 | 5 | + | + | + | + | |||
| DA6313 | 200 | 6 | >450 | − | −/+ | − | + | ptsI deletion | glpT |
| DA6326 | 500 | 6 | 900 | − | −/+ | − | + | ptsI deletion | gtpT |
| DA6396 | 50 + G6P | 12 | >1,000 | − | + | − | + | cyaA deletion | glpT, cyaA, uhpT, uhpA |
| DA6328 | 500 | 48 | 900 | + | + | − | + | glpT missense mutation | glpT |
| DA6400 | 50 + G6P | 48 | >1,000 | − | − | − | + | ND | glpT, cyaA, uhpT, uhpA |
| DA6399 | 50 + G6P | 128 | >1,000 | − | −/+ | − | − | cyaA insertion | uhpT, uhpA, cyaA |
| DA6395 | 50 + G6P | >1,024 | >1,000 | − | + | − | − | uhpT and cyaA nonsense mutation | glpT, cyaA, uhpT, uhpA |
| DA6398 | 50 + G6P | >1,024 | >1,000 | − | + | − | − | cyaA frameshift mutation | glpT, cyaA, uhpT, uhpA |
| DA6402 | 50 + G6P | >1,024 | >1,000 | − | + | − | − | cyaA insertion, uhpT deletion | glpT, cyaA, uhpA |
| DA6401 | 50 + G6P | >1,024 | >1,000 | − | − | − | − | uhpT missense mutation | glpT, cyaA, uhpT, uhpA |
| DA6393 | 50 + G6P | >1,024 | >1,000 | + | + | + | + | ND | glpT, cyaA, uhpT, uhpA |
| DA6394 | 50 + G6P | >1,024 | >1,000 | + | + | + | + | ND | glpT, uhpT |
| DA6397 | 50 + G6P | >1,024 | >1,000 | + | + | + | + | ND | glpT, uhpT, uhpA |
When included, the concentration of G6P was 50 mg/liter.
MIC determined by E-test with G6P or by streaking bacteria on LA plates with different concentrations of fosfomycin.
α-GP (used at 0.2%) is the preferred substrate of the GlpT transporter, and G6P (used at 0.2%) is the preferred substrate of the UhpT transporter system.
See Table 3 for details regarding type and site of mutations. ND, not determined.
Ten strains failed to grow on α-GP, and their resistance was likely caused by inability to take up the antibiotic via GlpT (Table 1). Expression of the glpT gene is positively regulated by cAMP, and mutations in the cyaA and ptsI genes that lower the cAMP level in the cell can cause resistance (1, 45). Such mutations are also associated with defects in uptake and metabolism of certain sugars, and this defect is to some extent reversible by addition of exogenous cAMP. Nine of the 10 strains with a GlpT− phenotype (in several cases they also had a UhpT− phenotype) failed to grow on one or more of the sugars tested, and their negative phenotypes were reversible to different extents by addition of cAMP, suggesting that they were mutated in, for example, the cyaA or ptsI gene. Only one strain had a defect in growth on α-GP and not on any other carbohydrates. In summary, these results indicated that most fosfomycin resistance mutations were located in genes that caused a decrease in cAMP levels, and as a result they impaired both glpT and uhpT expression.
We analyzed the resistant mutants by PCR and sequencing (Table 1). Only one strain had a mutation in the glpT gene. Two strains had deletions in the ptsI gene, and five strains had other loss-of-function mutations in the cyaA gene. In one strain with a GlpT− phenotype, no mutations were identified in either the glpT, uhpT, uhpA, or cyaA gene. Since 10 of the 13 fosfomycin-resistant strains were isolated in the presence of G6P, we also sequenced the uhpT and uhpA genes in these strains. Three strains carried mutations in the uhpT gene (in two strains it was accompanied by a mutation in the cyaA gene). Three strains had mutations in only the cyaA gene. One mutant was defective in carbohydrate and α-GP growth, but this defect was not reversible by cAMP, and in this strain no mutations were identified in either glpT, cyaA, uhpT, or uhpA gene. Finally, three strains had GlpT+ and UhpT+ phenotypes, no defect in sugar utilization, and no mutations in either glpT, uhpT, or uhpA. For these four strains, the mechanism of resistance is unknown.
Identification of mechanisms of fosfomycin resistance in clinical isolates.
Thirteen resistant clinical isolates were investigated (Table 2). All isolates except one grew on α-GP but not on G6P. In four isolates, gene-inactivating mutations were found in the uhpT and/or uhpA gene. One strain carried a mutation in glpT as well as a deletion in the uhpA gene, and this strain also had the highest resistance level among the clinical isolates. The remaining eight isolates could grow on α-GP but not on G6P, strongly indicating that they were defective in UhpT-mediated transport. None of these eight strains were defective in growth on sugars, and they had no mutations in the cyaA gene, indicating that cAMP levels were normal. Thus, these strains were likely to be defective in uhpT, uhpA, uhpB, or uhpC, even though the sequencing did not reveal any gene-inactivating mutations (i.e., deletion, insertion, frameshift, or nonsense mutations) in the uhpT or uhpA gene. As the parental strains of the clinical isolates were not available, the effect of any missense mutations could not be evaluated.
TABLE 2.
Characteristics of fosfomycin-resistant E. coli mutants isolated in vivo (clinical isolates)
| Strain | MIC (mg/liter)
|
Growth on carbohydrates | Growth on α-GP | Growth on G6P | Resistance mutation | Genes sequenced | |
|---|---|---|---|---|---|---|---|
| With G6P | Without G6P | ||||||
| DA6886 | 16 | 15 | + | + | − | ND | glpT, uhpT, uhpA, cyaA |
| DA6883 | 32 | 15 | + | + | − | uhpT insertion and uhpA deletion | glpT, cyaA |
| DA6887 | 48 | 15 | + | + | − | ND | glpT, uhpT, uhpA, cyaA |
| DA6884 | 48 | 15 | + | + | − | ND | glpT, uhpT, uhpA, cyaA |
| DA6874 | 48 | 10 | + | + | − | ND | glpT, uhpT, uhpA, cyaA |
| DA6885 | 48 | 15 | + | + | − | uhpT duplication | glpT, uhpT, uhpA, cyaA |
| DA6890 | 48 | 20 | + | + | − | ND | glpT, uhpT, uhpA, cyaA |
| DA6439 | 64 | 20 | + | + | − | ND | glpT, uhpT, uhpA, cyaA |
| DA6891 | 64 | 20 | + | + | − | ND | glpT, uhpT, uhpA, cyaA |
| DA6435 | 96 | 15 | + | + | − | ND | glpT, uhpT, uhpA, cyaA |
| DA6896 | 96 | 25 | + | + | − | ND | glpT, uhpT, uhpA, cyaA |
| DA6440 | 96 | 25 | + | + | − | uhpT and uhpA deletion | glpT |
| DA6894 | 192 | >50 | + | − | − | uhpA deletion and glpT stop codon | glpT, uhpT, uhpA, cyaA |
See Table 1, footnotes, b, c, and d.
Sequence changes in fosfomycin-resistant strains isolated in vitro and clinically.
The identified deletion, duplication, and insertion mutations are shown in Table 3. Half of the mutations causing resistance were due to deletions (9 of 18 mutations, Tables 1 and 2). Among these nine mutants, we identified the deletion endpoints in three cases. In addition, we found five base-pair substitutions, three insertion sequence element insertions, and one duplication. This distribution of loss-of-function mutations is similar to what has been found previously in other genes (39, 46).
TABLE 3.
Fosfomycin resistance mutations identified in laboratory and clinical isolatesa
| Strain | Gene | Mutation |
|---|---|---|
| DA6313 | ptsI | Large deletiona |
| DA6326 | ptsI | Large deletiona |
| DA6396 | cyaA | CCTTCG−289 [del 371 bp]113GCCGACb |
| DA6328 | glpT | ATGGCCTTGTGAT, Pro (CC865T)→ Leu (CT865T) |
| DA6399 | cyaA | Insertion IS1, insertion pointcGCA536 |
| DA6395 | cyaA | TTGCTATAACGTA, Gln (C400AA)→ stop (T400AA) |
| uhpT | GCGTTCTAGGAAC, Gln (C862AG)→ stop (T862AG) | |
| DA6398 | cyaA | AAGTCA454[del 1 bp]456CTTCTTb |
| DA6402 | cyaA | GCTGGG430[del 7 bp]438TGTGGAb |
| uhpT | Insertion IS1, insertion pointc628CTG | |
| DA6401 | uhpT | CGTTACAGCAGCG, Gly (G637GC)→ Ser (A637GC) |
| DA6883 | uhpT | Insertion IS200 AAAc1434 (insertion point 42 bp downstream of stop codon for uhpT) |
| uhpA | Large deletiona | |
| DA6885 | uhpT | GCCGCG1299[CTGGATATCGCCGCG]1300ATTGGTb 15-bp duplication |
| DA6440 | uhpT | Large deletiona |
| uhpA | Large deletiona | |
| DA6894 | uhpA | GTGCGG140[del 8 bp]149TGTGTAb |
| glpT | CCTCCTAGGCCTA, Trp (TG878G)→ stop (TA878G) |
Deletions identified by PCR and Southern hybridization. Endpoints have not been determined.
Subscript numbers indicate the nucleotide within the gene (+1 designates the first nucleotide in the respective start codon).
Only one side of the insertion point has been determined by sequencing.
Growth rates of resistant bacteria in absence and presence of fosfomycin.
For uropathogenic E. coli, the growth rate is of particular importance because it has to exceed a certain threshold level to allow bacterial maintenance in the bladder (14). We determined growth rates in LB and urine in both the absence and presence of fosfomycin. For the in vitro-isolated fosfomycin-resistant strains, the growth rate in LB and urine in the absence of fosfomycin was between 10 and 25% slower than that of the susceptible parental strain NU14 (Fig. 3). For the clinically isolated resistant strains, we had no access to the isogenic susceptible parental strain, and therefore the growth rates of the resistant mutants were compared to that of a randomly selected set of fully susceptible clinical E. coli isolates (Fig. 4). No significant difference in growth rate could be detected between the susceptible and resistant strains in the absence of fosfomycin. One explanation for the apparent lack of a decreased growth rate in the clinical isolates could be that the nonisogeneity of the strains obscured the cost conferred by the resistance. Alternatively, the costs could already have been ameliorated by compensatory mutations, as has been observed in both experimental and clinical settings for other resistances (8, 11, 33, 37, 43).
FIG. 3.

Relative growth rate (tgen susceptible parent/tgen resistant mutant) in urine (open triangles) and LB (solid triangles) of fosfomycin-resistant mutants isolated in vitro. Note that the solid triangle for strain DA6402 is hidden under the open triangle.
FIG. 4.

Relative growth rates of clinically isolated fosfomycin-resistant and fosfomycin-susceptible strains in urine (open triangles) and LB (solid triangles). A relative growth rate of 1 is set to correspond to a tgen of 20 min.
We also measured growth rates in the presence of different concentrations of fosfomycin in three in vitro-isolated strains with identified resistance mutations and in all the clinically isolated fosfomycin-resistant strains. Growth rates were measured in LB and urine with concentrations of fosfomycin ranging from 5 to 64 mg per liter. The concentration of fosfomycin in urine normally exceeds 128 mg per liter during treatment (7). In the in vitro-isolated fosfomycin-resistant strains DA6313, DA6328, and DA6395, the presence of fosfomycin reduced the growth rate in both LB and urine (Fig. 5). Similarly, the in vivo isolates were inhibited in both LB and urine when fosfomycin was added (data not shown). Thus, all clinical strains, with one exception, were severely disturbed in their growth at concentrations of fosfomycin above 8 mg per liter. With such a reduction in growth rate, the model showed that the resistant strains cannot be maintained in the bladder.
FIG. 5.

Relative growth rates (tgen susceptible parent without fosfomycin/tgen resistant mutant with fosfomycin) in urine (open symbols) and LB (solid symbols) of the susceptible parent strain (NU14) and resistant mutants isolated in vitro as a function of fosfomycin concentration; NU14 (circles), DA6313 (squares), DA6328 (diamonds), and DA6395 (triangles). Note that strain DA6313 does not grow in urine at >64 mg of fosfomycin per liter.
DISCUSSION
Fosfomycin resistance can be conferred by a number of different chromosomal mutations. Among the in vitro-derived and clinical isolates examined here, we identified mutations in at least five genes that can interfere with the GlpT and/or UhpT transport system and thereby result in fosfomycin resistance. The mutants found clinically represent a subset of the mutants found in vitro, suggesting that certain types of mutants are counterselected in vivo. Thus, no cyaA mutations were found in vivo. These mutations confer resistance by lowering cAMP levels and thereby expression of the GlpT and UhpT transporters. One potential explanation for the absence of these mutants in vivo is that they could be defective for growth in feces (or outside the host) because of their disturbed carbon metabolism. This might prohibit them from establishment in a host, and therefore they can never be transmitted from the intestine and cause a UTI.
As shown here, resistance to fosfomycin is easily acquired in vitro, and the high mutation frequency results from the large mutational target (several genes) and the fact that loss-of-function mutations in any of these genes can confer resistance. Mutation rates for inactivation of other genes (e.g., lacI and tonB) in E. coli are in a similar range (39, 46). In spite of this high mutation frequency in vitro, fosfomycin resistance in clinical isolates is rare. To quantitatively examine this phenomenon, we modeled the probability of resistance development in the bladder. Using the mutation frequency determined here, the number of bacteria present in the bladder during infection, and the flow dynamics of the bladder, the model suggested that resistance should develop often during UTI treatment unless the resistance mutation has a fitness cost. If this cost is sufficiently high, the resistant bacteria that appear will not increase in number and go to fixation in the bladder. The theoretical analysis by Gordon and Riley (14) demonstrated that a threshold level of growth is required for bacteria to establish in the bladder. This growth rate is near the maximum growth rate that E. coli may attain in urine (14).
Since most of the in vitro-selected mutants showed a decreased growth rate in both the absence and presence of fosfomycin, this provides a conceivable explanation for why most of the resistant bacteria have difficulty becoming established in the bladder. Clinical data also support this notion. It was shown in a treatment study that resistant strains with a MIC of >128 mg per liter in vitro could still be eradicated by fosfomycin treatment, suggesting that growth is decreased sufficiently by fosfomycin to prevent establishment even of these highly resistant strains (34). This reasoning applies irrespective of whether the fosfomycin-resistant mutants were selected in the bladder during treatment or were transmitted to the bladder from feces. An important general inference from these results is that the mutation frequency to resistance may not be the most relevant parameter (it might even be misleading) to consider when evaluating antibiotics with respect to how rapidly resistance will develop. Instead, biological fitness (in the absence and presence of antibiotic) is an additional significant parameter that ought to be analyzed when assessing the risk of drug resistance development (2, 11, 26)
Our hypothesis that fosfomycin-resistant mutants are unable to establish themselves in the bladder due to their lowered fitness is compatible with the experimental data and the model. Another factor (not included in the model) that could contribute to reducing resistance development is the effect of fosfomycin on bacterial adhesion to epithelial cells. Many strains of E. coli can adhere to the bladder epithelium, and as a result they could be maintained in the bladder even though their growth rate is below the threshold required to prevent washout. Thus, if the antibiotic also decreases adhesion, this might further prevent bacterial establishment. Indeed, it has been shown that fosfomycin decreases bacterial adhesion (27), and conceivably this effect could also reduce resistance development. Furthermore, some of the resistance mutations (e.g., cyaA and ptsI) will also decrease adhesion because they result in lowered cAMP levels, and as a consequence pilus biosynthesis is reduced (6). As shown previously, many fosfomycin-resistant mutants exhibit reduced adhesion to epithelial cells (12, 13, 21). Finally, an explanation for the limited human spread of the rare existing fosfomycin-resistant strains could be that they are disturbed for growth in feces and therefore their spread among people is hampered.
Acknowledgments
This work was supported by grants to D.I.A. from the Swedish Research Council (VR), the Swedish Institute for Infectious Disease Control (SMI), the Swedish Strategic Research Foundation (SSF), the AFA Research Fund, and the European Union 5th Framework Programme.
We thank Lars G. Burman, Sophie Maisnier-Patin, and Cecilia Dahlberg for critical reading of the manuscript.
REFERENCES
- 1.Alper, M. D., and B. N. Ames. 1978. Transport of antibiotics and metabolite analogs by systems under cyclic AMP control: positive selection of Salmonella typhimurium cya and crp mutants. J. Bacteriol. 133:149-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson, D. I., and B. R. Levin. 1999. The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 2:489-493. [DOI] [PubMed] [Google Scholar]
- 3.Arca, P., C. Hardisson, and J. E. Suarez. 1990. Purification of a glutathione S-transferase that mediates fosfomycin resistance in bacteria. Antimicrob. Agents Chemother. 34:844-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arca, P., G. Reguera, and C. Hardisson. 1997. Plasmid-encoded fosfomycin resistance in bacteria isolated from the urinary tract in a multicentre survey. J. Antimicrob. Chemother. 40:393-399. [DOI] [PubMed] [Google Scholar]
- 5.Arca, P., M. Rico, A. F. Brana, C. J. Villar, C. Hardisson, and J. E. Suarez. 1988. Formation of an adduct between fosfomycin and glutathione: a new mechanism of antibiotic resistance in bacteria. Antimicrob. Agents Chemother. 32:1552-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Båga, M., M. Göransson, S. Normark, and B. E. Uhlin. 1985. Transcriptional activation of a pap pilus virulence operon from uropathogenic Escherichia coli. EMBO J. 4:3887-3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergan, T. 1995. Pharmacokinetics of fosfomycin. Rev. Contemp. Pharmacother. 6:55-62. [Google Scholar]
- 8.Björkholm, B., M. Sjölund, P. G. Falk, O. G. Berg, L. Engstrand, and D. I. Andersson. 2001. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc. Natl. Acad. Sci. USA 98:14607-14612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Björkman, J., and D. I. Andersson. 2000. The cost of antibiotic resistance from a bacterial perspective. Drug Resist. Update 3:237-245. [DOI] [PubMed] [Google Scholar]
- 10.Björkman, J., D. Hughes, and D. I. Andersson. 1998. Virulence of antibiotic-resistant Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 95:3949-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Björkman, J., I. Nagaev, O. G. Berg, D. Hughes, and D. I. Andersson. 2000. Effects of environment on compensatory mutations to ameliorate costs of antibiotic resistance. Science 287:1479-1482. [DOI] [PubMed] [Google Scholar]
- 12.Carlone, N. A., M. Borsotto, A. M. Cuffini, and D. Savoia. 1987. Effect of fosfomycin trometamol on bacterial adhesion in comparison with other chemotherapeutic agents. Eur. Urol. 13:86-91. [DOI] [PubMed] [Google Scholar]
- 13.Gismondo, M. R., L. Drago, C. Fassina, M. L. Garlaschi, M. Rosina, and A. Lombardi. 1994. Escherichia coli: effect of fosfomycin trometamol on some urovirulence factors. J. Chemother. 6:167-172. [DOI] [PubMed] [Google Scholar]
- 14.Gordon, D. M., and M. A. Riley. 1992. A theoretical and experimental analysis of bacterial growth in the bladder. Mol Microbiol. 6:555-562. [DOI] [PubMed] [Google Scholar]
- 15.Hultgren, S. J., W. R. Schwan, A. J. Schaeffer, and J. L. Duncan. 1986. Regulation of production of type 1 pili among urinary tract isolates of Escherichia coli. Infect. Immun. 54:613-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kadner, R. J. 1995. Expression of the Uhp sugar-phosphate transport system of Escherichia coli, p. 263-274. In J. A. Hoch and T. J. Silhavy (ed.), Two-component signal transduction. ASM Press, Washington, D.C.
- 17.Kadner, R. J., and H. H. Winkler. 1973. Isolation and characterization of mutations affecting the transport of hexose phosphates in Escherichia coli. J. Bacteriol. 113:895-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahan, F. M., J. S. Kahan, P. J. Cassidy, and H. Kropp. 1974. The mechanism of action of fosfomycin (phosphonomycin). Ann. N.Y. Acad. Sci. 235:364-386. [DOI] [PubMed] [Google Scholar]
- 19.Kahlmeter, G. 2000. The ECO.SENS Project: a prospective, multinational, multicentre epidemiological survey of the prevalence and antimicrobial susceptibility of urinary tract pathogens—interim report. J. Antimicrob. Chemother. 46(Suppl. 1):15-22; discussion, 63-65. [PubMed] [Google Scholar]
- 20.Kahlmeter, G., D. F. J. Brown, F. W. Goldestein, A. P. MacGowan, J. W. Mouton, A. österlund, A. Rodloff, M. Steinbakk, P. Urbaskova, and A. Vatopoulos. European harmonization of MCI breakpoints for antimicrobial susceptibility testing of bacteria. J. Antimicrob. Chemother., in press. [DOI] [PubMed]
- 21.Klein, U., M. Pawelzik, and W. Opferkuch. 1985. Influence of beta-lactam antibiotics, fosfomycin and vancomycin on the adherence (hemagglutination) of Escherichia coli-containing different adhesins. Chemotherapy 31:138-145. [DOI] [PubMed] [Google Scholar]
- 22.Kronvall, G. G. Kahlmeter, E. Myhre, and M. F. Galas. 2003. A new method for normalized interpretation of antimicrobial resistance from disk test results for comparative purposes. Clin, Microbiol, Infect. 9:120-132. [DOI] [PubMed] [Google Scholar]
- 23.Langermann, S., S. Palaszynski, M. Barnhart, G. Auguste, J. S. Pinkner, J. Burlein, P. Barren, S. Koenig, S. Leath, C. H. Jones, and S. J. Hultgren. 1997. Prevention of mucosal Escherichia coli infection by FimH-adhesin-based systemic vaccination. Science 276:607-611. [DOI] [PubMed] [Google Scholar]
- 24.Lea, D. E., and C. A. Coulson. 1949. The distribution of the number of mutants in bacterial populations. J. Genet. 49:264-285. [DOI] [PubMed] [Google Scholar]
- 25.Levin, B. R. 2001. Minimizing potential resistance: a population dynamics view. Clin. Infect. Dis. 33(Suppl. 3):S161-S169. [DOI] [PubMed] [Google Scholar]
- 26.Levin, B. R., M. Lipsitch, V. Perrot, S. Schrag, R. Antia, L. Simonsen, N. M. Walker, and F. M. Stewart. 1997. The population genetics of antibiotic resistance. Clin. Infect. Dis. 24(Suppl. 1):S9-S16. [DOI] [PubMed] [Google Scholar]
- 27.Li Pira, G., C. Pruzzo, and G. C. Schito. 1987. Monuril and modification of pathogenicity traits in resistant microorganisms. Eur. Urol. 13:92-97. [PubMed] [Google Scholar]
- 28.Luria, S. E., and M. Delbrück. 1943. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28:491-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maisnier-Patin, S., O. G. Berg, L. Liljas, and D. I. Andersson. 2002. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol. Microbiol. 46:355-366. [DOI] [PubMed] [Google Scholar]
- 30.Merkel, T. J., J. L. Dahl, R. H. Ebright, and R. J. Kadner. 1995. Transcription activation at the Escherichia coli uhpT promoter by the catabolite gene activator protein. J. Bacteriol. 177:1712-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minassian, M. A., and J. D. Williams. 1995. The clinical pharmacology of fosfomycin trometamol. Rev. Contemp. Pharmacother. 6:45-53. [Google Scholar]
- 32.Mulvey, M. A., J. D. Schilling, J. J. Martinez, and S. J. Hultgren. 2000. Bad bugs and beleaguered bladders: interplay between uropathogenic Escherichia coli and innate host defenses. Proc. Natl. Acad. Sci. USA 97:8829-8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagaev, I., J. Björkman, D. I. Andersson, and D. Hughes. 2001. Biological cost and compensatory evolution in fusidic acid-resistant Staphylococcus aureus. Mol. Microbiol. 40:433-439. [DOI] [PubMed] [Google Scholar]
- 34.Neuman, M., and F. Rufin. 1987. Activity of Monuril in lower urinary tract infections due to fosfomycin-resistant bacteria. Eur. Urol. 13:105-107. [DOI] [PubMed] [Google Scholar]
- 35.Olekhnovich, I. N., J. L. Dahl, and R. J. Kadner. 1999. Separate contributions of UhpA and CAP to activation of transcription of the uhpT promoter of Escherichia coli. J. Mol. Biol. 292:973-986. [DOI] [PubMed] [Google Scholar]
- 36.Österberg, E., H. O. Hallander, A. Kallner, A. Lundin, S. B. Svensson, and H. Åberg. 1990. Female urinary tract infection in primary health care: bacteriological and clinical characteristics. Scand. J. Infect. Dis. 22:477-484. [DOI] [PubMed] [Google Scholar]
- 37.Reynolds, M. G. 2000. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156:1471-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts, F. J. 1986. Quantitative urine culture in patients with urinary tract infection and bacteremia. Am. J. Clin. Pathol. 85:616-618. [DOI] [PubMed] [Google Scholar]
- 39.Schaaper, R. M., and R. L. Dunn. 1991. Spontaneous mutation in the Escherichia coli lacI gene. Genetics 129:317-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schrag, S. J., and V. Perrot. 1996. Reducing antibiotic resistance. Nature 381:120-121. [DOI] [PubMed] [Google Scholar]
- 41.Schrag, S. J., V. Perrot, and B. R. Levin. 1997. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc. R. Soc. Lond. B Biol. Sci. 264:1287-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schumacher, G., and K. Bussmann. 1978. Cell-free synthesis of proteins related to sn-glycerol-3-phosphate transport in Escherichia coli. J. Bacteriol. 135:239-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sherman, D. R., K. Mdluli, M. J. Hickey, T. M. Arain, S. L. Morris, C. E. Barry 3rd, and C. K. Stover. 1996. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272:1641-1643. [DOI] [PubMed] [Google Scholar]
- 44.Stamm, W. E., K. F. Wagner, R. Amsel, E. R. Alexander, M. Turck, G. W. Counts, and K. K. Holmes. 1980. Causes of the acute urethral syndrome in women. N. Engl. J. Med. 303:409-415. [DOI] [PubMed] [Google Scholar]
- 45.Tsuruoka, T., A. Miyata, and Y. Yamada. 1978. Two kinds of mutants defective in multiple carbohydrate utilization isolated from in vitro fosfomycin-resistant strains of Escherichia coli K-12. J. Antibiot. (Tokyo) 31:192-201. [DOI] [PubMed] [Google Scholar]
- 46.Yamamura, E., T. Nunoshiba, M. Kawata, and K. Yamamoto. 2000. Characterization of spontaneous mutation in the oxyR strain of Escherichia coli. Biochem. Biophys. Res. Commun. 279:427-432. [DOI] [PubMed] [Google Scholar]