

Abstract
Endogenous factors, including hormones, growth factors and cytokines, play an important role in the regulation of hepatic drug metabolizing enzyme expression in both physiological and pathophysiological conditions. Alterations of hepatic drug metabolizing enzymes gene and protein expression, observed in diabetes, fasting, obesity, protein-calorie malnutrition and long-term alcohol consumption alters the metabolism of xenobiotics, including procarcinogens, carcinogens, toxicants, and therapeutic agents and may also impact the efficacy and safety of therapeutic agents, as well as result in drug-drug interactions. Although the mechanisms by which xenobiotics regulate drug metabolizing enzymes have been studied intensively, less is known regarding the cellular signaling pathways and components which regulate drug metabolizing enzyme gene and protein expression in response to hormones and cytokines. Recent findings, however, have revealed that several cellular signaling pathways are involved in hormone- and growth factor-mediated regulation of drug metabolizing enzymes. Our laboratory, and others, have demonstrated that insulin and growth factors regulate drug metabolizing enzyme gene and protein expression, including cytochromes P450, glutathione S-transferases and microsomal epoxide hydrolase, through receptors which are members of the large receptor tyrosine kinase family, and by downstream effectors such as phosphatidylinositol 3-kinase, the mitogen activated protein kinase, Akt/protein kinase B, mTOR, and the p70S6 kinase. Here, we review current knowledge of the signaling pathways implicated in regulation of drug metabolizing enzyme gene and protein expression in response to insulin and growth factors, with the goal of increasing our understanding of how chronic disease affects these signaling pathways, components, and ultimately gene expression and translational control.
Keywords: Drug metabolizing enzyme, Insulin, Growth factor, Signaling pathway, Kinase, Phosphatase
1. Introduction
1.1 General
The hepatic expression of drug metabolizing enzymes may be altered in response to development, age, gender, genetic factors, nutrition, pregnancy and pathophysiological conditions such as diabetes, long-term alcohol consumption, inflammation, protein-calorie malnutrition, therapeutic agents, and environmental chemicals. Although the transcriptional and post-transcriptional mechanisms by which xenobiotics regulate drug metabolizing enzymes have been intensively studied, relatively less is known regarding the cellular mechanisms by which endogenous factors, including hormones and growth factors, regulate drug metabolizing enzyme gene and protein expression. Recent research, however, has revealed that hormones and growth factors play a critical role in regulating the expression of drug metabolizing enzyme genes and proteins. These regulatory events proceed through various signaling pathways, many of which are involved in cell growth, proliferation, transformation and tumorigenesis. The mechanism(s) by which these signaling pathway kinases, and phosphatases, regulate gene expression in response to hormone and growth factor agonists are being actively examined with a view that the ultimate functional endpoint involves effects on transcription factors in regulation of gene transcription as well as mRNA translation in the production of proteins. Thus, signaling pathways regulate not only gene transcription, but also mRNA translation. Furthermore, these signaling pathways have multiple arms, fingers and family member components, which provides for cross-communication and essentially a three dimensional platform for regulating transcriptional and/or translational processes.
Xenobiotic metabolism occurs predominantly in the liver, although substantial biotransformation activity also exists in nasal mucosa, intestine, kidney, lung, placenta and skin. Hepatic drug metabolizing enzymes involved in phase I and II reactions may be markedly altered in diabetes, fasting, obesity and long-term alcohol consumption, resulting in the altered metabolism of xenobiotics, procarcinogens, carcinogens, toxicants and therapeutic agents. Such differences in drug metabolizing enzyme expression and levels can also lead to altered toxicity and changes in the efficacy and safety of therapeutic agents and drug-drug interactions. In view of the expression of these genes and enzymes in different tissues and cell types, it is important to recognize that intracellular signaling pathways and components involved in the regulation of these genes may differ in a tissue and cell-specific manner. Hence, most of this presentation will focus on insulin signaling in the regulation of hepatic gene and protein expression.
Diabetes has been reported to result in increased expression of hepatic cytochromes P450 (CYP) 1A1, 2B, 3A, 4A, 2E1 and bilirubin UDP-glucuronosyltransferase (UGT1A1), whereas decreased expression of CYP2C11, microsomal epoxide hydrolase (mEH) and sulfotransferases (SULTs), such as hydroxysteroid SULT-a (SULT2A1) and aryl SULT IV (SULT1A1), has been reported, (summarized in Table 1; Bellward et al., 1988; Barnett et al., 1990; Donahue et al., 1991; Song et al., 1990; Shimojo et al., 1993; Thomas et al., 1989; Runge-Morris and Vento 1995; Braun et al., 1998; Hong et al., 1987; Ronis et al., 1993; Van de Wiel et al., 1993; Kardon et al., 2000; Duvaldestin et al., 1975; Visser et al., 1996; Chaudhary et al., 1993; Abernethy et al., 1983; Woodcroft, et al., 2002; Woodcroft and Novak, 1997; Woodcroft and Novak, 1999a). In contrast, studies on the glutathione S-transferase (GST) gene expression and metabolic activity during diabetes report both increased and decreased GST expression or activity reported in vivo (Rouer et al., 1981; Agius and Gidari, 1985; Grant and Duthie, 1987; Thomas et al., 1989; Mukherjee et al., 1994; Raza et al., 1996). This difference however, may reflect competing factors in vivo and/or oxidative stress, which may be present during diabetes. The change in the cellular redox state in conjunction with oxidative stress has been reported to result in the transcriptional activation of some GST genes (Wasserman and Fahl, 1997; Kang et al., 2001a). Glutathione (GSH) synthesis can also be altered in response to pathophysiologic conditions such as diabetes, protein-calorie malnutrition and alcohol consumption through regulation of gamma-glutamylcysteine ligase (GCL) expression, the enzyme which catalyzes the first step of GSH synthesis, and by the availability of cysteine for GSH synthesis (Yoshida et al., 1995; Lu et al., 1999; Kim et al., 2003c).
Table 1.
Effect of diabetes, insulin and glucagon on drug-metabolizing enzyme expression and/or activity.
| Enzyme | Substrate | Diabetes | Insulin | Restored by Insulin | Glucagon | Refs |
|---|---|---|---|---|---|---|
| CYP2B | Cyclophosphamide, Mephenytoin, | ↑ | ↓ | Yes | ND | Yamazoe et al., 1989b; Donahue and Morgan, 1990; Truong et al., 2005; Woodcroft and Novak, 1997 |
| CYP3A | Nifedipine, Midazolam, Erythromycin, Cyclosporine, Testostreone | ↑ | ← → | Yes | ND | Barnett et al., 1990; Shimojo et al., 1993; Woodcroft and Novak, 1997 |
| CYP4A | Fatty acids and derivatives | ↑ | Marginal
↑ |
Yes | ND | Barnett et al., 1990; Shimojo et al., 1993; Woodcroft and Novak, 1997 |
| CYP2E1 | Chlorzoxazone, 4-Nitrophenol, Ethanol, Nitrosamines, Benzene, Halogenated hydrocarbons | ↑ | ↓ | Yes | ↑ | Bellward et al., 1988; Song et al., 1990; Woodcroft and Novak, 1997, 1999a, 1999b; Woodcroft et al., 2002 |
| mEH | Alkene epoxides (styrene 7,8-epoxide), Arene oxides (naphthalene 1,2-oxide) | ↓ | ↑ | Yes | ↓ | Thomas et al., 1989; Kim et al., 2003b |
| UGT1A1 | Oripavine opioids, Anthraquinones, Catechol estrogens, Bilirubin, 7-Hydroxy coumarines | ↑ | ND | Yes | ↑ | Braun et al., 1998; Tunon et al., 1991 |
| SULT2A1 | Dehydroepiandrosterone, Estradiols | ↓ | ND | Yes | ND | Runge-Morris and Vento 1995 |
| GST alpha | Androstene dione, Lipid peroxides, 4-Hydroxyalkenals, 1-Chloro-2,4-dinitrobenzene, 7-Chloro-4-nitrobenzo-2-oxa-1,3-dizaone, Busulfan, Chlorambucil, Ethacrynic acid, Polyaromatic hydrocarbon diol epoxides | ND | ↑ | ND | ↓ | Kim et al., 2003a |
| GST mu | trans-Stilbene oxide, 1,2-Dichloro-4-nitrobenzene, Prostaglandin H2, 1,3-bis-(2- Chloroethyl)-1-nitrosourea, Benzo[a]pyrene diol epoxide | ND | ← → | ND | ← → | Kim et al., 2003a |
| GST pi | Acrolein, Ethacrynic acid, Prostaglandin H2, Benzo[a]pyrene diol epoxide, Chlorambucil, 1-Chloro-2,4-dinitrobenzene | ND | ← → | ND | ↓ | Kim et al., 2003a |
| GCL | ↓ | ↑ | Yes | ↓ | Yoshida et al., 1995; Lu et al., 1999; Kim et al., 2004 |
ND is not determined.
Because diabetes, obesity and starvation can result in altered hormone (insulin, glucagon, growth hormone) secretion, sensitivity (i.e. resistance) and/or levels, these hormones may alter the expression of hepatic drug-metabolizing enzymes. Insulin or growth hormone administration to chemically-induced or spontaneously diabetic rats has been reported to restore drug-metabolizing enzyme activity and expression to control values (Table 1) (Dong et al., 1988; Favreau and Schenkman, 1988; Yamazoe et al., 1989a and 1989b; Thomas et al., 1989; Donahue and Morgan, 1990; Thummel and Schenkman 1990; Runge-Morris and Vento 1995; Donahue et al., 1991; Tunon et al., 1991; Morrison and Hawksworth, 1984; Woodcroft et al., 2002; Woodcroft and Novak, 1997; Woodcroft et al. 1999a). Glucagon, as a physiologic antagonist of insulin, plays a critical role in regulating glucose and the expression of drug metabolizing enzyme genes and activity (Okuda et al., 1987; Jiang and Zhang, 2003; Woodcroft and Novak, 1999b; Kim et al., 2003a, b). We have demonstrated that insulin and glucagon regulate, in an opposing manner, the expression of CYP2E1 (Woodcroft and Novak, 1999b), alpha-class GSTs (Kim et al., 2003a) and mEH (Kim et al., 2003b) in primary cultured rat hepatocytes. Glucagon prevents the expression of pi class GSTs in primary cultured hepatocytes (Kim et al., 2003a). Table 1 shows the insulin- and glucagon-mediated regulation of the activity and/or expression of hepatic drug metabolizing enzymes genes and protein such as CYP2B, CYP2E1, CYP2A5, GCL, GST alpha class, GST pi class, UGT and mEH (De Waziers et al., 1995; Constantopoulos and Matsaniotis, 1978; Ricci and Fevery, 1988; Viitala et al., 2001; Carrillo et al., 1995; Iber et al., 2001; Woodcroft and Novak, 1997 and 1999a; Woodcroft et al., 2002; Lu et al., 1991 and 1992; Kim et al., 2003a and 2003b; Truong et al., 2005). These results show that changes in drug-metabolizing enzyme gene expression or protein levels may be attributed to alterations in insulin levels. The primary question is how does this occur, since neither insulin nor growth factors directly function as transcription factors. The answer resides in the signaling pathways, which communicate information to the nucleus for regulation of gene transcriptional and concomitantly regulate the various processes and translational machinery which controls translational activity.
Although expression of drug metabolizing enzyme genes and proteins are altered in response to pathophysiologic conditions, drug metabolizing enzyme gene expression and protein levels are also changed during development and aging. This alteration in expression occurs in an organ-, sex- and species-specific manner. Hence, cell and organ context are important considerations for the expression of drug metabolizing enzymes and their regulation by hormones or growth factors.
Hepatic GSTA1, GSTA5, GSTM1 and GSTM2 gene expression in the male rat and the hypophysectomized female rat is down regulated by growth hormone (Srivastava and Waxman, 1993; Staffas et al., 1998). It has been reported that treatment of hypophysectomized rats with growth hormone results in decreased mEH and UGT1A1 activity, and SULT1E2 mRNA level (Inoue et al., 1995; Liu and Klaassen, 1996; Gueraud et al., 1997; Yokota et al., 2002). Also, growth hormone has been reported to regulate gender-dependent differences in CYP expression (Shapiro et al., 1995).
Growth factors, including epidermal growth factor (EGF) and hepatocyte growth factor (scatter factor) (HGF), however, also regulate drug metabolizing enzyme gene expression. HGF results in decreased CYP2C11 expression in primary cultured rat hepatocytes (Iber and Morgan, 1998), and decreased CYP1A1/2, 2A6, 2B6 and 2E1 activities in primary cultured human hepatocytes (Donato et al., 1998). In primary cultured rat hepatocytes, EGF addition results in the suppression of constitutive and xenobiotic-inducible CYP2C11, CYP2C12, CYP1A1, and CYP2B1/2 expression (De Smet et al., 2001; Garcia et al., 2001; Ching et al., 1996) and CYP2E1 (Abdelmageed et al., unpublished observation). In contrast, EGF treatment has been reported to increase alpha and pi class GST expression (Matsumoto et al., 2000; Desmots et al., 2002).
In this review, the insulin- and growth factor-mediated receptor tyrosine kinase signaling pathways are reviewed in the regulation of drug metabolizing enzyme expression in response to insulin and growth factors. An overview of the major insulin and growth factor signaling cascade is provided in Figure 1A.
Fig. 1.
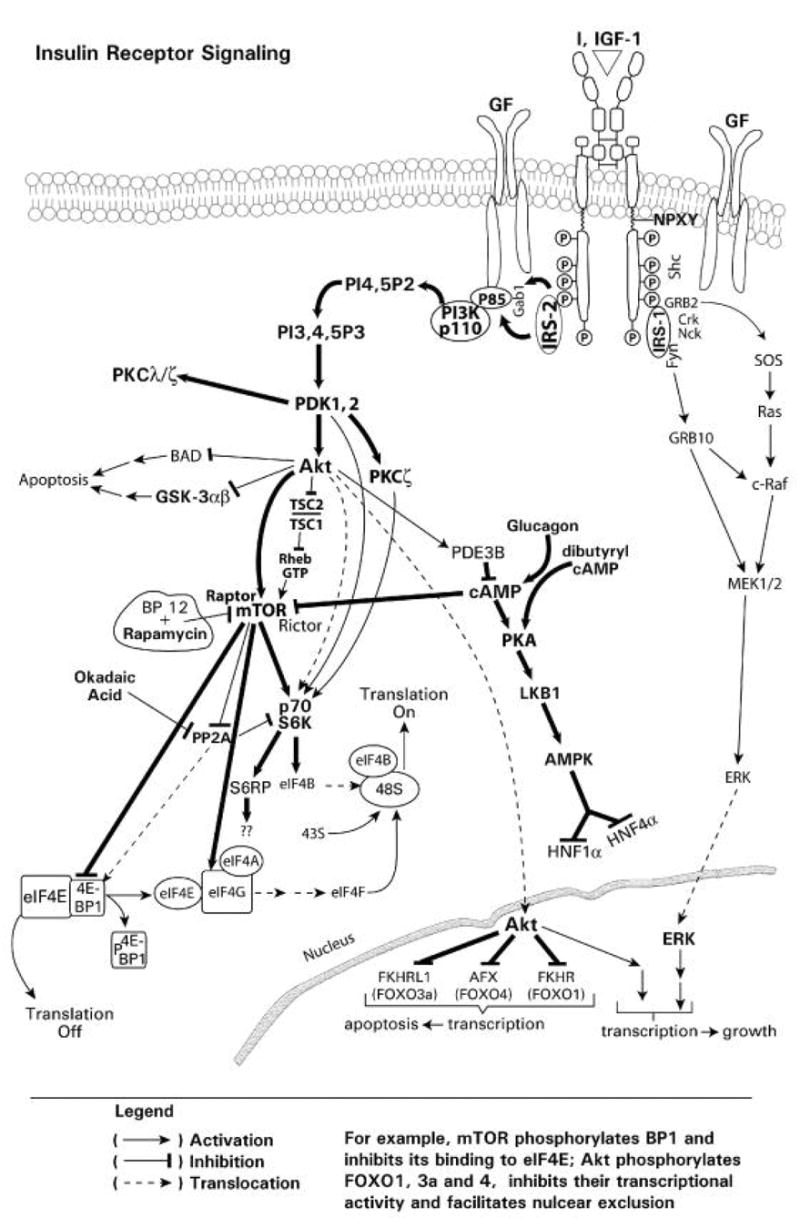
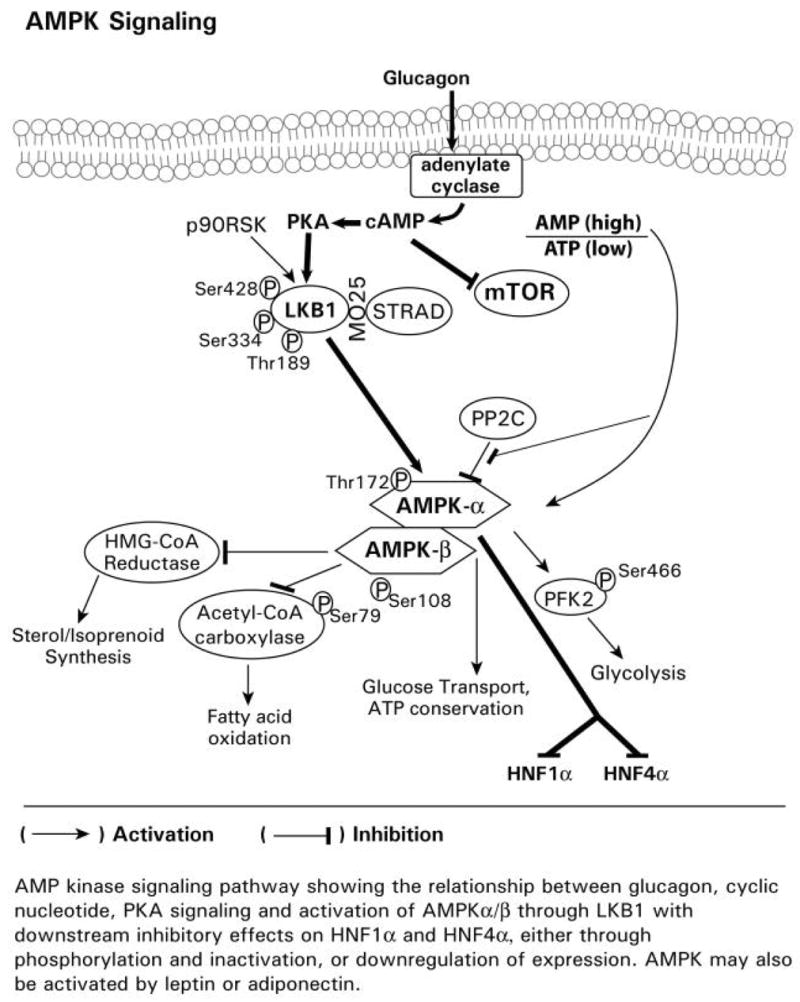
Fig. 1A Insulin-mediated signaling pathways. A diagrammatic representation of insulin and growth factor receptor mediated signaling with effects on gene transcription, through phosphorylation of the FOXO1, 3a and 4 transcription factors, and mRNA translation, through 4E-PB1, p70 S6 kinase, eIF4B, S6 ribosomal protein, and eIF4G.
Fig. 1B Glucagon-mediated signaling pathways. A diagrammatic representation of glucagon mediated signaling through the adenylate cyclase, cAMP, PKA, AMPK pathway with inhibitory effects on mTOR and the transcription factors HNF1alpha and HNF4alpha. The opposing effects of glucagon on insulin-mediated alterations in gene expression may occur through glucagon repression of insulin signaling through mTOR, which is associated with activation of PKA, the phosphorylation of LKB1, and activation of AMPK. Glucagon also represses activation of the phosphorylation of 4E-B P1 and the p70S6 kinase.
1.2 Overview of Insulin and Growth Factor Signaling
Insulin and growth factors produce their effects through receptor tyrosine kinases (RTKs) (Cantley, 2002; Cobb, 1999; Porter and Vaillancourt, 1998; Ullrich and Schlessinger, 1990; Vivanco and Sawyers, 2002) which have cell surface, transmembrane and cytosolic domains. The cell surface domain binds the agonist and the cytosolic domain, which contains bound ATP, exhibits inherent tyrosine kinase activity and is activated in response to agonist binding. The insulin receptor (IR) is a α2β2 heterotetramer, which consists of an extracellular agonist binding domain, a transmembrane domain and a cytoplasmic domain (Fig. 1A). Binding of insulin (or IGF1) results in a conformational change in the juxtapositioned cytosolic β subunits, which results in autoactivation of the cytosolic subunit tyrosine kinase activity and transphosphorylation of the tyrosine resides in one cytosolic β subunit by another β subunit resulting in increased kinase activity and additional phosphorylation at the juxta-membrane regions and intracellular tail. This phosphorylation produces conformational changes, which present binding sites for recruitment of SH2 (Src homology 2) or phosphotyrosine binding (PTB) domain-containing proteins, including insulin receptor substrates IRS1-6 and Shc (Backer et al., 1993; Myers et al., 1994; Vivanco and Sawyers, 2002) (Fig. 1A). IRS (Insulin Receptor Substrate) proteins, following recruitment and phosphorylation, amplify signaling through their recruitment of numerous other adapter, activator proteins (i.e. GTP/GDP exchangers), and kinases via their phosphorylated tyrosine motifs (Cantley 2002; Myers et al., 1994; Vivanco and Sawyers, 2002). These tyrosine phosphorylated motifs (i.e. SH2 domains) serve as docking sites for numerous downstream substrate effector proteins, including the lipid kinase phosphatidylinositol 3-kinase (PI3K), Grb2, and the adaptor proteins Crk, Nck, Fyn (Cantley, 2002; Myers et al., 1994; Vivanco and Sawyers, 2002) (Fig. 1A). The IR, through Shc and Grb2, also recruits SOS (Son of Sevenless), the GTP/GDP exchange which recruits and activates Ras which in turn recruits and activates Raf, with subsequent recruitment and activation of MEK and ERK. Thus, insulin, via the IR and IRS, exerts its downstream metabolic and mitogenic effects through multiple downstream signaling proteins, with both pathways stimulating cell proliferation.
Insulin signaling leads to a number of effects, including stimulation of glucose transport, protein and glycogen synthesis, inhibition of lipolysis, regulation of gene transcription and translation, and cell growth and proliferation (Brazil et al., 2002; Cantley 2002; Myers et al., 1994; Vivanco and Sawyers, 2002). PI3K is activated following the binding of its p85 regulatory subunit Src homology-2 (SH2) domain to the tyrosine-phosphorylated IRS-1 or-2, resulting in activation of the p100 catalytic subunit of PI3K (Backer et al., 1993; Brazil et al., 2002; Cantley, 2002; Cobb, 1999; Myers et al., 1994; Monostory et al., 2004, Vivanco and Sawyers, 2002) (Fig. 1A). The activation of PI3K results in the production of 3-phosphorylated phosphatidylinositides (e.g. PI(3,4,5)P3) (Fig. 1A), which then activate the downstream protein serine/threonine kinase Akt/PKB, via 3-phosphoinositide-dependent protein kinase-1 and -2 (PDK1, 2) (Alessi et al., 1997b; Brazil et al., 2002; Cantley, 2002; Stokoe et al., 1997; Vivanco and Sawyers, 2002) (Fig. 1A). Akt phosphorylates and inhibits TSC1,2 (Tuberous Sclerous Complex 1 and 2), which inhibits Rheb, and results in the phosphorylation and activation of mTOR. mTOR, in turn, phosphorylates and inactivates 4E-BP1 (Binding Protein 1) of the eIF4E/BP1 complex, thereby releasing BP1 from eIF4E (eukaryotic Initiation Factor 4E) and stimulating the initiation of translation (Fig. 1A). PDK1, 2 directly, or through PKCζ, as well as Akt, or Akt through mTOR, phosphorylates and activates the p70S6 kinase and the ribosomal S6 protein RS6P (Beugnet et al., 2003; Brazil et al., 2002; Cantley 2002; Cheatham et al., 1994; Chung et al., 1994; Vivanco and Sawyers, 2002), which also regulates translation. Thus, Akt is a central transducer of insulin signaling which regulates mRNA translation (Fig. 1A).
Akt is a major regulator of the activity of the downstream mammalian target of rapamycin (mTOR) and tuberous sclerosis complex 1 and 2 (TSC1 and 2) (Fig. 1A). Akt activates mTOR through phosphorylation. Under nonstimulated conditions, TSC1/2 act as negative regulators of mTOR. TSC1 and 2 are hamartin and tuberin, respectively (Michalet et al., 1997; Tee et al., 2003). TSC2 phosphorylation by Akt results in mTOR activation (Hay and Sonenberg, 2004). TSC1 and 2 form heterodimers which exhibit GTPase activity that inhibits the GTPase Rheb which is required for mTOR activation (Hay and Sonenberg, 2004). Thus, TSC1 and 2 constitute a functional complex that inhibits mTOR activity, resulting in inhibition of the phosphorylation of downstream targets, 4E-BP1, p70S6K and RS6P, and hence translation (Goncharova et al., 2002; Inoki et al., 2002; Manning et al., 2002, Hay and Sonenberg, 2004). Phosphorylation of TSC1/2 by Akt reverses the inhibitory effects of TSC1/2 on mTOR and results in mTOR activation (Carraway and Hidalgo, 2004) (Fig. 1A). The regulatory associated protein of mTOR (Raptor) is required for mTOR-mediated phosphorylation of BP-1(binding protein 1) which results in the release of eIF4E and the initiation of translation (Nojima et al., 2003; Oshiro et al., 2004) as well as activation of the 40S ribosomal protein S6 kinase (p70S6K) and ribosomal S6 protein RS6P (Fig. 1A). The inhibition of mTOR by rapamycin, occurs through the binding of rapamycin to FKBP12 (FK506 binding protein 12), which results in the binding of this protein complex to mTOR, and inhibition of the mTOR-Raptor interaction (Nojima et al., 2003; Oshiro et al., 2004). This inactivation of mTOR inhibits translation by preventing the phosphorylation and release of BP1 from eIF4E, and the activation p70S6K and RS6P (Fig. 1A). Thus, Akt, through mTOR, regulates mRNA translation, a process which is inhibited by rapamycin (Fig. 1A). An additional downstream target of Akt phosphorylation is glycogen synthase kinase 3α,β (GSK3α,β), which is involved in glucose metabolism, and is used to monitor Akt activation and inhibition (Hajduch et al., 2001) (Fig. 1A). Rictor is a rapamycin-insensitive companion of mTOR and defines a distinct pathway regulating PKCα and Akt signaling (Sarbassov and Sabatini, 2005). Akt also activates phosphodiesterase 3B, which inhibits the production of cAMP required for PKA activation (Fig. 1A).
In addition to translation, Akt also regulates gene transcription. Upon activation by PI3K signaling, Akt translocates to the nucleus (Andjelkovic et al., 1997), where it regulates the expression of genes by catalyzing the phosphorylation of the transcription factors FOXO1, 3a and 4 (Fig. 1A), which results in inactivation through export from the nucleus and inhibition of FOXO-regulated transcription, although recent reports suggest that insulin effects through Akt can occur by direct mechanisms and do not necessarily involve altered subcellular distribution (Du and Montminy, 1998; Zhang et al., 2002a; Kwon et al., 2004; Tsai et al., 2003; Matsuzaki et al., 2005). Thus, Akt activation results in the downregulation of genes for which FOXO 1, 3a and 4 serve as transcription factors. Akt is also able to effect signaling through PKA, via inhibition of phosphodiesterase 3β and AMPK activity (Hahn-Windgassen et al., 2005).
1.3 Overview of Glucagon, Cyclic Nucleotides, PKA, AMPK Signaling
Glucagon increases cAMP levels through adenylate cyclase. Elevated cAMP levels have been reported to attenuate mTOR signaling (Fig. 1 A, B), and hence translational control (Graves et al., 1995; Monfar et al., 1995; Harris and Lawrence, 2003). The glucagon, cyclic nucleotide activated PKA signaling cascade is also complex and appears to involve activation of LKB1 and AMP kinase (AMPK), which phosphorylates other targets, including HNF1α, the transcription factor reported for regulating CYP2E1 gene transcription (Cheung et al., 2003), and HNF4α (Fig. 1 A, B). The opposing effects of glucagon on insulin in CYP2E1 gene expression may occur through glucagons/cyclic nucleotide repression of signaling through mTOR, which is associated with activation of PKA, the phosphorylation of LKB1, and activation of AMPK. Glucagon also represses activation of the phosphorylation of 4E-BP1 and the p70S6 kinase (Kimball, et al., 2004). The glucagon, cyclic nucleotide activated PKA signaling cascade is also complex and appears to involve activation of LKB1 and AMP kinase (AMPK), which phosphorylates other targets, including HNF1α, the transcription factor reported for regulating CYP2E1 gene transcription (Cheung et al., 2003), and HNF4α (Fig. 1 A, B). AMPK is inhibited by Akt (Hahn-Windgassen et al., 2005) and by phosphatase 2C (Fig. 1 A, B). Recent reports show that AMPK activation results in downregulation of both HNF1α and HNF4α levels and target gene expression in hepatocytes (Leclerc et al., 2001). AMPK was also reported to phosphorylate HNFα1, increase degradation, and decrease target gene expression in kidney cells (Hong et al., 2003). Thus, insulin and glucagon signaling pathways cross-communicate, with each pathway exerting regulatory effects. The exact mechanism(s) which regulate glucagon-mediated CYP2E1 expression and that of the other xenobiotic metabolizing enzyme genes remains to be established.
The individual components of the signaling cascade will be discussed in subsequent sections in greater detail and their role in the signaling cascade identified. The various chemical inhibitors or biological approaches which may be use to disrupt the signaling cascade are also presented. In addition to regulation of gene transcription and/or translation by tyrosine, serine or threonine kinase-mediated phosphorylation, it is also important to recognize that phosphatases play an equally critical role in maintaining a dynamic balance in the signaling cascade. The mechanisms by which the various signaling cascades and components regulate gene transcription and translation are discussed. Finally, examples of various experiments in which have used chemical inhibitors or biochemical approaches to assess the role of the different pathways and components in the regulatory process are also presented to provide a tangible perspective on the role of signaling in the regulation of drug metabolizing enzyme gene and protein expression.
2. Insulin- and Growth Factor-Mediated Cellular Signaling
Insulin, and growth factors, following activation of the TRK (section 1.2), stimulate the recruitment of a family of lipid kinases, known as class I phosphatidylinositol 3-kinases (PI3Ks), to the plasma membrane. Recent evidence indicates that serine/threonine protein kinase Akt/PKB (protein kinase B), atypical protein kinase C (PKC ζ/λ), eukaryotic translation initiation factor 4E (eIF4E)/eIF4E binding protein 1 (4E-BP1) and the p70 ribosomal protein S6 kinase (p70S6 kinase) mediate many of the downstream events in response to PI3K activation (Fig. 2).
Fig. 2.
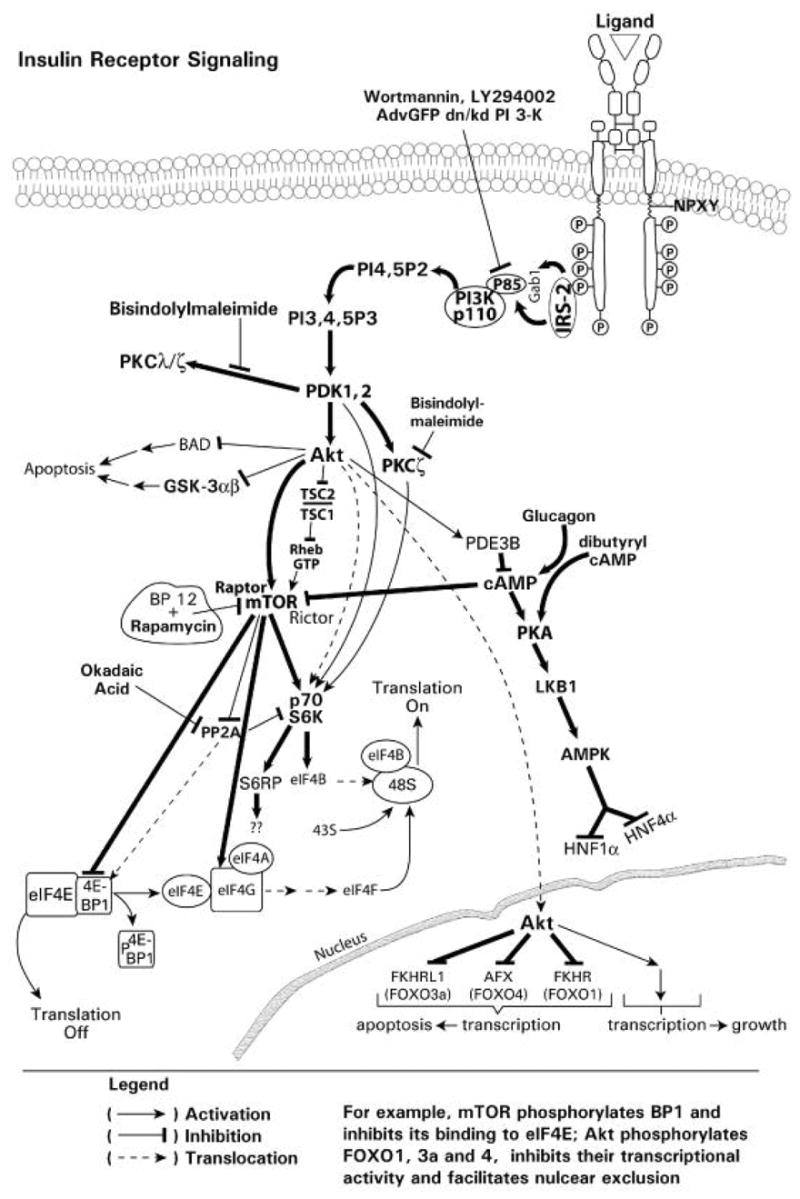
Insulin-mediated activation of PI3K signaling pathways. A diagrammatic representation of insulin/insulin receptor-mediated signaling through the PI3K, PDK1/2 and Akt signaling pathway showing downstream effects on gene transcription and mRNA translation.
Insulin, and growth factors, also lead to activation of the mitogen activated protein kinases (MAPK) signaling cascade (Fig. 3). Following tyrosine phosphorylation of the cytoplasmic domain of the receptor, recruitment of adaptor proteins such as Grb2, a growth factor receptor binding protein 2 (Grb2) which is constitutively associated with the SOS through binding of the SH3 domain of Grb2 to a SOS proline-rich region, and Ras occurs. Ras is activated by SOS, Son-of Sevenless, a guanine nucleotide exchange factor, which converts inactive Ras-GDP to activated Ras-GTP. The small guanosine triphosphatase G-protein Ras subsequently recruits and activates Raf, which leads to phosphorylation and activation of a downstream serine threonine signaling cascade involving recruitment and activation of MEK (MAP Kinase Kinase) and MAPKs/ERKs (MAP Kinase). RTK signaling is regulated not only by a cascade of phosphorylation via protein kinases but also by dephosphorylation via tyrosine and serine/threonine phosphatases and lipid phosphatases.
Fig. 3.
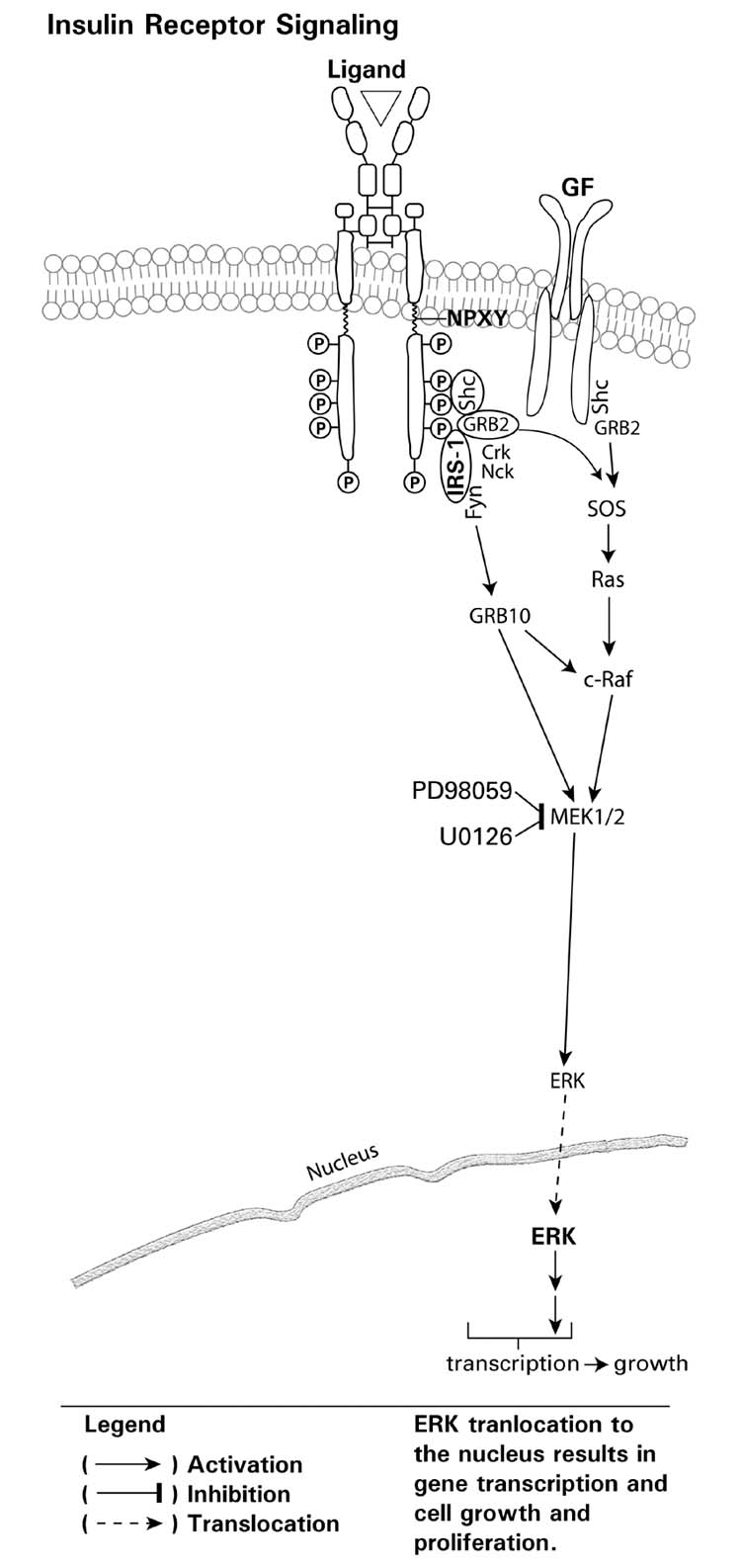
A diagrammatic representation of insulin on growth factor signaling through the Ras-Raf, MEK-ERK signaling pathway with downstream effects on gene transcription through ERK translocation.
2.1 Insulin Receptor and IRS
The insulin receptor is encoded by a single gene located on the short arm of chromosome 19 and contains 22 exons and 21 introns (Fig. 4) (Seino et al., 1989; Perz and Torlinska, 2001). The short exon 11 is alternatively spliced, resulting in two isoforms that differ slightly in affinity for insulin (Seino et al., 1989). Exon 11 codes for 12 amino acids that are inserted the alpha-subunit of the insulin receptor B isoform. The insulin receptor A isoform lacks these 12 amino acids. Although both isoforms have similar affinity for insulin, the A isoform binds insulin-like growth factor-II with more than a 10-fold greater affinity than the B isoform (De Meyts, 2004). Following the binding of agonist (insulin, IGF) to the receptor, the receptor undergoes internalization. The A isoform exhibits a more rapid internalization and higher recycling rate than the B isoform. Processing of the insulin receptor occurs during transport from the golgi to the cell membrane. Proteolytic cleavage of the single high molecular weight proreceptor occurs at a tetrabasic amino acid sequence (arginine-lysine-arginine-arginine), which is located at the alpha and beta subunit junction. Oligosaccharide chains are added at specific sites of glycosylation. Disulfide bridges stabilize the interactions between the two alpha subunits, and between the alpha and beta subunits (Sparrow et al., 1997). Consequently, the single disulfide bond, which occurs between residues cysteine-647 in the alpha subunit and cysteine-872 in the beta subunit, provides a covalent link between the subunits (Cheatham and Kahn, 1992). Thus, the insulin receptor exists as a heterotetramer (Fig. 4).
Fig. 4.
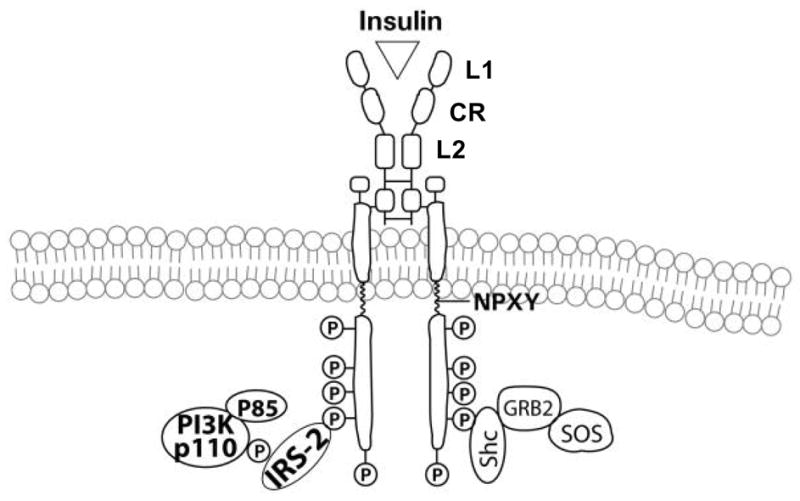
A diagrammatic representation of the insulin receptor, activation of the cytosolic kinase, with subsequent phosphorylation of the kinase domain and tail, followed by the recruitment of the insulin receptor substrate(s) (e.g. IRS-2) and PI3K.
The N-terminal half of the receptor consists of two large homologous globular domains, L1 and L2, separated by a cysteine-rich region (CR) and the C-terminal half consists of three fibronectin type III domains. The second fibronectin type III domain contains a large insert domain of unknown structure containing the site of cleavage between alpha- and beta-subunits. The alpha subunits bind the agonist, e.g. insulin or IGF, and hence represent the critical component in regulating activation. Unoccupied alpha-subunits on the cell surface, in the absence of agonist, inhibit the intrinsic tyrosine kinase activity of the cytoplasmic domain of the beta subunit and hence, function as regulatory subunits of the catalytic intracellular subunits (Kahn, 1994). The beta subunit is composed of a short extracellular domain, a transmembrane domain (TM), and an intracellular domain which contains the ATP binding site and autophosphorylation sites and hence, exhibits tyrosine kinase activity when the receptor is activated through agonist binding. The intracellular portion of the beta-subunit contains the tyrosine kinase domain (KM) which is flanked by two regulatory regions, a juxtamembrane region (JM) and a C-terminal tail (CT). Also the intracellular domain contains the ATP binding site and autophosphorylation sites.
Various lines of evidence suggest that a single molecule of insulin makes asymmetric contacts with both alpha-subunits within a native receptor (De Meyts, 2004). In the two-step bivalent receptor cross-linking model, insulin binds asymmetrically to two discrete sites in the surface alpha-subunits. Binding of the agonist, insulin or IGF to the alpha subunits of the receptor results in conformational changes that allosterically regulate the intracellular beta-subunit tyrosine kinase domain. Subsequently, the beta-subunits undergo a series of intermolecular trans-autophosphorylation reactions that generate multiple phosphotyrosine sites, some of which serve distinct functional roles. Tyrosine phosphorylation at residues 1146, 1150, and 1151 in the kinase domain relieves pseudosubstrate inhibition, further enhancing the receptor’s tyrosine kinase activity. Phosphorylation of tyrosine 960 at the juxtamembrane is necessary for appropriate substrate recognition. The phosphorylated tyrosine residues of the beta subunits of the receptor become binding sites for recruitment of other signaling adaptor proteins and downstream signaling molecules. The insulin receptor, through these sites, is thus able to recruit and utilize a family of soluble adaptors or scaffolding molecules, such as the insulin receptor substrates (IRSs) and Shc, in order to initiate and amplify its signaling cascade through other downstream effectors (Fig. 4).
As many as six members of the IRS family exist (Uchida et al., 2000; Cai et al., 2003). At least three IRS proteins (IRS1, 2 and 4) exist in both humans and mice, with IRS1 and IRS2 widely expressed in tissues, whereas IRS4 expression appears limited to the thymus, brain and kidney (Uchida et al., 2000). IRS3 is expressed exclusively in rodent adipose tissue (Bjornholm et al., 2002), and IRS5 and 6 were recently reported (Cai et al., 2003). The IRSs lack intrinsic catalytic activity. However, they contain pleckstrin homology (PH) and phosphotyrosine binding (PTB) domains, and multiple phosphorylation motifs. The PH domains consist of globular protein domains of ~100 to 120 amino acids, and are primarily involved in the binding of lipids, although they may also mediate, albeit weakly, protein-protein interactions. Residues in PH domains essential for high-affinity binding to phosphoinositides have been identified (Fruman et al., 1999; Isakoff et al., 1998). The IRS PTB domain binds other phosphotyrosine-containing protein motifs. Moreover the PTB domain, similar to the PH domain, also binds phosphoinositides. The PTB domain of IRS binds to the phosphorylated NPXP motifs of the cytoplasmic beta-subunit domain of the activated insulin receptor, and the activated insulin receptor subsequently phosphorylates IRS on multiple tyrosine residues. This activation of IRS results in amplification of insulin receptor signaling. Following phosphorylation, IRS attracts and binds additional effector molecules to the receptor, thereby serving to increase and amplify the extent and diversity of insulin receptor signaling (Cheatham and Kahn, 1995; White and Kahn, 1994; White, 2002). In mice, IRS1 and IRS2 are major mediators of insulin action, as insulin resistance is observed in both IRS1-deficient and IRS2-deficient mice (White, 2002), although functional differences between these molecules have been reported. For example, IRS1 exerts its greatest effect on metabolism by regulating insulin signaling in muscle and adipose tissue. In contrast, IRS-deficient mice display disregulated lipolysis, peripheral glucose uptake, and hepatic gluconeogenesis (Previs et al., 2000).
One of the primary downstream signaling effectors that binds to IRSs following insulin receptor activation is the lipid kinase PI3K. PI3K produces the 3-hydroxylated lipid product PI(3,4,5)P3 from PI4, 5P2 which subsequently activates Akt, and the downstream targets mTOR, p70S6 kinase, eIF4E and PKCs through PDK1, 2 (Fig. 2). While the IRS tyrosine phosphorylation is required for insulin-mediated signaling, the phosphorylation of IRS on serine residues (serine-307, serine-612 and serine-632 in murine IRS1) may disrupt the association between IRS1 and insulin receptor or PI3K and hence, result in resistance to insulin signaling (Gual et al., 2005). In summary, the binding of insulin or IGF to the insulin receptor results in activation via tyrosine phosphorylation of the cytosolic domain and the recruitment of other effectors, including IRS family members, which results in the activation and amplification of transmitted information though the recruitment and activation of downstream effectors in signaling cascades which ultimately regulate gene transcription and mRNA translation.
2.2 PI3K Signaling
The family of PI3Ks in mammalian cells can be divided into three classes, designated Class I through III; class I is also subdivided into Ia and Ib subsets (Vanhaesebroeck et al., 2005). The different classes of PI3Ks catalyze phosphorylation of the 3’-hydroxyl position of phosphatidyl myo-inositol lipids, generating different 3’-phosphorylated lipid products. These 3’-phosphorylated myo-inositol lipids then act as second messengers. Class Ia PI3Ks are activated in response to insulin and growth factors and produce 3’-hydroxylated phosphoinositides (Fruman et al., 1998). Class Ia enzymes exist as heterodimers and consist of a 110-kDa catalytic subunit which is associated noncovalently with either an 85-, 55-, or 50-kDa regulatory subunit. The subclass 1a of the class I PI3K catalytic subunit is subdivided into p110 alpha, beta and delta forms. The 85-, 55- or 50-kDa regulatory subunit maintains the catalytic subunit in a low-activity state in quiescent cells. The regulatory subunit also mediates the activation of the 110 kDa catalytic subunit through interactions between the SH2 domains of the regulatory subunit, and phosphotyrosine residues of adaptor proteins, such as IRSs or those of activated growth factor receptors (Cantley, 2002). Direct binding of p110 to activated Ras has been reported to have an important role in the activation of PI3K in response to growth factors (Shields et al., 2000). The physiological significance of this interaction, however, for insulin-mediated PI3K signaling is not entirely clear. The single class Ib PI3K consists of a p110gamma catalytic subunit complexed to a p101 or p87 regulatory subunit, and signals downstream of G-protein-coupled receptors (Wymann et al., 2003).
A general diagrammatic model illustrating the PI3K recruitment and initiation of the PI3K signaling cascade is illustrated in Fig. 5. In resting cells, PI3K is a cytoplasmic enzyme, the substrate of which is membrane phosphatidylinositol and its phosphorylated derivatives. Insulin receptor activation, and IRS binding recruits PI3K from the cytoplasm to the membrane, primarily via interactions involving the regulatory subunit. Following PI3K recruitment to the plasma membrane, and activation, the lipid kinase phosphorylates the 3’-hydroxyl position of the inositol ring to generate the phosphoinositides, PI(3,4,5)P3, PI(3,4)P2 and PI(3)P with PI(3,4)P2 being the preferred substrate for class I PI3Ks. These events occur rapidly following activation of the receptor. These lipids subsequently function to promote further assembly of signaling complexes at the membrane by recruiting specific proteins with domains (e.g. pleckstrin homology domains) that selectively bind the 3’-hydroxyphosphoinositides. This membrane-targeting signal, however, is reversible and is opposed by specific lipid phosphatases, including PTEN (phosphatase and tensin homolog deleted on chromosome ten). The rapid increase in PI(3,4,5)P3 concentration in response to insulin activates several downstream protein kinases, such as PDK1, Akt, PKC isoforms, p70S6 kinase and the RS6P (Bellacosa et al., 1991; Alessi et al., 1997a; Le Good et al., 1998; Deprez et al., 2000; Vanhaesebroeck and Alessi, 2000), which function to regulate gene transcription and translational control, through phosphorylation of transcription factors, and regulation of mRNA translation and translational machinery. One of the critical and pivotal kinases in this cascade is Akt, which also regulates cell proliferation and survival (Downward, 1998).
Fig. 5.
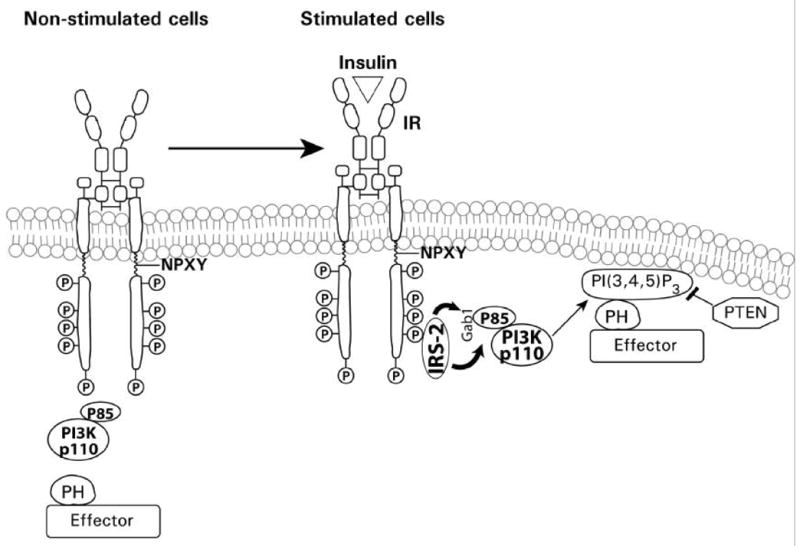
Insulin receptor activation and recruitment. An illustration of insulin binding, insulin receptor activation, phosphorylation, recruitment of PI3K, production of PI(3,4,5)P3, and recruitment of Akt with potential inhibition of Akt by the tumor suppressor PTEN. PH refers to the Pleckstrin homology domain of Akt.
2.3 Akt (Protein Kinase B) Signaling
Hence, among the PI(3,4,5)P3-dependent kinases, Akt has been the focus of much research. Akt/PKB is the cellular homologue of the viral oncoprotein v-Akt, and exhibits a high degree of homology with protein kinases A and C. Akt is a 57 kDa serine/threonine kinase with a PH domain, and three known isoforms (Akt1/PKB alpha, Akt2/PKB beta and Akt3/PKB beta). The Akts are widely expressed in tissues, including the liver (Chan et al., 1999; Song et al., 2005). All three Akt isoforms (Akt1/PKBalpha, Akt2/PKBbeta and Akt3/PKBbeta) consist of a conserved domain structure, an N-terminal PH domain, a central kinase domain (KD; T-loop) and a C-terminal regulatory domain (RD) that contains the hydrophobic motif (Song et al., 2005). In unstimulated cells, Akt is cytosolic and exhibits a low basal activity. Upon activation of PI3K, the association of PI(3,4,5)P3 at the membrane results in the recruitment of Akt and PDK1 into proximity through their PH domains. This results in phosphorylation of Akt at threonine-308 by PDK1 (Fig. 6). Wortmannin and LY294002 are cell-permeable inhibitors of PI3K (Vlahos et al., 1994; Wymann et al., 1996). Wortmannin, an irreversible inhibitor, alkylates a lysine residue at the putative ATP binding site of p110 alpha of PI3K; in contrast, LY294002 is a pure competitive inhibitor of ATP binding. Activation of Akt, and phosphorylation of both threonine-308 and sereine-473 residues, is inhibited by wortmannin and LY294002 (Alessi et al., 1996). Hence, inhibition of PI3K by wortmannin, LY294002 or through the use of dominant-negative/kinase dead constructs, results in downstream inhibition of Akt activation and signaling. The phosphorylation of Akt by PDK1 is regulated by the conformation of Akt. The PH domain masks the activation loop site and its release is required for the phosphorylation by PDK1. Hence, PI(3,4,5)P3/PI(3,4)P2 binding to the PH domain relieves autoinhibition of the active site, allowing PDK1 to access and phosphorylate threonine-308 (Stokoe et al., 1997). Although phosphorylation of threonine-308 partially activates Akt, full activation of Akt requires phosphorylation of serine-473 (Alessi et al., 1996). The phosphorylation of threonine-308 and serine-473 residues can occur independently from each other. The mechanism mediating serine-473 phosphorylation, however, remains controversial. Initial studies proposed that an upstream kinase, also in the PI3K pathway, but distinct from PDK1, is responsible for Akt serine-473 phosphorylation (Alessi et al., 1997b). However, it has also been reported that autophosphorylation may be the mechanism by which serine-473 site is phosphorylated in Akt (Toker and Newton, 2000). This may occur by displacing PDK1 from the C-terminal site of Akt, thus making this site accessible and subject to autophosphorylation. The identity of the protein(s) that mediate the release of PDK1 from the C-terminal site, however, remains to be determined. Alternatively, a putative PDK2 has been proposed, and several other kinases, including the integrin-linked kinase, MAPK-activated protein kinase 2, protein kinase C-related kinase 2, mammalian target of rapamycin (mTOR) and others, have been implicated in the phosphorylation of serine-473 (Brazil and Hemmings, 2001; Sarbassov et al., 2005).
Fig. 6.
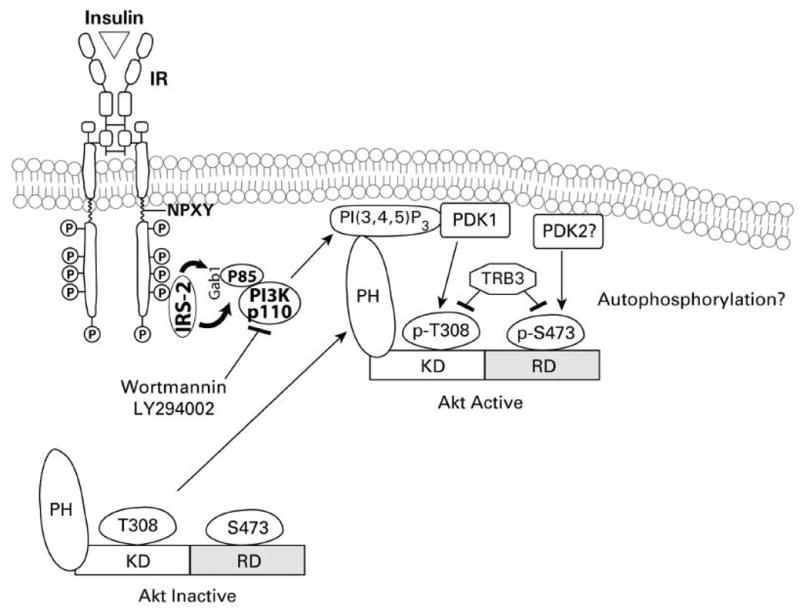
Akt recruitment and activation. This figure showes the process of insulin receptor activation, PI3K recruitment to the IRS, production of PI(3,4,5)P3, recruitment of Akt to the membrane and subsequent activation. PH refers to the Pleckstrin homology domain of Akt.
An additional mechanism for regulating Akt activity has recently been identified. TRB3, a mammalian homologue of Drosophila tribbles, has been identified, and shown to inhibit hepatic Akt activation by insulin (Du et al., 2003). Moreover, it was suggested that functional polymorphism(s) of TRB3 might be associated with insulin resistance and related clinical outcomes (Prudente et al., 2005). Thus, the regulation of Akt activity is complex. The recent identification of a novel and specific inhibitor of Akt, A-443654, by Luo et al., 2005, may facilitate additional experiments on the complex regulation of the kinase and its downstream targets.
Despite its key role as an upstream activator of enzymes such as Akt, the mechanism regulating PDK1 activity remains controversial. Some studies have shown that PDK1, constitutively active in resting cells, is not further activated by growth factor stimulation, and several serine sites (25, 241, 393, 396, 410) are phosphorylated on PDK1 in unstimulated cells (Pullen et al., 1998; Casamayor et al., 1999). However, three groups have demonstrated that insulin, hydrogen peroxide and pervanadate induce an increase in kinase activity and tyrosine phosphorylation of PDK1 (Prasad et al., 2000; Grillo et al., 2000; Park et al., 2001). The phosphorylation of tyrosine residues in PDK1 can be mediated by a Src kinase family member, suggesting Src kinase may also play a role in insulin-mediated PI3K signaling via activation of PDK1 (Park et al., 2001). This and earlier reports constituted a basis for examining a potential role of Src in regulating CYP2E1 expression, although subsequent experiments using chemical inhibitors of Src kinase suggested that it did not play a role in the signaling cascade that regulates CYP2E1 expression.
2.4 Atypical PKC Signaling
Atypical PKC isoforms ζ (rat) and λ (mouse) are also downstream targets for PI3K (Farese, 2001; Farese et al., 2005)(Fig. 7). Increased activity of PKCζ/λ results from PDK1-dependent phosphorylation of the catalytic domain, via threonine 410 in rat PKCζ and threonine 411 in mouse PKCλ, followed by autophosphorylation of threonine 560 in rat PKCζ and threonine 563 in mouse PKCλ (Le Good, et al., 1998; Standaert et al., 1999). PI(3,4,5)P3 may interact with the N-terminal lipid-binding domain of PKCζ to facilitate the interaction of threonine-410 with the catalytic site of PDK1 (Standaert et al., 1999). PI(3,4,5)P3 also stimulates autophosphorylation of PKCζ and relieves the autoinhibition exerted by the N-terminal pseudosubstrate sequence on the C-terminal catalytic domain of PKCζ (Standaert et al., 1999 and 2001). Insulin-stimulated glucose transport and protein synthesis are dependent on PI3K/PKCζ activity (Farese, 2001; Mendez et al., 1997). The latter is consistent with the observation that dominant negative PKCζ antagonizes activation of p70S6 kinase (Romanelli et al., 1999). However, it is not known whether PKCζ can directly phosphorylate p70S6 kinase or which residue(s) is/are involved.
Fig. 7.
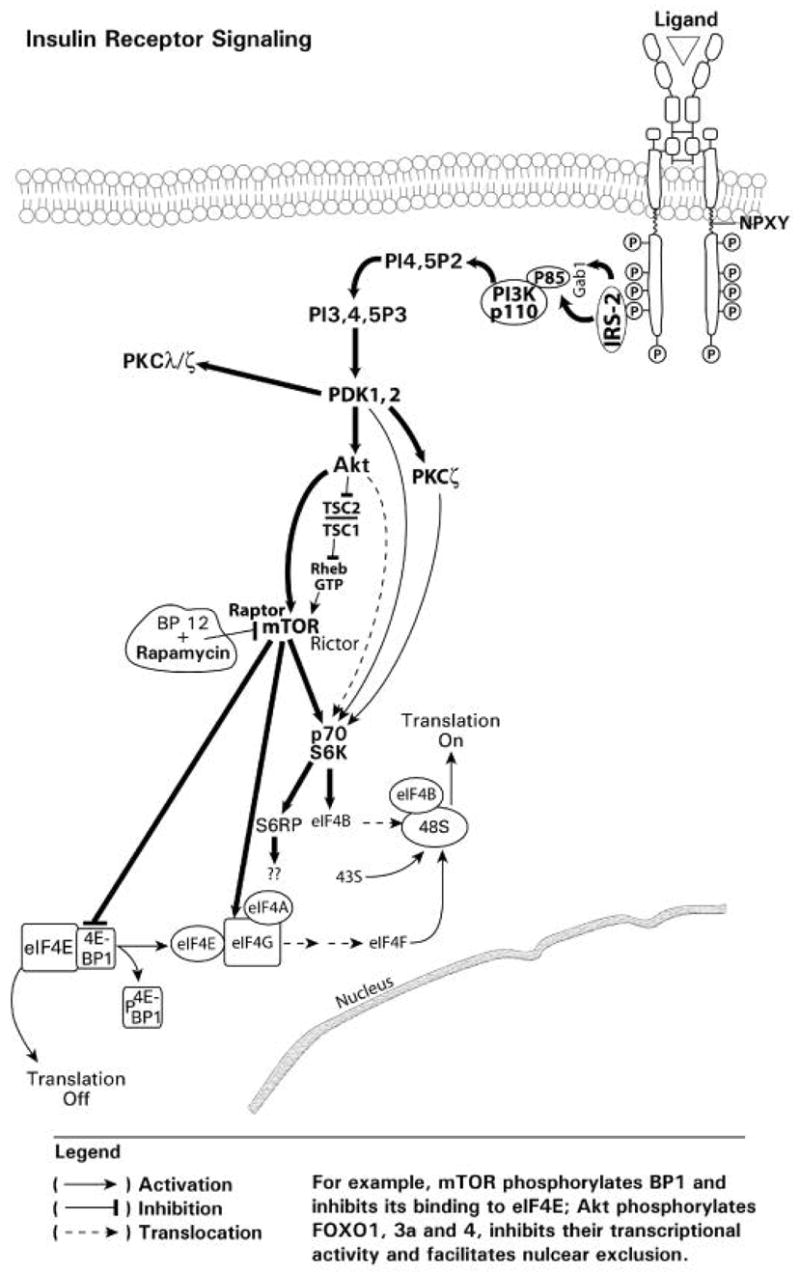
The role of Akt and mTOR in translational control. The activation of mTOR results in phosphorylation of 4E-BP1, release of 4E-BP1 from the eIF4E-BP1 complex and initiation of translaton through formation of the eIF4E/4G/4A complex. mTOR also activated the p70S6K with effects on translational machinery.
2.5 mTOR Signaling
mTOR (Mammalian Target of Rapamycin) is a serine/threonine protein kinase that controls cell growth and proliferation in response to nutrient availability and growth factor stimulation (Richter and Sonenberg, 2005) (Fig. 7). mTOR plays a critical role in regulating translation through phosphorylation of inhibitory proteins (i.e. eIF4E-BP1) that bind the rate limiting eukaryotic initiative factor eIF4E. This protein, 4E-BP1, binds to a cleft in eIF4E and prevents eIF4E from binding to the scaffold protein eIF4G, thereby inhibiting the formation of the active translational complex, eIF4F. mTOR phosphorylates 4E-BP1, which is bound to inactive eIF4E, and relieves the inhibitory effect of 4E-BP1, thereby releasing eIF4E, the rate-limiting step in mRNA translation (Fig. 7). The p70S6 kinase phosphorylates the S6 ribosomal protein (S6RP), which is a component of the 40S subunit of eukaryotic ribosomes. The p70S6 kinase thus plays a critical role in regulating protein synthesis (Jefferies et al., 1997; Kawasome et al., 1998). The p70S6 kinase and S6RP also participate in the translational control of mRNA transcripts that, for the most part, encode components of the translational apparatus (Fig. 7).
The p70S6 kinase contains a highly acidic N-terminus, a serine/threonine kinase catalytic domain and a regulatory C-terminal tail. The C-terminal tail contains an auto-inhibitory domain, and four phosphorylation sites (serine 411, 418, 424 and threonine 421) have been identified in the inhibitory domain. The activation of the p70S6 kinase appears to occur as a result of a phosphorylation-induced conformational change in the C-terminal domain. This conformational change exposes additional phosphorylation sites. Phosphorylation of these newly exposed sites (threonine 229, 389 and serine 371) then occurs, and based on inhibition by wortmannin (i.e. PI3K inhibition) and by rapamycin (i.e. inhibition of mTOR), appears to be dependent on PI3K and mTOR signaling respectively (Saitoh et al., 2002). Threonine 229 is in the kinase catalytic domain, and PDK1 has been shown to be the upstream kinase responsible for the p70S6 kinase threonine 229 phosphorylation (Pullen et al., 1998). Unlike phosphorylation of Akt, the phosphorylation of threonine 229 by PDK1 occurs independent of PI(3,4,5)P3, suggesting that this phosphorylation step may not require membrane association of either PDK1 or the p70S6 kinase. The phosphorylation of threonine 389 in the linker domain is required to disrupt the interaction of the N- and C-termini in order to allow PDK1 access for phosphorylation of threonine 229 (Vanhaesebroeck and Alessi, 2000). The process by which the threonine 389 residue is phosphorylated is less clear. Balendran et al. (1999) reported that PDK1 activity was required for the insulin-like growth factor 1-induced phosphorylation of threonine 389, however, whether PDK1 directly phosphorylates this residue in cells remains unknown.
Expression of a constitutively membrane-anchored and active Akt variant has been reported to phosphorylate and activate the p70S6 kinase (Kohn et al., 1996). However, Akt does not appear to be the kinase for activation of the p70S6 kinase. Rather, activation of the p70S6 kinase may occur independent of Akt (Conus et al., 1998). Subsequent research revealed that a constitutively active wortmannin-resistant form of Akt was able to phosphorylate glycogen synthase kinase-3 and BP-1 in the 4E-BP1 complex, but did not phosphorylate the p70S6 kinase (Dufner et al., 1999). The data suggest that phosphorylation of the p70S6 kinase may be associated with, or an artifact of, membrane localization. Akt, however, can regulate p70S6 kinase activity through activation of mTOR. Recent findings indicate that TSC1 and 2, tuberous sclerosis complex 1 and 2, are hamartin and tuberin, respectively, and constitute a functional complex that inhibits mTOR, resulting in inhibition of the phosphorylation of downstream targets, 4E-BP1 and p70S6 kinase, and hence mRNA translation (Manning, 2004) (Fig. 7). Akt-mediated phosphorylation of tuberin inhibits the TSC1, 2 complex and allows Rheb to bind to GTP, resulting in the activation of mTOR (Manning, 2004). Recently Sarbassov et al. (2004 and 2005) have shown that mTOR can associate with a rapamycin-insensitive companion of mTOR referred to as Rictor, forming a rapamycin-insensitive complex capable of phosphorylating Akt at serine 473. These results suggest that mTOR can be found both upstream and downstream of Akt in the insulin-stimulated PI3K pathway. The regulatory associated protein of mTOR (Raptor) was also identified as an mTOR binding partner that mediated mTOR signaling to downstream targets (Hara et al., 2002; Kim et al., 2002). The binding of Raptor to mTOR is necessary for the mTOR-catalyzed phosphorylation of 4E-BP1, and it strongly enhances the mTOR kinase activity towards the p70S6 kinase. The inhibition of mTOR by rapamycin, occurs through the binding of rapamycin to FKBP12, FK506 binding protein 12, which results in the binding of this complex to mTOR, and inhibition of the mTOR-Raptor interaction (Kim and Sabatini, 2004). This inactivation of mTOR inhibits translation by preventing the phosphorylation and release of BP1 from the eIF4E-BP1 complex, and the activation p70S6 kinase (Fig. 7).
3. MAPK Signaling
3.1 Overview
Many RTKs activate intracellular protein serine/threonine kinases, referred to as the MAPK family, which phosphorylate various downstream targets, including other kinases and transcription factors (Kolch, 2000). The signaling cascade begins with agonist (e.g. insulin, growth factor) induced activation of the receptor followed by recruitment of Ras and the subsequent activation of downstream kinases, which ultimately regulate gene transcription (Fig. 8). The MAPK family consists of several subfamilies, each of which contain multiple members: the extracellular signal-regulated kinase1/2 (ERK1/2), the Jun N-terminal kinases (JNKs), the p38 mitogen activated protein kinase (p38 MAPK) and ERK5. Each MAPK (e.g. ERK1/2, JNK, p38 MAPK, ERK5) is the terminal component of a three-protein kinase cascade, which proceed through a step-wise activation process which involves a MAPK kinase kinase (MKKK) phosphorylating a MKK (MAPK kinase), which then phosphorylates the MAPK. The basic ERK1/2 signaling cascade proceeds through Ras, Raf (MKKK), MEK1/2 (MKK) and ERK1/2 (MAPK) (Fig. 8). Activation of these kinases occurs through agonist binding or stress (e.g. oxidative stress, UV, etc.) and can be monitored via antibodies directed towards the specific kinase and phosphorylated serine/threonine sites in the kinase.
Fig. 8.
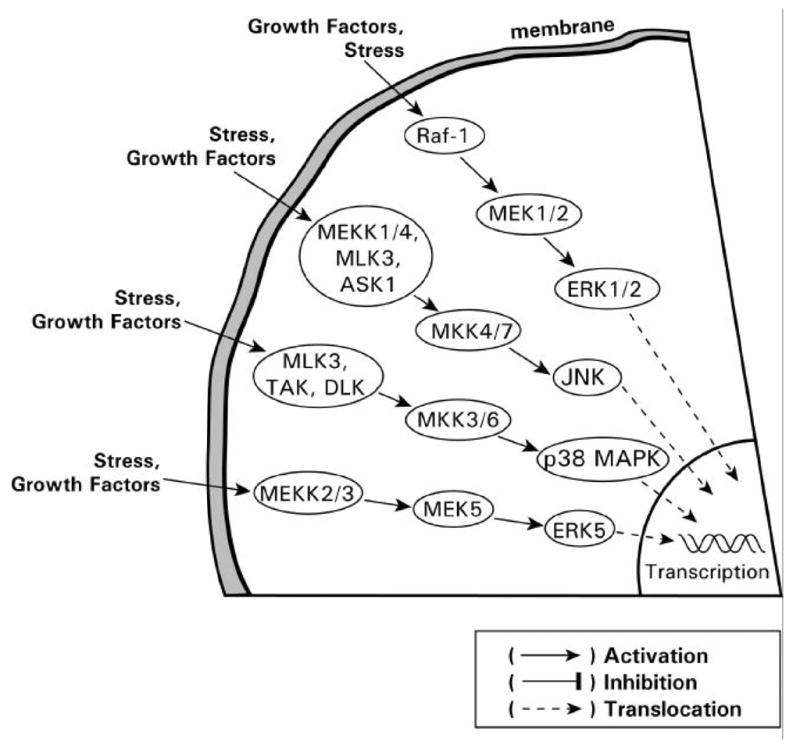
The MAPK family members. The different categories and factors which activate the members of the MAPK family.
3.2 Ras Activation and Signaling
The Ras superfamily of small GTPases comprises over 150 members in human cells and the Ras proteins are the founding members of this superfamily, which is divided into five major branches on the basis of sequence and functional similarities: Ras, Rho, Rab, Ran and Arf (Wennerberg et al., 2005). Three different Ras genes are present in mammalian cells and these genes result in the expression of four Ras small GTPases; H-Ras, N-Ras, KA-Ras and KB-Ras. These Ras proteins are key regulators of critical intracellular signal pathways, which control cell growth proliferation, differentiation, survival and apoptosis (Khosravi-Far et al., 1998; Chong et al., 2003; Giehl, 2005). In the Ras family, amino acid residues 1–85 are identical in each of the proteins, residues 86–166 exhibit 85% identity and residues 167–185 represent a variable region. This variable region contains the CAAX-motif (C is cysteine; A is any aliphatic amino acid; X is methionine or serine), which signals for post-translational lipid modification. The lipid modifications, such as farnesylation and geranylgeranylation increase the hydrophobicity of Ras proteins, which is obligatory for Ras protein insertion into the membrane and subsequent activation (Mangues et al., 1998). Inhibition of Ras farnesylation or geranylgeranylation results in an absence of membrane insertion and rapid degradation, which thereby results in decreased Ras signaling. Consequently, inhibition of this lipid modification of Ras has been a therapeutic target for inhibition of Ras signaling which is important in tumors, such as breast, which overexpress Ras or those in which Ras mutations have been implicated (e.g. pancreatic cancer).
Ras proteins may be activated by a large variety of extracellular stimuli, including hormones, growth factors, cell-extracellular matrix interactions and oxidative stress (Fig. 9). Upon activation of the RTKs, the association between the RTK and Ras occurs through the GTP exchange factor SOS, which exists in the cytosol in a complex with the adaptor protein Grb2, through binding of the Grb2 SH3 domain to a SOS proline-rich region. Activated RTKs with phosphorylated tyrosine residues serve as recruitment docking sites for Grb2 with SH2 domains. In addition, the association between Grb2/SOS, and the tyrosine phosphorylated domain in the receptor, can also be mediated by adaptor proteins such as Shc or IRS, which undergo tyrosine phosphorylation following recruitment to the activated receptor. This process brings SOS to the plasma membrane in close proximity to Ras. Activation of Ras proteins occurs by exchange of GDP/GTP through the nucleotide exchange factor SOS, which converts Ras-GDP to Ras-GTP, and thereby results in activated Ras (Fig. 9). Ras cycles between an inactive GDP-bound state and an active GTP-bound state which occurs through the regulated activity of the GTP nucleotide exchange factor (i.e. SOS) and GTPase activating proteins. Activated Ras then recruits and activates three main classes of effector proteins, Raf kinases, PI3K and RalGDS (Shields et al., 2000) (Fig. 9).
Fig. 9.
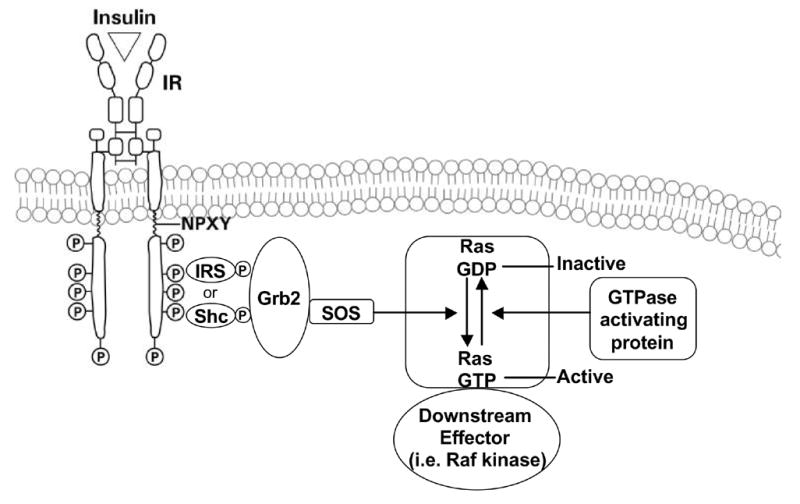
Ras activation. Diagrammatic representation showing the recruitment of the guanine nucleotide exchanger SOS, and the exchange of GDP for GTP resulting in the activation of Ras.
3.3 Raf Kinase Signaling
The Raf family of serine/threonine kinases, A-Raf, B-Raf and Raf-1 (c-Raf) is encoded by three genes. All Raf kinases contain three conserved regions, CR1, CR2 and CR3 (Morrison and Cutler, 1997; Baccarini, 2005) (Fig. 10). The N-terminus (CR1 and CR2), domain which represses Raf activity, and the CR3 catalytic domain of Raf, are all regulated by phosphorylation. The interaction of the active GTP-bound Ras with the Ras binding domain and cysteine rich domain of CR1, recruits Raf to the membrane for activation (Fig. 10). Raf activation occurs at the plasma membrane and involves many steps, including lipid binding, binding to other proteins, conformational changes and phosphorylation. Raf activation thus results in the subsequent recruitment of downstream kinases.
Fig. 10.
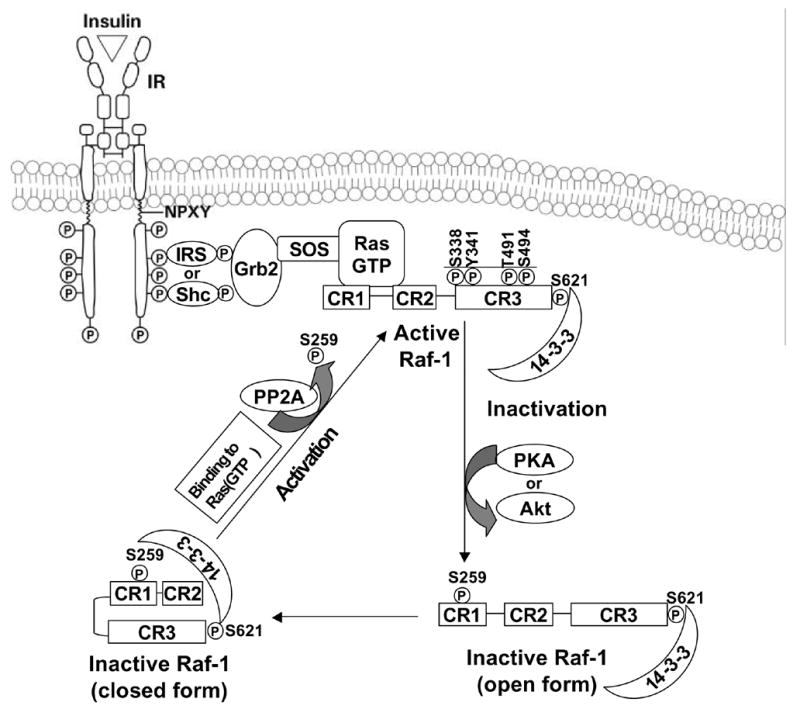
Ras and Raf activation. The process for activation and recruitment of Raf kinase and subsequent inactivation by Akt or PKA.
The majority of research directed towards understanding the role of Raf in ERK1/2 activation has focused on Raf-1 (Fig. 10). Under basal conditions, Raf-1 is phosphorylated at serines 259 and 261 and is cytosolic. Under this condition, Raf-1 is stabilized by binding the 14-3-3 scaffold protein dimer. The 14-3-3 scaffold protein binds Raf-1 phosphorylated serines 259 and 621 (Tzivion et al., 1998). Phosphorylated serine 43, 233 and 259 sites in Raf1, appear to have an inhibitory function, and their phosphorylation is increased further by PKA and Akt activation (Wu et al., 1993; Rommel et al., 1999; Dhillon et al., 2002; Dumaz et al., 2002). Thus, activation of Akt or of PKA, through adenylate cyclase and cyclic nucleotides, can limit Ras signaling and activation of downstream targets. This exemplifies the type of cross-talk that occur between seemingly distinct signaling pathways.
The binding of Raf to Ras and concomitant translocation to the plasma membrane results in the displacement of 14-3-3 from phosphoserine 259 (Fig. 10). This makes phosphoserine 259 accessible for activation by protein phosphatase 2A dephosphorylation (PP2A) (Kubicek et al., 2002), although the role of serine 259 dephosphorylation in Raf-1 activation has been questioned (Light et al., 2002). The phosphorylation stoichiometry of serine 259 can be as high as 0.8 mol/mol in preparations of Raf-1 activated by coexpression of oncogenic Ras and Src, suggesting that the rephosphorylation of serine 259 is rapidly induced by activated Ras following Raf activation (Dhillon et al., 2002). Nonetheless, the recruitment of Raf-1 to Ras and subsequent activation of Raf-1 by relief from autoinhibition at the plasma membrane is obligatory for subsequent multistep events to occur. Following activation of the RTK by agonists, such as insulin, growth factors or stress, the phosphorylation of several residues, including serine 338, tyrosine 341, threonine 491 and serine 494 is increased (Mason et al., 1999). Serine 338 and tyrosine 341 phosphorylation is critical for Raf activation (Mason et al., 1999). Phosphorylation of serine 338 may be used as a relative index of Raf-1 activation.
Both p21 activated kinase (PAK) 1 and 3 have been reported to be Raf-1 serine 338 kinases, and act in a PI3K-dependent manner (King et al., 1998); however, others have argued against the role of PAK1 and 3 as Raf-1 kinases (Chiloeches et al., 2001). Two phosphorylation sites, threonine 491 and serine 494, in the activation loop of Raf-1 are phosphorylated in a mitogen-induced manner and contribute to Raf-1 activation (Chong et al., 2001). Although overexpression of Src tyrosine kinases induces phosphorylation of tyrosine 341 (Mason et al., 1999), the role of Src tyrosine kinases in the phosphorylation of this site in response to mitogens has not yet been conclusively demonstrated.
3.4 MAPK Signaling
The fidelity of signaling among MAPKs is ensured by scaffold and adaptor proteins (Morrison and Davis, 2003) (Fig. 11). A “Kinase Suppressor of Ras” (KSR) has been identified in C. Elegans and Drosophila. KSR functions as a positive regulator and is proposed to act downstream of, or in parallel to, Ras (Therrien et al., 1995; Sundaram and Han, 1995; Kornfeld et al., 1995). KSR serves as a scaffold protein for the ERK pathway based on its ability to interact with Raf-1, MEK, ERK, 14-3-3 and various heat shock proteins (HSPs) (Morrison, 2001). A two-hybrid screen using MEK1 as bait identified MEK partner 1 as a scaffold protein that specifically binds MEK1 and ERK1 to the exclusion of MEK2 and ERK2 (Schaeffer et al., 1998). Thus, MEK partner 1 can induce the activation of ERK1, but not that of ERK2, although in most cases ERK1 and ERK2 appear to be functionally equivalent. Raf kinase inhibitor protein (RKIP) is a scaffolding protein that inhibits MEK phosphorylation by Raf-1 (Yeung et al., 1999). The Raf and MEK binding sites on RKIP overlap and exclude each other from binding by steric interference (Yeung et al., 2000). RKIP seems to be a physiological regulator of ERK signaling. RKIP binding to Raf-1, but not to MEK, is controlled by growth factors, probably via modification of Raf-1. In a recent study by Matheny et al. (2004), a Ras effector protein referred to as IMP (Impedes Mitogenic signal Propagation) is identified as a Ras-binding protein that inhibits signal transmission through the Raf/MEK/ERK cascade. IMP appears to uncouple signal transmission from Raf to MEK through the inactivation of KSR. In addition, a number of chaperones have been found to associate with Raf-1, including Bag1, FKBP and HSP 50 and 90 (Kolch, 2000). Chaperones can maintain and assist the folding of many signaling proteins, including Raf-1. Recent results in our laboratory have shown that insulin upregulates, while glucagon downregulates, a number of HSP genes as determined by microarray analysis and confirmed by RT-PCR (Dombkowski and Novak, unpublished observation).
Fig. 11.
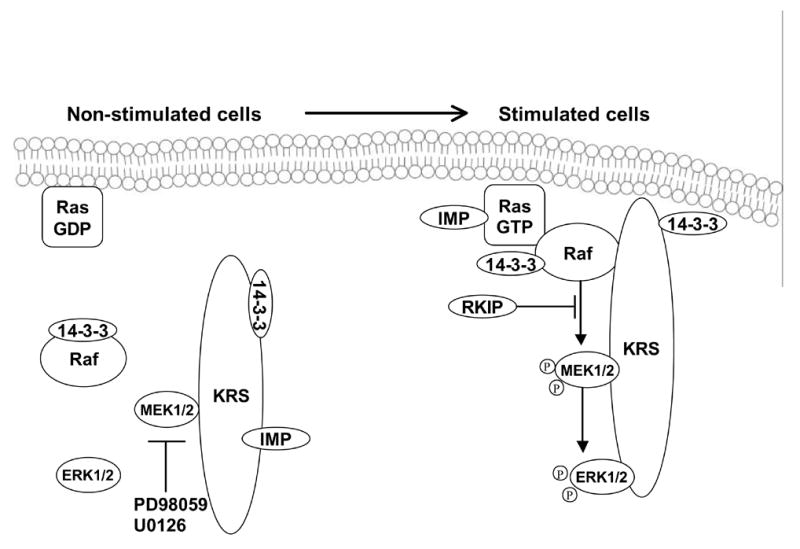
Scaffold proteins involved in MAPK signaling pathways. A drawing showing the binding of MAPKs and 14-3-3 to the KRS scaffold protein and subsequent activation.
A proline-rich sequence of MEK1 (MKK1) and MEK2 (MKK2) is required for interaction with Raf-1 (Catling et al., 1995). MEKs are phosphorylated by Raf-1 on two serine residues (serine 217, 221), which are obligatory for complete activation. MEK1 and MEK2 activate ERK1 (p44 MAPK) and ERK2 (p42 MAPK) via phosphorylation of a threonine-glutamate-tyrosine-motif in the activation loop. ERK is a proline-directed serine/threonine kinase at the end of this pathway with more than 50 identified substrates, including transcription factors, MAPK-activated protein kinase-2, and the p90 ribosomal S6 kinase (p90RSK) (Lewis et al., 1998). ERK activation occurs in response to a number of stimuli, including hydrogen peroxide, metals, xenobiotics and oxidative stress, and has traditionally been associated primarily with cell proliferation. The inhibitors PD98059 and U0126 bind to the inactive form of MEK, preventing its activation by Raf-1 and other upstream activators (Alessi et al., 1995; Favata et al, 1998). These inhibitors do not compete with ATP and do not inhibit the phosphorylation of MEK, and thus are likely to have a distinct inhibitory binding site on MEK. Quantitative evaluation of the steady-state kinetics of MEK inhibition by these compounds shows that U0126 has higher affinity than PD98059 (Favata et al., 1998). The use of PD98059 to examine the role of MEK signaling in the regulation of CYP2E1 showed that it exerted no effect on the insulin-mediated downregulation of CYP2E1 (Woodcroft et al., 2002).
The stress-activated protein kinases (SAPKs) such as JNK, p38 MAPK and ERK5 are generally only slightly activated in response to insulin or growth factors. In contrast, the SAPKs are quite markedly activated in response to stress (UV irradiation, heat- or cold-shock, osmotic stress, mechanical shear stress, oxidative stress), cytokines, or G-protein-coupled receptor agonists (Kyriakis and Avruch, 2001) (Fig. 8). The SAPKs regulate growth arrest, apoptosis and proliferation. SAPKs activation occurs through a similar kinase cascade as ERK, although some distinct differences do exist. For example, MKK4/SEK1 and MKK7 phosphorylate and activate JNK; in contrast p38 MAPK is activated by MKK3 and MKK6 (Fig. 8). MKK4 preferentially phosphorylates tyrosine 185, while MKK7 targets only threonine 183 of JNK (Wada et al., 2001). For MKKK, many kinases have been implicated in the activation of JNK and/or p38 MAPK (Hagemann and Blank, 2001) (Fig. 8).
SB203580, [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole], and SB202190, [4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole], members of a class of pyridinyl imidazoles, are relatively specific inhibitors of p38 MAPK alpha and beta, but not p38 MAPK gamma and delta, at a concentration of 10 μM (Cuenda et al., 1995; Eyers et al., 1998). These compounds bind the ATP-binding cleft of the low-activity p38 MAPK, which binds ATP poorly. As a consequence of binding the unphosphorylated form, these inhibitors appear to interfere with the activation of p38 MAPK (Frantz et al., 1998). SP600125, an anthrapyrazolone inhibitor of JNK1, JNK2 and JNK3, has been reported to inhibit JNKs through a reversible ATP-competition (Bennett et al., 2001). A number of studies have reported that the compound prevents the expression of several anti-inflammatory genes in cell-based assays and the activation of AP1 in synoviocytes (Bennett et al., 2001; Han et al., 2001). Experiments employing SB202190 and SP600125 have failed to establish a role for the p38 MAPK alpha and beta or JNK1, 2 or 3 in regulating the insulin-mediated downregulation of CYP2E1 (Woodcroft et al., 2002) or other genes, such as class alpha GSTs, mEH or gamma-glutamylcysteine ligase (Kim et al., 2003b, 2004 and 2006a).
4. Phosphatases
4.1 Overview
Protein phosphorylation is one of the most important, widespread and consequential post-translational modifications that occurs in cells. Protein phosphorylation results in a rapid and activity-dependent signal transduction process that regulates gene expression and cellular function. The ability of phosphoproteins to act as loci and conduits for information processing and ultimately, the regulation of transcriptional, translational and functional events, requires an element of 'reversibility' of protein phosphorylation. A simple hydrolytic reaction catalyzed by protein phosphatases enables phosphoproteins to be rapidly restored to their original physical and functional states. Inhibition of phosphatase activity may result in effects comparable to stimulation of kinase activity since the dynamic of protein phosphorylation/dephosphorylation is disrupted. Decades of intensive research have led to the molecular characterization of four families of protein tyrosine phosphatases (PTP) and two families of protein serine/threonine phosphatases.
4.2 Protein-tyrosine Phosphatases
The level of receptor tyrosine phosphorylation and, hence subsequent phosphorylation of their downstream targets, is dynamically and precisely regulated by two types of enzymes – namely the protein tyrosine kinases, which catalyze the phosphorylation of tyrosine residues, and the PTPs, which dephosphorylate phosphotyrosine residues (Zhang et al., 2002b; Asante-Appiah and Kennedy, 2003; Alonso et al., 2004; Stoker, 2005). Disregulated PTP activity can result in aberrant tyrosine phosphorylation, with the consequent elevated signaling activity contributing to various diseases, including cancer and diabetes (Wu et al., 2003; Elchebly et al., 2000). The recent sequencing of the human genome, in conjunction with a comprehensive amino acid sequence analysis of conserved catalytic motifs and whole catalytic domains of all previously identified PTPs, revealed the presence of 107 phosphatase genes (Alonso et al., 2004). Based on the amino acid sequences of their catalytic domains, the PTPs can be grouped into four separate families, class I cysteine-base PTPs (99 genes), class II cysteine-based PTPs (1 gene), class III cysteine-based PTPs (3 genes), and aspartate-based PTPs (4 genes). Also, PTPs, based on their substrate specificities, may be classified as tyrosine-specific and dual-specificity families, with the later subfamily recognizing phosphotyrosine, phosphothreonine and phosphoserine residues. Based on domain architecture and the degree of homology between catalytic domains, the 99 class I cysteine-based family of PTPs can be further classified into several subfamilies. These include the classical PTPs, dual-specific phosphatases, atypical dual-specific phosphatases, proteins of regenerating liver, cdc14s, slingshots, PTENs and myotubularines. The 38 strictly tyrosine-specific "classical PTPs" can be divided into transmembrane, receptor-like enzymes and the intracellular, nonreceptor PTPs (Andersen et al., 2004). Intracellular PTPs have a single conserved catalytic domain, with the N- or C-terminus exerting a regulatory or targeting role. PTP-1B and SHP-2, an SH2-containing PTP-2, are members of this subfamily and regulate intracellular phosphotyrosine levels. Receptor-like PTPs, (e.g. CD45 and PTP alpha), have either one, or two, cytoplasmic catalytic domains, a single transmembrane domain, and an extracellular domain. The extracellular domains, which have structures present in cell-adhesion molecules, suggest that this subfamily of PTPs may have a role in mediating cell-cell and/or cell-extracellular matrix interactions.
The class II cysteine-based PTPs are represented in the human genome by a single gene, referred to as ACP1, which encodes the 18 kDa low molecular phosphatase. Although this PTP can dephosphorylate a number of tyrosine kinases, its physiological function remains unclear. The class III cysteine-based PTPs is comprised of three cell cycle regulators, CDC25A, CDC25B, and CDC25C, in human cells. Their function is to dephosphorylate cyclin-dependent kinases at their inhibitory dually phosphorylated N-terminal threonine-tyrosine motifs, a reaction that is required for activation of these kinases to drive the progression of cells through the cell cycle (Honda et al., 1993). Recently the four Eya genes, as members of the aspartate-based PTPs, were reported to have tyrosine and serine phosphatase activity (Tootle et al. 2003; Rayapureddi et al. 2003 and Li et al., 2003).
PTP-1B is widely expressed in tissue and cells. It is primarily localized to the endoplasmic reticulum (ER) through a proline-rich C-terminal segment, which may be cleaved, thereby releasing the phosphatase from the ER and increasing its activity (Charbonneau et al., 1989; Frangioni et al., 1993). A deficiency of PTP-1B has been reported to result in enhanced insulin sensitivity, which is reflected by increased insulin receptor phosphorylation in muscle and liver in response to insulin as well as improved glucose clearance, and a significant reduction in fed glucose levels (Elchebly et al., 1999). These data suggest that PTP-1B is a negative regulator of insulin-stimulated signaling and exerts its regulatory function by dephosphorylating the phosphotyrosine residues of the cytosolic domain of the insulin receptor (Salmeen et al., 2000). Insulin receptor and PTP-1B association has been demonstrated using substrate trapping, coimmunoprecipitation, and immunoblot analysis (Seely et al., 1996; Bandyopadhyay et al., 1997). The mechanism(s) regulating PTP-1B activity are unclear, although oxidation and phosphorylation have been implicated. Insulin-stimulated intracellular hydrogen peroxide production may reversibly oxidize PTP-1B, resulting in inhibition of PTP-1B and enhancement of the insulin signaling cascade (Mahadev et al., 2001; Wu et al., 2001), whereas PTP-1B phosphorylation by the insulin receptor, and other protein kinases, has also been shown to alter PTP-1B enzyme activity (Dadke et al., 2001; Ravichandran et al., 2001; Tao et al., 2001). In addition to the insulin receptor, PTP-1B has also been reported to target the EGF receptor and Src kinase (Liu and Chernoff, 1997; Bjorge et al., 2000).
The SHP-2 phosphatase is ubiquitously expressed in various tissues and contains two tandem SH2 domains that mediate SHP-2 binding to phosphotyrosine residues on proteins. Under normal conditions, SHP-2 is inactive. However, its mechanism of activation is unclear. It has been suggested that phosphorylation of tyrosine 542 results in the interaction of this residue intramolecularly with the N-terminal SH2 domain to relieve basal inhibition of SHP-2, whereas phosphorylation of tyrosine 580 stimulates SHP-2 PTP activity through interaction of this residue with the C-terminal SH2 domain (Lu et al., 2001). SHP-2 may play a positive or negative role in RTK signaling. For example, catalytically inactive SHP-2 markedly inhibited ERK activation in response to the agonist EGF and insulin (Milarski and Saltiel, 1994; Bennett et al., 1996). Overexpression of a SHP-2 dominant negative produced only a modest inhibition of insulin-stimulated glucose transporter 4 (Glut4) translocation, suggesting that SHP-2 may have only a minor effect in positively modulating the metabolic effects of insulin (Chen et al., 1997). In contrast, SHP-2 expression in cells produced a negative effect on insulin-mediated IRS-1 phosphorylation, PI3K activation and stimulation of glycogen synthesis (Ouwens et al., 2001). Discrepancies in the role of SHP-2 in insulin-mediated metabolic effects between knockout and dominant-negative transgenic models have been reported. Since homozygous SHP-2 knockout mice die during embryonic development, hemizygous mice which express half the levels of SHP-2 protein, were used to study insulin signaling (Arrandale et al., 1996). In this study, plasma insulin levels, glucose uptake and tyrosine phosphorylation of the IR and IRS were unaffected. In another study, the overexpression of a dominant negative SHP-2 in transgenic mice induced insulin resistance, suggesting a positive role for SHP-2 in insulin signaling (Maegawa et al., 1999). Thus, the role of SHP-2 in insulin signaling remains to be established.
4.3 Dual Specificity MAPK Phosphatases
The dual specificity MAPK phosphatases (MKPs) are a family of protein phosphatases that inactivate MAPKs through dephosphorylation of phosphotyrosine and phosphothreonine residues in the MAPKs activation loop. In mammalian cells, a minimum of 10 MKPs have been identified. Each of these MKPs display selectivity towards different MAPK family members (Nichols et al., 2000; Masuda et al., 2001; Groom et al., 1996). The distinct substrate specificity of the MPKs appears to be mediated by specific protein-protein interactions through the non-catalytic domain of the MKP. Some of the MPK family members have been reported to be expressed only in a subset of tissue types and may be localized either in the nucleus or the cytoplasm (Camps et al., 2000).
4.4 Serine/Threonine Phosphatases
The serine/threonine protein phosphatases also constitute a diverse family, with the families classified as the phosphoprotein phosphatase (PPP), and the Mg2+-dependent protein phosphatase (PPM) gene families, based on the primary amino acid sequence identity between the different catalytic subunits (Sim et al., 2003). The PPP family is the most abundant protein phosphatase family, and includes PP1, PP2A and PP2B (calcineurin), as well as PP4 (also known as PPX), PP5, PP6 and PP7. The PPM family consists of the five PPC2 isoforms along with pyruvate dehydrogenase phosphatase. There are four isoforms of the PP1 catalytic subunit that are inhibited by the membrane-permeable inhibitors, okadaic acid and calyculin A, and also by microcystin which is membrane-impermeable (Cohen, 1989; Winder and Sweatt, 2001). PP2A exhibits constitutive activity and is also inhibited by the inhibitors of PP1. In contrast, PP2B, a Ca2+-dependent protein phosphatase, is unaffected by okadaic acid, calyculin or microcystin (Klumpp and Krieglstein, 2002). PP1 is activated in response to insulin and catalyzes the dephosphorylation of metabolic enzymes. The glycogen-associated form of PP1 has been reported to dephosphorylate both glycogen synthase (resulting in enzyme activation) and glycogen phosphorylase (resulting in inactivation) to provide insulin-mediated coordination of glycogen metabolism (Brady and Saltiel, 2001). PP2A is active in the dephosphorylation, and consequently in the regulation of the MAPKs (Keyse, 2000).
5. Insulin Signaling in the Regulation of Drug Metabolizing Enzyme Gene and Protein Expression
5.1 General
The normal physiologic rat hepatic insulin concentration has been reported to range between 0.4 and 2 nM with an average value of 1.2 nM (Balks and Jungermann, 1984). The average rat plasma insulin levels are ~0.2 nM under normal conditions with these levels markedly decreased in diabetic rats treated with streptozotocin (Hwang et al., 2005). In contrast, elevated plasma insulin levels have been observed in animals with type II (insulin-resistant) diabetes compared with normal rats (Alemzadeh and Tushaus, 2004; Bitar et al., 2005). These insulin concentrations are directly relevant to those employed in our research to examine insulin effects on drug metabolizing enzyme gene and protein expression. In order to facilitate understanding of the following sections on drug metabolizing enzyme gene and protein expression, a composite signaling diagram showing the major pathways, components and specific inhibitors is provided in Fig. 12.
Fig. 12.
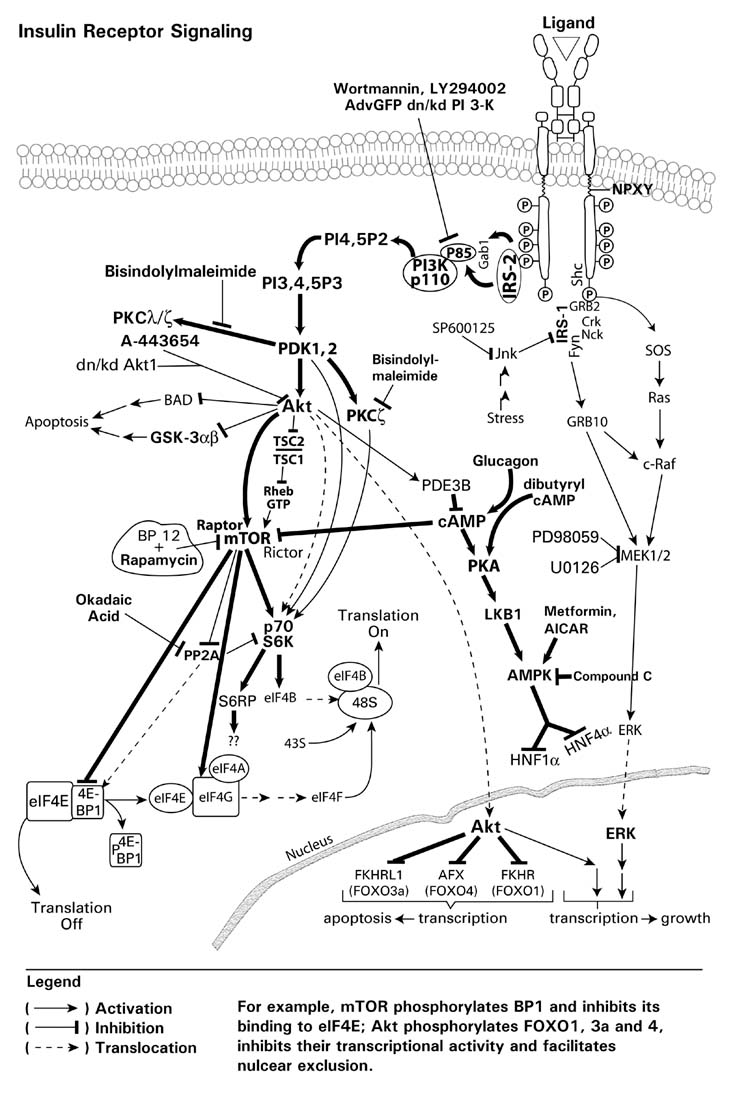
Insulin/growth factor receptor signaling pathways. An insulin-signlaing diagram showing the various inhibitors which may be used to examine the role of individual kinases in the signaling process.
5.2 Cytochrome P450
Our laboratory has demonstrated that the expression of CYP2E1 is downregulated by insulin (and EGF) and enhanced by glucagon in primary cultured rat hepatocytes (Woodcroft and Novak, 1997; Woodcroft and Novak, 1999b). Insulin concentrations ≥1 nM in primary cultured hepatocytes resulted in a greater than 55% decrease in CYP2E1 mRNA levels, in a concentration-dependent manner (Fig. 13) with continued downregulation occurring at supraphysiologic concentrations up to 1 μM. Examination of the immunoprecipitated insulin receptor showed that tyrosine phosphorylation could be detected at insulin concentrations of 1, 10 and 1,000 nM (Fig. 13) and corresponded to the concentrations at which CYP2E1 mRNA levels decreased. These data show a correspondence between insulin-mediated activation of the insulin receptor and the decrease in CYP2E1 mRNA levels in primary cultured rat hepatocytes. The insulin level in primary cultured hepatocytes has been typically 1 μM, a supraphysiological level, and likely the reason for limited expression of CYP2E1 under these conditions. Insulin is necessary, however, to maintain CYP2B mRNA levels and facilitates the expression and phenobarbital-mediated induction of CYP2B (Woodcroft et al., 1999a; Woodcroft and Novak, 1997). Insulin has only a modest down-regulatory effect on CYP3A, although it decreases CYP4A expression by ~40% (Woodcroft and Novak, 1997). Thus, insulin exerts different effects on the various P450 subfamily members. In Fao rat hepatoma cells, De Waziers et al. (1995) showed that 100 nM insulin produced a 60% and 80% decrease in the steady state levels of CYP2B and CYP2E1 proteins, respectively, within 24 hr. Moreover these authors suggested that decreased CYP2E1 and CYP2B mRNA levels in Fao hepatoma cells, which occurred as a result of insulin treatment, were primarily the result of decreased mRNA turnover. In rat primary cultured hepatocytes, however, we have found that CYP2E1 and CYP2B expression is differentially regulated by insulin (Fig. 14). An elevated insulin concentration in primary cultured rat hepatocytes decreased CYP2E1 mRNA half-life; in contrast CYP2B1 mRNA half-life was actually increased, while insulin was without a significant effect on CYP3A mRNA half-life (Fig. 14). This difference in insulin-mediated regulation of CYP2E1 and CYP2B expression likely reflects differences associated with cell context (i.e. tumor cell lines versus primary cultured hepatocytes) and the levels/activity of the regulatory signaling pathways.
Fig. 13.
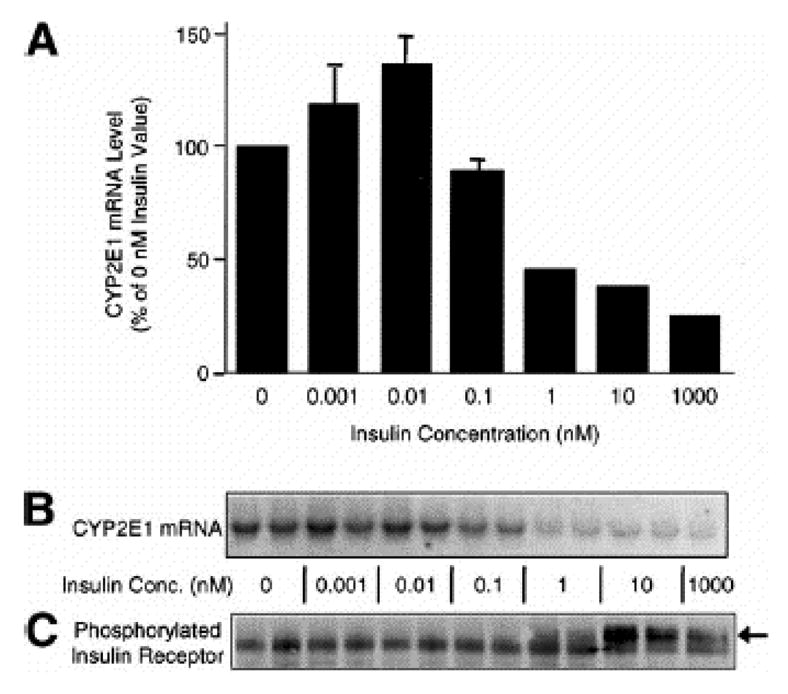
CYP2E1 mRNA levels and phosphorylation status of immunoprecipitated insulin receptor in response to insulin. After the 4-hr plating period in the presence of 1 μmol/L insulin, hepatocytes were maintained in the absence of insulin for 48 hr. Hepatocytes were then treated for 24 hr with 0 to 1,000 nmol/L insulin. CYP2E1 mRNA levels were monitored by Northern blot analysis and band density normalized to 7S RNA. (A) CYP2E1 mRNA levels plotted as a percentage of the CYP2E1 mRNA level monitored in hepatocytes cultured in the absence of insulin. Data are means ± range of Northern blot band densities of 2 preparations of total RNA. (B) Northern blot of CYP2E1 mRNA levels in hepatocytes treated with 0 to 1,000 nmol/L insulin. (C) Phosphotyrosine immunoblot of phosphorylated insulin receptor in hepatocytes treated with 0 to 1,000 nmol/L insulin. Phosphorylated insulin receptor is indicated by an arrow. This band comigrated with insulin receptor as detected with anti-insulin receptor antibody. Insulin receptor levels were similar in all samples (data not shown). The identity of the additional phosphotyrosine cross-reactive band below the phosphorylated insulin receptor is unknown. Reprinted from Woodcroft, KJ, Hafner, MS and Novak, RF, Hepatology, 35, 263–273, 2002, with permission.
Fig. 14.
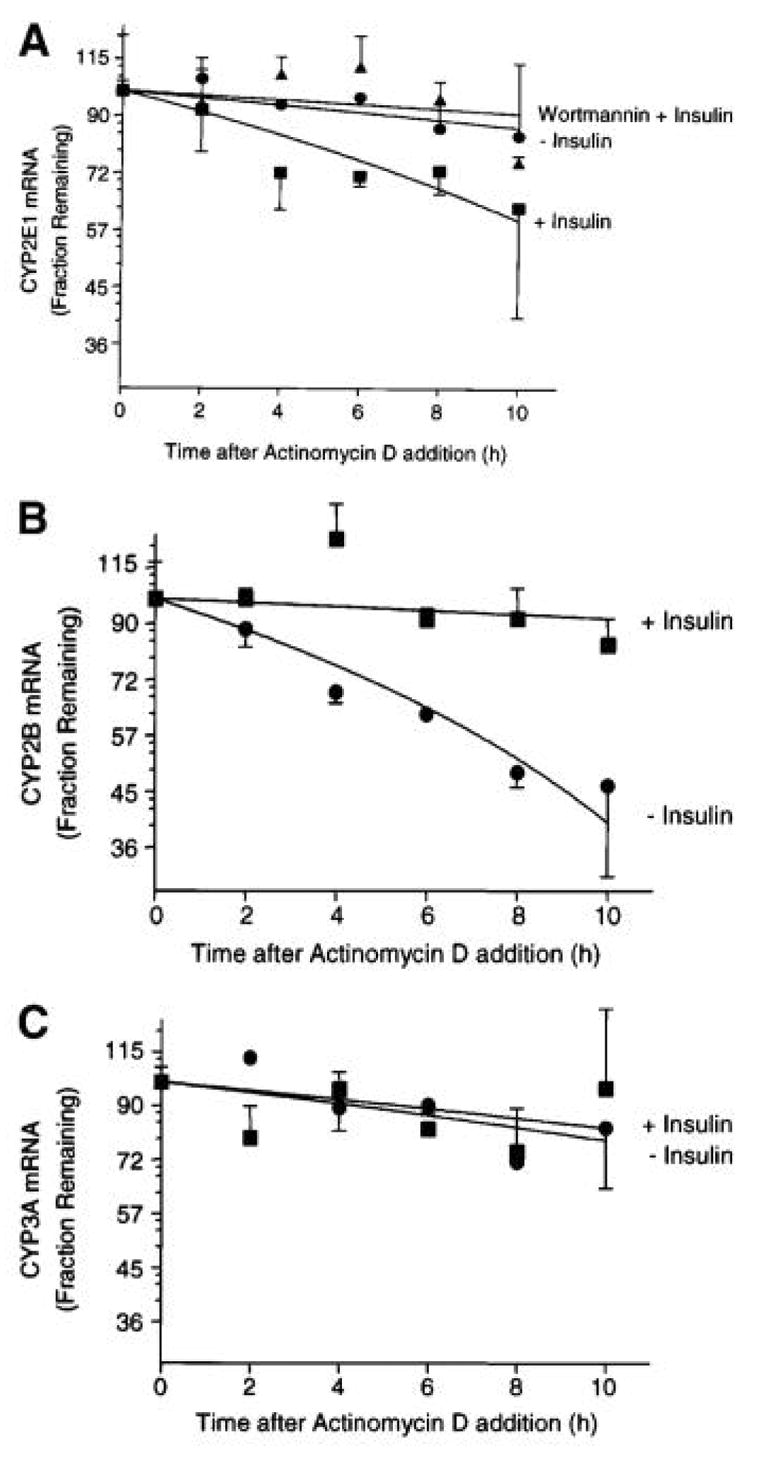
Effect of insulin on CYP2E1, CYP2B, and CYP3A mRNA turnover. After the 4-h plating period in the presence of 1 μM insulin, hepatocytes were maintained in the absence of insulin for 48 h. Hepatocytes were then treated for 12 h with either medium alone (,), 10 nM insulin (!), or wortmannin (10 μM) plus 10 nM insulin(7). Actinomycin D (10 μg/mL) was added to hepatocytes without change of medium, and total RNA was isolated at the times indicated. CYP2E1 (A), CYP2B (B), and CYP3A (C) mRNA levels were monitored by Northern blot analysis and band density normalized to 7 S RNA. mRNA levels are plotted as a fraction of time zero mRNA levels for each treatment. Data are means ± SEM of Northern blot densities of 3 preparations of total RNA. First-order decay rate constants were derived and used to calculate half-life values.
Reprinted from Woodcroft, KJ, Hafner, MS and Novak, RF, Hepatology, 35, 263–273, 2002, with permission.
We have observed that the treatment of primary cultures of rat hepatocytes with EGF results in a decrease in hepatocyte CYP2E1 mRNA levels and that this decrease was coincidental with the EGF-mediated tyrosine phosphorylation of the EGF receptor (Abdelmageed et al., unpublished observation). In addition, we have observed that the pretreatment of cells with AG1478, an EGF receptor tyrosine kinase inhibitor, or with inhibitors of PI3K such as wortmannin or LY294002, resulted in the amelioration of both the EGF-mediated increase in EGF receptor tyrosine phosphorylation levels (AG1478) and the EGF-mediated suppression of CYP2E1 mRNA levels (AG1478, wortmannin or LY294002), suggesting a crucial role of EGF receptor tyrosine phosphorylation and PI3K in mediating the EGF effect on CYP2E1 mRNA levels (Abdelmageed et al., unpublished observation).
As discussed previously, the phosphorylation state of signaling molecules is regulated through signaling cascades and involves both protein/lipid kinases and phosphatases. Our laboratory has demonstrated that protein phosphatase inhibitors, such as okadaic acid, an inhibitor of the serine/threonine phosphatases PP1 and PP2A, and sodium orthovanadate, an inhibitor of tyrosine phosphatase, decreased CYP2E1 mRNA levels by greater than 95% in hepatocytes (Woodcroft et al., 2002). The inhibition of these phosphatases may be likened to the insulin-mediated stimulation of downstream kinases and hence, in this context, these data are consistent with that of insulin-signaling effects. The exact mechanism(s) regulating these processes and the phosphatase(s) involved, however, remain to be established. Thus, these results further support the involvement of kinase/phosphatase signaling pathways in the insulin-mediated decrease in CYP2E1 mRNA levels.
We used chemical inhibitors of the kinases, as identified in previous sections, to implicate cellular signaling pathways involved in regulation of CYP2E1 gene expression in response to insulin and EGF (Woodcroft et al., 2002; Abdelmageed et al., unpublished observation). The PI3K inhibitors, wortmannin and LY294002, ameliorated both the insulin-mediated and EGF-mediated decrease in CYP2E1 mRNA levels, which implicates PI3K as an obligatory component in the suppression of CYP2E1 expression by insulin. In contrast, neither the MEK inhibitor, PD98059, nor the p38 MAPK inhibitor SB202190, had any significant effect on the insulin-mediated (or the EGF-mediated) decrease in CYP2E1 mRNA expression in primary cultured rat hepatocytes (Woodcroft and Novak, 1999b; Woodcroft et al., 2002; Abdelmageed et al., unpublished observation). Rapamycin, an inhibitor of mTOR, effectively inhibited the 1 nM insulin-mediated decrease in CYP2E1 mRNA expression and partially prevented the effect of 10 nM insulin in a typical agonist-antagonist concentration-dependent effect (Woodcroft et al., 2002). The broad-spectrum PKC inhibitor bisindolylmaleimide I (GF109203X) at 10 μM (sufficient to inhibit PKCs) failed to affect the insulin-mediated decrease in CYP2E1 mRNA levels (Woodcroft et al., 2002). These results suggest that the regulation of CYP2E1 mRNA expression by either insulin, or EGF, does not appear to involve Ras-Raf, MEK-ERK signaling. In contrast, our results suggest that the PI3K, Akt, mTOR and p70S6 kinase may be involved, but not PKC, all of which are downstream targets of PI3K.
Initial studies using geldanamycin suggested that Src kinase may be involved in the insulin-mediated decrease in CYP2E1 mRNA levels in primary cultured rat hepatocytes (Woodcroft et al., 2002). Geldanamycin, however, functions to inhibit Hsp90 and Hsp70, which function as chaperonins, and are required to protect a number of kinases and proteins from ubiquitination and proteasomal degradation (Pratt, 1998). Although geldanamycin inhibited the insulin suppression of CYP2E1 mRNA expression and increased CYP2E1 levels to levels in control (time 0) hepatocytes and hepatic tissue, it is our belief that this likely reflected the overall effect of geldanamycin on the chaperonin protection of several signaling kinases which effectively truncated IR signaling in hepatocytes. Subsequent studies using SU6656, the Src kinase inhibitor, however, failed to show that Src kinase was involved in the regulation of CYP2E1 expression (Woodcroft et al., unpublished observation).
CYP2E1 mRNA half-life decreased from approximately 48 hours in the absence of insulin to approximately 15 hours at 10 nM insulin, and this decrease was prevented by the PI3K inhibitor wortmannin (Fig. 14A). Analysis of CYP2E1 gene transcription using heterogeneous nuclear RNA showed that insulin also suppressed CYP2E1 gene transcription (Fig. 15). The addition of wortmannin (10 μM) prior to the addition of 10 nM insulin resulted in an~40% recovery of CYP2E1 gene transcription activity relative to that in untreated hepatocytes (Woodcroft et al., 2002). Thus, our results show involvement of transcriptional and posttranscriptional mechanisms in the insulin-mediated regulation of CYP2E1 expression and implicate PI3K and downstream targets such as Akt, mTOR and the p70S6 kinase in mediating these effects (Woodcroft et al., 2002) (Fig. 15).
Fig. 15.
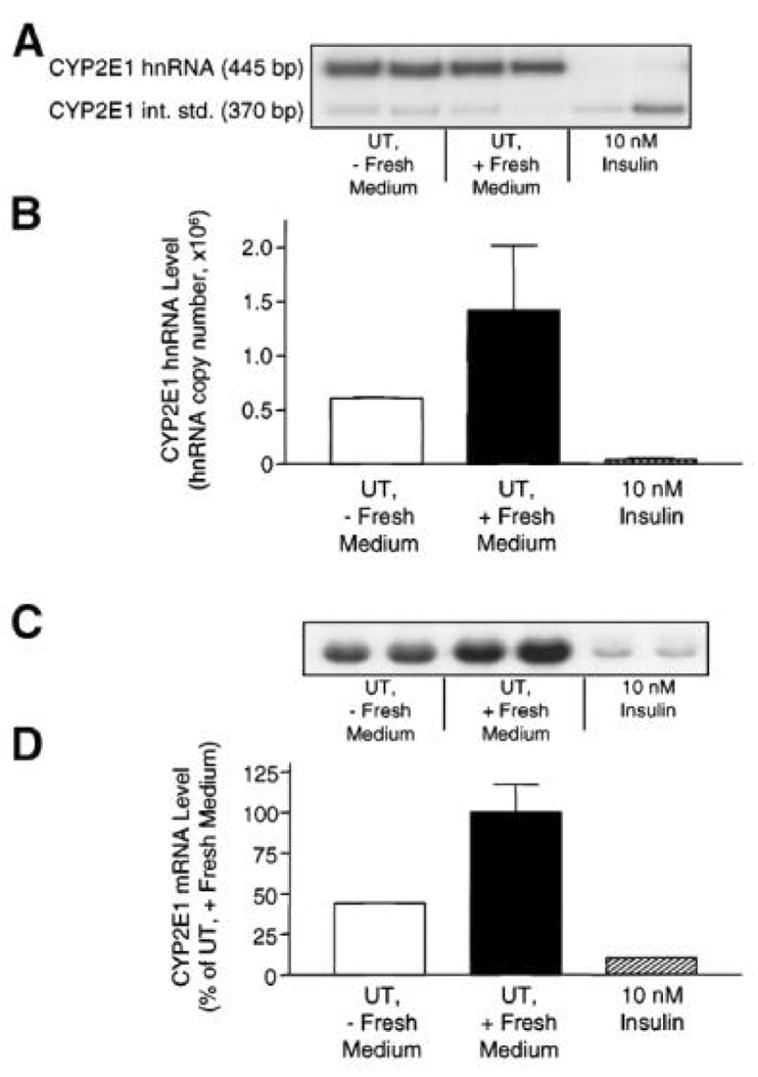
Effect of insulin on CYP2E1 gene transcription monitored by CYP2E1 hnRNA analysis. After the 4-hour plating period in the presence of 1μmol/L insulin, hepatocytes were maintained in the absence of insulin for 48 hours. Hepatocytes were then treated for 20 hours without medium change (UT, - Fresh Medium), with fresh medium alone (UT, + Fresh Medium), or with 10 nmol/L insulin in fresh medium (10 nmol/L Insulin). (A) Polymerase chain reaction products of CYP2E1 hnRNA and CYP2E1 internal standard. (B) CYP2E1 hnRNA levels plotted as hnRNA copy number, normalized to CYP2E1 internal standard band density. Data are mean ± range of hnRNA band densities from 2 preparations of total RNA. (C) Northern blot of CYP2E1 mRNA levels in identical samples used for hnRNA analysis. (D) CYP2E1 mRNA levels in C plotted as a percentage of the CYP2E1 mRNA level monitored in hepatocytes treated with fresh medium (UT, + Fresh Medium), normalized to 7S RNA band density.
Reprinted from Woodcroft, KJ, Hafner, MS and Novak, RF, Hepatology, 35, 263–273, 2002, with permission.
Because hyperketonemia is a well established event in diabetes, our laboratory has also studied the effect of the two major ketone bodies acetoacetate (AA) and DL-beta-hydroxybutyrate on the expression of CYP2E1 mRNA and protein (Woodcroft et al., 2002; Abdelmegeed et al., 2005). We showed that AA significantly decreased CYP2E1 mRNA levels in primary cultured rat hepatocytes mainly through the inhibition of CYP2E1 gene transcription as monitored by the heterogeneous nuclear RNA analysis (Fig. 16). Our results implicate PI3K and PKC signaling in mediating AA effects on CYP2E1 mRNA expression, as both PI3K inhibitors LY294002 and wortmannin, and the broad spectrum PKC inhibitor bisindolylmaleimide, ameliorated the suppressive effect of AA on CYP2E1 mRNA expression (Fig. 16). In addition, our results also suggest that mTOR and downstream p70S6 kinase may effect AA-mediated inhibition of CYP2E1 mRNA expression (Abdelmegeed et al., 2005). In contrast, neither p38 MAPK nor ERK signaling pathways appear to play an active role in mediating the downregulation of CYP2E1 mRNA levels by AA as shown by the use of the MEK inhibitor PD98059 and the p38 MAPK inhibitor SB202190. Neither inhibitor had any significant effect in reversing the AA-mediated decrease in CYP2E1 mRNA expression in primary cultured rat hepatocytes. Although AA decreased CYP2E1 mRNA, it significantly increased CYP2E1 protein levels which appears to be associated with increased mRNA translation and may reflect activation of eIF4E/4E-BP1 phosphorylation, as well as activation of the p70S6 kinase. This is supported in part, by the inhibitory effect of rapamycin, and inhibitor of mTOR, on AA-mediated downregulation of CYP2E1 expression (Abdelmegeed et al., 2005).
Fig. 16.
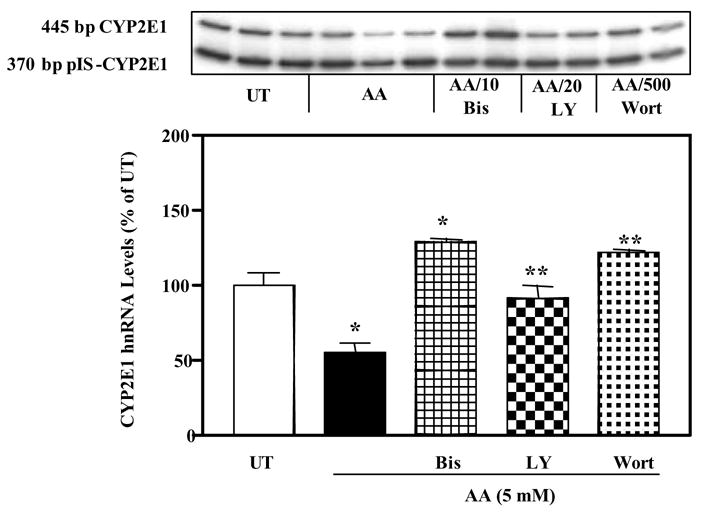
The effect of acetoacetate (AA) on CYP2E1 gene transcription. Hepatocytes were treated with PI3K inhibitors, LY294002 (LY) (10–20 μM) or wortmannin (Wort) (100–500 nM), or the PKC inhibitor, bisindolylmaleimide (Bis) (10 μM), for 1.5 h before treatment for 24 h with 5 mM AA. Untreated hepatocytes (UT) were cultured in the absence of AA, PI-3K inhibitors, and the PKC inhibitor. CYP2E1 gene transcription was monitored by hnRNA analysis and band density normalized to the 370bp CYP2E1 internal standard. CYP2E1 hnRNA levels are plotted as a percentage of the CYP2E1 hnRNA levels monitored in untreated hepatocytes. Data are means ± SEM of band densities of 2 or 3 preparations of total RNA *Significantly different from UT, ** significantly different from hepatocytes treated with AA only (p < 0.05).
Reprinted from Abdelmegeed MA, Carruthers NJ, Woodcroft KJ, Kim SK and Novak RF, J Pharmacol Exp Ther. 315, 203–213, 2005, with permission.
5.3 Phase II Enzymes
The expression of alpha-class GSTs (Kim et al., 2003a; Kim et al., 2006a) and mEH (Kim et al., 2003b) is enhanced by insulin and decreased by glucagon. Treatment of primary cultured rat hepatocytes with glucagon also inhibits the expression of pi-class GST (Kim et al., 2003a). In contrast, neither insulin nor glucagon alters the expression of mu-class GSTs, suggesting that each class of GSTs is differentially regulated by insulin and glucagon (Kim et al, 2003a).
Inhibition of the insulin-mediated elevation in alpha-class GSTs expression (Kim et al., 2006a) by either wortmannin or LY294002 occurred in a concentration-dependent manner implicating PI3K signaling. Consistent with these results, Kang et al. (2001a) reported that the activation of PI3K by tert-butylhydroquinone-induced oxidative stress was an essential step for the induction of GSTA2 through activation of the antioxidant response element. Moreover, these authors demonstrated that the activation of PI3K by oxidative stress and insulin resulted in nuclear translocation of NF-E2-related factor 2, which is involved in the activation of antioxidant response element (Kang et al., 2002a). These studies raise the possibility that PI3K serves as an essential pathway for the regulation of drug metabolizing enzyme expression in response to insulin and oxidative stress. The possible involvement of PKC, mTOR and p70S6 kinase, all downstream targets of PI3K in the insulin-mediated regulation of alpha-class GSTs was examined in hepatocytes using bisindolylmaleimide or rapamycin, respectively (Kim et al., 2006a). The increase in expression of alpha-class GSTs in hepatocytes treated with insulin was attenuated by rapamycin, but not by bisindolylmaleimide. These results suggest that mTOR and translation may also play a role in the insulin-mediated increase in alpha-class GSTs protein levels, whereas the PKCs do not appear to be involved in this process.
To determine the effect of inhibition of Akt on insulin-mediated elevation of alpha-class GST expression, a dominant negative/kinase-dead mutant of Akt1 (dn/kd Akt) was expressed by adenoviral infection of primary cultured hepatocytes. The dn/kd Akt has a point mutation (K179M) resulting in loss of kinase activity. Immunoblot analysis of hepatocytes infected with adenovirus containing the dn/kd Akt indicated expression of a higher molecular weight Akt protein in addition to the endogenous Akt owing to the additional Myc/His tags present on the adenovirally introduced Akt (Fig. 17). To confirm that the overexpressed dn/kd Akt construct was functional, Akt activation, via phosphoAkt levels, was examined in hepatocytes treated with 10 nM insulin for 30 min (Fig. 17). Akt phosphorylation was increased ~10-fold in response to 10 nM insulin and this increase was inhibited ~65% by the dn/kd Akt. Overexpression of the dn/kd Akt in hepatocytes resulted in a decline in the insulin-mediated increase in alpha-class GSTs (Kim et al., 2006a), suggesting that Akt is involved in the insulin-mediated increase in expression of alpha-class GSTs.
Fig. 17.
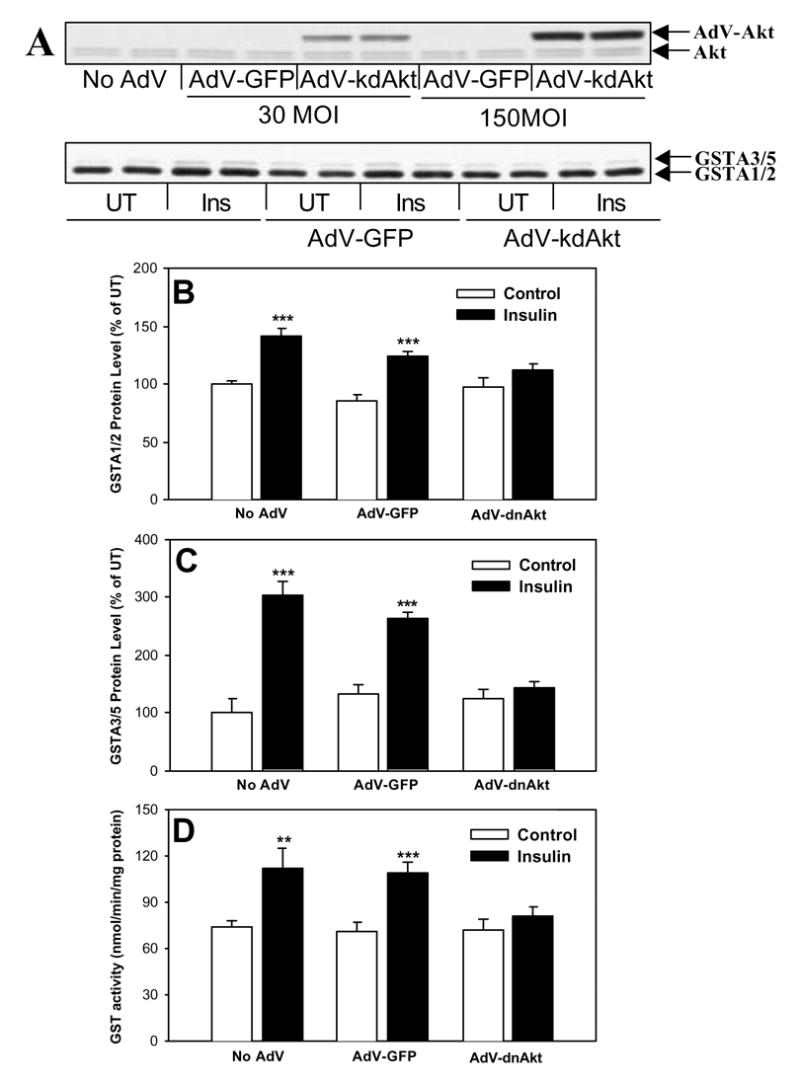
Effect of dn/kd Akt expression on insulin-mediated increased in GSTA1/2, GSTA3/5 and GST activity in primary cultured rat hepatocytes. A, hepatocytes were infected with 30 or 150 MOI AdVGFP-Akt and 24 h later cells were harvested for determination of Akt protein level. B, C and D, 24 h after infection with 150 MOI AdVGFP dn/kd Akt or AdV-GFP, hepatocytes were treated with 10 nM insulin for 2 days. Data are means ± SD of 3–4 preparations of cell lysates from a single hepatocyte preparation. **,*** Significantly different than levels monitored in corresponding control hepatocytes, p<0.01 or p<0.001, respectively.
Reprinted from Kim SK, Abdelmegeed MA and Novak RF, J Pharmacol Exp Ther. 316, 1255–1261, 2006, with permission.
It has been reported that the MAPKs play a critical role in oxidative stress-induced expression of alpha-class GSTs (Kang et al., 2002b; Nguyen et al., 2003). However, it is unclear whether MAPK signaling pathways are involved in the insulin-mediated regulation of alpha class GST expression. Neither the MEK inhibitor PD98059, the p38 MAPK inhibitor SB203580, nor the JNK inhibitor SP600125 inhibited the insulin-mediated increase in alpha-class GST protein levels or GST activity toward 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole, a selective substrate for the alpha-class GSTs (Kim et al., 2006a). These results suggest that MAPK signaling pathways are not primarily involved in the insulin-mediated increase in alpha-class GST expression.
Mechanistic signaling research was also conducted on insulin-mediated effects on mEH. Both wortmannin and LY294002 pretreatment resulted in a concentration-dependent inhibition of the insulin-mediated increase in mEH protein, with complete inhibition of the insulin effect observed at 0.5 μM wortmannin or 10 μM LY294002 (Kim et al., 2003b). These results implicate PI3K as an obligatory component in the stimulation of mEH protein expression by insulin. Consistent with these results, Kang et al. (2001b) showed that PI3K was essential for the induction of mEH by oxidative stress following sulfur amino acid deprivation in H4IIE cells. Also, wortmannin or LY294002 addition to hepatocytes cultured in the absence of insulin produced a ~30 to 40% decrease in mEH protein levels and a marked inhibition of basal Akt phosphorylation. These results suggest that basal activity of PI3K in the absence of insulin plays an important role in regulating the expression of mEH protein in hepatocytes under these conditions. The mTOR inhibitor rapamycin produced a concentration-dependent decline in the insulin-mediated increase in mEH protein levels, resulting in a maximal 50% inhibition of the insulin effect, suggesting that mTOR and downstream signaling via eIF4E-BP1, and p70S6 kinase may play a role in the insulin-mediated increase in mEH protein expression (Kim et al., 2003b). Neither bisindolylmaleimide (10 μM) nor SU6656 (1 μM) inhibited the insulin-mediated increase in mEH protein levels (Kim et al., 2003b), providing evidence for a lack of PKC or JNK involvement in the regulatory process.
Interestingly however, the insulin-mediated increase in mEH protein levels was also inhibited by the p38 MAPK inhibitors, SB202190 or SB203580, but not by the MAPK inhibitor PD98059 or the JNK inhibitor SP600125 (Kim et al., 2003b). These data suggest that p38 MAPK may also play a role in regulating the insulin-mediated increase in mEH protein expression. It was reported that SB203580 abolished interleukin-1-induced Akt phosphorylation with an IC50 value of 3 to 5 μM and activation of p70S6 kinase with an IC50 value above 10 μM by inhibiting PDK1 (Lali et al., 2000). Also Wang et al. (2001) observed that the p38 MAP kinase inhibitors, SB203580 (10 μM) and SB202190 (10 μM), markedly inhibited the phosphorylation of Akt induced by 10 nM insulin in adult rat ventricular cardiomyocytes. These results suggest that the activities of Akt and p70S6 kinase, downstream targets of PDK1, may be inhibited by SB203580 and SB202190 at concentrations above 1 μM. SB203580 and SB202190, however, failed to inhibit the phosphorylation of Akt (Kim et al., 2003b), indicating that the inhibitory effect of SB203580 or SB202190 on the insulin-mediated increase in mEH protein expression is not due to inhibition of PDK1 or its downstream effectors in primary cultured hepatocytes under the conditions employed. A number of studies have reported that the phosphorylation of MAPKs, through activation of a variety of receptors, is regulated by PI3K (Assefa et al., 1999; Sasaoka et al., 1999; Smalley et al., 1999; Hirasawa et al., 2000). The PI3K inhibitors, however, failed to inhibit insulin-induced phosphorylation of p38 MAPK (Kim et al., 2003b), in our studies in primary cultured hepatocytes, suggesting that p38 MAPK may represent a distinct pathway responsible for the induction of mEH in response to insulin treatment.
Our laboratory has also studied the effect of insulin on GSH synthesis in primary cultured rat hepatocytes (Kim et al., 2004) by monitoring gamma-glutamylcysteine ligase expression and activity, the rate limiting component for cellular GSH synthesis. GCL catalytic subunit, GCL catalytic subunit mRNA, GCL activity, and GSH levels were increased in an insulin concentration-dependent manner and were increased maximally 210, 270, 165, and 135%, respectively, relative to primary cultured hepatocytes that did not contain insulin (Fig. 18A). Significantly increased levels of GCL catalytic subunit, GCL activity, and GSH were observed at the normal physiologic level of 1 nM insulin, and a further increase was observed in hepatocytes cultured in the presence of 10 nM insulin. To assess short-term insulin effects on GSH synthesis, insulin (10 nM) was added to hepatocytes for 2, 4, 12, or 24 h (Fig. 18B). Twenty-four hours following initiation of each insulin treatment, GCL catalytic subunit, GCL activity, and GSH levels were monitored. Insulin treatment for 2 h, followed by 22 h of culture in the absence of insulin, resulted in a significant increase in GCL catalytic subunit levels. GCL activity was significantly elevated at 4 h of insulin treatment, and the elevation of GSH levels was observed following 12 or 24 h of insulin treatment. These results suggest that the insulin-mediated elevation of GSH levels is associated with increased synthesis of this tripeptide through the induction of GCL catalytic subunit, and a short treatment with insulin (2 h) is sufficient to initiate this process
Fig. 18.
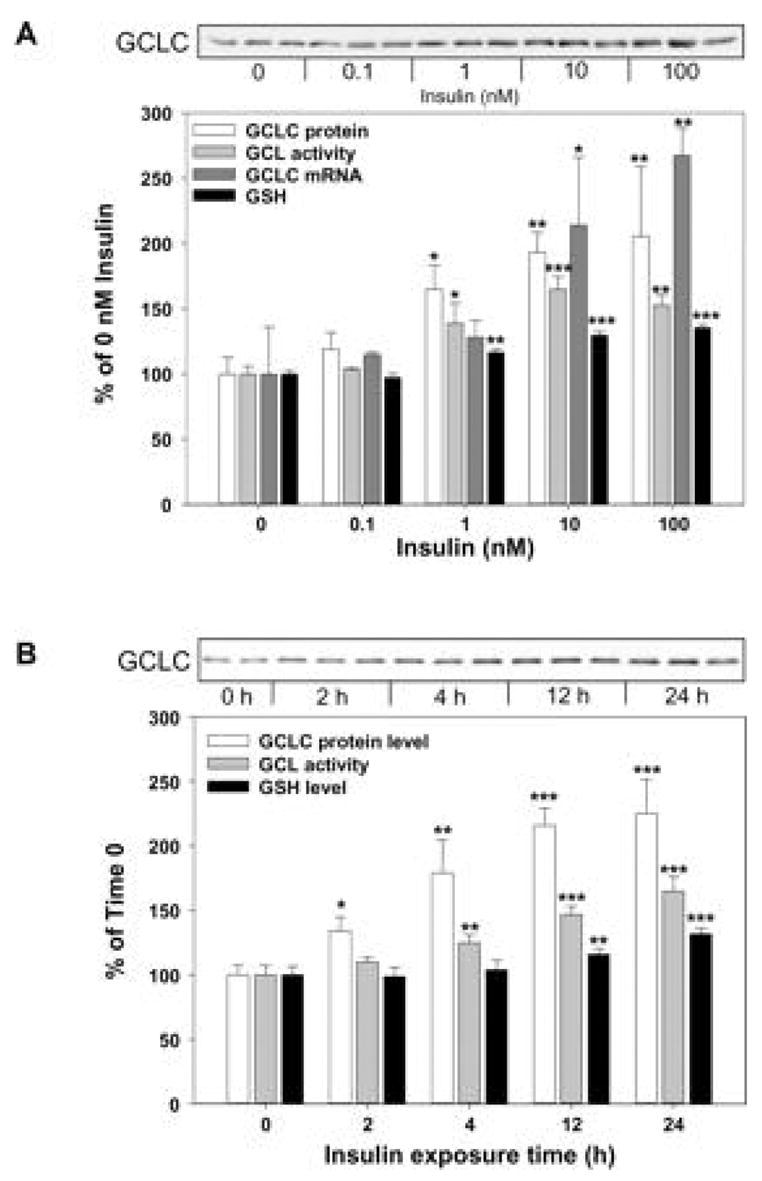
Insulin effect on GCLC protein (A), GCLC mRNA (B), GCL activity (C), and GSH (D) levels in primary cultured rat hepatocytes. Hepatocytes were maintained in the presence or absence of 100 nM insulin for 1 to 4 days. GCLC protein and mRNA levels are plotted as a percentage of the level monitored in freshly isolated hepatocytes (0 day, 100%). Data are means ± SD of three to five preparations of cell lysates from a single hepatocyte preparation. **,***, significantly different from levels monitored in corresponding hepatocytes maintained in the absence of insulin, p < 0.01 or p < 0.001, respectively.
Reprinted from Kim SK, Woodcroft KJ, Khodadadeh SS and Novak RF, J Pharmacol Exp Ther. 311, 99–108, 2004, with permission.
The insulin-mediated increase in GCL catalytic subunit protein and GSH levels were also completely inhibited by pretreatment of cells with wortmannin or LY294002 (Kim et al., 2004). These results implicate PI3K as an obligatory component in the stimulation of GSH synthesis by insulin. Pretreatment of cells with the mTOR inhibitor rapamycin, but not the PKC inhibitor bisindolylmaleimide, inhibited the insulin-mediated increase in GCL catalytic subunit protein and GSH levels, suggesting that mTOR and downstream targets, but not PKC, play a role in the insulin-mediated increase in GCL catalytic subunit protein and GSH levels (Kim et al., 2004). Also overexpression of dn/kd Akt resulted in a decline in the insulin-mediated increase in GCL catalytic subunit protein and GSH levels, resulting in a ~50% inhibition of the insulin effect (Fig. 19). These data suggest that the insulin signaling pathway involving PI3K/Akt/mTOR/p70S6K are active in the insulin-mediated regulation of GSH synthesis via increased GCL catalytic subunit expression.
Fig. 19.
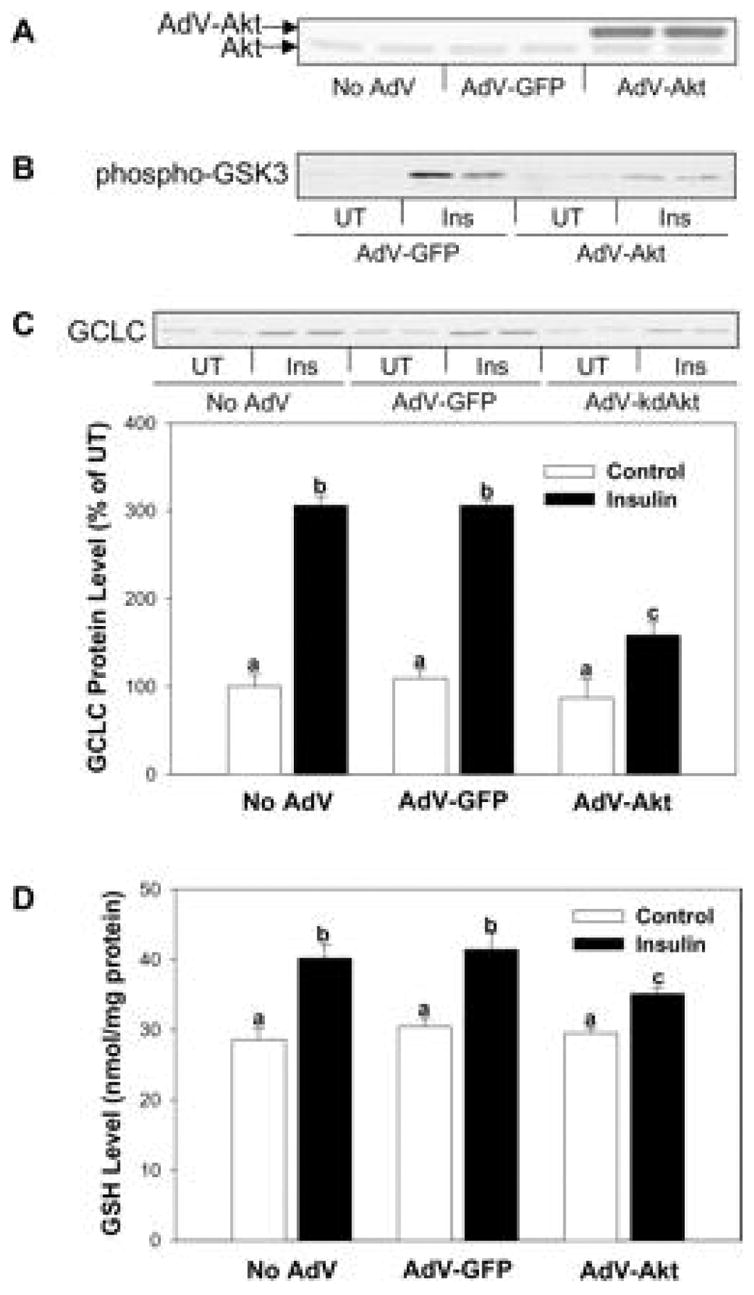
Effect of AdvGFP dn/kd Akt expression on insulin-mediated GCLC protein and GSH levels in primary cultured rat hepatocytes. A, hepatocytes were infected with 150 MOI AdVGFP dn/kd Akt1 or 15 MOI AdV-GFP, and cells were harvested 24 h later for determination of Akt protein levels. B, following 24-h infection with AdV-Akt or AdV-GFP, hepatocytes were treated with 10 nM insulin for 30 min and assayed for Akt activity by maintaining the Akt-mediated phosphorylation of GSKα/β. C and D, 24 h after infection with AdV-Akt or AdV-GFP hepatocytes were treated with 10 nM insulin for 2 days. The AdvGFP dn/kd Akt infection diminished the insulin-mediated increase in GCLC protein as compared to controls (no AdvGFP treatment or treatment with the AdvGFP vector alone). Cellular GSH levels were also decreased in response to the dn/kd Akt infection. Data are means ± S.D. of three and four preparations of cell lysates from a single hepatocyte preparation. Values with different letters are significantly different from each other, p < 0.05.
Reprinted from Kim SK, Woodcroft KJ, Khodadadeh SS and Novak RF, J Pharmacol Exp Ther. 311, 99–108, 2004, with permission.
The p38 MAPK inhibitor SB203580 was without effect on the insulin-mediated elevation of GSH levels (Kim et al., 2004). Interestingly, we found that the JNK inhibitor SP600125 and MAPK inhibitor PD98059 actually enhanced the insulin-mediated increase in GSH levels (Kim et al, 2004). JNK activity is elevated in obesity, and an absence of JNK1 results in decreased adiposity, significantly improved insulin sensitivity and enhanced insulin receptor signaling capacity in two different models of mouse obesity (Hirosumi et al., 2002). Also Lee et al. (2003) showed that insulin-stimulated JNK associated with IRS1 and phosphorylated IRS1 at serine-307 in mouse embryo fibroblasts and 3T3-L1 adipocytes, and that this interaction inhibited insulin signaling. These results raise that possibility that activation of JNK attenuates the insulin-mediated elevation of GSH synthesis through inhibition of IRS-associated PI3K activity. However, Bain et al. (2003) recently reported that SP600125 was a relatively weak inhibitor of JNK isoforms and also inhibited other protein kinases with similar or greater potency. PD98059 (2’-amino-3’-methoxyflavone), the first synthetic inhibitor of the MAPK pathway (Alessi et al., 1995), is a derivative of flavone, which belongs to the chemical class of flavonoids that have attracted attention as potential chemopreventive and chemotherapeutic agents in inflammatory disease, cardiovascular disease and cancer. Thus, we examined whether MEK inhibition is responsible for the PD98059-mediated elevation of GSH levels. The MEK inhibitor U0126, which does not have a flavone structure, failed to affect GSH levels, whereas flavone elevated GSH levels, but was without effect on ERK phosphorylation (Kim et al., 2006b). Moreover, PD98059 increased GSH levels in rat hepatocytes cultured in the absence of insulin. Recent data show that PD98059 increases cysteine levels in rat primary cultured hepatocytes (Kim et al., 2006b). These results suggest that PD98059 produces dramatic changes in GSH homeostasis in hepatocytes, through a mechanism(s) unrelated to MEK inhibition. The above discussion on the use of chemical inhibitors, and differential effects which may be observed with their use, highlights the importance of cell context, concentration-dependent effects and potential contributions of specificity and structure when using these inhibitors to examine signaling pathways in the regulation of gene expression.
6. Conclusion
Studies of intracellular signaling events involved in the regulation of drug metabolizing enzyme expression in response to endogenous factors such as hormones, growth factors and cytokines yield important insights into the regulation of drug metabolism in pathophysiological conditions. Consequently, understanding the role of signaling pathways and components in the regulation of xenobiotic metabolizing gene expression will advance our understanding of differences in xenobiotic metabolism associated with disease, their role in disease initiation and/or promotion, as well as the consequent effects on efficacy and safety of many therapeutic agents targeted to components of these pathways. Our laboratory has used phospho-specific antibodies, chemical inhibitors, and dominant negative/kinase-dead constructs of protein kinases to define the signaling pathways involved in insulin-, EGF- and glucagon-mediated regulation of several drug metabolizing enzymes. Our results provide evidence that insulin and glucagon can serve as physiological regulators of the expression of drug metabolizing enzymes and suggest that the alterations in xenobiotic metabolism and/or cellular oxidative stress these conditions of low insulin may be attributed to altered intracellular signaling (Woodcroft and Novak, 1999b; Kim et al., 2003a Kim et al., 2003b and 2004). From a cellular perspective, CYP2E1 is considered of particular interest due to its poor coupling with NADPH-cytochrome P450 reductase, with NADPH oxidase activity generating reactive oxygen species (Lieber et al., 1997). In contrast alpha-class GSTs, mEH and GCL catalytic subunits constitute the major components of the antioxidant system which provides protection against oxidative stress, a major factor contributing to the extent of complications associated with diabetes (Ceriello and Motz, 2004). Our studies demonstrating that normal levels of insulin increase alpha-class GSTs, mEH and GCL catalytic subunit expression while suppressing CYP2E1 expression, suggest that impairment of the antioxidant defense system may be a contributing factor to the increased oxidative stress and incidence of hepatic disease observed during diabetes. Diabetes has been identified as a risk factor for development of chronic non-alcoholic liver disease, non-alcoholic steatohepatitis and hepatocellular carcinoma (Shimada et al., 2002; Taylor-Robinson et al., 1997; Younossi et al., 2004; Zen et al., 2001; El-Serag et al., 2001; El-Serag et al., 2004; Hassan et al., 2002). The results of our studies suggest that diet, including anti-oxidant supplements and maintenance of cellular GSH levels, may be important in preventing the development of chronic non-alcoholic liver disease (CNLD), non-alcoholic steatohepatitis, and hepatocellular carcinoma. The importance of such protection was recently emphasized by a report showing that glutathione-enhancing agents provided protection against steatohepatitis in a rat dietary model of this disease (Oz et al., 2006). Our studies implicate insulin signaling pathways involving PI3K and its downstream effectors in the insulin-mediated regulation of drug metabolizing enzymes including CYP2E1, alpha-class GSTs, mEH and GCL catalytic subunit. These results suggest that the insulin-mediated PI3K signaling pathways play a critical role in protection of hepatocytes against oxidative stress through induction of the antioxidant defense system, although identification of the effects of these kinases and the transcriptional factors involved in the regulation of drug metabolizing enzyme gene expression in response to insulin and growth factors remains to be determined and is currently under investigation in our laboratory.
Acknowledgments
This research was supported by National Institutes of Health grant ES03656 (R.F. Novak) and, in part, by the Environmental Health Sciences Center grant P30 ES06639 from the National Institutes of Environmental Health Sciences, and by the ERC program of KOSEF (grant R11-2002-100-00000-0)(S.K.K.). We thank Ms. Jennifer Ortwine for assistance in the preparation of the manuscript, figures and artwork.
Abbreviations
- 4E-BP1
eIF4E binding protein 1
- AA
acetoacetate
- dn/kd Akt
dominant negative/kinase-dead mutant of Akt1
- CYP
cytochrome P450
- EGF
epidermal growth factor
- eIF4E
eukaryotic translation initiation factor 4E
- ERK1/2
extracellular signal-regulated kinase1/2
- Gab
Grb2 associated binder
- GCL
gamma-glutamylcysteine ligase
- Grb2
Growth factor receptor binding protein 2
- GSH
glutathione
- GST
Glutathione S-transferase
- HGF
hepatocyte growth factor
- HSPs
heat shock proteins
- IMP
impedes mitogenic signal propagation
- IRS
insulin receptor substrate
- JNK
jun N-terminal kinase
- KSR
kinase suppressor of Ras
- MAPK
mitogen activated protein kinase
- mEH
microsomal epoxide hydrolase
- MEK
MAPK kinase
- MKKK
MAPK kinase kinase
- MKPs
MAPK phosphatases
- mTOR
mammalian target of rapamycin
- p38 MAPK
p38 Mitogen activated protein kinase
- p70S6 kinase
p70 ribosomal protein S6 kinase
- PAK
p21 Activated kinase
- PDK1
3-Phosphoinositide-dependent protein kinase-1
- PH
Pleckstrin homology
- PI
phosphatidylinositol
- PI(3,4,5)P3
PI 3,4,5-triphosphate
- PI3K
phosphatidylinositol-3-kinase
- PKB
protein kinase B
- PKC
protein kinase C
- PP2A
protein phosphatase 2A
- PPM
Mg2+-dependent protein phosphatase
- PPP
phosphoprotein phosphatase
- PTB
phosphotyrosine binding
- PTEN
phosphatase and tensin homologue deleted on chromosome ten
- PTPs
protein tyrosine phosphatases
- RKIP
Raf kinase inhibitor protein
- RTK
receptor tyrosine kinase
- SAPKs
stress-activated protein kinases
- SH2
Src homology-2
- SOS
Son of Sevenless
- SULT
sulfotransferase
- UGT
UDP-glucuronosyltransferase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdelmegeed MA, Carruthers NJ, Kim SK, Woodcroft KJ, Novak RF. Acetoacetate induces cytochrome P450 (CYP) 2E1 protein and suppresses CYP2E1 gene transcription in primary cultured rat hepatocytes. J Pharmacol Exp Ther. 2005;315:203–213. doi: 10.1124/jpet.105.084608. [DOI] [PubMed] [Google Scholar]
- Abernethy DR, Greenblatt DJ, Divoll M, Shader RI. Enhanced glucuronide conjugation of drugs in obesity: studies of lorazepam, oxazepam, and acetaminophen. J Lab Clin Med. 1983;101:873–880. [PubMed] [Google Scholar]
- Agius C, Gidari AS. Effect of streptozotocin on the glutathione S-transferases of mouse liver cytosol. Biochem Pharmacol. 1985;34:811–819. doi: 10.1016/0006-2952(85)90761-0. [DOI] [PubMed] [Google Scholar]
- Alemzadeh R, Tushaus KM. Modulation of adipoinsular axis in prediabetic zucker diabetic fatty rats by diazoxide. Endocrinology. 2004;145:5476–84. doi: 10.1210/en.2003-1523. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol. 1997a;7:776–789. doi: 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997b;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Andersen JN, Jansen PG, Echwald SM, Mortensen OH, Fukada T, Del Vecchio R, Tonks NK, Moller NP. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J. 2004;18:8–30. doi: 10.1096/fj.02-1212rev. [DOI] [PubMed] [Google Scholar]
- Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- Arrandale JM, Gore-Willse A, Rocks S, Ren JM, Zhu J, Davis A, Livingston JN, Rabin DU. Insulin signaling in mice expressing reduced levels of Syp. J Biol Chem. 1996;271:21353–21358. doi: 10.1074/jbc.271.35.21353. [DOI] [PubMed] [Google Scholar]
- Asante-Appiah E, Kennedy BP. Protein tyrosine phosphatases: the quest for negative regulators of insulin action. Am J Physiol Endocrinol Metab. 2003;284:E663–E670. doi: 10.1152/ajpendo.00462.2002. [DOI] [PubMed] [Google Scholar]
- Assefa Z, Valius M, Vantus T, Agostinis P, Merlevede W, Vandenheede JR. JNK/SAPK activation by platelet-derived growth factor in A431 cells requires both the phospholipase C- and the phosphatidylinositol 3-kinase signaling pathways of the receptor. Biochem Biophys Res Commun. 1999;261:641–645. doi: 10.1006/bbrc.1999.1090. [DOI] [PubMed] [Google Scholar]
- Baccarini M. Second nature: biological functions of the Raf-1 "kinase". FEBS Lett. 2005;579:3271–3277. doi: 10.1016/j.febslet.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Backer JM, Myers MG, Jr, Sun XJ, Chin DJ, Shoelson SE, Miralpeix M, White MF. Association of IRS-1 with the insulin receptor and the phosphatidylinositol 3′-kinase. Formation of binary and ternary signaling complexes in intact cells. J Biol Chem. 1993;268:8204–8212. [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balendran A, Currie R, Armstrong CG, Avruch J, Alessi DR. Evidence that 3-phosphoinositide-dependent protein kinase-1 mediates phosphorylation of p70S6 kinase in vivo at Thr-412 as well as Thr-252. J Biol Chem. 1999;274:37400–37406. doi: 10.1074/jbc.274.52.37400. [DOI] [PubMed] [Google Scholar]
- Balks HJ, Jungermann K. Regulation of peripheral insulin/glucagon levels by rat liver. Eur J Biochem. 1984;141:645–650. doi: 10.1111/j.1432-1033.1984.tb08240.x. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay D, Kusari A, Kenner KA, Liu F, Chernoff J, Gustafson TA, Kusari J. Protein-tyrosine phosphatase 1B complexes with the insulin receptor in vivo and is tyrosine-phosphorylated in the presence of insulin. J Biol Chem. 1997;272:1639–1645. doi: 10.1074/jbc.272.3.1639. [DOI] [PubMed] [Google Scholar]
- Barnett CR, Gibson GG, Wolf CR, Flatt PR, Ioannides C. Induction of cytochrome P450III and P450IV family proteins in streptozotocin-induced diabetes. Biochem J. 1990;268:765–769. doi: 10.1042/bj2680765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- Bellward GD, Chang T, Rodrigues B, McNeill JH, Maines S, Ryan DE, Levin W, Thomas PE. Hepatic cytochrome P-450j induction in the spontaneously diabetic BB rat. Mol Pharmacol. 1988;33:140–143. [PubMed] [Google Scholar]
- Bennett AM, Hausdorff SF, O'Reilly AM, Freeman RM, Neel BG. Multiple requirements for SHPTP2 in epidermal growth factor-mediated cell cycle progression. Mol Cell Biol. 1996;16:1189–1202. doi: 10.1128/mcb.16.3.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beugnet A, Wang X, Proud CG. Target of rapamycin (TOR)-signaling and RAIP motifs play distinct roles in the mammalian TOR-dependent phosphorylation of initiation factor 4E-binding protein 1. J Biol Chem. 2003;278:40717–40722. doi: 10.1074/jbc.M308573200. [DOI] [PubMed] [Google Scholar]
- Bitar MS, Al-Saleh E, Al-Mulla F. Oxidative stress--mediated alterations in glucose dynamics in a genetic animal model of type II diabetes. Life Sci. 2005;77:2552–2573. doi: 10.1016/j.lfs.2005.01.033. [DOI] [PubMed] [Google Scholar]
- Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem. 2000;275:41439–41446. doi: 10.1074/jbc.M004852200. [DOI] [PubMed] [Google Scholar]
- Bjornholm M, He AR, Attersand A, Lake S, Liu SC, Lienhard GE, Taylor S, Arner P, Zierath JR. Absence of functional insulin receptor substrate-3 (IRS-3) gene in humans. Diabetologia. 2002;45:1697–1702. doi: 10.1007/s00125-002-0945-z. [DOI] [PubMed] [Google Scholar]
- Brady MJ, Saltiel AR. The role of protein phosphatase-1 in insulin action. Recent Prog Horm Res. 2001;56:157–173. doi: 10.1210/rp.56.1.157. [DOI] [PubMed] [Google Scholar]
- Braun L, Coffey MJ, Puskas F, Kardon T, Nagy G, Conley AA, Burchell B, Mandl J. Molecular basis of bilirubin UDP-glucuronosyltransferase induction in spontaneously diabetic rats, acetone-treated rats and starved rats. Biochem J. 1998;336:587–592. doi: 10.1042/bj3360587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci. 2001;26:657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell. 2002;111:293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- Cai D, Dhe-Paganon S, Melendez PA, Lee J, Shoelson SE. Two new substrates in insulin signaling, IRS5/DOK4 and IRS6/DOK5. J Biol Chem. 2003;278:25323–25330. doi: 10.1074/jbc.M212430200. [DOI] [PubMed] [Google Scholar]
- Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14:6–16. [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Carraway H, Hidalgo M. New targets for therapy in breast cancer: mammalian target of rapamycin (mTOR) antagonists. Breast Cancer Res. 2004;6:219–224. doi: 10.1186/bcr927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo MC, Monti JA, Favre C, Carnovale CE. Acute regulation of hepatic glutathione S-transferase by insulin and glucagon. Toxicol Lett. 1995;76:105–111. doi: 10.1016/0378-4274(94)03203-j. [DOI] [PubMed] [Google Scholar]
- Casamayor A, Morrice NA, Alessi DR. Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochem J. 1999;342:287–292. [PMC free article] [PubMed] [Google Scholar]
- Catling AD, Schaeffer HJ, Reuter CW, Reddy GR, Weber MJ. A proline-rich sequence unique to MEK1 and MEK2 is required for raf binding and regulates MEK function. Mol Cell Biol. 1995;15:5214–5225. doi: 10.1128/mcb.15.10.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceriello A, Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol. 2004;24:816–823. doi: 10.1161/01.ATV.0000122852.22604.78. [DOI] [PubMed] [Google Scholar]
- Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- Charbonneau H, Tonks NK, Kumar S, Diltz CD, Harrylock M, Cool DE, Krebs EG, Fischer EH, Walsh KA. Human placenta protein-tyrosine-phosphatase: amino acid sequence and relationship to a family of receptor-like proteins. Proc Natl Acad Sci U S A. 1989;86:5252–5256. doi: 10.1073/pnas.86.14.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary IP, Tuntaterdtum S, McNamara PJ, Robertson LW, Blouin RA. Effect of genetic obesity and phenobarbital treatment on the hepatic conjugation pathways. J Pharmacol Exp Ther. 1993;265:1333–1338. [PubMed] [Google Scholar]
- Cheatham B, Kahn CR. Cysteine 647 in the insulin receptor is required for normal covalent interaction between alpha- and beta-subunits and signal transduction. J Biol Chem. 1992;267:7108–7115. [PubMed] [Google Scholar]
- Cheatham B, Kahn CR. Insulin action and the insulin signaling network. Endocr Rev. 1995;16:117–142. doi: 10.1210/edrv-16-2-117. [DOI] [PubMed] [Google Scholar]
- Cheatham B, Vlahos CJ, Cheatham L, Wang L, Blenis J, Kahn CR. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol Cell Biol. 1994;14:4902–4911. doi: 10.1128/mcb.14.7.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Wertheimer SJ, Lin CH, Katz SL, Amrein KE, Burn P, Quon MJ. Protein-tyrosine phosphatases PTP1B and syp are modulators of insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. J Biol Chem. 1997;272:8026–8031. doi: 10.1074/jbc.272.12.8026. [DOI] [PubMed] [Google Scholar]
- Cheung C, Akiyama TE, Kudo G, Gonzalez FJ. Hepatic expression of cytochrome P450s in hepatocyte nuclear factor 1-alpha (HNF1alpha)-deficient mice. Biochem Pharmacol. 2003;66:2011–2020. doi: 10.1016/s0006-2952(03)00586-0. [DOI] [PubMed] [Google Scholar]
- Chiloeches A, Mason CS, Marais R. S338 phosphorylation of Raf-1 is independent of phosphatidylinositol 3-kinase and Pak3. Mol Cell Biol. 2001;21:2423–2434. doi: 10.1128/MCB.21.7.2423-2434.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching KZ, Tenney KA, Chen J, Morgan ET. Suppression of constitutive cytochrome P450 gene expression by epidermal growth factor receptor ligands in cultured rat hepatocytes. Drug Metab Dispos. 1996;24:542–546. [PubMed] [Google Scholar]
- Chong H, Lee J, Guan KL. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 2001;20:3716–3727. doi: 10.1093/emboj/20.14.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell Signal. 2003;15:463–469. doi: 10.1016/s0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- Chung J, Grammer TC, Lemon KP, Kazlauskas A, Blenis J. PDGF- and insulin-dependent pp70S6k activation mediated by phosphatidylinositol-3-OH kinase. Nature. 1994;370:71–75. doi: 10.1038/370071a0. [DOI] [PubMed] [Google Scholar]
- Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- Cohen P. The structure and regulation of protein phosphatases. Annu Rev Biochem. 1989;58:453–508. doi: 10.1146/annurev.bi.58.070189.002321. [DOI] [PubMed] [Google Scholar]
- Constantopoulos A, Matsaniotis N. Augmentation of uridine diphosphate glucuronyltransferase activity in rat liver by adenosine 3′,5′-monophosphate. Gastroenterology. 1978;75:486–491. [PubMed] [Google Scholar]
- Conus NM, Hemmings BA, Pearson RB. Differential regulation by calcium reveals distinct signaling requirements for the activation of Akt and p70S6k. J Biol Chem. 1998;273:4776–4782. doi: 10.1074/jbc.273.8.4776. [DOI] [PubMed] [Google Scholar]
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- Dadke S, Kusari A, Kusari J. Phosphorylation and activation of protein tyrosine phosphatase (PTP) 1B by insulin receptor. Mol Cell Biochem. 2001;221:147–154. doi: 10.1023/a:1010909031310. [DOI] [PubMed] [Google Scholar]
- De Meyts P. Insulin and its receptor: structure, function and evolution. Bioessays. 2004;26:1351–1362. doi: 10.1002/bies.20151. [DOI] [PubMed] [Google Scholar]
- De Smet K, Loyer P, Gilot D, Vercruysse A, Rogiers V, Guguen-Guillouzo C. Effects of epidermal growth factor on CYP inducibility by xenobiotics, DNA replication, and caspase activations in collagen I gel sandwich cultures of rat hepatocytes. Biochem Pharmacol. 2001;61:1293–1303. doi: 10.1016/s0006-2952(01)00612-8. [DOI] [PubMed] [Google Scholar]
- De Waziers I, Garlatti M, Bouguet J, Beaune PH, Barouki R. Insulin down-regulates cytochrome P450 2B and 2E expression at the post-transcriptional level in the rat hepatoma cell line. Mol Pharmacol. 1995;47:474–479. [PubMed] [Google Scholar]
- Deprez J, Bertrand L, Alessi DR, Krause U, Hue L, Rider MH. Partial purification and characterization of a wortmannin-sensitive and insulin-stimulated protein kinase that activates heart 6-phosphofructo-2-kinase. Biochem J. 2000;347:305–312. [PMC free article] [PubMed] [Google Scholar]
- Desmots F, Rissel M, Gilot D, Lagadic-Gossmann D, Morel F, Guguen-Guillouzo C, Guillouzo A, Loyer P. Pro-inflammatory cytokines tumor necrosis factor alpha and interleukin-6 and survival factor epidermal growth factor positively regulate the murine GSTA4 enzyme in hepatocytes. J Biol Chem. 2002;277:17892–17900. doi: 10.1074/jbc.M112351200. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Pollock C, Steen H, Shaw PE, Mischak H, Kolch W. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol Cell Biol. 2002;22:3237–3246. doi: 10.1128/MCB.22.10.3237-3246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue BS, Morgan ET. Effects of vanadate on hepatic cytochrome P450 expression in streptozotocin-diabetic rats. Drug Metab Dispos. 1990;18:519–526. [PubMed] [Google Scholar]
- Donahue BS, Skottner-Lundin A, Morgan ET. Growth hormone-dependent and -independent regulation of cytochrome P-450 isozyme expression in streptozotocin-diabetic rats. Endocrinology. 1991;128:2065–2076. doi: 10.1210/endo-128-4-2065. [DOI] [PubMed] [Google Scholar]
- Donato MT, Gomez-Lechon MJ, Jover R, Nakamura T, Castell JV. Human hepatocyte growth factor down-regulates the expression of cytochrome P450 isozymes in human hepatocytes in primary culture. J Pharmacol Exp Ther. 1998;284:760–767. [PubMed] [Google Scholar]
- Dong Z, Hong J, Ma Q, Li D, Bullock J, Gonzalez FJ, Park SS, Gelboin HV, Yang CS. Mechanism of induction of cytochrome P450ac (P450j) in chemically induced and spontaneously diabetic rats. Arch Biochem Biophys. 1988;263:29–35. doi: 10.1016/0003-9861(88)90610-8. [DOI] [PubMed] [Google Scholar]
- Downward J. Signal transduction. New exchange, new target. Nature. 1998;396:416–417. doi: 10.1038/24743. [DOI] [PubMed] [Google Scholar]
- Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- Dufner A, Andjelkovic M, Burgering BM, Hemmings BA, Thomas G. Protein kinase B localization and activation differentially affect S6 kinase 1 activity and eukaryotic translation initiation factor 4E-binding protein 1 phosphorylation. Mol Cell Biol. 1999;19:4525–4534. doi: 10.1128/mcb.19.6.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumaz N, Light Y, Marais R. Cyclic AMP blocks cell growth through Raf-1-dependent and Raf-1-independent mechanisms. Mol Cell Biol. 2002;22:3717–3728. doi: 10.1128/MCB.22.11.3717-3728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvaldestin P, Mahu J-L, Berthelot P. Effect of fasting on substrate specificity of rat liver UDP-glucuronosyltransferase. Biochim Biophys Acta. 1975;384:81–86. doi: 10.1016/0005-2744(75)90097-2. [DOI] [PubMed] [Google Scholar]
- Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- Elchebly M, Cheng A, Tremblay ML. Modulation of insulin signaling by protein tyrosine phosphatases. J Mol Med. 2000;78:473–482. doi: 10.1007/s001090000141. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, Richardson PA, Everhart JE. The role of diabetes in hepatocellular carcinoma: a case-control study among United States Veterans. Am J Gastroenterol. 2001;96:2462–2467. doi: 10.1111/j.1572-0241.2001.04054.x. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology. 2004;126:460–468. doi: 10.1053/j.gastro.2003.10.065. [DOI] [PubMed] [Google Scholar]
- Eyers PA, Craxton M, Morrice N, Cohen P, Goedert M. Conversion of SB 203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem Biol. 1998;5:321–328. doi: 10.1016/s1074-5521(98)90170-3. [DOI] [PubMed] [Google Scholar]
- Farese RV. Insulin-sensitive phospholipid signaling systems and glucose transport. Update II. Exp Biol Med (Maywood) 2001;226:283–295. doi: 10.1177/153537020122600404. [DOI] [PubMed] [Google Scholar]
- Farese RV, Sajan MP, Standaert ML. Insulin-Sensitive Protein Kinases (Atypical Protein Kinase C and Protein Kinase B/Akt): Actions and Defects in Obesity and Type II Diabetes. Exp Biol Med (Maywood) 2005;230:593–605. doi: 10.1177/153537020523000901. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Favreau LV, Schenkman JB. Composition changes in hepatic microsomal cytochrome P450 during onset of streptozotocin-induced diabetes and during insulin treatment. Diabetes. 1988;37:577–584. doi: 10.2337/diab.37.5.577. [DOI] [PubMed] [Google Scholar]
- Frangioni JV, Oda A, Smith M, Salzman EW, Neel BG. Calpain-catalyzed cleavage and subcellular relocation of protein phosphotyrosine phosphatase 1B (PTP-1B) in human platelets. EMBO J. 1993;12:4843–4856. doi: 10.1002/j.1460-2075.1993.tb06174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz B, Klatt T, Pang M, Parsons J, Rolando A, Williams H, Tocci MJ, O'Keefe SJ, O'Neill EA. The activation state of p38 mitogen-activated protein kinase determines the efficiency of ATP competition for pyridinylimidazole inhibitor binding. Biochemistry. 1998;37:13846–13853. doi: 10.1021/bi980832y. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Rameh LE, Cantley LC. Phosphoinositide binding domains: embracing 3-phosphate. Cell. 1999;97:817–820. doi: 10.1016/s0092-8674(00)80792-8. [DOI] [PubMed] [Google Scholar]
- Garcia MC, Thangavel C, Shapiro BH. Epidermal growth factor regulation of female-dependent CYP2A1 and CYP2C12 in primary rat hepatocyte culture. Drug Metab Dispos. 2001;29:111–120. [PubMed] [Google Scholar]
- Giehl K. Oncogenic Ras in tumour progression and metastasis. Biol Chem. 2005;386:193–205. doi: 10.1515/BC.2005.025. [DOI] [PubMed] [Google Scholar]
- Goncharova EA, Goncharov DA, Eszterhas A, Hunter DS, Glassberg MK, Yeung RS, Walker CL, Noonan D, Kwiatkowski DJ, Chou MM, Panettieri RA, Jr, Krymskaya VP. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation. A role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM) J Biol Chem. 2002;277:30958–30967. doi: 10.1074/jbc.M202678200. [DOI] [PubMed] [Google Scholar]
- Grant MH, Duthie SJ. Conjugation reactions in hepatocytes isolated from streptozotocin-induced diabetic rats. Biochem Pharmacol. 1987;36:3647–3655. doi: 10.1016/0006-2952(87)90015-3. [DOI] [PubMed] [Google Scholar]
- Graves LM, Bornfeldt KE, Argast GM, Krebs EG, Kong X, Lin TA, Lawrence JC., Jr cAMP- and rapamycin-sensitive regulation of the association of eukaryotic initiation factor 4E and the translational regulator PHAS-I in aortic smooth muscle cells. Proc Natl Acad Sci USA. 1995;92:7222–7226. doi: 10.1073/pnas.92.16.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo S, Gremeaux T, Casamayor A, Alessi DR, Le Marchand-Brustel Y, Tanti JF. Peroxovanadate induces tyrosine phosphorylation of phosphoinositide-dependent protein kinase-1 potential involvement of src kinase. Eur J Biochem. 2000;267:6642–6649. doi: 10.1046/j.1432-1327.2000.01759.x. [DOI] [PubMed] [Google Scholar]
- Groom LA, Sneddon AA, Alessi DR, Dowd S, Keyse SM. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. EMBO J. 1996;15:3621–3632. [PMC free article] [PubMed] [Google Scholar]
- Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Gueraud F, Masmoudi T, Goudonnet H, Paris A. Differential effect of hypophysectomy and growth hormone treatment on hepatic glucuronosyltransferases in male rats: evidence for an action at a pretranslational level for isoforms glucuronidating bilirubin. Biochem Pharmacol. 1997;53:1637–1647. doi: 10.1016/s0006-2952(97)82452-5. [DOI] [PubMed] [Google Scholar]
- Hagemann C, Blank JL. The ups and downs of MEK kinase interactions. Cell Signal. 2001;13:863–875. doi: 10.1016/s0898-6568(01)00220-0. [DOI] [PubMed] [Google Scholar]
- Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- Hajduch E, Litherland GJ, Hundal HS. Protein kinase B (PKB/Akt)--a key regulator of glucose transport? FEBS Lett. 2001;492:199–203. doi: 10.1016/s0014-5793(01)02242-6. [DOI] [PubMed] [Google Scholar]
- Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- Harris TE, Lawrence JC., Jr TOR signaling. Sci STKE. 2003;212:re15. doi: 10.1126/stke.2122003re15. [DOI] [PubMed] [Google Scholar]
- Hassan MM, Hwang LY, Hatten CJ, Swaim M, Li D, Abbruzzese JL, Beasley P, Patt YZ. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206–1213. doi: 10.1053/jhep.2002.36780. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hirasawa N, Sato Y, Fujita Y, Ohuchi K. Involvement of a phosphatidylinositol 3-kinase-p38 mitogen-activated protein kinase pathway in antigen-induced IL-4 production in mast cells. Biochim Biophys Acta. 2000;1456:45–55. doi: 10.1016/s0005-2728(99)00104-8. [DOI] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Honda R, Ohba Y, Nagata A, Okayama H, Yasuda H. Dephosphorylation of human p34cdc2 kinase on both Thr-14 and Tyr-15 by human cdc25B phosphatase. FEBS Lett. 1993;318:331–334. doi: 10.1016/0014-5793(93)80540-b. [DOI] [PubMed] [Google Scholar]
- Hong JY, Pan JM, Gonzalez FJ, Gelboin HV, Yang CS. The induction of a specific form of cytochrome P-450 (P-450j) by fasting. Biochem Biophys Res Commun. 1987;142:1077–1083. doi: 10.1016/0006-291x(87)91525-7. [DOI] [PubMed] [Google Scholar]
- Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278:27495–27501. doi: 10.1074/jbc.M304112200. [DOI] [PubMed] [Google Scholar]
- Hwang SL, Liu IM, Tzeng TF, Cheng JT. Activation of imidazoline receptors in adrenal gland to lower plasma glucose in streptozotocin-induced diabetic rats. Diabetologia. 2005;48:767–775. doi: 10.1007/s00125-005-1698-2. [DOI] [PubMed] [Google Scholar]
- Iber H, Morgan ET. Regulation of hepatic cytochrome P450 2C11 by transforming growth factor-beta, hepatocyte growth factor, and interleukin-11. Drug Metab Dispos. 1998;26:1042–1044. [PubMed] [Google Scholar]
- Iber H, Li-Masters T, Chen Q, Yu S, Morgan ET. Regulation of hepatic cytochrome P450 2C11 via cAMP: implications for down-regulation in diabetes, fasting, and inflammation. J Pharmacol Exp Ther. 2001;297:174–180. [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Inoue N, Fujiwara K, Iwata T, Imai K, Aimoto T. Involvement of pituitary hormone in the sex-related regulation of hepatic epoxide hydrolase activity in mice. Biol Pharm Bull. 1995;18:536–539. doi: 10.1248/bpb.18.536. [DOI] [PubMed] [Google Scholar]
- Isakoff SJ, Cardozo T, Andreev J, Li Z, Ferguson KM, Abagyan R, Lemmon MA, Aronheim A, Skolnik EY. Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. EMBO J. 1998;17:5374–5387. doi: 10.1093/emboj/17.18.5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5'TOP mRNA translation through inhibition of p70s6k. EMBO J. 1997;16:3693–3704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Zhang BB. Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab. 2003;284:E671–678. doi: 10.1152/ajpendo.00492.2002. [DOI] [PubMed] [Google Scholar]
- Kahn CR. Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes. 1994;43:1066–1084. doi: 10.2337/diab.43.8.1066. [DOI] [PubMed] [Google Scholar]
- Kang KW, Cho MK, Lee CH, Kim SG. Activation of phosphatidylinositol 3-kinase and Akt by tert-butylhydroquinone is responsible for antioxidant response element-mediated rGSTA2 induction in H4IIE cells. Mol Pharmacol. 2001a;59:1147–1156. doi: 10.1124/mol.59.5.1147. [DOI] [PubMed] [Google Scholar]
- Kang KW, Ryu JH, Kim SG. Activation of phosphatidylinositol 3-kinase by oxidative stress leads to the induction of microsomal epoxide hydrolase in H4IIE cells. Toxicol Lett. 2001b;121:191–197. doi: 10.1016/s0378-4274(01)00337-x. [DOI] [PubMed] [Google Scholar]
- Kang KW, Lee SJ, Park JW, Kim SG. Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2-related factor 2 through actin rearrangement in response to oxidative stress. Mol Pharmacol. 2002a;62:1001–1010. doi: 10.1124/mol.62.5.1001. [DOI] [PubMed] [Google Scholar]
- Kang KW, Novak RF, Lee CH, Kim SG. Induction of microsomal epoxide hydrolase by sulfur amino acid deprivation via the pathway of C-Jun N-terminal kinase and its extracellular exposure during cell death. Free Radic Biol Med. 2002b;32:1017–32. doi: 10.1016/s0891-5849(02)00788-8. [DOI] [PubMed] [Google Scholar]
- Kardon T, Coffey MJ, Banhegyi G, Conley AA, Burchell B, Mandl J, Braun L. Transcriptional induction of bilirubin UDP-glucuronosyltransrase by ethanol in rat liver. Alcohol. 2000;21:251–257. doi: 10.1016/s0741-8329(00)00095-1. [DOI] [PubMed] [Google Scholar]
- Kawasome H, Papst P, Webb S, Keller GM, Johnson GL, Gelfand EW, Terada N. Targeted disruption of p70(s6k) defines its role in protein synthesis and rapamycin sensitivity. Proc Natl Acad Sci U S A. 1998;95:5033–5038. doi: 10.1073/pnas.95.9.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- Khosravi-Far R, Campbell S, Rossman KL, Der CJ. Increasing complexity of Ras signal transduction: involvement of Rho family proteins. Adv Cancer Res. 1998;72:57–107. doi: 10.1016/s0065-230x(08)60700-9. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sabatini DM. Raptor and mTOR: subunits of a nutrient-sensitive complex. Curr Top Microbiol Immunol. 2004;279:259–270. doi: 10.1007/978-3-642-18930-2_15. [DOI] [PubMed] [Google Scholar]
- Kim SK, Abdelmegeed MA, Novak RF. Identification of the Insulin Signaling Cascade in the Regulation of Alpha-Class Glutathione S-Transferase Expression in Primary Cultured Rat Hepatocytes. J Pharmacol Exp Ther. 2006a;316:1255–1261. doi: 10.1124/jpet.105.096065. [DOI] [PubMed] [Google Scholar]
- Kim SK, Abdelmegeed MA, Novak RF. The mitogen-activated protein kinase kinase (mek) inhibitor pd98059 elevates primary cultured rat hepatocyte glutathione levels independent of inhibiting mek. Drug Metab Dispos. 2006b;34:683–689. doi: 10.1124/dmd.105.007666. [DOI] [PubMed] [Google Scholar]
- Kim SK, Woodcroft KJ, Novak RF. Insulin and glucagon regulation of glutathione S-transferase expression in primary cultured rat hepatocytes. J Pharmacol Exp Ther. 2003a;305:353–361. doi: 10.1124/jpet.102.045153. [DOI] [PubMed] [Google Scholar]
- Kim SK, Woodcroft KJ, Kim SG, Novak RF. Insulin and Glucagon Signaling in Regulation of Microsomal Epoxide Hydrolase Expression in Primary Cultured Rat Hepatocytes. Drug Metab Dispos. 2003b;31:1260–1268. doi: 10.1124/dmd.31.10.1260. [DOI] [PubMed] [Google Scholar]
- Kim SK, Woodcroft KJ, Khodadadeh SS, Novak RF. Insulin signaling regulates gamma-glutamylcysteine ligase catalytic subunit expression in primary cultured rat hepatocytes. J Pharmacol Exp Ther. 2004;311:99–108. doi: 10.1124/jpet.104.070375. [DOI] [PubMed] [Google Scholar]
- Kim YG, Kim SK, Kwon JW, Park OJ, Kim SG, Kim YC, Lee MG. Effects of cysteine on amino acid concentrations and transsulfuration enzyme activities in rat liver with protein-calorie malnutrition. Life Sci. 2003c;72:1171–1181. doi: 10.1016/s0024-3205(02)02366-4. [DOI] [PubMed] [Google Scholar]
- Kimball SR, Siegfried BA, Jefferson LS. Glucagon represses signaling through the mammalian target of rapamycin in rat liver by activating AMP-activated protein kinase. J Biol Chem. 2004;279:54103–54109. doi: 10.1074/jbc.M410755200. [DOI] [PubMed] [Google Scholar]
- King AJ, Sun H, Diaz B, Barnard D, Miao W, Bagrodia S, Marshall MS. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature. 1998;396:180–183. doi: 10.1038/24184. [DOI] [PubMed] [Google Scholar]
- Klumpp S, Krieglstein J. Serine/threonine protein phosphatases in apoptosis. Curr Opin Pharmacol. 2002;2:458–462. doi: 10.1016/s1471-4892(02)00176-5. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Takeuchi F, Roth RA. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem. 1996;271:21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351:289–305. [PMC free article] [PubMed] [Google Scholar]
- Kornfeld K, Hom DB, Horvitz HR. The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell. 1995;83:903–913. doi: 10.1016/0092-8674(95)90206-6. [DOI] [PubMed] [Google Scholar]
- Kubicek M, Pacher M, Abraham D, Podar K, Eulitz M, Baccarini M. Dephosphorylation of Ser-259 regulates Raf-1 membrane association. J Biol Chem. 2002;277:7913–7919. doi: 10.1074/jbc.M108733200. [DOI] [PubMed] [Google Scholar]
- Kwon HS, Huang B, Unterman TG, Harris RA. Protein kinase B-alpha inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes. 2004;53:899–910. doi: 10.2337/diabetes.53.4.899. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Lali FV, Hunt AE, Turner SJ, Foxwell BM. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J Biol Chem. 2000;275:7395–7402. doi: 10.1074/jbc.275.10.7395. [DOI] [PubMed] [Google Scholar]
- Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- Lee YH, Giraud J, Davis RJ, White MF. c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem. 2003;278:2896–2902. doi: 10.1074/jbc.M208359200. [DOI] [PubMed] [Google Scholar]
- Leclerc I, Lenzner C, Gourdon L, Vaulont S, Kahn A, Viollet B. Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes. 2001;50:1515–1521. doi: 10.2337/diabetes.50.7.1515. [DOI] [PubMed] [Google Scholar]
- Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, Rosenfeld MG. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426(6964):247–254. doi: 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- Lieber CS. Cytochrome P-4502E1: its physiological and pathological role. Physiol Rev. 1997;77:517–544. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- Light Y, Paterson H, Marais R. 14-3-3 antagonizes Ras-mediated Raf-1 recruitment to the plasma membrane to maintain signaling fidelity. Mol Cell Biol. 2002;22:4984–4996. doi: 10.1128/MCB.22.14.4984-4996.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Chernoff J. Protein tyrosine phosphatase 1B interacts with and is tyrosine phosphorylated by the epidermal growth factor receptor. Biochem J. 1997;327:139–145. doi: 10.1042/bj3270139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Klaassen CD. Ontogeny and hormonal basis of male-dominant rat hepatic sulfotransferases. Mol Pharmacol. 1996;50:565–572. [PubMed] [Google Scholar]
- Lu SC, Kuhlenkamp J, Garcia-Ruiz C, Kaplowitz N. Hormone-mediated down-regulation of hepatic glutathione synthesis in the rat. J Clin Invest. 1991;88:260–269. doi: 10.1172/JCI115286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC, Ge JL, Kuhlenkamp J, Kaplowitz N. Insulin and glucocorticoid dependence of hepatic gamma-glutamylcysteine synthetase and glutathione synthesis in the rat. Studies in cultured hepatocytes and in vivo. J Clin Invest. 1992;90:524–532. doi: 10.1172/JCI115890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC, Huang ZZ, Yang JM, Tsukamoto H. Effect of ethanol and high-fat feeding on hepatic gamma-glutamylcysteine synthetase subunit expression in the rat. Hepatology. 1999;30:209–214. doi: 10.1002/hep.510300134. [DOI] [PubMed] [Google Scholar]
- Lu W, Gong D, Bar-Sagi D, Cole PA. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol Cell. 2001;8:759–769. doi: 10.1016/s1097-2765(01)00369-0. [DOI] [PubMed] [Google Scholar]
- Luo Y, Shoemaker AR, Liu X, Woods KW, Thomas SA, de JR, Han EK, Li T, Stoll VS, Powlas JA, Oleksijew A, Mitten MJ, Shi Y, Guan R, McGonigal TP, Klinghofer V, Johnson EF, Leverson JD, Bouska JJ, Mamo M, Smith RA, Gramling-Evans EE, Zinker BA, Mika AK, Nguyen PT, Oltersdorf T, Rosenberg SH, Li Q, Giranda VL. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol Cancer Ther. 2005;4:977–986. doi: 10.1158/1535-7163.MCT-05-0005. [DOI] [PubMed] [Google Scholar]
- Maegawa H, Hasegawa M, Sugai S, Obata T, Ugi S, Morino K, Egawa K, Fujita T, Sakamoto T, Nishio Y, Kojima H, Haneda M, Yasuda H, Kikkawa R, Kashiwagi A. Expression of a dominant negative SHP-2 in transgenic mice induces insulin resistance. J Biol Chem. 1999;274:30236–30243. doi: 10.1074/jbc.274.42.30236. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- Mangues R, Corral T, Kohl NE, Symmans WF, Lu S, Malumbres M, Gibbs JB, Oliff A, Pellicer A. Antitumor effect of a farnesyl protein transferase inhibitor in mammary and lymphoid tumors overexpressing N-ras in transgenic mice. Cancer Res. 1998;58:1253–1259. [PubMed] [Google Scholar]
- Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167:399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999;18:2137–2148. doi: 10.1093/emboj/18.8.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda K, Shima H, Watanabe M, Kikuchi K. MKP-7, a novel mitogen-activated protein kinase phosphatase, functions as a shuttle protein. J Biol Chem. 2001;276:39002–39011. doi: 10.1074/jbc.M104600200. [DOI] [PubMed] [Google Scholar]
- Matheny SA, Chen C, Kortum RL, Razidlo GL, Lewis RE, White MA. Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature. 2004;427(6971):256–260. doi: 10.1038/nature02237. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Imagawa M, Aoki Y. Epidermal growth factor regulation of glutathione S-transferase gene expression in the rat is mediated by class Pi glutathione S-transferase enhancer I. Biochem J. 2000;349:225–230. doi: 10.1042/0264-6021:3490225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci USA. 2005;102:11278–11283. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R, Kollmorgen G, White MF, Rhoads RE. Requirement of protein kinase C zeta for stimulation of protein synthesis by insulin. Mol Cell Biol. 1997;17:5184–5192. doi: 10.1128/mcb.17.9.5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalet X, Ekong R, Fougerousse F, Rousseaux S, Schurra C, Hornigold N, van J, Wolfe SM, Povey S, Beckmann JS, Bensimon A. Dynamic molecular combing: stretching the whole human genome for high-resolution studies. Science. 1997;277:1518–1523. doi: 10.1126/science.277.5331.1518. [DOI] [PubMed] [Google Scholar]
- Milarski KL, Saltiel AR. Expression of catalytically inactive Syp phosphatase in 3T3 cells blocks stimulation of mitogen-activated protein kinase by insulin. J Biol Chem. 1994;269:21239–21243. [PubMed] [Google Scholar]
- Monfar M, Lemon KP, Grammer TC, Cheatham L, Chung J, Vlahos CJ, Blenis J. Activation of pp70/85 S6 kinases in interleukin-2-responsive lymphoid cells is mediated by phosphatidylinositol 3-kinase and inhibited by cyclic AMP. Mol Cell Biol. 1995;15:326–337. doi: 10.1128/mcb.15.1.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monostory K, Hazai E, Vereczkey L. Inhibition of cytochrome P450 enzymes participating in p-nitrophenol hydroxylation by drugs known as CYP2E1 inhibitors. Chem Biol Interact. 2004;147:331–340. doi: 10.1016/j.cbi.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Morrison DK. KSR: a MAPK scaffold of the Ras pathway? J Cell Sci. 2001;114:1609–1612. doi: 10.1242/jcs.114.9.1609. [DOI] [PubMed] [Google Scholar]
- Morrison DK, Cutler RE. The complexity of Raf-1 regulation. Curr Opin Cell Biol. 1997;9:174–179. doi: 10.1016/s0955-0674(97)80060-9. [DOI] [PubMed] [Google Scholar]
- Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- Morrison MH, Hawksworth GM. Glucuronic acid conjugation by hepatic microsomal fractions isolated from streptozotocin-induced diabetic rats. Biochem Pharmacol. 1984;33:3833–3838. doi: 10.1016/0006-2952(84)90048-0. [DOI] [PubMed] [Google Scholar]
- Mukherjee B, Mukherjee JR, Chatterjee M. Lipid peroxidation, glutathione levels and changes in glutathione-related enzyme activities in streptozotocin-induced diabetic rats. Immunol Cell Biol. 1994;72:109–114. doi: 10.1038/icb.1994.17. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Sun XJ, White MF. The IRS-1 signaling system. Trends Biochem Sci. 1994;19:289–293. doi: 10.1016/0968-0004(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- Nichols A, Camps M, Gillieron C, Chabert C, Brunet A, Wilsbacher J, Cobb M, Pouyssegur J, Shaw JP, Arkinstall S. Substrate recognition domains within extracellular signal-regulated kinase mediate binding and catalytic activation of mitogen-activated protein kinase phosphatase-3. J Biol Chem. 2000;275:24613–24621. doi: 10.1074/jbc.M001515200. [DOI] [PubMed] [Google Scholar]
- Nojima H, Tokunaga C, Eguchi S, Oshiro N, Hidayat S, Yoshino K, Hara K, Tanaka N, Avruch J, Yonezawa K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278:15461–15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- Okuda Y, Kawai K, Yamashita K. Age-related change in ketone body metabolism: diminished glucagon effect on ketogenesis in adult rats. Endocrinology. 1987;120:2152–2157. doi: 10.1210/endo-120-5-2152. [DOI] [PubMed] [Google Scholar]
- Oshiro N, Yoshino K, Hidayat S, Tokunaga C, Hara K, Eguchi S, Avruch J, Yonezawa K. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells. 2004;9:359–366. doi: 10.1111/j.1356-9597.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- Ouwens DM, van der Zon GC, Maassen JA. Modulation of insulin-stimulated glycogen synthesis by Src Homology Phosphatase 2. Mol Cell Endocrinol. 2001;175:131–140. doi: 10.1016/s0303-7207(01)00389-6. [DOI] [PubMed] [Google Scholar]
- Oz HS, Im HJ, Chen TS, de Villiers WJ, McClain CJ. Glutathione-enhancing agents protect against steatohepatitis in a dietary model. J Biochem Mol Toxicol. 2006;20:39–47. doi: 10.1002/jbt.20109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Hill MM, Hess D, Brazil DP, Hofsteenge J, Hemmings BA. Identification of tyrosine phosphorylation sites on 3-phosphoinositide-dependent protein kinase-1 and their role in regulating kinase activity. J Biol Chem. 2001;276:37459–37471. doi: 10.1074/jbc.M105916200. [DOI] [PubMed] [Google Scholar]
- Perz M, Torlinska T. Insulin receptor--structural and functional characteristics. Med Sci Monit. 2001;7:169–177. [PubMed] [Google Scholar]
- Porter AC, Vaillancourt RR. Tyrosine kinase receptor-activated signal transduction pathways which lead to oncogenesis. Oncogene. 1998;17:1343–1352. doi: 10.1038/sj.onc.1202171. [DOI] [PubMed] [Google Scholar]
- Prasad N, Topping RS, Zhou D, Decker SJ. Oxidative stress and vanadate induce tyrosine phosphorylation of phosphoinositide-dependent kinase 1 (PDK1) Biochemistry. 2000;39:6929–6935. doi: 10.1021/bi000387i. [DOI] [PubMed] [Google Scholar]
- Pratt WB. The hsp90-based chaperone system: involvement in signal transduction from a variety of hormone and growth factor receptors. Proc Soc Exp Biol Med. 1998;217:420–434. doi: 10.3181/00379727-217-44252. [DOI] [PubMed] [Google Scholar]
- Previs SF, Withers DJ, Ren JM, White MF, Shulman GI. Contrasting effects of IRS-1 vs IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J Biol Chem. 2000;275:38990–38994. doi: 10.1074/jbc.M006490200. [DOI] [PubMed] [Google Scholar]
- Prudente S, Hribal ML, Flex E, Turchi F, Morini E, De Cosmo S, Bacci S, Tassi V, Cardellini M, Lauro R, Sesti G, Dallapiccola B, Trischitta V. The Functional Q84R Polymorphism of Mammalian Tribbles Homolog TRB3 Is Associated With Insulin Resistance and Related Cardiovascular Risk in Caucasians From Italy. Diabetes. 2005;54:2807–2811. doi: 10.2337/diabetes.54.9.2807. [DOI] [PubMed] [Google Scholar]
- Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279:707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- Ravichandran LV, Chen H, Li Y, Quon MJ. Phosphorylation of PTP1B at Ser(50) by Akt impairs its ability to dephosphorylate the insulin receptor. Mol Endocrinol. 2001;15:1768–1780. doi: 10.1210/mend.15.10.0711. [DOI] [PubMed] [Google Scholar]
- Rayapureddi JP, Kattamuri C, Steinmetz BD, Frankfort BJ, Ostrin EJ, Mardon G, Hegde RS. Eyes absent represents a class of protein tyrosine phosphatases. Nature. 2003;426:295–298. doi: 10.1038/nature02093. [DOI] [PubMed] [Google Scholar]
- Raza H, Ahmed I, Lakhani MS, Sharma AK, Pallot D, Monague W. Effect of bitter melon (Momordica charantia) fruit juice on the hepatic cytochrome P450-dependent monooxygenases and glutathione S-transferases in streptozotocin-induced diabetic rats. Biochem Pharmacol. 1996;52:1639–1642. doi: 10.1016/s0006-2952(96)00526-6. [DOI] [PubMed] [Google Scholar]
- Ricci GL, Fevery J. Treatment of rats with glucagon, vasointestinal peptide or secretin has a different effect on bilirubin and p-nitrophenol UDP-glucuronyltransferase. Biochem Pharmacol. 1988;37:3526–3528. doi: 10.1016/0006-2952(88)90707-1. [DOI] [PubMed] [Google Scholar]
- Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–480. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- Romanelli A, Martin KA, Toker A, Blenis J. p70S6 kinase is regulated by protein kinase Czeta and participates in a phosphoinositide 3-kinase-regulated signalling complex. Mol Cell Biol. 1999;19:2921–2928. doi: 10.1128/mcb.19.4.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738–1741. doi: 10.1126/science.286.5445.1738. [DOI] [PubMed] [Google Scholar]
- Ronis MJ, Huang J, Crouch J, Mercado C, Irby D, Valentine CR, Lumpkin CK, Ingelman-Sundberg M, Badger TM. Cytochrome P450 CYP 2E1 induction during chronic alcohol exposure occurs by a two-step mechanism associated with blood alcohol concentrations in rats. J Pharmacol Exp Ther. 1993;264:944–950. [PubMed] [Google Scholar]
- Rouer E, Mahu JL, Dansette P, Leroux JP. UDP-glucuronosyltransferase, epoxide hydrolase and glutathione S-transferase activities in the liver of diabetic mice. Biochim Biophys Acta. 1981;676:274–277. doi: 10.1016/0304-4165(81)90197-5. [DOI] [PubMed] [Google Scholar]
- Runge-Morris M, Vento C. Effects of streptozotocin-induced diabetes on rat liver sulfotransferase gene expression. Drug Metab Dispos. 1995;23:455–459. [PubMed] [Google Scholar]
- Saitoh M, Pullen N, Brennan P, Cantrell D, Dennis PB, Thomas G. Regulation of an activated S6 kinase 1 variant reveals a novel mammalian target of rapamycin phosphorylation site. J Biol Chem. 2002;277:20104–20112. doi: 10.1074/jbc.M201745200. [DOI] [PubMed] [Google Scholar]
- Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J Biol Chem. 2005;280:39505–39509. doi: 10.1074/jbc.M506096200. [DOI] [PubMed] [Google Scholar]
- Sasaoka T, Wada T, Ishihara H, Takata Y, Haruta T, Usui I, Ishiki M, Kobayashi M. Synergistic role of the phosphatidylinositol 3-kinase and mitogen-activated protein kinase cascade in the regulation of insulin receptor trafficking. Endocrinol. 1999;140:3826–3834. doi: 10.1210/endo.140.8.6904. [DOI] [PubMed] [Google Scholar]
- Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- Seely BL, Staubs PA, Reichart DR, Berhanu P, Milarski KL, Saltiel AR, Kusari J, Olefsky JM. Protein tyrosine phosphatase 1B interacts with the activated insulin receptor. Diabetes. 1996;45:1379–1385. doi: 10.2337/diab.45.10.1379. [DOI] [PubMed] [Google Scholar]
- Seino S, Seino M, Nishi S, Bell GI. Structure of the human insulin receptor gene and characterization of its promoter. Proc Natl Acad Sci U S A. 1989;86:114–118. doi: 10.1073/pnas.86.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro BH, Agrawal AK, Pampori NA. Gender differences in drug metabolism regulated by growth hormone. Int J Biochem Cell Biol. 1995;27:9–20. doi: 10.1016/1357-2725(94)00056-5. [DOI] [PubMed] [Google Scholar]
- Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: 'it ain't over 'til it's over'. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- Shimada M, Hashimoto E, Taniai M, Hasegawa K, Okuda H, Hayashi N, Takasaki D, Ludwig J. Hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J Hepatol. 2002;37:154–160. doi: 10.1016/s0168-8278(02)00099-5. [DOI] [PubMed] [Google Scholar]
- Shimojo N, Ishizaki T, Imaoka S, Funae Y, Fujii S, Okuda K. Changes in amounts of cytochrome P450 isozymes and levels of catalytic activities in hepatic and renal microsomes of rats with streptozocin-induced diabetes. Biochem Pharmacol. 1993;46:621–627. doi: 10.1016/0006-2952(93)90547-a. [DOI] [PubMed] [Google Scholar]
- Sim AT, Baldwin ML, Rostas JA, Holst J, Ludowyke RI. The role of serine/threonine protein phosphatases in exocytosis. Biochem J. 2003;373:641–659. doi: 10.1042/BJ20030484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley KSM, Feniuk W, Sellers LA, Humphrey PPA. The pivotal role of phosphoinositide-3 kinase in the human somatostatin sst4 receptor-mediated stimulation of p44/p42 mitogen-activated protein kinase and extracellular acidification. Biochem Biophys Res Commun. 1999;263:239–243. doi: 10.1006/bbrc.1999.1351. [DOI] [PubMed] [Google Scholar]
- Song BJ, Veech RL, Saenger P. Cytochrome P450IIE1 is elevated in lymphocytes from poorly controlled insulin-dependent diabetics. J Clin Endocrinol Metab. 1990;71:1036–1040. doi: 10.1210/jcem-71-4-1036. [DOI] [PubMed] [Google Scholar]
- Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow LG, McKern NM, Gorman JJ, Strike PM, Robinson CP, Bentley JD, Ward CW. The disulfide bonds in the C-terminal domains of the human insulin receptor ectodomain. J Biol Chem. 1997;272:29460–29467. doi: 10.1074/jbc.272.47.29460. [DOI] [PubMed] [Google Scholar]
- Srivastava PK, Waxman DJ. Sex-dependent expression and growth hormone regulation of class alpha and class mu glutathione S-transferase mRNAs in adult rat liver. Biochem J. 1993;294:159–165. doi: 10.1042/bj2940159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staffas L, Ellis EM, Hayes JD, Lundgren B, Depierre JW, Mankowitz L. Growth hormone- and testosterone-dependent regulation of glutathione transferase subunit A5 in rat liver. Biochem J. 1998;332:763–768. doi: 10.1042/bj3320763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standaert ML, Bandyopadhyay G, Kanoh Y, Sajan MP, Farese RV. Insulin and PIP3 activate PKC-zeta by mechanisms that are both dependent and independent of phosphorylation of activation loop (T410) and autophosphorylation (T560) sites. Biochemistry. 2001;40:249–255. doi: 10.1021/bi0018234. [DOI] [PubMed] [Google Scholar]
- Standaert ML, Bandyopadhyay G, Perez L, Price D, Galloway L, Poklepovic A, Sajan MP, Cenni V, Sirri A, Moscat J, Toker A, Farese RV. Insulin activates protein kinases C-zeta and C-lambda by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J Biol Chem. 1999;274:25308–25316. doi: 10.1074/jbc.274.36.25308. [DOI] [PubMed] [Google Scholar]
- Stoker AW. Protein tyrosine phosphatases and signalling. J Endocrinol. 2005;185:19–33. doi: 10.1677/joe.1.06069. [DOI] [PubMed] [Google Scholar]
- Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- Sundaram M, Han M. The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell. 1995;83:889–901. doi: 10.1016/0092-8674(95)90205-8. [DOI] [PubMed] [Google Scholar]
- Tao J, Malbon CC, Wang HY. Insulin stimulates tyrosine phosphorylation and inactivation of protein-tyrosine phosphatase 1B in vivo. J Biol Chem. 2001;276:29520–29525. doi: 10.1074/jbc.M103721200. [DOI] [PubMed] [Google Scholar]
- Taylor-Robinson SD, Foster GR, Arora S, Hargreaves S, Thomas HC. Increase in primary liver cancer in the UK, 1979–94. Lancet. 1997;350:1142–1143. doi: 10.1016/S0140-6736(05)63789-0. [DOI] [PubMed] [Google Scholar]
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- Therrien M, Chang HC, Solomon NM, Karim FD, Wassarman DA, Rubin GM. KSR, a novel protein kinase required for RAS signal transduction. Cell. 1995;83:879–888. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- Thomas H, Schladt L, Knehr M, Oesch F. Effect of diabetes and starvation on the activity of rat liver epoxide hydrolases, glutathione S-transferases and peroxisomal beta-oxidation. Biochem Pharmacol. 1989;38:4291–4297. doi: 10.1016/0006-2952(89)90528-5. [DOI] [PubMed] [Google Scholar]
- Thummel KE, Schenkman JB. Effects of testosterone and growth hormone treatment on hepatic microsomal P450 expression in the diabetic rat. Mol Pharmacol. 1990;37:119–129. [PubMed] [Google Scholar]
- Toker A, Newton AC. Akt/protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J Biol Chem. 2000;275:8271–8274. doi: 10.1074/jbc.275.12.8271. [DOI] [PubMed] [Google Scholar]
- Tootle TL, Silver SJ, Davies EL, Newman V, Latek RR, Mills IA, Selengut JD, Parlikar BE, Rebay I. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature. 2003;426(6964):299–302. doi: 10.1038/nature02097. [DOI] [PubMed] [Google Scholar]
- Truong NT, Moncion A, Barouki R, Beaune P, de Waziers I. Regulatory sequence responsible for insulin destabilization of cytochrome P450 2B1 (CYP2B1) mRNA. Biochem J. 2005;388:227–235. doi: 10.1042/BJ20041510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WC, Bhattacharyya N, Han LY, Hanover JA, Rechler MM. Insulin inhibition of transcription stimulated by the forkhead protein Foxo1 is not solely due to nuclear exclusion. Endocrinology. 2003;144:5615–5622. doi: 10.1210/en.2003-0481. [DOI] [PubMed] [Google Scholar]
- Tunon MJ, Gonzalez P, Garcia-Pardo LA, Gonzalez J. Hepatic transport of bilirubin in rats with streptozotocin-induced diabetes. J Hepatol. 1991;13:71–77. doi: 10.1016/0168-8278(91)90866-a. [DOI] [PubMed] [Google Scholar]
- Tzivion G, Luo Z, Avruch J. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature. 1998;394:88–92. doi: 10.1038/27938. [DOI] [PubMed] [Google Scholar]
- Uchida T, Myers MG, Jr, White MF. IRS-4 mediates protein kinase B signaling during insulin stimulation without promoting antiapoptosis. Mol Cell Biol. 2000;20:126–138. doi: 10.1128/mcb.20.1.126-138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- Van de Wiel JA, Fijneman PH, Teeuw KB, Van Ommen B, Noordhoek J, Bos RP. Influence of long-term ethanol treatment on rat liver biotransformation enzymes. Alcohol. 1993;10:397–402. doi: 10.1016/0741-8329(93)90027-l. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346:561–576. [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci. 2005;30:194–204. doi: 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Viitala P, Posti K, Lindfors A, Pelkonen O, Raunio H. cAMP mediated upregulation of CYP2A5 in mouse hepatocytes. Biochem Biophys Res Commun. 2001;280:761–767. doi: 10.1006/bbrc.2000.4195. [DOI] [PubMed] [Google Scholar]
- Visser TJ, van Haasteren GA, Linkels E, Kaptein E, van Toor H, de Greef WJ. Gender-specific changes in thyroid hormone-glucuronidating enzymes in rat liver during short-term fasting and long-term food restriction. Eur J Endocrinol. 1996;135:489–497. doi: 10.1530/eje.0.1350489. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- Wada T, Nakagawa K, Watanabe T, Nishitai G, Seo J, Kishimoto H, Kitagawa D, Sasaki T, Penninger JM, Nishina H, Katada T. Impaired synergistic activation of stress-activated protein kinase SAPK/JNK in mouse embryonic stem cells lacking SEK1/MKK4: different contribution of SEK2/MKK7 isoforms to the synergistic activation. J Biol Chem. 2001;276:30892–30897. doi: 10.1074/jbc.M011780200. [DOI] [PubMed] [Google Scholar]
- Wang L, Gout I, Proud CG. Cross-talk between the ERK and p70S6 kinase (S6K) signaling pathways. J Biol Chem. 2001;276:32670–32677. doi: 10.1074/jbc.M102776200. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci U S A. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- White MF, Kahn CR. The insulin signaling system. J Biol Chem. 1994;269:1–4. [PubMed] [Google Scholar]
- White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–E422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- Woodcroft KJ, Novak RF. Insulin differentially affects xenobiotic-enhanced, cytochrome P-450 (CYP)2E1, CYP2B, CYP3A, and CYP4A expression in primary cultured rat hepatocytes. J Pharmacol Exp Ther. 1999a;289:1121–1127. [PubMed] [Google Scholar]
- Woodcroft KJ, Novak RF. Insulin effects on CYP2E1, 2B, 3A, and 4A expression in primary cultured rat hepatocytes. Chem Biol Interact. 1997;107:75–91. doi: 10.1016/s0009-2797(97)00075-6. [DOI] [PubMed] [Google Scholar]
- Woodcroft KJ, Novak RF. The role of phosphatidylinositol 3-kinase, Src kinase, and protein kinase A signaling pathways in insulin and glucagon regulation of CYP2E1 expression. Biochem Biophys Res Commun. 1999b;266:304–307. doi: 10.1006/bbrc.1999.1817. [DOI] [PubMed] [Google Scholar]
- Woodcroft KJ, Hafner MS, Novak RF. Insulin signaling in the transcriptional and posttranscriptional regulation of CYP2E1 expression. Hepatology. 2002;35:263–273. doi: 10.1053/jhep.2002.30691. [DOI] [PubMed] [Google Scholar]
- Wu C, Sun M, Liu L, Zhou GW. The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene. 2003;306:1–12. doi: 10.1016/s0378-1119(03)00400-1. [DOI] [PubMed] [Google Scholar]
- Wu J, Dent P, Jelinek T, Wolfman A, Weber MJ, Sturgill TW. Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3′,5′-monophosphate. Science. 1993;262:1065–1069. doi: 10.1126/science.7694366. [DOI] [PubMed] [Google Scholar]
- Wu X, Hoffstedt J, Deeb W, Singh R, Sedkova N, Zilbering A, Zhu L, Park PK, Arner P, Goldstein BJ. Depot-specific variation in protein-tyrosine phosphatase activities in human omental and subcutaneous adipose tissue: a potential contribution to differential insulin sensitivity. J Clin Endocrinol Metab. 2001;86:5973–5980. doi: 10.1210/jcem.86.12.8109. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, Panayotou G. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol. 1996;16:1722–1733. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann MP, Zvelebil M, Laffargue M. Phosphoinositide 3-kinase signalling--which way to target? Trends Pharmacol Sci. 2003;24:366–376. doi: 10.1016/S0165-6147(03)00163-9. [DOI] [PubMed] [Google Scholar]
- Yamazoe Y, Murayama N, Shimada M, Imaoka S, Funae Y, Kato R. Suppression of hepatic levels of an ethanol-inducible P-450DM/j by growth hormone: relationship between the increased level of P-450DM/j and depletion of growth hormone in diabetes. Mol Pharmacol. 1989a;36:716–722. [PubMed] [Google Scholar]
- Yamazoe Y, Murayama N, Shimada M, Yamauchi K, Kato R. Cytochrome P450 in livers of diabetic rats: Regulation by growth hormone and insulin. Arch Biochem Biophys. 1989b;268:567–575. doi: 10.1016/0003-9861(89)90324-x. [DOI] [PubMed] [Google Scholar]
- Yeung K, Janosch P, McFerran B, Rose DW, Mischak H, Sedivy JM, Kolch W. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol Cell Biol. 2000;20:3079–3085. doi: 10.1128/mcb.20.9.3079-3085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–177. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- Yokota H, Kunimasa Y, Shimoyama Y, Kobayashi T, Matsumoto J, Yuasa A. Effects on extrahepatic UDP-glucuronosyltransferases in hypophysectomized rat. J Biochem (Tokyo) 2002;132:265–270. doi: 10.1093/oxfordjournals.jbchem.a003220. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Hirokawa J, Tagami S, Kawakami Y, Urata Y, Kondo T. Weakened cellular scavenging activity against oxidative stress in diabetes mellitus: regulation of glutathione synthesis and efflux. Diabetologia. 1995;38:201–210. doi: 10.1007/BF00400095. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Gramlich T, Matteoni CA, Boparai N, McCullough AJ. Nonalcoholic fatty liver disease in patients with type 2 diabetes. Clin. Gastroenterol. Hepatol. 2004;2:262–265. doi: 10.1016/s1542-3565(04)00014-x. [DOI] [PubMed] [Google Scholar]
- Zen Y, Katayanagi K, Tsuneyama K, Harada K, Araki I, Nakanuma Y. Hepatocellular carcinoma arising in non-alcoholic steatohepatitis. Pathol Int. 2001;51:127–131. doi: 10.1046/j.1440-1827.2001.01174.x. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gan L, Pan H, Guo S, He X, Olson ST, Mesecar A, Adam S, Unterman TG. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J Biol Chem. 2002a;277:45276–45284. doi: 10.1074/jbc.M208063200. [DOI] [PubMed] [Google Scholar]
- Zhang ZY, Zhou B, Xie L. Modulation of protein kinase signaling by protein phosphatases and inhibitors. Pharmacol Ther. 2002b;93:307–317. doi: 10.1016/s0163-7258(02)00199-7. [DOI] [PubMed] [Google Scholar]