

Abstract
Tricyclodecan-9-yl-xanthogenate (D609) has in vivo and in vitro antioxidant properties. D609 mimics glutathione (GSH), has a free thiol group, which upon oxidation forms a disulfide. The resulting dixanthate is a substrate for glutathione reductase, regenerating D609. Recent studies have also shown that D609 protects brain in vivo and neuronal cultures in vitro against the potential Alzheimer’s disease (AD) causative factor, Aβ (1-42)-induced oxidative stress and cytotoxicity. Mitochondria are important organelles with both pro- and anti-apoptotic factor proteins. The present study was undertaken to test the hypothesis that i.p. injection of D609 would provide neuroprotection against free radical-induced, mitochondria-mediated apoptosis in vitro. Brain mitochondria were isolated from gerbils 1 h post injection intraperitonially (i.p.) with D609 and subsequently treated in vitro with the oxidants Fe2+/H2O2 (hydroxyl free radicals), 2,2-azobis-(2-amidinopropane) dihydrochloride (AAPH, alkoxyl and peroxyl free radicals) and AD-relevant amyloid β-peptide 1-42 [Aβ (1-42)]. Brain mitochondria isolated from the gerbils previously injected i.p. with D609 and subjected to these oxidative stress inducers, in vitro, showed significant reduction in levels of protein carbonyls, protein-bound hydroxynonenal (HNE) [a lipid peroxidation product], 3-nitrotyrosine (3-NT), and cytochrome-c release compared to oxidant-treated brain mitochondria isolated from saline-injected gerbils. D609 treatment significantly maintains the GSH/GSSG ratio in oxidant-treated mitochondria. Increased activity of glutathione-S-transferase (GST), glutathione peroxidase (GPx) and glutathione reductase (GR) in brain isolated from D609-injected gerbils is consistent with the notion that D609 acts like GSH. These anti-apoptotic findings are discussed with reference to the potential use of this brain-accessible glutathione mimetic in the treatment of oxidative stress-related neurodegenerative disorders, including AD.
Keywords: Oxidative stress, mitochondria, cytochrome-c, Alzheimer’s disease, D609
Introduction
Oxidative stress has been implicated in many neurodegenerative disorders, including Alzheimer’s disease (AD) [1-4], AD is characterized clinically by progressive dementia and pathologically by extracellular amyloid protein deposits, intracellular neurofibrilary tangles (NFTs) (composed mostly of hyperphosphorylated tau protein), loss of synapses, mitochondrial dysfunction and programmed cell death (PCD) [5, 6]. The reduced energy metabolism in AD may be due to oxidative dysfunction of some of the key metabolic or mitochondrial enzymes [7-12], which may lead to increased reactive oxygen species (ROS) production.
It is well known that mitochondria are the major cellular site of energy production, and these organelles also play a key role in the ROS generation, resulting in oxidative damage to neurons. Amyloid deposition, oxidative stress, mitochondria DNA deletion and mitochondrial structural and functional abnormalities are prominent in AD [5, 13, 14]. Many pro-apoptotic signals and anti-apoptotic defenses converge in the mitochondria [15]. Protein factors (cytochrome-C, Apaf1, AIF and SMAC/DIBLO) and Ca2+ released from mitochondria during oxidative stress activate caspase dependent and/or caspase independent mechanisms that lead to apoptotic cell death [15]. The role of mitochondria is not only as ATP producers but also as regulators of intracellular Ca2+ homeostasis and endogenous producers of ROS. Increased mitochondrial Ca2+ overload has been associated with the generation of superoxide and the release of pro-apoptotic mitochondrial proteins leading to cell death [16, 17]. The alterations in Ca2+ homeostasis and ROS generation lead to increased susceptibility to cell death under circumstances that are otherwise not ordinarily toxic [17].
In AD brain ROS lead to protein oxidation [1, 7-12], lipid peroxidation [2, 3], DNA and RNA oxidation [18-20], and neuronal dysfunction or death. Recent studies indicate that protein oxidation and lipid peroxidation in brain from mild cognitive impairment subjects [21, 22], suggesting oxidative stress is an early event in the pathogenesis of AD. ROS generation from mitochondria and its impact on neuronal systems hence becomes important in understanding oxidative stress and oxidative stress-related disorders, including AD.
Mitochondrial electron transport is a potential source of ROS production [23]. It is now well known that ROS such as superoxide anion (O2-), hydroxyl radical (OH), hydrogen peroxide (H2O2), and peroxynitrite (ONOO-) contribute to neurodegeneration [21, 24-28]. Mitochondrial membrane potential depolarization induces cytochrome-c release into the cytoplasm and elevates the activity of caspase-3, suggesting a role for mtDNA-derived mitochondrial dysfunction in AD degeneration [5].
Glutathione is widely recognized as an endogenous nonenzymatic antioxidant, an oxyradical scavenger, thereby useful in protecting against oxidative damage by free radicals and inhibiting lipid peroxidation and DNA damage [29-33]. Glutathione has been implicated in a wide range of metabolic processes, including cell division, DNA repair, regulation of enzyme activity, activation of transcription factors, modulation of anion and cation homeostasis and protection against oxidative damage [34]. The nervous system is particularly susceptible to oxidative insults, and dependent on its glutathione defense.
Tricyclodecan-9-yl-xanthogenate (D609) exhibits a variety of potent biological functions, including antiviral [35] and anti-inflammatory [36-38] activities. Most of these activities have been linked to the inhibitory effect of D609 on phosphatidylcholine-specific phospholipase C (PC-PLC) [35, 39]. Such inhibition decreases production of the secondary messenger diacylglycerol (DAG) that activates protein kinase C (PKC) and acidic sphingomyelinase (aSMase) [40]. However, with a free thiol group, D609 may also possess strong antioxidant activity [41] with in vitro and in vivo radical scavenging properties and inhibition of free radial-induced oxidative stress [42-46].
The particular species of ROS that D609 can effectively scavenge is not clear, but this xanthate has ability to scavenge hydroxyl radicals [42-46]. The reaction with other ROS is also possible since xanthates generally have high reductive potential [41]. D609 may protect intracellular GSH, which is important intracellular defense molecule against oxidative stress in neurons and has been shown to play an important role in radiation protection [43, 47, 48]. Recently, we showed that D609, a glutathione mimetic [44], protects primary neuronal culture against amyloid β-peptide (1-42) [Aβ (1-42)]-induced oxidative stress and neurotoxicity in vitro [49] and in synaptosomes in vivo [46].
We performed the current study to test the hypothesis that in vivo loading with D609 had and antioxidant effect on isolated brain mitochondria exposed to various oxidants, including Aβ (1-42).
Materials and Methods
Animals
For all studies male Mongolian gerbils (2-3 months of age), approximately 100 g in size, housed in the University of Kentucky Central Animal Facility under 12-h light/dark conditions and fed standard Purina rodent laboratory chow ad libitum, were used. The animal protocols were approved by the University of Kentucky Animal Care and Use Committee.
Materials
D609 was purchased from Biomol (Plymouth Meeting, PA, USA) and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fresh D609 (50 mg/kg body wt) was prepared in phosphate-buffered saline (PBS). The primary anti body for 4-hydroxy-nonenal (HNE) and 3-nitrotyrosine (3-NT) were purchased from Chemicon International.
Preparation of mitochondria
Brain mitochondria were isolated from gerbils previously injected i.p. with saline (control) or with D609 (50 mg/kg body wt), 60 min after injection. This time and level of D609 were chosen based on previous [42] dose-response experiments. The brain mitochondria were isolated according to the procedure of Sims et al. [49] with minor modifications. Gerbils were decapitated and the whole brain was isolated on ice. Whole brain was homogenized in ice-cold isolation buffer (250 mM sucrose, 10 mM HEPES, and 1 mM potassium EDTA, pH 7.2, 4 μg/ml leupeptin, 4 μg/ml pepstatin, 5 μg/ml aprotinin 20 μg/ml trypsin inhibitor) with 6 passes of a Wheaton tissue homogenizer. The homogenate was centrifuged for 3 min at 1,330× g at 4°C, and the resulting pellet was resuspended in isolation buffer and centrifuged at 1,330× g for 3 min. The supernatants from both spins were combined and spun at 21,200x g for 10 min at 4°C. The pellet was resuspended in 15% Percoll solution (v/v in isolation buffer) and layered onto discontinuous Percoll gradients of 23 and 40% Percoll (v/v in isolation buffer). Gradients were centrifuged at 30,700× g for 5 min at 4°C. At the 23-40% Percoll interface, mitochondria were isolated and resuspended in respiration buffer (250 mM sucrose, 2 mM magnesium chloride, 20 mM HEPES, and 2.5 mM phosphate buffer, pH 7.2) and centrifuged at 16,700× g for 10 min at 4°C. The pellet was resuspended in respiration buffer, centrifuged at 6,900× g for 10 min at 4°C, and the resulting pellet was washed in PBS at 6,900× g for 10 min at 4°C.
Protein estimation and treatment
The pellet was resuspended in 250 μl PBS and protein concentration determined by the Pierce BCA method [50], using bovine serum albumin (BSA) as a standard. The mitochondrial samples were divided into six aliquots and incubated at 37 °C. The first sample set was incubated without treatment of any oxidant for 1 h; the second set of mitochondria was incubated with 30 μM Fe SO4 and 2.0 mM H2O2, a process that induces hydroxyl radical formation [51], the third and fourth set of samples were also incubated in same manner with/or without the oxidant AAPH (1 mM) for 1 h at 37 °C. The fifth sample set was incubated without treatment of any oxidant for 6 h and the sixth sample set was incubated with Aβ(1-42) for 6 h. The mitochondrial samples were washed after incubation and resuspended in PBS/Tween.
Protein carbonyls
Protein carbonyls are markers of protein oxidation and were assessed by following the standard protocol described previously [42]. Samples (5 μl) (normalized to 4 mg/ml), 5 μl of 12% sodium dodecyl sulfate (SDS), and 10 μl of 10 times diluted 2,4-dinitrophenyl hydrazine (DNPH) from 200 mM stock were incubated at room temperature for 20 min. Samples were neutralized with 7.5 μl neutralization solution (2 M Tris in 30% glycerol). The resulting solution was loaded into each well on nitrocellulose membrane under vacuum using a slot-blot apparatus. The membrane was blocked in blocking buffer (3% bovine serum albumin) in PBS/Tween for 1 h and incubated with a 1:100 dilution of anti-DNP polyclonal antibody in PBS/Tween for 1 h. The membrane was washed three times in PBS/Tween and was incubated for 1 h with an anti-rabbit IgG alkaline phosphatase secondary antibody diluted in PBS/Tween in a 1:8000 ratio. The membrane was washed three times in PBS/Tween for 5 min and developed in Sigma Fast tablets (BCIP/NBT substrate). Blots were dried, scanned with Adobe PhotoShop, and quantified with Scion Image (PC version of Macintosh-compatible NIH Image). No non-specific binding of antibody to the membrane was observed.
3-Nitrotyrosine (3-NT)
Samples (5 μl) (normalized to 4 mg/ml), 5 μl of 12% SDS, and 5 μl of modified Laemmli buffer containing 0.125 M Tris base, pH 6.8, 4% (v/v) SDS, and 20% (v/v) glycerol were incubated for 20 min at room temperature. Sample (250 ng) was loaded into each well on a nitrocellulose membrane in a slot blot apparatus under vacuum. The membrane was blocked in blocking buffer (3% bovine serum albumin) in PBS/Tween for 1 h and incubated with a 1:2000 dilution of anti-3-N T polyclonal antibody in PBS/Tween for 1 h 30 min. The membrane was washed in PBS/Tween for 5 min three times after incubation. The membrane was incubated for 1 h, after washing, with an anti-rabbit IgG a lkaline phosphatase secondary anti body diluted in PBS/Tween in a 1:8000 ratio. The membrane was washed three times in PBS/Tween for 5 min and developed in Sigma Fast tablets. Blots were dried, scanned with Adobe PhotoShop, and quantified with Scion Image as above. No non-specific binding of antibody to the membrane was observed.
4-Hydroxynonenal (HNE, an index of lipid peroxidation)
Sample (5 μl) (normalized to 4 mg/ml), 5 μl of 12% SDS, and 5 μl of modified Laemmli buffer containing 0.125 M Tris base, pH 6.8, 4% (v/v) SDS, and 20% (v/v) glycerol were incubated for 20 min at room temperature. Sample (250 ng) was loaded into each well on a nitrocellulose membrane in a slot blot apparatus under vacuum. The membrane was blocked in blocking buffer (3% bovine serum albumin) in PBS/Tween for 1 h and incubated with a 1:5000 dilution of anti-HNE polyclonal antibody in PBS/Tween for 1 h 30 min. The membrane was washed in PBS/Tween for 5 min three times after incubation. The membrane was incubated for 1 h, after washing, with an anti-rabbit IgG alkaline phosphatase secondary antibody diluted in PBS/Tween in a 1:8000 ratio. The membrane was washed three times in PBS/Tween for 5 min and developed in Sigma Fast tablets. Blots were dried, scanned with Adob PhotoShop, and quantified with Scion Image as above. A faint background staining due to the antibody alone was observed, but since each sample had a control, this minor effect was well controlled.
Estimation of cytochrome-c release
Cytochrome c release was detected by the method of Yang et al., (1997) [52]with slight modification. After incubation and spun of mitochondrial samples, supernatant was used for Western blot analysis to cytochrome c release. The membrane was blocked in blocking buffer (3% bovine serum albumin) in PBS/Tween for 1 h and incubated with a 1:2000 dilution of anti-cytochrome-c polyclonal antibody (C-5723; anti-sheep; Sigma) in PBS/Tween for 1 h 30 min. The membrane was washed in PBS/Tween for 5 min three times after incubation. The membrane was incubated for 1 h, after washing, with an anti-sheep IgG alkaline phosphatase secondary anti body diluted in PBS/Tween in a 1:8000 ratio. The membrane was washed three times in PBS/Tween for 5 min and developed in Sigma Fast tablets. Blots were dried, scanned with Adobe PhotoShop, and quantified with Scion Image as above.
Estimation of reduced glutathione (GSH)
Determination of GSH was performed by the method of Hissin and Hilf [53]. The reaction mixture containing 0.1 M sodium phosphate buffer (pH-8.0), 5.0 mM EDTA, 10 μl O-phthaldehyde (1.0 mg/ml) and 10 μl of sample. After incubation for 15 min at room temperature, fluorescence at emission 420 nm was recorded by excitation at 350nm.
Estimation of oxidized glutathione (GSSG)
The estimation of GSSG was performed by the method of Hissin and Hilf [53]. The samples were incubated first with 0.04 M N-ethyleimide (NEM) for 30 min to interact with GSH present in sample. The reaction mixture containing 0.1 N NaOH, 5.0 mM EDTA, 10 μl O-phthaldehyde (1.0 mg/ml) and 10 μl of sample. After incubation for 15 min at room temperature, fluorescence at emission 420 nm was recorded by excitation at 350nm.
Estimation of glutathione-S-transferase activity
GST (EC 2.5.1.18) activity was measured in a 96-well plate reader, with the reaction mixture consisting of 0.1 M phosphate buffer (pH 6.5), 1.0 mM reduced glutathione, 1.0 mM CDNB and 0.1 μl of PMS in a total volume of 200 μl[54]. The changes in absorbance were recorded at 340 nm, and the enzymatic activity was calculated as nmol CDNB conjugate formed min-1 mg-1 protein.
Estimation of glutathione peroxidase activity
GPx (EC 1.11.1.9) activity was measured in a 96-well plate reader at 37 °C by a coupled assay system [55]. The reaction mixture consisted of 0.2 mM H2O2, 1.0 mM GSH, 0.14 U of GR, 1.5 mM NADPH, 1.0 mM sodium azide and 0.1M phosphate buffer (pH 7.4) and 10 μl PMS in a total volume of 200 μl. The enzyme activity was calculated as nmol NADPH oxidized min-1 mg-1 protein.
Estimation of glutathione reductase activity
The assay system to estimate GR (EC 1.6.4.2) activity consisted of 0.1 M phosphate buffer (pH 7.6), 0.5 mM EDTA, 1.0 mM oxidized glutathione, 0.1 mM NADPH and 10 μl PMS in a total volume of 200 μl [56]. The enzyme activity was assayed in a 96-well plate reader by measuring the disappearance of NADPH at 340 nm and was calculated as nmol NADPH oxidized min-1 mg-1 protein.
Statistical analysis
ANOVA was used for statistical evaluation of data followed by Student’s t-test. Results are presented as mean ± SEM. P-values less than 0.05 were considered significant.
Results
Protein Oxidation and Lipid Peroxidation
Fig. 1, 2 and 3 show the levels of protein carbonyl, 3NT and HNE, respectively, in mitochondria isolated from brain of gerbils that had been previously injected i.p. with saline or D609. These isolated brain mitochondrial samples were subsequently treated with Fe2+/H2O2, AAPH or Aβ (1-42) in vitro. The concentrations of these oxidants were chosen based on prior dose-response studies of the agents in in vivo investigations [42, 46]. The levels of protein carbonyl, 3NT and HNE were found significantly increased in mitochondria isolated from saline-injected gerbils brain and subsequently treated with oxidants, in vitro, compared to control (without treatment of oxidants). Brain mitochondria isolated from gerbils previously injected i.p. with D609 and subsequent in vitro treatment with Fe2+/H2O2, AAPH and Aβ (1-42) showed significantly decreased protein carbonyl, 3NT and HNE levels as compared to brain mitochondria isolated from gerbils previously injected i.p. with saline and subsequently treated with Fe2+/H2O2, AAPH and Aβ (1-42), in vitro. The D609-alone sample had no significant effect on the levels of protein carbonyls, 3NT or HNE in brain mitochondria. It is clear that less protein oxidation and lipid peroxidation product, HNE was found in Fe2+/H2O2, AAPH and Aβ (1-42) treated mitochondria isolated from gerbils previously injected with D609.
Figure 1.
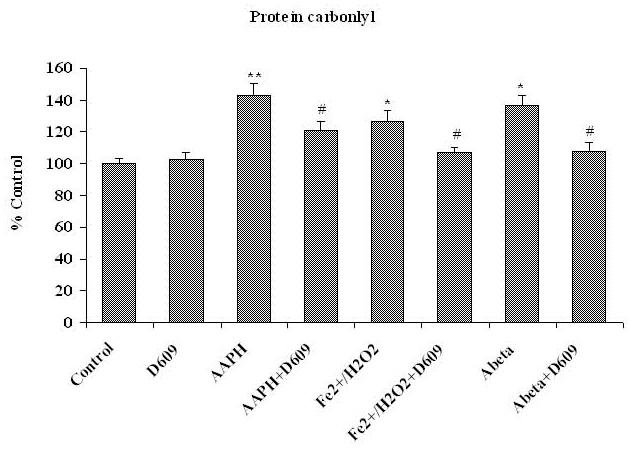
Shows the increment in protein carbonyl formation in brain mitochondria isolated from saline-injected gerbils and treated with various oxidants [AAPH, Fe2+/H2O2 or Aβ (1-42)] compared to control. The protective effects of D609 against protein carbonyl formation in brain mitochondria isolated from gerbils injected i.p. 1h before sacrifice with D609 and treated with AAPH, Fe2+/H2O2 and Aβ (1-42) also are shown. *p<0.01 and **p<0.001 compared to control and # p<0.01 compared to oxidant treatment. The data are presented as mean ± SEM expressed as percentage of control (n=6).
Figure 2.
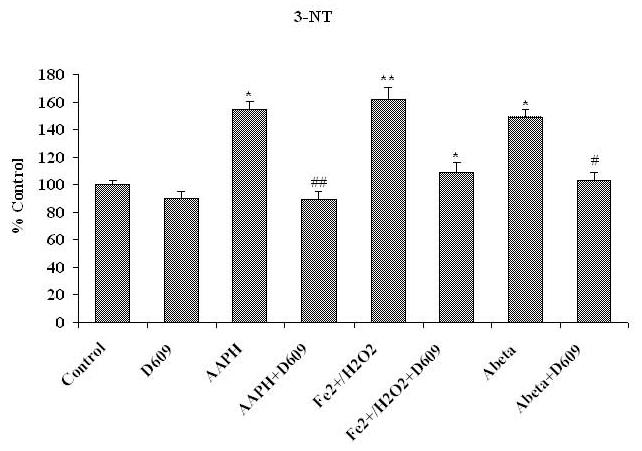
Shows the increment in 3-NT levels in brain mitochondria isolated from saline-injected gerbils and subsequently treated with AAPH, Fe2+/H2O2 or Aβ (1-42) compared to 3-NT levels in brain mitochondria isolated from saline-injected gerbil that received no treatment of any oxidant, *p<0.01 and **p<0.001. This figure also shows decreased 3-NT levels shows in brain mitochondria isolated from gerbils previously injected i.p. with D609 1 h before sacrifice and treated with AAPH, Fe2+/H2O2 or Aβ (1-42) compared to the oxidant treatment but no prior injection of D609, # p<0.01. The data are presented as mean ± SEM expressed as percentage of control (n=6).
Figure 3.
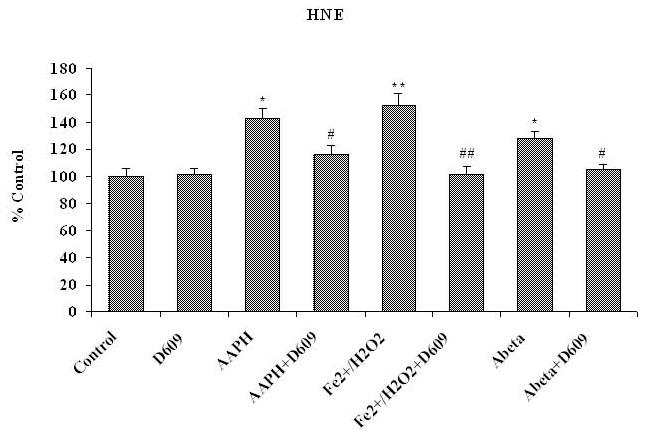
Shows the significantly elevated protein-bound HNE content in brain mitochondria isolated from saline-injected gerbils and treated with different oxidants [AAPH, Fe2+/H2O2 or Aβ (1-42)]. The protective effects of D609 against HNE formation of protein-bound HNE in brain mitochondria isolated from gerbil injected i.p. with D609 1 h before sacrifice and treated with AAPH, Fe2+/H2O2 or Aβ (1-42) also are shown. *p<0.01 and **p<0.001 compared to control, # p<0.01 and ## p<0.001 compared to oxidant treatment. The data are presented as mean ± SEM expressed as percentage of control (n=6).
Reduced glutathione (GSH)
Fig. 4A shows the levels of GSH in mitochondria isolated from gerbil brain from saline-as well as D609-injected rodents. These brain mitochondrial samples were subsequently treated with Fe2+/H2O2, AAPH or Aβ (1-42) in vitro. The levels of GSH were found significantly decreased in mitochondria isolated from saline-injected gerbils brain and treated in vitro with oxidants compared to control. In contrast, brain mitochondria isolated from D609-injected gerbils and subsequently treated in vitro with Fe2+/H2O2, AAPH or Aβ (1-42) showed increased levels of GSH.
Figure 4A.
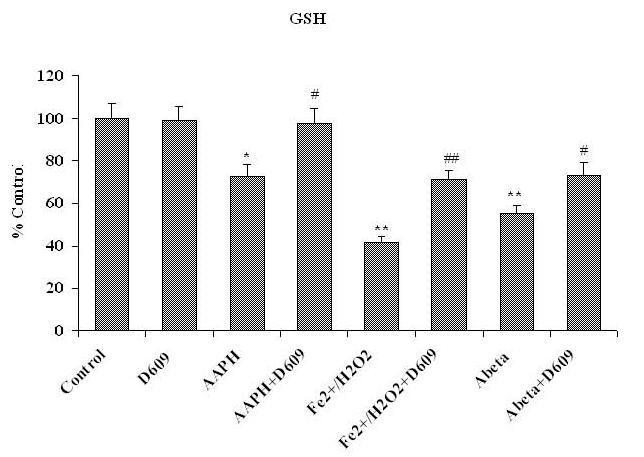
Shows a significant decrement in GSH levels in brain mitochondria isolated from saline-injected gerbils and subsequently treated with AAPH, Fe2+/H2O2 or Aβ (1-42) compared to GSH levels in brain mitochondria isolated from saline-injected gerbils not subjected to treatment of any oxidant. Also shown is the protection of GSH levels in brain mitochondria isolated from gerbils previously injected i.p. with D609 1 h before sacrifice and treated with AAPH, Fe2+/H2O2 or Aβ (1-42) compared to GSH levels in brain mitochondria isolated from saline-treated gerbils and then treated with oxidants. *p<0.01 and **p<0.001 compared to control, # p<0.01 and ## p<0.01 compared to oxidant treatment. The data are presented as mean ± SEM expressed as percentage of control (n=6).
Oxidized Glutathione (GSSG)
Fig. 4B shows significantly increased GSSG levels in brain mitochondria treated with different oxidants [Fe2+/H2O2, AAPH or Aβ (1-42)] that were isolated from saline-injected gerbils (control). In contrast, there were significantly decreased GSSG levels in mitochondria isolated from gerbils previously injected i.p. with D609 and treated with Fe2+/H2O2, AAPH or Aβ (1-42) in vitro.
Figure 4B.
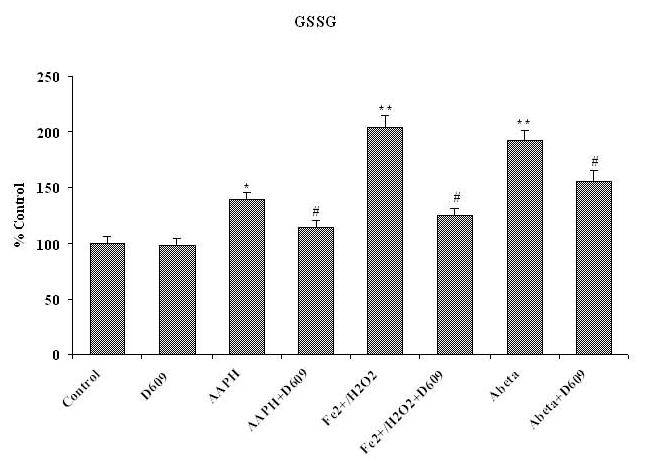
Shows the increased level of GSSG in brain mitochondria isolated from saline-injected gerbils and subsequently treated with AAPH, Fe2+/H2O2 or Aβ (1-42) as compared to GSSG levels in brain mitochondria isolated from saline-injected gerbils but not subjected to treatment of any oxidant. The reduction in GSSG level shows in brain mitochondria isolated from gerbils previously injected i.p. with D609 1 h before sacrifice and treated with AAPH, Fe2+/H2O2 or Aβ (1-42) compared to GSSG levels in brain mitochondria isolated from saline-treated gerbil and then treated with oxidants. *p<0.01 and **p<0.001 compared to control, # p<0.01 and ## p<0.01 compared to oxidant treatment. The data are presented as mean ± SEM expressed as percentage of control (n=6).
The Ratio of Reduced and Oxidized Glutathione (GSH/GSSG)
Fig.4C shows the significantly decreased GSH/GSSG ratio in brain mitochondria isolated from saline-injected gerbils (control) and treated with different oxidants in vitro. This GSH/GSSG ratio was significantly increased in brain mitochondria isolated from gerbils previously injected i.p. with D609 and treated with Fe2+/H2O2, AAPH or Aβ (1-42) in vitro.
Figure 4C.
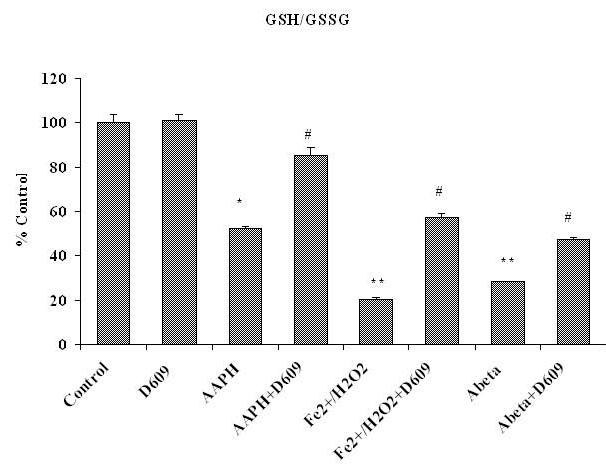
Shows the ratio of GSH/GSSG, decreased in brain mitochondria isolated from saline-injected gerbils and subsequently treated with AAPH, Fe2+/H2O2 or Aβ (1-42), compared to the GSH/GSSG ratio in brain mitochondria isolated from saline-injected gerbils but not subjected to treatment of any oxidant. The increment in the ratio of GSH/GSSG in brain mitochondria isolated from gerbils previously injected i.p. with D609 1 h before sacrifice and then treated with AAPH, Fe2+/H2O2 or Aβ (1-42) compared to this ratio determined in brain from mice treated with oxidant but no pre-injection of gerbils with D609 is also shown. *p<0.01 and **p<0.001 compared to control, # p<0.01 and ## p<0.01 compared to oxidant treatment. The data are presented as mean ± SEM expressed as percentage of control (n=6).
Cytochrome-c Release
Reactive oxygen species are produced by mitochondria that can cause the release of cytochrome-c from the mitochondrial membrane. Fig. 5 shows the level of cytochrome-c released from brain mitochondria isolated from saline-as well as D609-injected gerbils and subsequently treated with different oxidants in vitro. There was a significant reduction in cytochrome-c release from brain mitochondria by prior i.p. D609 treatment compared to that in brain mitochondria isolated from saline-injected gerbils. Increased cytochrome-c release from brain mitochondria isolated from control gerbils with subsequent in vitro treatment of Fe2+/H2O2, AAPH or Aβ (1-42) compared to control was discussed. Brain mitochondria isolated from D609-injected gerbils and subsequently treated with Fe2+/H2O2, AAPH or Aβ (1-42), in vitro, showed a significant decrease in cytochrome-c release compared to mitochondria isolated from saline-injected gerbils treated with oxidants (Fig.5).
Figure 5.
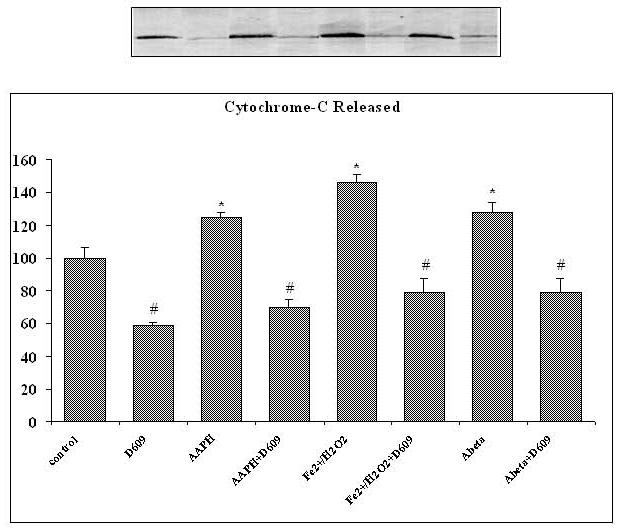
Shows the increased level of cytochrome-c released from brain mitochondria isolated from saline-injected gerbils and treated with various oxidants (AAPH, Fe2+/H2O2 or Aβ (1-42) as compared to cytochrome-c released from brain mitochondria isolated from saline-injected gerbils but not subjected to treatment of any oxidant. Also shown is the decrement of cytochrome-c release from brain mitochondria isolated from gerbils previously injected i.p. with D609 1 h before sacrifice and treated with AAPH, Fe2+/H2O2 or Aβ (1-42) compared to that released from brain mitochindria isolated from gerbils subjected to oxidant treatment. The D609 only treatment shows significantly less cytochrome-c release compared control. *p<0.01 as compared to control, # p<0.01 compared to oxidant treatment. The data are presented as mean ± SEM expressed as percentage of control (n=5).
The Activity of GSH-relevant Enzymes
Table I shows the activity of some GSH-relevant enzymes in gerbil brain from which mitochondria were isolated. The activities of glutathione-S-transferase (GST) and glutathione peroxidase (GPx) were increased significantly in post mitochondrial supernatant from D609-injected gerbils. The activity of glutathione reductase (GR) also was increased, but non-significantly. Thus, these results suggest that D609 has a role in the redox cycle of GSH (oxidation and reduction of GSH) and in the neuroprotection against free radicals.
Table I.
Activities of some GSH-related enzymes in brain homogenate obtained from gerbils that previously had been injected i.p. with saline (control) or D609
| Enzyme | Brain from Control Group | Brain from D609 Treated Group |
|---|---|---|
| GST (nmol CDNB conjugate formed min-1 mg-1 protein) | 38.4±3.44 | 61.5±4.74* |
| GPx (nmol NADPH oxidized min-1 mg-1 protein) | 34.5±2.87 | 43.1±3.41# |
| GR (nmol NADPH oxidized min-1 mg-1 protein) | 23.4±2.89 | 26.6±3.10 |
a. The protective effect of D609 on activity of glutathione -S-transferase, glutathione peroxidase and glutathione reductase isolated from gerbil barin from which mitochondria were obtained. The data are presented as mean ± SEM expressed as percentage of control (n=6).
p<0.01
p<0.05 compared to control.
Discussion
Oxidative stress reflects a marked imbalance between ROS and their removal by antioxidant systems. This imbalance may originate from an overproduction of ROS or from a reduction in antioxidant defenses or both [57]. An inverse relationship between lipid peroxidation and GSH and its dependent enzymes, along with the activities of catalase and superoxide dismutase is well known [31, 32]. A reduction in GSH may impair H2O2 clearance and promote hydroxyl radical formation, thus increasing the free radical load, which triggers oxidative stress. Conversely, GSH is converted to GSSG, a process that in conjunction with the cofactor NADPH can reduce lipid peroxides, free radicals and H2O2. NADPH also acts as a peroxynitrite reductant, thereby providing enzymatic defense against peroxynitrite [58]. Glutathione peroxidase (GPx) and glutathione reductase (GR) protect the neurons from oxidative stress by catalyzing the reduction of H2O2 at the expense of glutathione [31, 32]. Glutathine-S-transferase (GST) play a role in neuroprotection by catalyzing the formation of the GSH-HNE conjugate, which is then removed from neurons by the action of the multidrug resistant protein-1 [59] In AD brain, both GST and MRP-1 are oxidatively modified and likely dysfunctional [59]. In our results GST (Table I) and GPx have higher activities following in vitro oxidant treatment of brain homogenate isolated from gerbils previously injected with D609. GR activity in brain (Table I) of D609-injected animals was not significantly elevated following subsequent in vitro oxidant treatment to isolated mitochondria relative to control.
Reduced levels of GSH (Fig. 4A) have been observed in oxidative stress-related disorders [60] in specific regions of the central nervous system of AD patients, and thus reduced GSH may contribute to the neuronal cell dysfunction and/or loss. In AD, increased levels of GSSG [61] have been observed. Studies have shown that an increase in endogenous GSH levels by dietary or pharmacological intake of GSH precursors or GSH mimetics or substrates for GSH synthesis protects the brain against oxidative stress [47, 48, 62-66]. The increase in the content of GSH and decrease in the extent of GSSG in mitochondria isolated from gerbil brain previously injected with D609 in our study is in agreement with earlier reports [46].
We earlier showed that D609 could effectively scavenge hydroxyl radicals [43]. The identification of D609 as a potent antioxidant implies that D609 may exert some of the reported activities that have been largely attributed to the inhibition of phosphatidyl choline-specific phospholipase C (PC-PLC) by its antioxidant properties. Among these activities are inhibition of LPS-and TNF-induced NF-kB activation and inflammatory cytokine production [35, 39]. ROS activated NF-kB causes a differential change in gene expression between neurons and astrocytes in the AD brain [67, 68]. We earlier reported the anticarcinogenic activity of D609 might be due to its antioxidant property [43]. In vitro, D609 kills a variety of tumor cells but has limited toxic effects on normal cells [38]. Although D609 might be involved in alternate biochemical pathways, its glutathione mimetic property cannot be ignored. The free thiol group in D609 may act as substrate for hydroxyl radicals, or may act as electron acceptor from hydrogen peroxide to form a dixanthate, which forms the substrate for glutathione reductase to convert it back to xanthate. This property of D609 is yet to be explored.
Protein carbonyls, 3-NT and HNE levels are elevated in AD and MCI brains [3, 4, 7-12, 21, 22]. HNE is a highly reactive product of arachidonic acid metabolism that is believed to interfere with normal cellular functions [3]. During oxidative stress several lipid peroxidation products are formed, including HNE, one of the most abundant and toxic lipid-derived aldehydes, and which can induce oxidative stress [69, 70]. Lipid peroxidation products such as HNE and acrolein are known to cause damage to biomembranes, proteins and other biomolecules in AD brain [70-72]. These alkenals form an immediate substrate for GSH [73] and these lipid peroxidation products are known to be involved in apoptosis, which can be initiated as a consequence of GSH depletion [74].
GSH is known to detoxify HNE and protect cultured neurons against oxidative damage resulting from amyloid β-peptide, iron, and HNE [74]. GSH can also protect brain from damage by peroxynitrite, hydroxyl free radicals, or reactive alkenals [63-66, 75]. D609 may binds to reactive alkenals formed due to peroxidation of lipids and detoxifies their effects [44].
HNE can alter pyruvate dehydrogenase (PD) [76], decrease cell survival (decrease MTT reduction) and cause inhibition of Na+K+ ATPase [74]. The activity of the adenine nucleotide transporter in mitochondria is inhibited by HNE, which directly results in ATP depletion [77]. Aβ increases lipid derived free radical production [3, 4, 74, 77, 78]. HNE rapidly leads to decline in multiple sites of the mitochondrial respiratory chain and modified specific mitochondrial target proteins lead to apoptosis and cell death in AD [79].
Recently, we showed that the xanthate D609, a glutathione mimetic [44], protects primary neuronal culture against Aβ (1-42)-induced oxidative stress and neurotoxicity in vitro [45] and against Aβ (1-42) in vivo [46]. D609 has the ability to scavenge hydrogen peroxide and hydroxyl free radicals [44]. D609 can bind to reactive alkenals and detoxify their effect, thereby preventing these alkenals from damaging mitochondria [42, 44]. AAPH is known to induce protein carbonylation and nitration [80] in mitochondria. In the current study, we report that in vivo delivery of D609 inhibits the damage to brain mitochondria caused by production of hydroxyl free radicals, alkoxyl and peroxyl radicals and Aβ (1-42)-induced in vitro oxidative stress to decrease protein oxidation and lipid peroxidation.
Aβ disrupts Ca2+ homeostasis in neurons [81], and increased intracellular Ca2+ level can increase sphingomylinase activity to produce ceramide [82]. Activation of the apoptogenic sphingomyelin-dependent signaling pathway is mediated by ceramide [82] during oxidative stress to play role in the pathogenesis of neuronal disease [83]. Apoptosis induced by the membrane-permeable second messenger ceramide, followed by the release of cytochrome-c and Ca2+ from the mitochondria with the loss of mitochondrial transmembrane potential has been observed [83]. Both cytochrome-c release and rise of intracellular Ca2+ cause caspase-3 activation and nuclear condensation [83]. ROS-dependent and independent pathways are initiated by caspase-8 activation and contribute to ceramide formation via the activation of both neutral and acid sphingomyelinases (SMases) in TNF-alpha-induced apoptosis of human glioma cells [83]. In one study, D609 has also been shown to inhibit sphingomyelin synthase in SV40-transformed human lung fibroblasts, but ceramide and DAG levels evolved in opposite directions [84] and partially protected against apoptosis [85].
In conclusion, this study has demonstrated that, similar to the case with elevated cytosolic GSH protecting brain mitochondria [65], i.p. injection of D609 protects brain mitochondria (anti-apoptotic) against oxidative stress induced in vitro by different oxidants, such as AAPH, Fe2+/H2O2 or Aβ(1-42). In vivo delivery of D609 showed significant protection against protein oxidation, lipid peroxidation and cytochrome-c release in gerbil brain mitochondria. While a role for inhibition of PC-PLC by D609 in the protection of brain mitochondria cannot be excluded, based on the data from the present and previous studies [42, 46], the increment in the GSH level and the activity of its dependent enzymes (GST, GPx and GR), are strong indications to suggest that D609 has neuroprotective effects to brain mitochondria due in significant part to its antioxidant properties. Thus, in brain mitochondria the antioxidant properties of in vivo delivery of D609 conceivably could be beneficial in the treatment of diseases related to oxidative stress that involve mitochondria (AD, Huntington disease, Parkinson’s disease, for example). This xanthate compound protects against in vitro treatment of Aβ (1-42); consequently, we suggest D609 may be part of a promising therapeutic strategy for Alzheimer’s disease. Studies to test this notion in rodent models of AD are in progress.
Acknowledgement
This research was supported in part by grants from NIH [AG-10836; AG-05119].
References
- 1.Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65::2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 2.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging. 1998;19::33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 3.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32::1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 4.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7::548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 5.Bosetti F, Brizzi F, Barogi S, Mancuso M, Siciliano G, Tendi EA, Murri L, Rapoport SI, Solaini G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol Aging. 2002;23::371–376. doi: 10.1016/s0197-4580(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 6.Katzman R, Saitoh T. Advances in Alzheimer’s disease. Faseb J. 1991;5::278–286. [PubMed] [Google Scholar]
- 7.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33::562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 8.Castegna A, Thongboonkerd V, Klein JB, Lynn B, Markesbery WR, Butterfield DA. Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J Neurochem. 2003;85::1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 9.Castegna A, Thongboonkerd V, Klein J, Lynn BC, Wang YL, Osaka H, Wada K, Butterfield DA. Proteomic analysis of brain proteins in the gracile axonal dystrophy (gad) mouse, a syndrome that emanates from dysfunctional ubiquitin carboxyl-terminal hydrolase L-1, reveals oxidation of key proteins. J Neurochem. 2004;88::1540–1546. doi: 10.1046/j.1471-4159.2003.02288.x. [DOI] [PubMed] [Google Scholar]
- 10.Butterfield DA. Proteomics: a new approach to investigate oxidative stress in Alzheimer’s disease brain. Brain Res. 2004;1000::1–7. doi: 10.1016/j.brainres.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 11.Sultana R, Poon HF, Cai J, Pierce WM, Merchant M, Klein JB, Markesbery WR, Butterfield DA. Identification of nitrosatively modified proteins in Alzheimer’s disease brain using redox proteomics approach. NeuroBiol Dis. 2006 doi: 10.1016/j.nbd.2005.10.004. In press.: [DOI] [PubMed] [Google Scholar]
- 12.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2005.09.021. In press: [DOI] [PubMed] [Google Scholar]
- 13.Huang HM, Zhang H, Ou HC, Chen HL, Gibson GE. alpha-keto-beta-methyl-n-valeric acid diminishes reactive oxygen species and alters endoplasmic reticulum Ca(2+) stores. Free Radic Biol Med. 2004;37::1779–1789. doi: 10.1016/j.freeradbiomed.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Aliyev A, Chen SG, Seyidova D, Smith MA, Perry G, de la Torre J, Aliev G. Mitochondria DNA deletions in atherosclerotic hypoperfused brain microvessels as a primary target for the development of Alzheimer’s disease. J Neurol Sci. 2005;229-230::285–292. doi: 10.1016/j.jns.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 15.Antonsson B. Mitochondria and the Bcl-2 family proteins in apoptosis signaling pathways. Mol Cell Biochem. 2004;256-257::141–155. doi: 10.1023/b:mcbi.0000009865.70898.36. [DOI] [PubMed] [Google Scholar]
- 16.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182::367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheehan JP, Swerdlow RH, Miller SW, Davis RE, Parks JK, Parker WD, Tuttle JB. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s disease. J Neurosci. 1997;17::4612–4622. doi: 10.1523/JNEUROSCI.17-12-04612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993;34::609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- 19.Lovell MA, Gabbita SP, Markesbery WR. Increased DNA oxidation and decreased levels of repair products in Alzheimer’s disease ventricular CSF. J Neurochem. 1999;72::771–776. doi: 10.1046/j.1471-4159.1999.0720771.x. [DOI] [PubMed] [Google Scholar]
- 20.Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci. 1999;19::1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2005 doi: 10.1016/j.neulet.2005.12.017. In press: [DOI] [PubMed] [Google Scholar]
- 22.Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiol Dis. 2006 doi: 10.1016/j.nbd.2005.11.002. In press: [DOI] [PubMed] [Google Scholar]
- 23.Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85::357–363. doi: 10.1161/01.res.85.4.357. [DOI] [PubMed] [Google Scholar]
- 24.Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Lett. 2005 doi: 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 25.Rego AC, Oliveira CR. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochem Res. 2003;28::1563–1574. doi: 10.1023/a:1025682611389. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263::687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 27.Ansari MA, Ahmad AS, Ahmad M, Salim S, Yousuf S, Ishrat T, Islam F. Selenium protects cerebral ischemia in rat brain mitochondria. Biol Trace Elem Res. 2004;101::73–86. doi: 10.1385/BTER:101:1:73. [DOI] [PubMed] [Google Scholar]
- 28.Tangpong J, Cole MP, Sultana R, Joshi G, Estus S, Vore M, Clair WS, Ratanachaiyavong S, Clair DKS, Butterfield DA. Adriamycin-induced, TNF-á-mediated Central nervous System Toxicity. NeuroBiol Dis. 2006 doi: 10.1016/j.nbd.2006.02.013. In press: [DOI] [PubMed] [Google Scholar]
- 29.Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267::4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 30.Darley-Usmar V, Halliwell B. Blood radicals: reactive nitrogen species, reactive oxygen species, transition metal ions, and the vascular system. Pharm Res. 1996;13::649–662. doi: 10.1023/a:1016079012214. [DOI] [PubMed] [Google Scholar]
- 31.Hansen JM, Go YM, Jones DP. Nuclear and Mitochondrial Compartmentation of Oxidative Stress and Redox Signaling. Annu Rev Pharmacol Toxicol. 2005 doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- 32.Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med. 1999;27::916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 33.Meister A. Mitochondrial changes associated with glutathione deficiency. Biochim Biophys Acta. 1995;1271::35–42. doi: 10.1016/0925-4439(95)00007-q. [DOI] [PubMed] [Google Scholar]
- 34.Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52::711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 35.Amtmann E. The antiviral, antitumoural xanthate D609 is a competitive inhibitor of phosphatidylcholine-specific phospholipase C. Drugs Exp Clin Res. 1996;22::287–294. [PubMed] [Google Scholar]
- 36.Tschaikowsky K, Schmidt J, Meisner M. Modulation of mouse endotoxin shock by inhibition of phosphatidylcholine-specific phospholipase C. J Pharmacol Exp Ther. 1998;285::800–804. [PubMed] [Google Scholar]
- 37.Amtmann E, Sauer G. Selective killing of tumor cells by xanthates. Cancer Lett. 1987;35::237–244. doi: 10.1016/0304-3835(87)90125-x. [DOI] [PubMed] [Google Scholar]
- 38.Sauer G, Amtmann E, Hofmann W. Systemic treatment of a human epidermoid non-small cell lung carcinoma xenograft with a xanthate compound causes extensive intratumoral necrosis. Cancer Lett. 1990;53::97–102. doi: 10.1016/0304-3835(90)90200-h. [DOI] [PubMed] [Google Scholar]
- 39.Schutze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, Kronke M. TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell. 1992;71::765–776. doi: 10.1016/0092-8674(92)90553-o. [DOI] [PubMed] [Google Scholar]
- 40.Cifone MG, Roncaioli P, De Maria R, Camarda G, Santoni A, Ruberti G, Testi R. Multiple pathways originate at the Fas/APO-1 (CD95) receptor: sequential involvement of phosphatidylcholine-specific phospholipase C and acidic sphingomyelinase in the propagation of the apoptotic signal. Embo J. 1995;14::5859–5868. doi: 10.1002/j.1460-2075.1995.tb00274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao SR. In: Xanthates and Related Compounds. Dekker M, editor. New York: 1971. [Google Scholar]
- 42.Joshi G, Sultana R, Perluigi M, Allan Butterfield D. In vivo protection of synaptosomes from oxidative stress mediated by Fe2+/H2O2 or 2,2-azobis-(2-amidinopropane) dihydrochloride by the glutathione mimetic tricyclodecan-9-yl-xanthogenate. Free Radic Biol Med. 2005;38::1023–1031. doi: 10.1016/j.freeradbiomed.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 43.Zhou D, Lauderback CM, Yu T, Brown SA, Butterfield DA, Thompson JS. D609 inhibits ionizing radiation-induced oxidative damage by acting as a potent antioxidant. J Pharmacol Exp Ther. 2001;298::103–109. [PubMed] [Google Scholar]
- 44.Lauderback CM, Drake J, Zhou D, Hackett JM, Castegna A, Kanski J, Tsoras M, Varadarajan S, Butterfield DA. Derivatives of xanthic acid are novel antioxidants: application to synaptosomes. Free Radic Res. 2003;37::355–365. doi: 10.1080/1071576021000040664. [DOI] [PubMed] [Google Scholar]
- 45.Sultana R, Newman S, Mohmmad-Abdul H, Keller JN, Butterfield DA. Protective effect of the xanthate, D609, on Alzheimer’s amyloid beta-peptide (1-42)-induced oxidative stress in primary neuronal cells. Free Radic Res. 2004;38::449–458. doi: 10.1080/1071576042000206478. [DOI] [PubMed] [Google Scholar]
- 46.Perluigi M, Joshi G, Sultana R, Calabrese V, De Marco C, Coccia R, Butterfield DA. In vivo protection by the xanthate tricyclodecan-9-yl-xanthogenate against amyloid beta-peptide (1-42)-induced oxidative stress. Neuroscience. 2006;138::1161–1170. doi: 10.1016/j.neuroscience.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 47.Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18::685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- 48.Anderson ME, Luo JL. Glutathione therapy: from prodrugs to genes. Semin Liver Dis. 1998;18::415–424. doi: 10.1055/s-2007-1007174. [DOI] [PubMed] [Google Scholar]
- 49.Sims NR. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem. 1990;55::698–707. doi: 10.1111/j.1471-4159.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- 50.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72::248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 51.Halliwell B, Gutteridge JMC. Free Radical in Biology and Medicine. Oxford University Press; Oxford: 1999. [Google Scholar]
- 52.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275::1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 53.Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem. 1976;74::214–226. doi: 10.1016/0003-2697(76)90326-2. [DOI] [PubMed] [Google Scholar]
- 54.Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249::7130–7139. [PubMed] [Google Scholar]
- 55.Wheeler CR, Salzman JA, Elsayed NM, Omaye ST, Korte DW., Jr. Automated assays for superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase activity. Anal Biochem. 1990;184::193–199. doi: 10.1016/0003-2697(90)90668-y. [DOI] [PubMed] [Google Scholar]
- 56.Carlberg I, Mannervik B. Glutathione reductase. Methods Enzymol. 1985;113::484–490. doi: 10.1016/s0076-6879(85)13062-4. [DOI] [PubMed] [Google Scholar]
- 57.Butterfield DA, Stadtman ER. Protein oxidation processes in aging brain. Advanced Cell Aging and Gerontology. 1997;2::161–191. [Google Scholar]
- 58.Sies H, Sharov VS, Klotz LO, Briviba K. Glutathione peroxidase protects against peroxynitrite-mediated oxidations. A new function for selenoproteins as peroxynitrite reductase. J Biol Chem. 1997;272::27812–27817. doi: 10.1074/jbc.272.44.27812. [DOI] [PubMed] [Google Scholar]
- 59.Sultana R, Butterfield DA. Oxidatively modified GST and MRP1 in Alzheimer’s disease brain: implications for accumulation of reactive lipid peroxidation products. Neurochem Res. 2004;29::2215–2220. doi: 10.1007/s11064-004-7028-0. [DOI] [PubMed] [Google Scholar]
- 60.Bains JS, Shaw CA. Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Res Brain Res Rev. 1997;25::335–358. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- 61.Cooper JJL. Glutathione in the brain: disorders of glutathione metabolism. In: Barchi RL, Kunk LM, editors. The molecular and genetic basis of neurological disease. Vol. 35. Butterworth-Heinemann; Boston: 1997. [Google Scholar]
- 62.Fontaine M, Geddes J, Banks A, Butterfield D. Effect of exogenous and endogenous antioxidants on 3-nitropionic acid-induced in vivo oxidative stress and striatal lesions: insights into Huntington’s disease. Journal of Neurochemistry. 2000;75::1709–1715. doi: 10.1046/j.1471-4159.2000.0751709.x. [DOI] [PubMed] [Google Scholar]
- 63.Pocernich CB, La Fontaine M, Butterfield DA. In-vivo glutathione elevation protects against hydroxyl free radical-induced protein oxidation in rat brain. Neurochem Int. 2000;36::185–191. doi: 10.1016/s0197-0186(99)00126-6. [DOI] [PubMed] [Google Scholar]
- 64.Pocernich CB, Cardin AL, Racine CL, Lauderback CM, Butterfield DA. Glutathione elevation and its protective role in acrolein-induced protein damage in synaptosomal membranes: relevance to brain lipid peroxidation in neurodegenerative disease. Neurochem Int. 2001;39::141–149. doi: 10.1016/s0197-0186(01)00012-2. [DOI] [PubMed] [Google Scholar]
- 65.Drake J, Kanski J, Varadarajan S, Tsoras M, Butterfield DA. Elevation of brain glutathione by gamma-glutamylcysteine ethyl ester protects against peroxynitrite-induced oxidative stress. J Neurosci Res. 2002;68::776–784. doi: 10.1002/jnr.10266. [DOI] [PubMed] [Google Scholar]
- 66.Drake J, Sultana R, Aksenova M, Calabrese V, Butterfield DA. Elevation of mitochondrial glutathione by gamma-glutamylcysteine ethyl ester protects mitochondria against peroxynitrite-induced oxidative stress. J Neurosci Res. 2003;74::917–927. doi: 10.1002/jnr.10810. [DOI] [PubMed] [Google Scholar]
- 67.Tanaka S, Takehashi M, Matoh N, Iida S, Suzuki T, Futaki S, Hamada H, Masliah E, Sugiura Y, Ueda K. Generation of reactive oxygen species and activation of NF-kappaB by non-Abeta component of Alzheimer’s disease amyloid. J Neurochem. 2002;82::305–315. doi: 10.1046/j.1471-4159.2002.00958.x. [DOI] [PubMed] [Google Scholar]
- 68.Onyango IG, Bennett JP, Jr., Tuttle JB. Endogenous oxidative stress in sporadic Alzheimer’s disease neuronal cybrids reduces viability by increasing apoptosis through pro-death signaling pathways and is mimicked by oxidant exposure of control cybrids. Neurobiol Dis. 2005;19::312–322. doi: 10.1016/j.nbd.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 69.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11::81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 70.Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1-42. J Neurochem. 2001;78::413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 71.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68::2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 72.Lovell MA, Xie C, Markesbery WR. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology. 1998;51::1562–1566. doi: 10.1212/wnl.51.6.1562. [DOI] [PubMed] [Google Scholar]
- 73.Xie C, Lovell MA, Markesbery WR. Glutathione transferase protects neuronal cultures against four hydroxynonenal toxicity. Free Radic Biol Med. 1998;25::979–988. doi: 10.1016/s0891-5849(98)00186-5. [DOI] [PubMed] [Google Scholar]
- 74.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997;68::255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 75.Koppal T, Drake J, Yatin S, Jordan B, Varadarajan S, Bettenhausen L, Butterfield DA. Peroxynitrite-induced alterations in synaptosomal membrane proteins: insight into oxidative stress in Alzheimer’s disease. J Neurochem. 1999;72::310–317. doi: 10.1046/j.1471-4159.1999.0720310.x. [DOI] [PubMed] [Google Scholar]
- 76.Pocernich CB, Butterfield DA. Acrolein inhibits NADH-linked mitochondrial enzyme activity: implications for Alzheimer’s disease. Neurotox Res. 2003;5::515–520. doi: 10.1007/BF03033161. [DOI] [PubMed] [Google Scholar]
- 77.Chen JJ, Bertrand H, Yu BP. Inhibition of adenine nucleotide translocator by lipid peroxidation products. Free Radic Biol Med. 1995;19::583–590. doi: 10.1016/0891-5849(95)00066-7. [DOI] [PubMed] [Google Scholar]
- 78.Boyd-Kimball D, Mohmmad Abdul H, Reed T, Sultana R, Butterfield DA. Role of phenylalanine 20 in Alzheimer’s amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity. Chem Res Toxicol. 2004;17::1743–1749. doi: 10.1021/tx049796w. [DOI] [PubMed] [Google Scholar]
- 79.Picklo MJ, Amarnath V, McIntyre JO, Graham DG, Montine TJ. 4-Hydroxy-2(E)-nonenal inhibits CNS mitochondrial respiration at multiple sites. J Neurochem. 1999;72::1617–1624. doi: 10.1046/j.1471-4159.1999.721617.x. [DOI] [PubMed] [Google Scholar]
- 80.Kanski J, Lauderback C, Butterfield DA. 5-Aminosalicylic acid protection against oxidative damage to synaptosomal membranes by alkoxyl radicals in vitro. Neurochem Res. 2001;26::23–29. doi: 10.1023/a:1007620330168. [DOI] [PubMed] [Google Scholar]
- 81.Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, Rydel RE. beta-Amyloid precursor protein metabolites and loss of neuronal Ca2+ homeostasis in Alzheimer’s disease. Trends Neurosci. 1993;16::409–414. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- 82.Di Paola M, Zaccagnino P, Montedoro G, Cocco T, Lorusso M. Ceramide induces release of pro-apoptotic proteins from mitochondria by either a Ca2+ -dependent or a Ca2+ -independent mechanism. J Bioenerg Biomembr. 2004;36::165–170. doi: 10.1023/b:jobb.0000023619.97392.0c. [DOI] [PubMed] [Google Scholar]
- 83.Michel PP, Lambeng N, Ruberg M. Neuropharmacologic aspects of apoptosis: significance for neurodegenerative diseases. Clin Neuropharmacol. 1999;22::137–150. [PubMed] [Google Scholar]
- 84.Luberto C, Hannun YA. Sphingomyelin synthase, a potential regulator of intracellular levels of ceramide and diacylglycerol during SV40 transformation. Does sphingomyelin synthase account for the putative phosphatidylcholine-specific phospholipase C? J Biol Chem. 1998;273::14550–14559. doi: 10.1074/jbc.273.23.14550. [DOI] [PubMed] [Google Scholar]
- 85.Denis U, Lecomte M, Paget C, Ruggiero D, Wiernsperger N, Lagarde M. Advanced glycation end-products induce apoptosis of bovine retinal pericytes in culture: involvement of diacylglycerol/ceramide production and oxidative stress induction. Free Radic Biol Med. 2002;33::236–247. doi: 10.1016/s0891-5849(02)00879-1. [DOI] [PubMed] [Google Scholar]