

Abstract
Many experiments have been done to determine how far and how freely holes can move along the stack of base pairs in DNA. The results of these experiments are usually described in terms of a parameter β under the assumption that it describes an exponential decay with distance. The reported values range from β < 0.2/Å to β > 1.4/Å. For the larger values of β, the transport can be accounted for as single step superexchange-mediated hole transfer. To account for the smaller values, hopping models have been proposed, the simplest being nearest-neighbor hopping. This model assumes that, between hops, the hole is localized on a single base with no overlap to neighbors. Noting that an electron or hole added to a DNA stack, as to other essentially one-dimensional entities, should distort its structure to form a polaron, Schuster and coworkers [Henderson, P. T., Jones, D., Hampikian, G., Kan, Y. & Schuster, G. B. (1999) Proc. Natl. Acad. Sci. USA 96, 8353–8358 and Ly, D., Sanii, L. & Schuster, G. B. (1999) J. Am. Chem. Soc. 121, 9400–9410] proposed that transport occurs by polaron hopping between sites having approximately equal energies as a result of overlap. A recent experimental determination by Wan et al. [Wan, C., Fiebig, T., Kelley, S. O., Treadway, C. R., Barton, J. K. & Zewail, A. H. (1999) Proc. Natl. Acad. Sci. USA 96, 6014–6019] of the time required for an injected hole on DNA to travel a known distance leads to a large value of the diffusion constant. From this constant, a mobility of 0.2 cm2/V⋅s was deduced, orders of magnitude larger than typical hopping mobilities. We suggest that this ultrafast transport is due to polaron drift, which has been shown to lead to similar mobilities in chains of conjugated polymers. Using a simple model for the polaron, similar to that used for conjugated polymers such as polyacetylene, we show that, for reasonable values of the parameters, an injected electron or hole can form a polaron on a DNA stack.
It is generally, although perhaps not universally, believed that DNA has no free carriers in its thermal equilibrium state because of the gap of many eV (1 eV = 1.602 × 10−19 J) between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). Electrons (radical anions) or holes (radical cations) may be introduced by ionizing radiation, injection from contacts, photoinduced electron transfer involving an impurity, etc. Of particular interest has been the transport of electrons and holes along the stack of base pairs. The process is important, because radical migration through DNA may play a crucial role in mutagenesis and carcinogenesis. The suggestion that the π–π interaction between the base pairs could support extended transport was made almost 40 years ago (1). It has long been known that electrons and holes created by ionizing radiation can have a range of many base pairs (2). Recent results suggest that these carriers migrate over distances greater than 30 bases at room temperature (3). However, these results were apparently not considered evidence that electrons and holes introduced by other means could migrate relatively freely over the base-pair stack.
Particularly in the last decade, many different experiments have been carried out to determine the range of a hole on the base-pair stack. In a frequently used technique, photoexcitation of an acceptor associated with the stack in some fashion, sometimes intercalated, allows transfer of an electron, usually from a guanine (G) base, leaving a hole on the stack. In some experiments, the migrating hole is trapped at a GG step on the stack, and its range is determined by observing strand cleavage at the trap site (4, 5). In other experiments, the range is determined by observation of fluorescence quenching of a donor intercalated in the stack (6–8).
On the expectation of the electron or hole transfer decreasing exponentially with distance R according to exp(−βR), as predicted by Marcus theory, the observed penetration distance for the hole was described in terms of a parameter β. β values ranging from greater than 1.4 Å−1 to less than 0.1 Å−1 have been reported. Values from 1.2 Å−1 to 1.6 Å−1, similar to those found for proteins, are predicted for unistep superexchange-mediated hole transfer from acceptor to donor across the DNA bridge (9). The low values of β, on the other hand, prompted the assertion that DNA behaves like a “molecular wire” (10, 11).
Jortner et al. (12) suggested that the low values of β are due to hopping transport. The hopping process was envisioned as a series of nearest-neighbor charge-transfer steps with phonon assistance between approximately isoenergetic bases. This process could involve hopping of the hole to the complementary DNA strand. On this theory, if the series of bases included a pair separated by a base with higher oxidation potential, the transfer between the pair would take place by superexchange (12). It should be noted that, in many instances of long range and rapid transit, such as the one seen in ref. 8, the base sequence was such that a path involving isoenergetic bases was not available. Assuming that hole hopping must take place between G and multiple G units (even if they are not nearest neighbors), because they have the lowest ionization potentials, Meggers et al. (13) found that multiple AT base pairs between the G units greatly decreased the transfer rate. Some other experimenters also found transmission to depend on the base sequence (11), but others did not (14). Another significant finding was that the transmission is highly sensitive to stacking perturbations; insertion of a CA base pair into the duplex resulted in a large decrease in transmission (6).
In the hopping models mentioned above, it was assumed that, between hops, the hole occupies a discrete molecular orbital localized on a single base (12, 13). Henderson et al. (5) and Ly et al. (14) argued, however, that to minimize its energy, the charge would spread out over several bases, imposing a characteristic pattern of distortion. In other words, the radical ion would become a polaron, which, in their model, would propagate by thermally activated hopping. The experimental finding that led to this suggestion was a very long path for the hole (55 bp) independent of base sequence. To justify the suggestion of phonon-assisted hopping, Henderson et al. (5) invoked the calculations of Saito et al. (15). These ab initio calculations showed that the ionization potential of a group of stacked bases would be reduced, and, in particular, various sequences of three to five bases would have ionization potentials within a few times thermal energy of each other. These sequences, Henderson et al. (5) suggested, would constitute sites between which phonon-assisted hopping of the polarons could occur.
Most recently, the time scale has been measured for electron transfer between an intercalated electron acceptor (ethidium) and an electron donor (7-deazaguanine), both covalently bonded to the DNA, with a well defined distance between them (8). The result was a time constant of 5 ± 1 ps for a donor-acceptor spacing of 5 bp or 1.7 nm. However, the electron-transfer efficiency decreased with distance, from 57% at 1.0 nm to 23% at 1.7 nm. The decrease was attributed to stacking disorder of the base pairs, static caused by defects in the stacking, and dynamic caused by the motions of the DNA. For those holes that traveled 1.7 nm in 5 ps, we calculate a diffusion constant of 6 × 10−3 cm2/s. Of course this is a lower limit, because the holes might have traveled further if they had not been trapped by the donor. According to the Einstein relation, this diffusion constant would correspond to a mobility of 0.2 cm2/V⋅s in an electric field. This mobility is orders of magnitude larger than the usual hopping mobility, which typically takes place in a disordered medium. Mobilities in unoriented conjugated polymers that involve hopping from one chain or conjugation length to another are typically of the order of 10−5 cm2/V⋅s and may be smaller. However, mobility within a conjugation length on a single polymer chain has been measured as a few cm2/V⋅s in trans-polyacetylene (16) and 0.8 cm2/V⋅s in poly(phenylene vinylene) (17). It is established that the motions that give rise to the latter mobilities correspond to polaron drift along the chains. Thus, we suggest that the ultrafast motion found in DNA is due to polaron drift along the base-pair stacks. Note that slower transport found in DNA may be due to polaron hopping or even superexchange, as determined by the local order. As discussed above, the local order is a function of defects and of temperature. Thus, the mobility would be expected to be larger at lower temperatures.
Equations for the Polaron.
Polaron formation is expected when an electron or hole is added to a one-dimensional or quasi-one-dimensional chain or stack. The lack of constraint on the motion of atoms or molecules in one dimension compared with those bound in a three-dimensional configuration allows a distortion along the chain or stack that decreases the energy of the added carrier. The combination of free carrier and distortion, the polaron, has been studied extensively.
The essential ingredients of a Hamiltonian that gives rise to polarons are (i) wavefunction overlap of nearest neighbors, quantified by the transfer or resonance integral t; (ii) electron–phonon coupling to allow the formation of the distortion; and (iii) an elastic restoring force that keeps the distortion finite. A simple tight-binding Hamiltonian embodying these elements, which has been used extensively to study polarons in conducting polymers, is the Su–Schrieffer–Heeger Hamiltonian (18). Written for a situation in which there may be sites of different energies, each of which may be doubly occupied, the Hamiltonian is
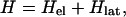 |
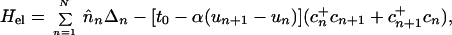 |
1 |
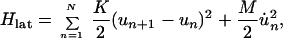 |
where cn+(cn) is the electron creation (annihilation) operator, n̂n = cn+cn, Δn is on-site energy, un is the displacement of the nth site from its thermal equilibrium position (sites evenly spaced), K the elastic constant, and M the mass of the entity at each site. The transfer integral has been broken up into the term t0, representing the transfer integral in thermal equilibrium, and a term representing the change in t caused by the displacements. The quantity α is the electron–phonon coupling. It is seen that Hlat treats the phonons classically. Minimization of the Hamiltonian (1) with respect to displacements yn = un+1 − un and electronic energies ɛn yields the following equations:
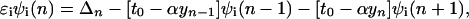 |
2 |
and
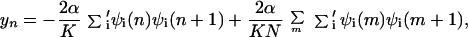 |
3 |
where the prime indicates that summation is over the occupied states only, and the last term in Eq. 3 is to ensure that the length of the stack does not change, which may be written
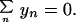 |
4 |
Parameters for DNA.
To apply these equations to DNA we may consider that each site is occupied by a single base, A, G, C or T, or a duplex pair, G/C or A/T. Although the wavefunction may be more concentrated at one member of the pair, e.g., G in G/C, because of its lower ionization energy, there is good evidence for wavefunction overlap in the pair. It is well documented that a hole inserted into one of the bases in the pair easily makes a transition to the other (19, 20). Also, the two bases in a duplex pair are mechanically joined because of the hydrogen bonds. If hole injection is due to an acceptor located on one strand and if the donor is a few bases away on the same strand, however, it is likely that the transfer takes place on this strand. In the experiments of Wan et al. (8), the acceptor was apparently intercalated so as to overlap both bases of the pair, and it may be that, under those circumstances, the polaron involves both stacks. We have calculated for the two cases and, as will be seen, they do not lead to very different properties for the polaron.
We assume that, in thermal equilibrium, the bases are uniformly stacked with a distance of 3.40 Å between them. As seen from Eq. 2, the motion relevant to the polaron formation is perpendicular to the bases. Fortunately, this motion is determined solely by the base-stacking interactions and is independent of the backbone (21).
The value of t0 is determined by interaction of the π-wavefunctions on adjacent bases. The energy levels resulting from two guanine bases exactly overlapped, with a distance d between them, has been calculated as a function of d (22). For d < 5.5 Å, the interaction is sufficient to split the HOMO into two levels. At 3.4 Å, the spacing between the two levels is 0.72 eV. On a simple Huckel model the splitting is 2t0, giving t0 = 0.36 eV. The splitting for 2 bp, say (GG)/(CC), aligned vertically and separated by this distance, would be larger because of the additional π-overlap. However, the base pairs may not be overlapped exactly in the stack. Based on these considerations, we chose, conservatively, 0.3 eV to represent the t0 value for a single pair or a duplex pair. The t0 value for a single pair, AA or TT, or for a duplex pair, (AA)/(TT), should be similar. It could be somewhat smaller for the cases where the purine and pyridine are overlapped.
The calculated splitting of the HOMO and the level below it, which we take as 2t, can also be used to obtain a value for α = ∂t/∂u. The value we obtain from the calculated values of these two levels for a pair of separated Gs, vertically overlapped (22), is 0.6 eV/Å. From the same considerations discussed above, for t0, we chose α = 0.6 eV/Å.
The value of the elastic constant can be calculated from the sound velocity in DNA. The sound velocity vs parallel to the stacks has been measured as 1.9 km/s (23). The elastic constant K = Mvs2/a2, where a is the interbase distance. We choose M as the mass of a duplex pair, because the pairs are joined by the hydrogen bonds. Because M differs by less than 0.5% between C/G and A/T, we will use the same mass for both, 4.35 × 10−22 g. The result is K = 0.85 eV/Å.
Calculated Properties of Polarons in DNA.
Stationary polaron properties were obtained by numerical solution of Eqs. 2 and 3 without the kinetic energy term. Solutions were obtained both for a periodic arrangement of bases and for a random arrangement of bases in the stack. The calculations were done for a stack of 100 sites with 199 electrons, thus providing for one hole. In Fig. 1, we show the resulting yn = un+1 − un vs. site number for a periodic arrangement, two bases or 2 bp alternating with each other, with the parameters specified above. Δ was taken as 0 for the lower energy site; 0.65 eV was taken for the upper. The latter value represents the difference between ionization energies of the base pairs G/C and A/T, 7.34 eV for the former, 7.99 eV for the latter, obtained by ab initio quantum chemical calculations at the 6-31G* level (22). For the calculation to apply to a polaron made up of individual bases rather than base pairs, the value of Δ must be changed to one appropriate for the pair of bases. That change makes only minor changes in the results, as will be seen below.
Figure 1.

●, change in interbase distance (yn) vs. site number characterizing a polaron on a periodic base stack with 2 different bp. *, |ψ|2 for the hole on the polaron in this case. The dashed lines indicate the symmetric polaron solution of the equations; the solid lines indicate the asymmetric polaron solution. Both polarons were calculated with the following parameters: t0 = 0.3 eV, α = 0.6 eV/Å, K = 0.85 eV/Å2, and Δ = 0.65 eV.
As is shown in Fig. 1, for this case of two different types of site, two different solutions are possible: one with wavefunction symmetric about the center of the polaron and the other with wavefunction asymmetric, the two with the same energy. It is seen that the polaron is approximately seven sites wide. The center of the polaron is on a G/C site, as expected, because that has higher energy. Within the polaron, the spacing of the bases is decreased to lower the polaron energy, as suggested by Henderson et al. (5). To keep the stack length constant, outside the polaron the spacing increases slightly. The spacing within the polaron decreases progressively with distance from the edge of the polaron, reaching a minimum around the center of the polaron. The energy levels for this situation are shown in Fig. 2. Because of overlap of the wavefunctions of G/C and A/T, the levels are spread into bands about 0.3 eV wide, the bands being separated by Δ. The presence of 199 electrons rather than 200 results in the appearance of two levels separated from the bands by 0.25 eV. Thus, the hole energy is lowered by 0.25 eV because of formation of the polaron.
Figure 2.

Energy levels for the two cases shown in Fig. 1. The zero of energy is the energy of an A/T base pair.
This polaron differs somewhat from that in polyacetylene, where the situation is rather different. In that case, what would be a half-filled band for evenly spaced sites (CHs) is split by dimerization (Peierls transition) into a filled and empty band with a gap of 1.4 eV between them. The polaron that results from removal of one electron from the band or addition of one electron has two empty levels in the gap, each separated by 0.2 eV from the closer band edge (24). The deformation that characterizes the polaron is a decrease in the dimerization over the region occupied by the hole or extra electron in the case of an electron polaron.
We have investigated the effects of changes in the various parameters. For t0 = 0.2 eV, α = 0.32 eV/Å, and K and Δ retain the values shown in Fig. 1; the polaron is wider and flatter, the width being 12 sites and the largest difference in interbase spacing being −0.12 Å. The polaron level is now only 0.03 eV above the top of the G/C band. |ψ|2 shows a series of peaks and valleys, being larger by at least a factor 2 on the G/C sites than on the A/T sites. For t0 = 0.1 eV and α = 0.16 eV/Å, the polaron is still wider, occupying 20 sites with a maximum yn value of −0.15Å. The polaron level for this case is only 0.001 eV above the top of the G/C band. Increase in K from 0.85 eV/Å2 to 1.2 eV/Å2, keeping t0 = 0.3 eV, α = 0.6 eV/Å, and Δ = 0.65 eV, resulted in a polaron of approximately the same width but, not surprisingly, with smaller minimum yn, −0.25 Å. The polaron level was reduced to 0.14 eV above the G/C band. Decrease in α from 0.6 eV/Å to 0.48 eV/Å, keeping the other parameters as listed in the Fig. 1 legend, resulted in a slightly wider polaron with minimum yn of only −0.3 Å.
To look at the situation for the case of a polaron made up of single bases rather than base pairs, we simulated a periodic arrangement of G alternating with T by choosing Δ = 1.4 eV, this value being the difference between their ionization energies (22). With the other parameters the same ones listed in Fig. 1, we obtain a polaron with about the same width as that in Fig. 1 but with minimum yn a little smaller, −0.3 Å. Corresponding to the decrease in the magnitude of the spacing, there was a lowering of the polaron level to 0.13 eV above the edge of the higher filled band. Because the difference between the ionization potentials of G and T represents the largest Δ that could be obtained for a pair of bases, we conclude that the value of Δ has only a minor effect on the properties of the polaron. To summarize, decrease in t0 and α and increase in K all make smaller the decrease in base spacing associated with the polaron as well as the gain in energy caused by its formation, as expected.
We consider now the results for random arrangements of the bases. Consider first the results for a random arrangement of the base pairs G/C and A/T. The random arrangement chosen when no restrictions were imposed had a sequence of five G/Cs. Although t0 was taken as only 0.2 eV, a five-site polaron resulted, centered at the middle of the five G/Cs. The polaron and the associated displacements were essentially symmetric, because almost all of its wavefunction was associated with a single base, G/C. The minimum yn was ≈−0.4 Å. As seen, despite the decrease in t0 from 0.3 to 0.2 eV, the polaron is narrower, and the displacements are larger. We believe that these differences are attributable to the sequence of five G/Cs. Realistically, such a sequence would not lead to a stable polaron, because the lower energy creates a site vulnerable to cleavage of the stack (22).
HOMO energies of G/C and A/T used above were apparently calculated for the G and A on the 5′ side and the C and T on the 3′ side (22). Values were not supplied for the base pairs interchanged (i.e., 5′ and 3′ sides reversed), although it seems that these values would be lower. Arbitrarily, we assumed that, with Δ for A/T taken as zero, Δ for T/A would be 0.15 eV, and Δ for C/G would be 0.35 eV; however, G/C remains 0.65 eV. For a random arrangement of A/T, T/A, C/G, and G/C, created, however, with the restriction that no G/C pairs be adjacent to each other, and the parameters chosen as shown in Fig. 1, we found a polaron quite similar to that shown in Fig. 1 with the magnitude of the minimum value of yn a little larger. The random arrangement led to a quasi-continuous distribution of energy levels, as shown in Fig. 3. The polaron level was a little higher than that found in the periodic case (Fig. 2), with the same parameters (0.34 eV as compared with 0.25 eV above the highest filled level), reflecting the greater displacements found in this case. It is clear that the details of the displacements, the wavefunctions, and energy levels will depend on the sequence of base pairs.
Figure 3.

Energy levels for a random arrangement of A/T, T/A, C/G, and G/C.
We have also carried out calculations of a polaron for a random arrangement of bases residing on a single stack. Again, the case of more than one G in a row was avoided. The differences between HOMO levels for the different bases were obtained from literature values for the differences in the ionization potentials for the single bases. As expected, the shape of the polaron depends on the sequence of bases. In Fig. 4, we show the results for the particular base sequence indicated. Although the flat top of |ψ|2 for this sequence indicates that the hole need not be as concentrated on one base as shown in Fig. 1, there were other sequences that resulted in a strongly peaked polaron. The energy levels for this case are shown in Fig. 5. The polaron level is 0.35 eV above the highest filled level.
Figure 4.

●, change in interbase distance (yn) vs. site number characterizing a polaron on a base stack of four different bases randomly arranged. *, |ψ|2 for the hole on the polaron. The following parameters were used in the calculation: t0 = 0.3 eV, α = 0.6 eV/Å, K = 0.85 eV/Å2, ΔG = 1.4 eV, ΔA = 0.9 eV, ΔC = 0.27 eV, and ΔT = 0.
Figure 5.

Energy levels for the case shown in Fig. 4. The zero of energy is the energy of an isolated T base.
In summary, we have shown, by means of a simple model used for polarons in conducting polymers, that injection of an electron or hole should result in formation of a polaron on the base stack of DNA under favorable circumstances. By favorable circumstances, we mean that the overlap of adjacent bases must not be cut off by a large fluctuation in their motion or by defects or obstacles along the stack. The polarons thus formed are robust, being stable over a fairly wide range of parameters within which those for DNA should lie. The polarons could move by hopping or by drifting along the stacks. Drift requires a base displacement of at most a few tenths of an angstrom along the stack. Although polaron hopping in other organics is too slow to account for the ultrafast diffusion found experimentally, polaron drift might be fast enough. The ultrafast diffusion constant means a relatively large mobility for a nonmetallic organic conductor, ≈0.2 cm2/V⋅s, and could be of interest for devices incorporating DNA.
Abbreviation
- HOMO
highest occupied molecular orbital
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050074497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050074497
References
- 1.Eley D D, Spivey D I. Trans Faraday Soc. 1962;58:411–415. [Google Scholar]
- 2.Spalletta R A, Bernhard W A. Radiat Res. 1992;130:7–14. [PubMed] [Google Scholar]
- 3.Razskazovskii Y, Swarts S, Falcone J, Taylor C, Sevilla M. J Phys Chem. 1997;101:1460–1467. [Google Scholar]
- 4.Hall D B, Holmlin R E, Barton J K. Nature (London) 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 5.Henderson P T, Jones D, Hampikian G, Kan Y, Schuster G B. Proc Natl Acad Sci USA. 1999;96:8353–8358. doi: 10.1073/pnas.96.15.8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelley S O, Holmlin R E, Stemp E D A, Barton J K. J Am Chem Soc. 1997;119:9861–9870. [Google Scholar]
- 7.Kelley S O, Barton J K. Science. 1999;283:375–381. doi: 10.1126/science.283.5400.375. [DOI] [PubMed] [Google Scholar]
- 8.Wan C, Fiebig T, Kelley S O, Treadway C R, Barton J K, Zewail A H. Proc Natl Acad Sci USA. 1999;96:6014–6019. doi: 10.1073/pnas.96.11.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Priyadarshy S, Risser S M, Beratan D N. J Phys Chem. 1996;100:17678–17682. [Google Scholar]
- 10.Murphy C J, Arkin M R, Jenkins Y, Ghatlia N D, Bossmann S H, Turro N J, Barton J K. Science. 1993;262:1025–1029. doi: 10.1126/science.7802858. [DOI] [PubMed] [Google Scholar]
- 11.Turro N J, Barton J K. J Biol Inorg Chem. 1998;3:201–209. [Google Scholar]
- 12.Jortner J, Bixon M, Langenbacher T, Michel-Beyerle M E. Proc Natl Acad Sci USA. 1998;95:12759–12765. doi: 10.1073/pnas.95.22.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meggers E, Michel-Beyerle M E, Giese B. J Am Chem Soc. 1998;120:12950–12955. [Google Scholar]
- 14.Ly D, Sanii L, Schuster G B. J Am Chem Soc. 1999;121:9400–9410. [Google Scholar]
- 15.Saito I, Nakamura T, Nakatani K, Yoshioka Y, Yamaguchi K, Sugiyama H. J Am Chem Soc. 1998;120:12686–12687. [Google Scholar]
- 16.Reichenbach J, Kaiser M, Roth S. Phys Rev B Condens Matter. 1993;48:14104–14112. doi: 10.1103/physrevb.48.14104. [DOI] [PubMed] [Google Scholar]
- 17.Hoofman R J O M, de Haas M P, Siebbeles L D A, Warman J M. Nature (London) 1998;392:54–56. [Google Scholar]
- 18.Su W P, Schrieffer J R, Heeger A J. Phys Rev B Condens Matter. 1980;22:2099–2111. [Google Scholar]
- 19.Osuka A, Nakajima S, Okada T, Taniguchi S, Nozaki K, Ohno T, Yamazaki I, Nishimura Y, Mataga N. Angew Chem Int Ed Engl. 1996;35:92–95. [Google Scholar]
- 20.Hayashi T, Miyahara T, Kumazaki S, Ogoshi H, Yoshihara K. Angew Chem Int Ed Engl. 1996;35:1964–1966. [Google Scholar]
- 21.Packer M J, Hunter C A. J Mol Biol. 1998;280:407–420. doi: 10.1006/jmbi.1998.1865. [DOI] [PubMed] [Google Scholar]
- 22.Sugiyama H, Saito I. J Am Chem Soc. 1996;118:7063–7068. [Google Scholar]
- 23.Hakim M B, Lindsay S M, Powell J. Biopolymers. 1984;23:1185–1192. doi: 10.1002/bip.360230704. [DOI] [PubMed] [Google Scholar]