

Abstract
To study the effect of leukemia inhibitory factor (LIF) on rat retinal vascular development, Sprague–Dawley rats at postnatal age 3 days (p3) were given intraperitoneal (IP) LIF and analysis performed at p6 (p3/6). p7 rats were given intravitreous (IV) LIF and analysis performed at p9 (p7/9). Control animals were PBS injected. At the time of analysis retinal flatmounts were prepared and stained with Griffonia lectin and activated caspase-3. The retinal peripheral avascular area was measured and number of apoptotic cells counted. In vitro, human retinal microvascular endothelial cells (RMVECs) were cultured in media containing LIF, with and without neutralizing antibody to LIF. Cells were stained with activated caspase-3 and apoptotic cells counted. Proliferation was measured by counting cell numbers, and cell cycle stage was determined using propidium iodide staining and FACS analysis. LIF injected either IP or IV had no effect on body weight or total retina area, but significantly increased the peripheral retinal avascular area. In both IP and IV injected groups there was no difference in the number of apoptotic cells between PBS-or LIF-injected groups; although in the p7/9 retinas, both injected groups had significantly more apoptotic cells than the non-injected group. In vitro, there was no effect of LIF on RMVEC apoptosis; however, cell counts were significantly lower in the LIF-treated group. Antibody to LIF restored the cell counts to untreated levels. LIF reduced the number of cells in S phase. LIF attenuates retinal vascular development in vivo through growth arrest, and not apoptosis, of endothelial cells.
Keywords: retina, blood vessel development, leukemia inhibitory factor
1. Introduction
Leukemia inhibitory factor (LIF) is a multifunctional, pleiotrophic cytokine (Metcalf, 2003) best known for its ability to maintain stem cell pluripotency in vitro (Williams et al., 1988). Although not normally expressed in adult tissue, LIF has a central role in human fertility (Senturk and Arici, 1998), especially during uterine blastocyst implantation (Stewart et al., 2003). It can be induced by ischemia-reperfusion injury in cortical neurons and neighboring astrocytes (Suzuki et al., 2000) and has been reported as a survival factor for neurons, astrocytes (Gadient et al., 1998), and oligodendrocytes (Butzkueven et al., 2002).
LIF is a member of the interleukin-6 family of cytokines (Heinrich et al., 2003). All members have their own specific receptors, but share the common feature of binding to the transmembrane receptor subunit, glycoprotein 130 (gp130) (Yamauchi-Takihara, 2002). LIF binding to its receptor LIFR causes tyrosine phosphorylation and formation of a heterodimer with gp130. This binding induces activation and signaling through Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathways (Auernhammer and Melmed, 2000).
The effect of LIF on angiogenesis is contradictory. It has been shown to cause proliferation (Gendron et al., 1996) and induce tube-formation (Paradis and Gendron, 2000) of an embryonic endothelial cell line. However, it had differential effects on bovine endothelial cells depending on the tissue origin of the cells. Growth (Ferrara et al., 1992), migration, and proliferation (Pepper et al., 1995) of aortic endothelial cells were strongly inhibited by LIF, whereas these properties were unaffected in adrenal cortex-derived capillary endothelial cells (Ferrara et al., 1992). LIF’s anti-angiogenic properties have been partly attributed to its ability to modulate integrin subunit expression (Collo and Pepper, 1999). LIF has also been demonstrated to arrest an epithelial tumor cell line in the G0/G1 phase (Park et al., 2003) and prevent entry of a murine pituitary cell line into S-phase (Stefana et al., 1996), although these effects have not been demonstrated in any type of endothelial cell.
Overexpression of LIF in the lens of transgenic mice resulted in a reduced hyaloid vasculature at birth and no retinal vasculature in the first 2 weeks postnatal (Ash et al., 2005). It was also shown to disrupt retinal neuronal development (Sherry et al., 2005) resulting in less cells and a loss of the normal structure. In culture, Muller-cell derived LIF was shown to arrest rod photoreceptor differentiation (Neophytou et al., 1997) and increase RPE survival (Gupta et al., 1997). Because LIF is inducible in adults and has been shown to be produced in retinal cells, we were interested in the effect of LIF on the development of the retinal vasculature. This may have implications for understanding LIF’s possible role in disease.
2. Methods
2.1. In vivo assays: animals
Sprague–Dawley rats (Charles River, MA) were treated in accordance with the ARVO statement for the Use of Animals in Ophthalmic and Vision Research.
2.2. Staining of retinal tissue for flatmounting
Rat pups were anesthetized by IP injection of ketamine (2.5 mg/kg) and xylazine (1 mg/kg). Paraformaldehyde (PFA) was then directly perfused (0.4 ml 0.5%) into the left ventricle, prior to euthanasia by intracardiac injection of 80 mg/kg Nembutol (NLS Animal Health, MD). Both eyes were enucleated, and whole eyes were fixed in 2% PFA for 2 h before being washed in 1 M PBS pH 7.4. The retinas were dissected using a modification of the method of Chan-Ling (1997). Briefly, under a dissecting microscope, an incision was made at the limbus, and the cornea was circumcised from the sclera. The lens was gently removed without disturbing the retina. The remaining eyecup was transferred to PBS, and the full extent of the retina with the ora serrata intact was eased from the sclera using fine forceps. The retina was then placed onto a microscope slide and flattened by making four incisions, each 90 degrees apart, beginning at the ora serrata and extending centrally from the equator stopping short of the optic nerve opening. As much vitreous as possible was removed using cellulose sponges and scissors.
2.3. Apoptosis staining
Flattened retinas were washed in 1 M PBS for 10 min, followed by pre-blocking in 1 M PBS with 3% bovine serum albumin (BSA, Sigma) for 30 min at room temperature. Primary activated caspase-3 antibody directly conjugated to FITC (Cell Signaling Technology, MA) was diluted 1:100 in PBS (3% BSA) and added to the slides that were incubated overnight at 4 °C. The retinas were washed three times in 1 M PBS for 5 min each. The retinas were then incubated overnight at 4 °C in TRITC conjugated Griffonia simplicifolia lectin (GSL, Sigma, MN) diluted to 20 μg/ml. Slides were washed three times in 1 M PBS for 10 min each. For nuclear staining, the retinas were covered for 30 min with Hoechst (Molecular Probes, OR) diluted 1:5000 in 1 M PBS. Slides were washed three times in PBS for 30 min, mounted in PBS:glycerol (1:2), and viewed using a confocal microscope.
2.4. Analysis of apoptotic cells
The blood vessels of the retina were viewed using a Leica SP2 laser confocal microscope. Three images were captured within each of the four retinal quadrants: one at the peripheral edge of the vasculature near the ora serrata, one close to the optic nerve head, and one equidistant between these two. Images were taken so as to avoid the major vessels. Within these areas all cells that were caspase-3 positive were counted. The sum of the 12 areas gave a total cell count in 1.7 mm2.
2.5. Analysis of avascular areas
For total retinal areas, images were captured using a Nikon TE2000U microscope with a 2× lens. Four images were captured to cover the total retinal area, and the images were montaged in Adobe Photoshop to give a single image of the retina. The total retinal area and peripheral avascular area in these images were measured using the freeware ImageTool (Version 3.0, University of Texas, TX), and the avascular area was expressed as a percentage of the total retinal area.
2.6. Tissue preparation for RT-PCR
Animals were euthanized with an overdose of pentobarbital (0.2 mg/ml IP). Brain tissue was removed using forceps, placed immediately into RNAlater (Ambion, CA), and frozen at − 20 °C until analysis. Both eyes were enucleated and the retinas were dissected under a dissecting microscope as described in the method for flatmounting retinas. However, the ora serrata was removed by gently pulling or cutting away from the retina before it was placed in RNAlater and frozen at − 20 °C until analysis.
2.7. Semi-quantitative RT-PCR
Samples were removed from RNAlater, and RNA extracted using Absolutely RNA Miniprep Kit (Stratagene, CA). DNA contamination was removed by using DNA-free (Ambion, TX), and RNA quantity was determined spectrophotometrically. Reverse transcription was done using Retroscript Kit (Ambion, TX). Briefly, 1 μg of RNA and 2 μl of random decamers were made up to a volume of 12 μl in nuclease free water. This was mixed, spun briefly, and heated at 75 °C for 3 min. To it, 2 μl 10× RT buffer (500 mM Tris–HCl, pH 8.3, 750 mM KCl, 30 mM MgCl2, and 50 mM DTT), 4 μl dNTP (2.5 mM each), 10 U RNase inhibitor, and 100 U MMLV-reverse transcriptase were added. The RT reactions were incubated at 42 °C for 60 min and terminated at 92 °C for 10 min. Samples were frozen at this stage until PCR.
PCR was performed using specific primers to rat LIF (forward 5′-tgt gcc cct act gct cat tct g; reverse atc cca ggt gat gtt ggt cag g-3′ annealing temperature 62 °C) or rat gp130 (forward 5′-ctt ctc acc ccg tag tgg atc tta and reverse gac tat ggc ttc gat ttc tcc tt -3′ annealing temperature 58 °C). Products were LIF at 343 bp and gp130 at 599 bp.
The linear range of each sample was determined empirically by increasing the number of cycles and resolving the products on a 2% agarose gel (USB Corporation, OH). Sample reactions were 2.5 μl cDNA, 5 μl 10× PCR complete buffer (100 mM TriseHCl, pH 8.3, 500 mM KCl, and 15 mM MgCl2), 2.5 μl dNTP mix (2.5 mM each), 1 μl primer mix (5 μM of each primer), 1 U superTaq polymerase (Ambion, TX).
The control gene was 18S ribosomal RNA and was amplified using a QuantumRNA primer:competimer set (Ambion, TX) yielding a band at 489 or 315 bp. Because the 18S is far more abundant than most other RNA, 18S amplification was reduced by adding competimers which compete with 18S primer for binding. The competimers are primers modified at their 3′ ends to block extension by DNA polymerase.
The ratio of primer:competimer was quantified empirically by increasing the ratio at the predetermined number of cycles (see above). 2.5 μl of cDNA was added to 5 μl 10× PCR complete buffer (100 mM Tris–HCl, pH 8.3, 500 mM KCl, and 15 mM MgCl2), 2.5 μl dNTP mix (2.5 mM each), 1 μl primer:competimer mix, 1 U superTaq polymerase (Ambion, TX). Samples were resolved on a 2% agarose gel, and the 18S rRNA band that had an intensity equal to that of the sample was selected for use in the relative-quantification.
For semi-quantitative analysis, a multiplex reaction with both specific primers and the 18S primers in the same sample was performed. Triplicates of each sample were run on a 2% gel and the bands were captured digitally using the UVP ChemiDoc System including a Chemi cooled CCD camera, PCI digitizing image acquisition board, EpiChemi II Darkroom with transilluminator, and LabWorks 4.0 Software. Data were exported to an Excel spreadsheet for data calculation where values were expressed relative to 18S within each sample. All PCR products were confirmed by gel extraction (Qiagen, CA) and sequence analysis (UNC Core Facility, http://152.19.68.152/gafsite/main.asp).
2.8. LIF injections
Sprague–Dawley rat pups (Charles River, MA) at postnatal age 3 days were injected intraperitoneally (IP) with 100 ng rat LIF (Chemicon, CA) in 0.1 ml sterile PBS. Controls were injected IP with 0.1 ml sterile PBS. For older pups, intravitreous injections of LIF were given at p7. These methods were chosen because p3 animals had exuberant wound healing responses after intravitreous injections that affected retinal dissection and the results. All intravitreous injections were done in the right eye with either LIF in PBS (5 ng/μL) or PBS. Controls were non-injected left eyes. Analysis of avascular areas were performed as described above.
2.9. Immunoprecipitation and Western blotting for phosphorylated gp130
Freshly dissected retinas were lysed in 500 μL of modified RIPA lysis buffer (10 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.1% SDS, 1% sodium deoxycholate plus protease inhibitor cocktail and sodium orthovanadate) in 1.5 mL centrifuge tubes, on ice for 20 min with frequent mixing. Lysates were clarified by spinning at 13,000 rpm for 25 min at 4 °C and supernatants were removed to new tubes. 10 μL from each sample were used to quantify protein by the Bradford method using standard protocols. 1.5 μg of anti-gp130 rabbit polyclonal antibody (Santa Cruz, CA) plus 30 μL of a 50% slurry of protein A-conjugated sepharose (Amersham, NJ) were added to each equalized sample. These were incubated overnight at 4 °C with continuous rocking, then washed three times with lysis buffer. Finally, an equal volume of sample buffer was added to the sepharose beads after the final wash and the samples were run on a 10% SDS-PAGE gel electrophoresis, followed by transfer to PVDF membranes. Western blotting was performed using a phosphotyrosine specific monoclonal antibody (BD Bioscience, CA), then anti-mouse HRP-conjugated secondary antibody (Jackson Laboratory), followed by enhanced chemiluminescence (Pierce, IL).
Radiographic films were scanned using a Cannon 4200F scanner and Adobe Photoshop 7.0 software. Band intensities were analyzed by densitometric analysis with UN-Scan-it digitizing software (Silken Scientific, Utah) and average ratios of phosphor-gp130 to total gp130 plus standard deviations as well as data graphs were generated using Microsoft Excel. One-way analysis of variance (ANOVA) and Tukey–Kramer multiple comparisons post test were preformed using Graph-Pad Instat 3.0.
2.10. In vitro assays: human retinal endothelial cells (RMVECs)
2.10.1. Effect of exogenous LIF on proliferation of RMVECs
Human retinal microvascular endothelial cells (RMVECs, Cell Systems, WA cat #: ACBRI-181; kindly provided by Gerard Lutty PhD, Wilmer Eye Institute) were plated at 5000/cm2 in low glucose DMEM (Invitrogen, CA) containing 120 μg/ml endothelial cell growth supplement (ECGS, Sigma, MO), 100 μg/ml heparin (Sigma, MO) and 10% fetal bovine serum (FBS, Sigma, MO). After 5 h, the media were removed and replaced with identical media except they contained only 5% FBS and a serial dilution of hLIF at 5, 10, 25 and 50 ng/ml (Chemicon, CA). Controls were cells cultured in 5% FBS medium without hLIF or 25 ng/ml hLIF plus 500 ng/ml neutralizing LIF antibody (R & D Systems, MN). After 4 days the cells were trypsinized and counted in a Z1 series Coulter Counter (Beckman-Coulter, CA). Results were expressed as fold increase compared to control that contained no LIF.
2.10.2. RMVECs apoptosis cell staining
RMVECs were seeded at 1 × 105 cells/cm2 in a CC2 chamber slide 8-well system (Nunc, NY) in DMEM supplemented with 120 μg/ml ECGS, 100 μg/ml heparin and 10% FBS. After 5 h, the media were removed and replaced with identical media except they contained only 5% FBS. hLIF was added at 25 ng/ml with or without LIF antibody at 500 ng/ml. Cells were grown for 2 days at 37 °C in 5% CO2. As a positive control for apoptosis, cells grown for 2 days but without LIF or LIF antibody were replaced with DMEM (supplemented with 5% FBS) containing 1 μg/ml staurosporine (Sigma, MO) and incubated for 6 h at 37 °C in 5% CO2. All wells were then washed three times with 1 M PBS containing 0.5% Triton X-100 (Sigma, MO) and fixed for 15 min in 2% paraformaldehyde (Sigma, MO) at room temperature. After three washes in 1 M PBS, the cells were permeabilized in ice cold methanol for 10 min. The cells were washed and pre-blocked in 1 M PBS containing 3% BSA (Sigma, MO) and Triton X-100 for 30 min. The cells were then incubated overnight at 4 °C in FITC-conjugated activated caspase-3 antibody diluted 1:250 in 1 M PBS containing 3% BSA. The cells were washed three times and incubated at 37 °C in 25 μg/ml propidium iodide (Sigma, MO) for 30 min. The cells were washed three times and mounted in PBS:glycerol (2:1) containing VectaShield (Vector Labs, CA). Images were captured using a Leica SP2 scanning laser confocal microscope.
2.11. Exogenous LIF and the cell cycle of RMVECs
RMVECs were plated in 6-well plates at 13,000/cm2 in low glucose DMEM containing 120 μg/ml ECGS, 100 μg/ml heparin and 10% fetal bovine serum. After 5 h, the media were removed and replaced with low glucose DMEM containing 5% FBS and 10 or 25 ng/ml of hLIF. Controls had no LIF. The cells were grown for 24 h after which they were trypsinized and spun at 200 × g for 5 min. The supernatant was removed and the cells stained with propidium iodide. Briefly, cells were resuspended in 100 μl ice cold 70% ethanol for 30 min. Cells were spun at 3000 × g for 5 min and resuspended in 1 ml of PBS with 2% FBS. Cells were spun again at 3000 × g for 5 min and resuspended in 100 μg/ml RNase A (Sigma, MO) in PBS and incubated at 37 °C for 30 min. Propidium iodide (20 μg/ml) was added and incubated for 30 min at room temperature. Total DNA content was measured within 24 h using a FACSCalibur Flow Cytometer (BD Biosciences, CA).
2.12. Statistics
Statistics were performed using SPSS for Windows v12.0 (Chicago, IL) or GraphPad Instat v3.0.
Experiments were performed at least in triplicate and results expressed as mean and standard deviations. A t-test was used to analyze two groups. For more than two groups, analysis was performed with an ANOVA. Post-hoc testing was done using the Bonferroni correction or Student–Newman–Keuls multiple comparisons. Significance was reported at p < 0.05.
3. Results
3.1. LIF and gp130 mRNA in developing brain and retina
Expression of LIF and its receptor subunit gp130 in developing brain and retina was measured at p2, 7, and 14. LIF mRNA expression was readily detectable in control brain tissue throughout early development (Fig. 1A). Densitometry results of LIF, normalized to 18S, showed mean values ± standard deviation (SD) at p2: 0.85 ± 0.11, p7: 0.84 ± 0.15, and p14: 0.41 ± 0.12. LIF mRNA was undetectable in retina (results not shown).
Fig. 1.
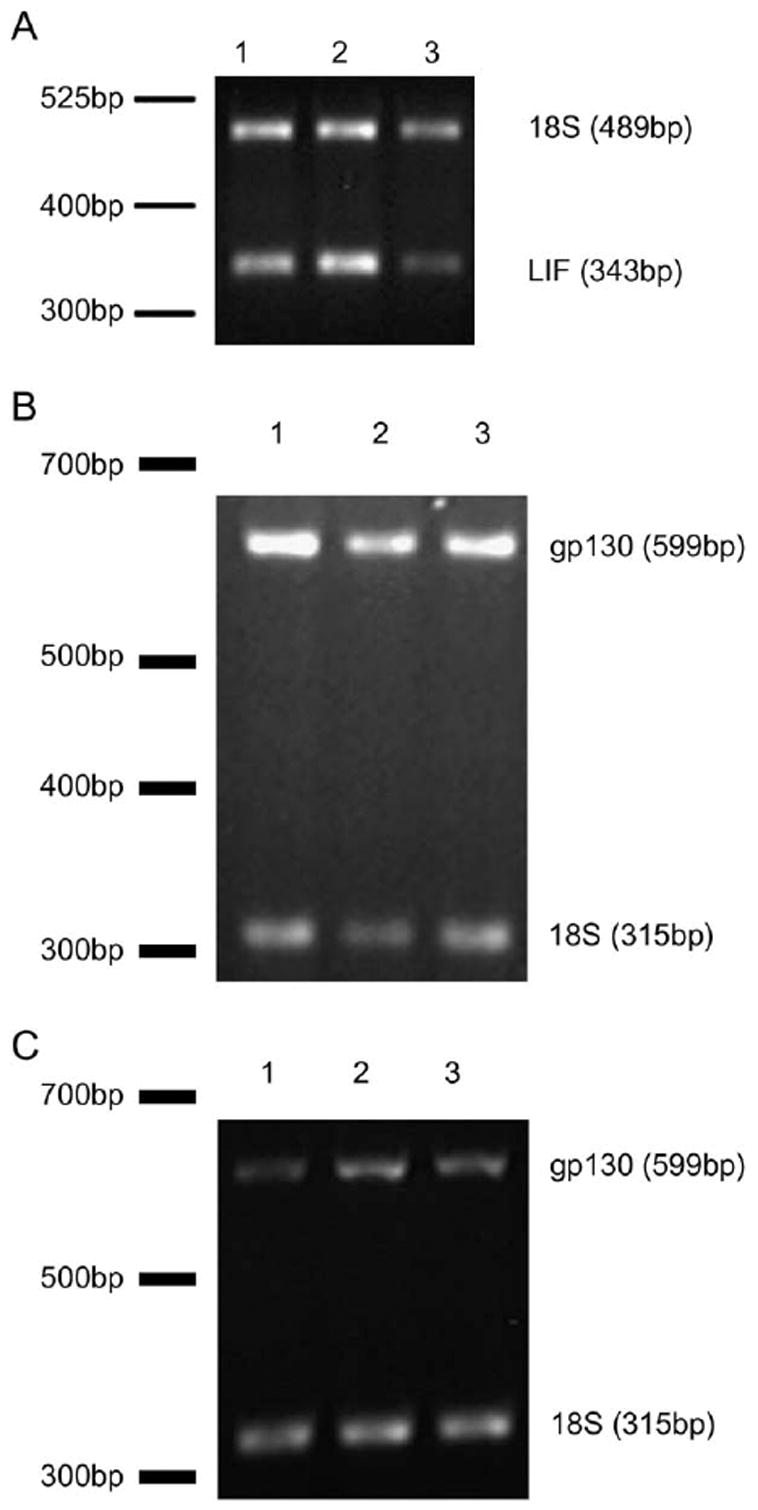
(A) LIF mRNA and (B) gp130 mRNA during brain development and (C) gp130 during retina development. Lane 1 postnatal age (p)2, lane 2 p7, and lane 3 p14.
gp130 mRNA was detectable in both brain (Fig. 1B) and retina (Fig. 1C) throughout development. Densitometry results of gp130 normalized to 18S in brain samples had mean values ± SD at p2: 2.09 ± 0.21; p7: 1.76 ± 0.56; and p14: 1.91 ± 0.44), suggesting that expression early on remained relatively constant through development. In the retina, densitometry results normalized to 18S showed mean values ± SD at p2: 2.39 ± 0.2, p7: 3.96 ± 0.24, and p14: 4.7 ± 0.03) demonstrating that expression significantly increased in the developing retina (p < 0.005 overall ANOVA, with each value significantly different from each other at p < 0.003 Bonferroni correction).
3.2. LIF attenuates retinal vascularization in developing rat pups when injected both locally and systemically
Previously, our group quantified rat retinal vascularization at time points during development (McColm et al., 2004) and found similar results to other groups (Ishida et al., 2003). At p6, 3 days after IP injection there was no difference in total weights or total retinal area of PBS-versus LIF-injected pups (Table 1). However, in LIF-injected animals, there was significantly greater peripheral avascular retina compared to PBS-injected pups (mean percent avascular ± SD, 39.7 ± 5.5% vs 36.8 ± 5.8% respectively, p < 0.045 t-test, Table 1 and Fig. 2).
Table 1.
Summary statistics for animals injected at postnatal age 3 and analyzed at p6 (p3/6) and animals injected at p7 and analyzed at p9 (p7/9)
| Injection group | N | Total retinal area (mm2) | Percent avascular peripheral retina | Weight (g) | |
|---|---|---|---|---|---|
| Intraperitoneal p3/6 | PBS | 28 | 33.0 ± 1.9 | 36.8 ± 5.8 | 11.4 ± 1.1d |
| LIF | 37 | 33.6 ± 3.1 | 39.7 ± 5.5b | 11.4 ± 0.8d | |
| Intravitreous p7/9 | Non-injecteda | 49 | 38.8 ± 3.8 | 4.7 ± 3.8 | – |
| PBS | 19 | 37.6 ± 3.3 | 5.7 ± 3.3 | 16.9 ± 1.8 | |
| LIF | 17 | 39.6 ± 4.1 | 9.1 ± 4.4c | 16.8 ± 1.6 |
The non-injected eyes of the PBS-and LIF-injected animals were not significantly different from each other and were therefore combined for the analysis. Each animal had one eye injected and the other non-injected; therefore, the weights of the non-injected eyes are analyzed with the injected pups data.
p < 0.045 t-test LIF-injected vs control.
p < 0.0001 ANOVA; significantly different from non-injected at p < 0.0001 and PBS-injected at p < 0.022, both Bonferroni correction.
Weights from a subset of nine animals within this group.
Fig. 2.
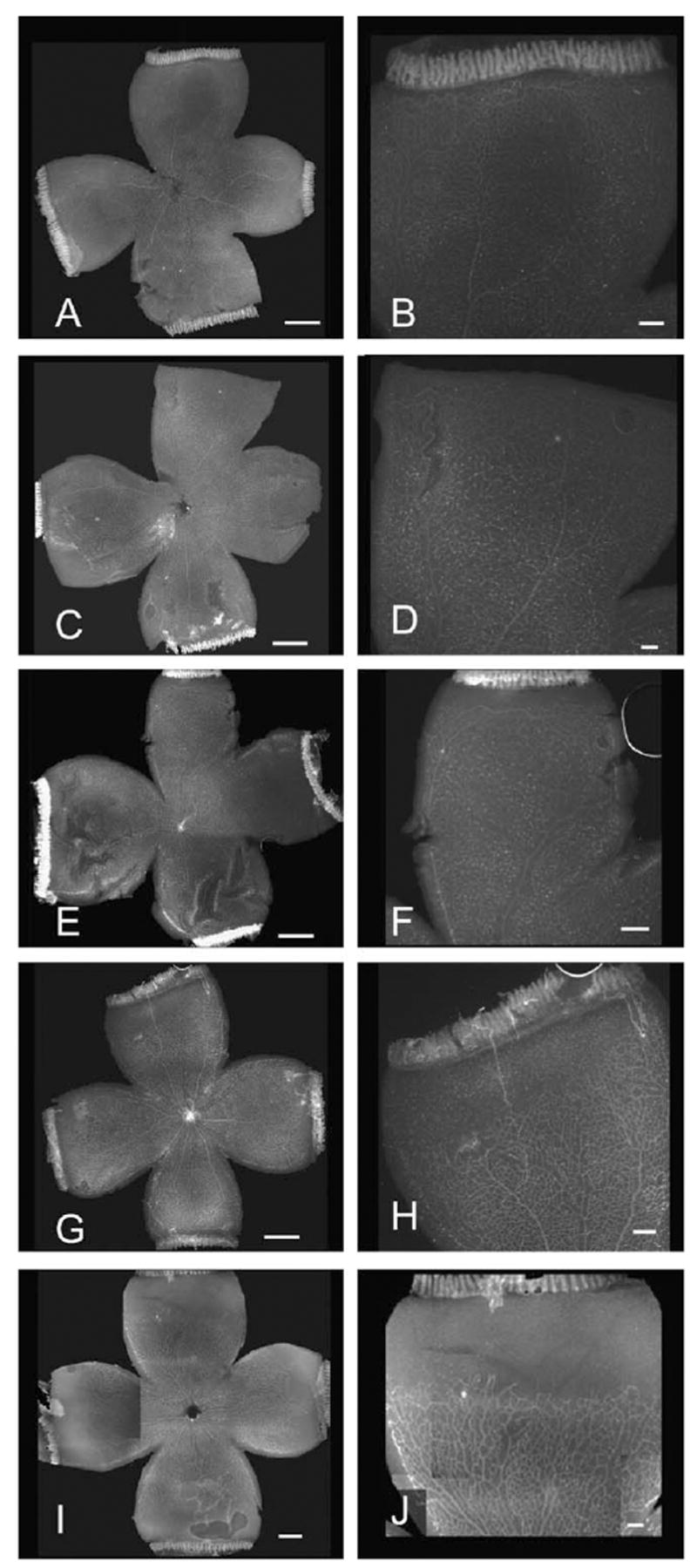
Retinal wholemounts stained with Griffonia lectin. Left side scale bar represents 1 mm; Right side is a higher magnification image of the top quadrant from respective image on right with scale bar representing 0.25 mm. p7 intravitreous injected rat pups analyzed at p9 (A and B, non-injected; C and D, PBS-injected; E and F, LIF-injected). G and I, p3 intraperitoneal injected rat pups analyzed at p6 (G and H, PBS-injected; I and J, LIF-injected).
Similar results were seen when LIF was injected directly into the vitreous. First, the non-injected left eye from the PBS-injected group and the non-injected left eye from the LIF-injected group were compared. There was no difference in body weight, total retinal area or avascular peripheral area (results not shown). Therefore, all of these data were grouped together into one group designated non-injected. There was no difference in total weight or total retinal area in PBS-injected and LIF-injected animals. However, there was a significantly larger peripheral retinal avascular area in LIF-injected (9.1 ± 4.4%) vs. PBS-injected (5.7 ± 3.3%) or non-injected pup eyes (4.7 ± 3.8%) (Fig. 2 and Table 1, p < 0.0001 overall ANOVA, LIF-vs. non-injected eyes p < 0.0001, LIF-vs. PBS-injected eyes p < 0.022, Bonferroni post-hoc testing). There was no significant difference in the peripheral retinal avascular area between non-injected and PBS-injected eyes. The numbers of animals in the PBS-and LIF-injected groups were similar at the start of the experiment. However, when intravitreous injection interfered with the ability to dissect complete retinal whole mounts necessary to analyze avascular/total retina, these eyes were not included in the analysis. For this reason, the number of samples for analysis differed among groups.
3.3. Intravitreous injection increases apoptosis in the inner capillary plexus, but IP injection does not
During development of the inner retinal capillary plexus, we found apoptosis from p2 through p10 (Fig. 3). This has been reported by others (Hughes and Chan-Ling, 2000) and is part of normal remodeling that occurs in the developing retinal vasculature. After p10, apoptosis was significantly reduced (overall ANOVA p < 0.0005; p2 vs. p12 and p14, p < 0.001 Bonferroni; Fig. 3).
Fig. 3.
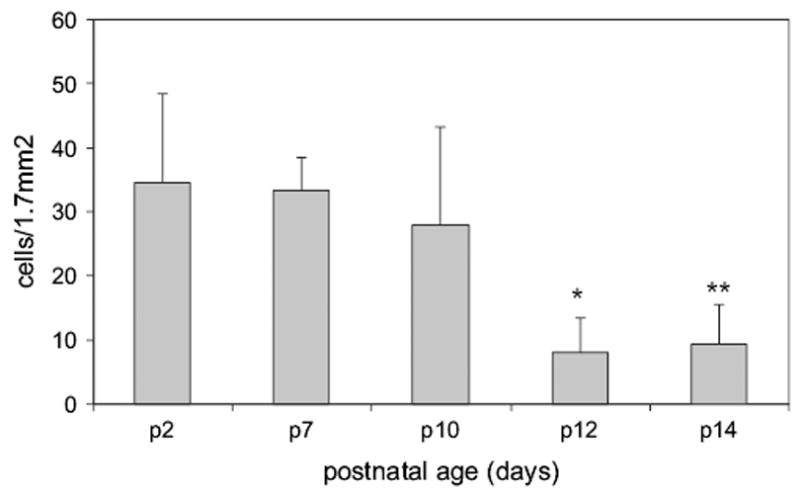
Total caspase-3 positive cells in the inner capillary layer during normal development. Overall ANOVA significant at p < 0.001. *p12 is significantly different from p2, p7 and p10 (p < 0.0005, p = 0.003 and p = 0.031 respectively). **p14 was significantly different from p2 and p7 (p = 0.001 and p = 0.006 respectively).
Analysis of apoptotic staining of the retina revealed that IP injection did not significantly increase apoptosis (mean cells counted/mm2 ± standard deviation, 9.7 ± 5.2 PBS-injected vs. 16.3 ± 7.9 LIF-injected, p = 0.08 t test). However, in the intravitreous-injected group there was a significant increase in caspase-3 positive positive cells in both injected groups compared to controls (PBS-vs non-injected p = 0.001, LIF-vs non-injected p = 0.0001, Fig. 4). There was no significant difference in the apoptosis counts between PBS-injected and LIF-injected. It was noted that caspase-3 positive cells when double stained with lectin, were within and not part of the capillary. As Griffonia isolectin B4 can also bind to other cell types, including microglia and leukocytes (Cho et al., 2002) it was unlikely these double stained lectin and caspase-3 cells were endothelial cells.
Fig. 4.
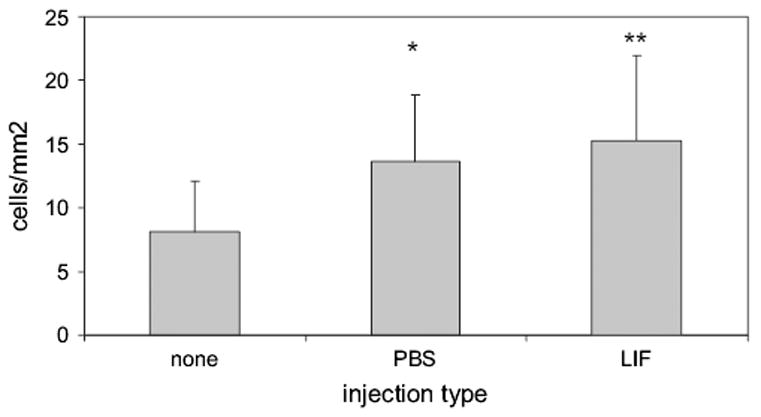
Apoptotic cells counted in the inner capillary plexus of p9 animals given intravitreous injection at p7. Overall ANOVA was significant at p < 0.0001. Post hoc testing with the Bonferroni correction *p = 0.001, **p = 0.0001 compared to non-injected. There was no difference between the PBS-and LIF-injected groups.
3.4. Effect of exogenous LIF on proliferation and apoptosis of cultured endothelial cells
Cell proliferation, measured by cell counts, was significantly decreased in the presence of LIF (p < 0.0001, overall ANOVA; Fig. 5). Addition of LIF antibody to media containing LIF restored cell proliferation to control levels, and antibody to LIF alone had no significant effect on cell proliferation (results not shown).
Fig. 5.
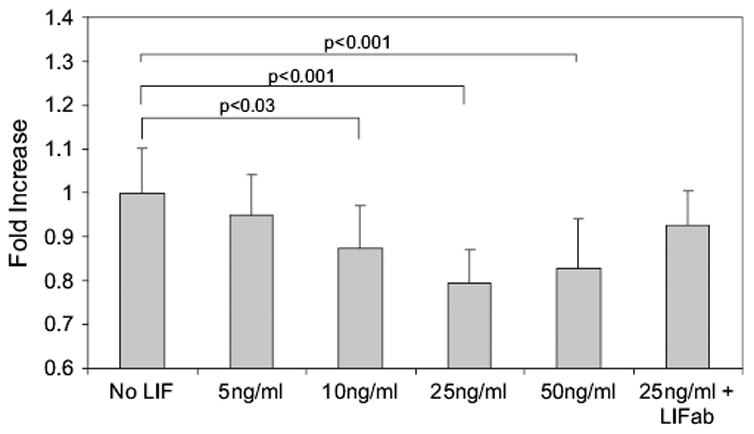
Effect of exogenous LIF on RMVEC proliferation in vitro. Overall ANOVA (p < 0.001). All p values in the figure represent the post-hoc Bonferroni test. ab, antibody; LIF, leukemia inhibitory factor.
LIF and LIF plus antibody had no effect on apoptosis in cultured RMVECs compared to control (Fig. 6B,C vs A respectively), whereas staurosporine caused apoptosis in 100% of cells (Fig. 6D, double stained yellow cells).
Fig. 6.
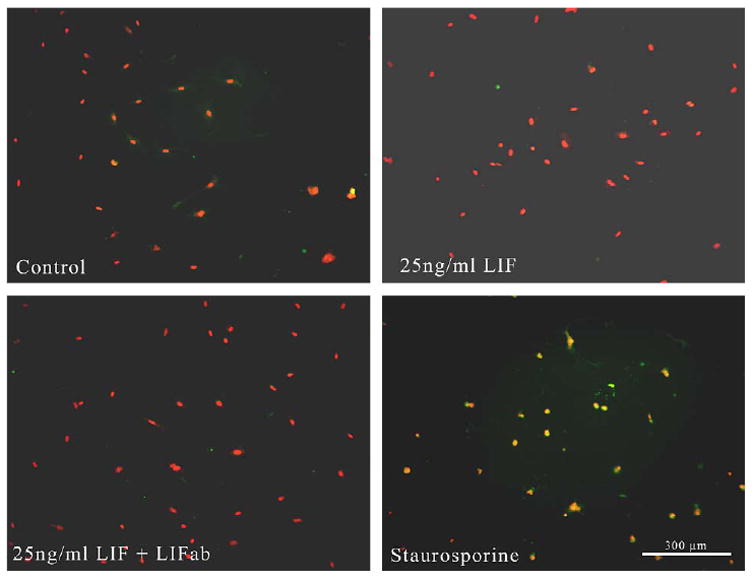
Retinal microvascular endothelial cells grown with LIF and stained with antibody to activated caspase-3 (green) and propidium iodide (red). (A) Controls without LIF. (B) LIF, (C) LIF plus antibody. LIF and LIF plus antibody had no effect on apoptosis compared to control (B, C vs A respectively). (D) RMVECs grown with staurosporine which caused apoptosis in all cells (double stained yellow cells).
3.5. Injection of intraocular LIF caused increased phosphorylation of gp130
Phosphorylation of the gp130 receptor was significantly increased by LIF injection, while uninjected and PBS injected eyes had similar results (Fig. 7).
Fig. 7.
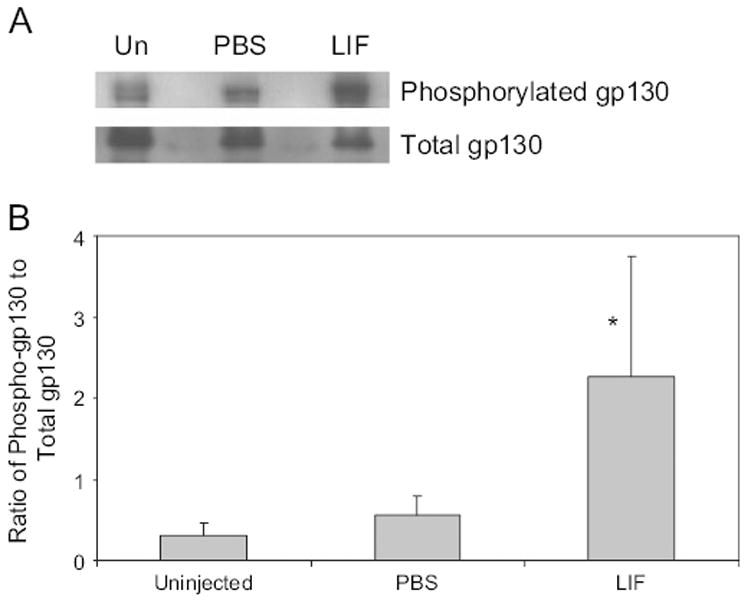
(A) Western blot of phosphorylated and total gp130 in uninjected eyes (Un), PBS injected or LIF injected. (B) Densitometry analysis of Western blot. Uninjected and PBS injected had similar amounts of gp130 phosphorylation, whereas eyes injected with LIF showed an increase in gp130 phosphorylation (overall ANOVA p = 0.0226, *p < 0.05 vs uninjected and PBS injected).
3.6. Cell cycle analysis
For each sample histograms were generated of cell number vs total fluorescence and then cell populations were assigned manually to a cell cycle stage according to quantity of total DNA. Results of nine separate runs were averaged and showed that there was less cells in S phase in the presence of LIF (Fig. 8, both concentrations p < 0.001 compared to controls).
Fig. 8.
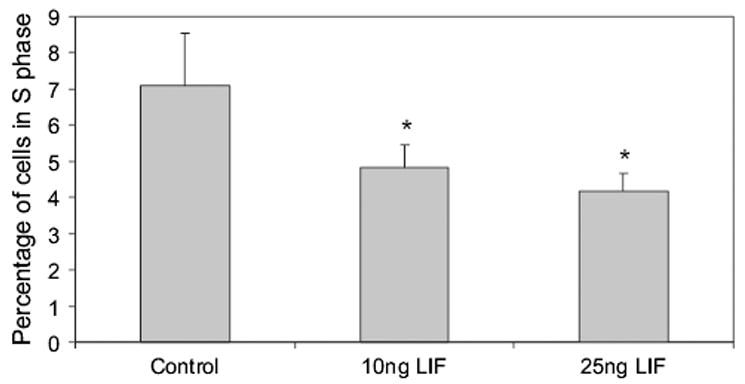
Cell cycle stages measured by flow cytometry analysis of total DNA staining by propidium iodide (overall ANOVA p < 0.001; *p < 0.001 compared to control).
4. Discussion
We found that exogenously administered LIF, given systemically or into the vitreous, caused an arrest in retinal vascular development. LIF did not cause significant growth retardation compared to the animals in the non-injected or PBS-injected groups. Both body weights and total retinal areas were comparable amongst all experimental groups.
We provide evidence that the effect of LIF was through growth arrest rather than apoptosis of endothelial cells. In vivo, there was no increase in the number of apoptotic cells in the IP-injected LIF group compared to PBS-injected group, even though there was a significantly larger peripheral avascular retinal area in the LIF-injected group. In p7 pups that had intravitreous injections, there was a greater number of apoptotic cells in both PBS- and LIF-injected groups compared to the non-injected group, but no difference between the two injected groups. The LIF-injected group had a significantly larger peripheral avascular retinal area compared to the PBS-injected group. This finding suggests that apoptosis was associated with effects from the injection, but that the increased avascularity seen with LIF administration was not mainly due to apoptosis or from the injection.
In vitro studies using RMVECs support the in vivo data and provide additional evidence that LIF does not increase apoptosis. When RMVECs were cultured in the presence of LIF, there was a significant decrease in cell proliferation compared to cells grown without LIF. This effect could be reversed when LIF antibody was added to cells exposed to LIF. LIF did not affect RMVECs apoptosis, but did reduce proliferation and entry into S phase. The in vitro and in vivo results provide evidence that LIF may prevent retinal vascularization through EC growth arrest and not through EC apoptosis. This is in agreement with Ash et al. (2005) who demonstrated that LIF overexpressed in the lens of mice resulted in absence of the retinal vasculature, rather than regression of an existing vascular network.
Although LIF was not detected in the retinal tissue during normal development, it was readily detectable in the brain. The receptor subunit, gp130, was also found in both brain and retina, so if LIF had access to the retina, signaling through gp130 would be possible.
gp130 belongs to the LIF/IL-6 family of receptors that are expressed predominantly on neuronal cells in the retina (Rhee and Yang, 2003), but also have been shown to be present in endothelial cells (Modur et al., 1997). Ligands of the LIF/IL-6 family all have their own receptors and form different heterodimers with gp130 (Yamauchi-Takihara, 2002). Ligands that signal through these receptors have contradictory effects on angiogenesis. LIF induced an embryonic cell line to proliferate and form capillary-like structures through STAT3 signaling (Paradis and Gendron, 2000). However, bovine aortic endothelial cells were strongly inhibited by LIF (Ferrara et al., 1992; Pepper et al., 1995) and adrenal cortex-derived capillary endothelial cells were unaffected (Ferrara et al., 1992). IL-6 has been shown to stimulate angiogenesis through STAT3-induced VEGF expression (Wei et al., 2003). One explanation of the different effects on angiogenesis in this family of cytokines and receptors might be that alternate signaling pathways are activated downstream of gp130.
We showed that systemically administered LIF produced a local effect on the developing retinal vasculature. Therefore, it is conceivable that soluble LIF produced in other tissues either during development (e.g. brain) or increased after ischemia-reperfusion (Ikegami et al., 2002; Suzuki et al., 2000) could access the retina and affect retinal vascular development. Retinal Muller cells in vitro have been stimulated to produce LIF (Neophytou et al., 1997); therefore, at least some cells in the retina are capable of LIF expression under the correct conditions. Previous studies in mice showed that LIF mRNA expression levels were very low, especially in postnatal tissue (Murphy et al., 1993). Thus, LIF may be present in the developing retina but undetectable by current methods.
Our data agrees with that of Ash et al. (2005) that showed that when overexpressed in mouse lens, LIF resulted in reduced retinal vasculature. Other studies show that LIF can arrest epithelial tumor cells in G0/G1 phase (Park et al., 2003). We now show that LIF can prevent entry of endothelial cells into S phase and that exogenous LIF delivered both systemically and locally attenuates retinal vascular development. These effects may have implications in certain diseases that occur during retinal vascular development.
Acknowledgments
We thank Drs Jason Vittitow and Terete Borràs for their help with RT-PCR, Dr Mohanish Deshmukh for assistance with the caspase-3 staining, Dr Silva Markovic-Plese for assistance with FACS analysis and Dr Gerard Lutty for supplying RMVECs. MEH is a member of the UNC Lineberger Comprehensive Cancer Center and the Carolina Cardiovascular Biology Center. Funding for parts of this study were through the University of North Carolina’s Research Council, Research to Prevent Blindness and the NIH R01 EY015130.
References
- Ash JD, McLeod DS, Lutty GA. Transgenic expression of leukemia inhibitory factor (LIF) blocks normal vascular development but not pathological neovascularization in the eye. Mol Vis. 2005;11:298–308. [PubMed] [Google Scholar]
- Auernhammer CJ, Melmed S. Leukemia-inhibitory factor – neuro-immune modulator of endocrine function. Endocr Rev. 2000;21:313–345. doi: 10.1210/edrv.21.3.0400. [DOI] [PubMed] [Google Scholar]
- Butzkueven H, Zhang JG, Soilu-Hanninen M, Hochrein H, Chionh F, Shipham KA, Emery B, Turnley AM, Petratos S, Ernst M, Bartlett PF, Kilpatrick TJ. LIF receptor signaling limits immune-mediated demyelination by enhancing oligodendrocyte survival. Nat Med. 2002;8:613–619. doi: 10.1038/nm0602-613. [DOI] [PubMed] [Google Scholar]
- Chan-Ling T. Glial, vascular and neuronal cytogenesis in whole-mounted cat retina. Microsc Res Tech. 1997;36:1–16. doi: 10.1002/(SICI)1097-0029(19970101)36:1<1::AID-JEMT1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Cho EY, Choi HL, Chan FL. Expression pattern of glycoconjugates in rat retina as analysed by lectin histochemistry. Histochem J. 2002;34:589–600. doi: 10.1023/a:1026032005521. [DOI] [PubMed] [Google Scholar]
- Collo G, Pepper MS. Endothelial cell integrin alpha5beta1 expression is modulated by cytokines and during migration in vitro. J Cell Sci. 1999;112:569–578. doi: 10.1242/jcs.112.4.569. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Winer J, Henzel WJ. Pituitary follicular cells secrete an inhibitor of aortic endothelial cell growth: identification as leukemia inhibitory factor. Proc Natl Acad Sci USA. 1992;89:698–702. doi: 10.1073/pnas.89.2.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadient RA, Lein P, Higgins D, Patterson PH. Effect of leukemia inhibitory factor (LIF) on the morphology and survival of cultured hippo-campal neurons and glial cells. Brain Res. 1998;798:140–146. doi: 10.1016/s0006-8993(98)00236-4. [DOI] [PubMed] [Google Scholar]
- Gendron RL, Tsai FY, Paradis H, Arceci RJ. Induction of embryonic vasculogenesis by bFGF and LIFin vitro and in vivo. Dev Biol. 1996;177:332–346. doi: 10.1006/dbio.1996.0167. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Jollimore CA, McLaren MJ, Inana G, Kelly ME. Mammalian retinal pigment epithelial cells in vitro respond to the neuro-kines ciliary neurotrophic factor and leukemia inhibitory factor. Biochem Cell Biol. 1997;75:119–125. [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes S, Chan-Ling T. Roles of endothelial cell migration and apoptosis in vascular remodeling during development of the central nervous system. Microcirculation. 2000;7:317–333. [PubMed] [Google Scholar]
- Ikegami T, Suzuki Y, Shimizu T, Isono K, Koseki H, Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene. Biochem Biophys Res Commun. 2002;296:729–736. doi: 10.1016/s0006-291x(02)00933-6. [DOI] [PubMed] [Google Scholar]
- Ishida S, Yamashiro K, Usui T, Kaji Y, Ogura Y, Hida T, Honda Y, Oguchi Y, Adamis AP. Leukocytes mediate retinal vascular remodeling during development and vaso-obliteration in disease. Nat Med. 2003;9:781–789. doi: 10.1038/nm877. [DOI] [PubMed] [Google Scholar]
- McColm JR, Geisen P, Hartnett ME. VEGF isoforms and their expression after a single episode of hypoxia or repeated fluctuations between hyperoxia and hypoxia: relevance to clinical ROP. Mol Vis. 2004;10:512–520. [PMC free article] [PubMed] [Google Scholar]
- Metcalf D. The unsolved enigmas of leukemia inhibitory factor. Stem Cells. 2003;21:5–14. doi: 10.1634/stemcells.21-1-5. [DOI] [PubMed] [Google Scholar]
- Modur V, Li Y, Zimmerman GA, Prescott SM, McIntyre TM. Retrograde inflammatory signaling from neutrophils to endothelial cells by soluble interleukin-6 receptor alpha. J Clin Invest. 1997;100:2752–2756. doi: 10.1172/JCI119821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Reid K, Brown MA, Bartlett PF. Involvement of leukemia inhibitory factor and nerve growth factor in the development of dorsal root ganglion neurons. Development. 1993;117:1173–1182. doi: 10.1242/dev.117.3.1173. [DOI] [PubMed] [Google Scholar]
- Neophytou C, Vernallis AB, Smith A, Raff MC. Muller-cell-derived leukaemiainhibitory factorarrests rod photoreceptor differentiationat a postmitotic pre-rod stage of development. Development. 1997;124:2345–2354. doi: 10.1242/dev.124.12.2345. [DOI] [PubMed] [Google Scholar]
- Paradis H, Gendron RL. LIF transduces contradictory signals on capillary outgrowth through induction of stat3 and (P41/43)MAP kinase. J Cell Sci. 2000;113:4331–4339. doi: 10.1242/jcs.113.23.4331. [DOI] [PubMed] [Google Scholar]
- Park JI, Strock CJ, Ball DW, Nelkin BD. The Ras/Raf/MEK/extracellular signal-regulated kinase pathway induces autocrine-paracrine growth inhibition via the leukemia inhibitory factor/JAK/STAT pathway. Mol Cell Biol. 2003;23:543–554. doi: 10.1128/MCB.23.2.543-554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper MS, Ferrara N, Orci L, Montesano R. Leukemia inhibitory factor (LIF) inhibits angiogenesis in vitro. J Cell Sci. 1995;108:73–83. doi: 10.1242/jcs.108.1.73. [DOI] [PubMed] [Google Scholar]
- Rhee KD, Yang XJ. Expression of cytokine signal transduction components in the postnatal mouse retina. Mol Vis. 2003;9:715–722. [PMC free article] [PubMed] [Google Scholar]
- Senturk LM, Arici A. Leukemia inhibitory factor in human reproduction. Am J Reprod Immunol. 1998;39:144–151. doi: 10.1111/j.1600-0897.1998.tb00346.x. [DOI] [PubMed] [Google Scholar]
- Sherry DM, Mitchell R, Li H, Graham DR, Ash JD. Leukemia inhibitory factor inhibits neuronal development and disrupts synaptic organization in the mouse retina. J Neurosci Res. 2005;82:316–332. doi: 10.1002/jnr.20619. [DOI] [PubMed] [Google Scholar]
- Stefana B, Ray DW, Melmed S. Leukemia inhibitory factor induces differentiation of pituitary corticotroph function: an immuno-neuroendocrine phenotypic switch. Proc Natl Acad Sci USA. 1996;93:12502–12506. doi: 10.1073/pnas.93.22.12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CL, Kaspar P, Brunet LJ, Bhatt H, Gadi I, Kontgen F, Abbondanzo SJ. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature. 2003;359:76–79. doi: 10.1038/359076a0. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Tanaka K, Nogawa S, Ito D, Dembo T, Kosakai A, Fukuchi Y. Immunohistochemical detection of leukemia inhibitory factor after focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2000;20:661–668. doi: 10.1097/00004647-200004000-00003. [DOI] [PubMed] [Google Scholar]
- Wei LH, Kuo ML, Chen CA, Chou CH, Lai KB, Lee CN, Hsieh CY. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene. 2003;22:1517–1527. doi: 10.1038/sj.onc.1206226. [DOI] [PubMed] [Google Scholar]
- Williams RL, Hilton DJ, Pease S, Willson TA, Stewart CL, Gearing DP, Wagner EF, Metcalf D, Nicola NA, Gough NM. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature. 1988;336:684–687. doi: 10.1038/336684a0. [DOI] [PubMed] [Google Scholar]
- Yamauchi-Takihara K. Gp130-mediated pathway and left ventricular remodeling. J Card Fail. 2002;8:S374–S378. doi: 10.1054/jcaf.2002.129254. [DOI] [PubMed] [Google Scholar]