

Abstract
This study tested the hypothesis that genetic variants of phase II detoxification enzymes and DNA repair proteins affect individual response to DNA damage from alkylating agents. In 171 healthy individuals, an immunoslot blot assay was used to measure O6-ethylguanosine (O6-EtGua) adduct levels in peripheral blood lymphocytes treated with N-ethyl-N-nitrosourea (ENU) in vitro. The genotypes of GSTM1, GSTT1, GSTP1 I105V and A114V, MGMT L84F and I143V, XPD D312N and K751Q, and XRCC3 T241M were determined. Demographic and exposure information was collected by in-person interview. Student’s t test, analysis of (co)variance, and multiple linear regression models were used in statistical analyses. The mean and median (range) O6-EtGua levels were 94.6 and 84.8 (3.2–508.1) fmol/g DNA, respectively. The adduct level was significantly lower in people who smoked ≥ 25 years than that in never-smokers (square-root transformed mean values 8.20 versus 9.37, P = 0.03). Multiple linear regression models revealed that GSTT1 (β = −2.36, P = 0.009) polymorphism was a significant predictor of the level of adducts in 82 never-smokers, whereas the number of years smoked (β = −0.08, P = 0.005) and XRCC3 T241M (β = 2.22, P = 0.007) in 89 ever-smokers. The association between GSTP1 I105V, MGMT I143V, and XPD D312N with the level of adducts was not conclusive. Each polymorphism could explain 2% to 10% of the variation of the adduct level. These observations suggest that GSTT1 null and XRCC3 T241M polymorphism may have some functional significance in modulating the level of ENU-induced DNA damage and these effects are smoking-dependent. Results from this exploratory study need to be confirmed in other experimental systems.
Keywords: single nucleotide polymorphism (SNP), phase II xenobiotic-metabolizing enzyme, DNA Repair protein, N-ethyl-N-nitrosourea (ENU), O6-ethylguanosine (O6-EtGua), human lymphocytes
1. Introduction
Exogenous and endogenous exposure to N-nitroso compounds is extensive in human beings [1]. N-nitroso compounds and their metabolites can react with cellular DNA to form a broad spectrum of alkylating DNA damage, which can be a critical event in the induction of cancer. O6-alkylguanine is a major premutagenic lesion in cells exposed to N-nitroso compounds and constitutes a significant genotoxic burden for the general population [2]. In view of the constant alkylation pressure imposed on cells, the protective mechanisms against alkylating agents deserve special attention. A growing number of studies have shown that subtle differences in carcinogen metabolism and DNA repair capacity (DRC) conferred by the inheritance of polymorphisms may predispose individuals to cancer [3]. However, molecular epidemiological findings on gene polymorphisms and cancer risk have often been inconsistent. Lack of knowledge on the functional significance of the polymorphisms makes the data interpretation more difficult. Understanding the functional significance of the polymorphisms may help to better define the study population and the data interpretation in molecular epidemiology study.
In vitro genotoxicity studies with peripheral blood lymphocytes offer an experimental tool to investigate genotypic and phenotypic associations because they allow assessment of the toxicological response to xenobiotics under controlled experimental conditions [4]. By using different challenging agents and endpoints in a limited cell system, such assays can be applied to examine genotype and exposure interactions and to predict individual response to carcinogens. We have previously established an assay for measuring benzo(a)pyrene diol epoxide (BPDE)-induced DNA adducts in cultured peripheral lymphocytes as a marker for DRC [5] and hence a predictor for the risk of developing smoking-related human cancers [6]. In the current study, the N-nitroso carcinogen N-ethyl-N-nitrosourea (ENU) was used as the challenging agent and O6-ethylguanosine (O6-EtGua) level was evaluated as the endpoint biomarker. ENU is a potent monofunctional ethylating agent that is mutagenic in a wide variety of test systems and carcinogenic in various mammalian organs [7]. O6-EtGua is one of a dozen ethylation products formed in DNA upon exposure to ENU [8]. However, factors that influencing the formation and repair of O6-EtGua adduct have not been fully elucidated.
The aim of the present study was to examine the association between several polymorphisms in genes encoding phase II xenobiotic-metabolizing enzymes and DNA repair proteins and the level of in vitro-induced O6-EtGua in cultured human lymphocytes. Because ENU is a direct alkylating agent, we focused on genes that are involved in the phase II detoxification (GSTM1, GSTT1, and GSTP1) and DNA repair pathways (MGMT, XPD and XRCC3). Information on the selected single nucleotide polymorphisms (SNPs) is summarized in Table 1. Homozygous deletion of the GSTM1 and GSTT1 genes (null genotype) has been associated with the loss or lack of enzyme activity and increased vulnerability to cytogenetic damage [9]. The GSTP1 Ile105Val and Ala114Val variant alleles have been associated with altered enzyme activities towards different drug or carcinogens [10,11]. The selected DNA repair SNPs all reside in the coding regions of the genes and result in amino acid changes. To our knowledge, this study is the first to examine the association between these genotypes and in vitro-induced DNA adduct levels in human lymphocytes. We found that genetic polymorphisms of GSTT1 contribute to a subtle proportion of the variance in adduct levels in cultured lymphocytes of never-smokers, whereas the number of years smoked and XRCC3 T241M polymorphism in ever-smokers.
Table 1.
Information on the polymorphisms under investigation
| Gene | GeneID | Chromosome | Polymorphism | Amino Acid Change | Reference SNP ID | Genotyping assay |
|---|---|---|---|---|---|---|
| GSTM1 | 2944 | 1p13.3 | Deletion | Multiplex PCRa | ||
| GSTT1 | 2952 | 22q11.23 | Deletion | Multiplex PCRa | ||
| GSTP1 | 2950 | 11q13.2 | Exon 5–24 A>G | Codon 105 Ile (I) → Val(V) | 947894 | MassCodeb |
| GSTP1 | 2950 | 11q13.2 | Exon 6+5 C>T | Codon 114 Ala (A) → Val (V) | 1799811 | MassCodeb |
| MGMT | 4255 | 10q26 | Exon 2–25 C>T | Codon 84 Leu (L) → Phe (F) | 12917 | MassCodeb |
| MGMT | 4255 | 10q26 | Exon 5+13 A>G | Codon 143 Ile (I) → Val (V) | 2308321 | PCR-SSCPa |
| XPD | 2608 | 19q13.3 | Exon10–16 G>A | Codon 312 Asp (D) → Asn (N) | 1799793 | MassCodeb |
| XPD | 2608 | 19q13.3 | Exon23+61 A>C | Codon 751 Lys (K) → Gln (Q) | 13181 | PCR-RFLPa |
| XRCC3 | 7517 | 14q32.3 | Exon 8–53 C>T | Codon 241 Thr (T) → Met (M) | 861539 | MassCodeb |
From the SNP500Cancer database of the U.S. National Cancer Institute. PCR, polymerase chain reaction; SSCP, single-strand conformation polymorphism; RFLP, restriction fragment length polymorphism. GST, glutathione-S-transferase; MGMT, O6-methylguanine-DNA-methyltransferase; XPD, xeroderma pigmentosum group D; XRCC3, X-ray repair cross-complementing group 3.
Methods are described in reference 12 (for the GSTM1 and GSTT1 polymorphisms), reference 13 (for the MGMT I143V polymorphism) and reference 14 (for the XPD K751Q polymorphism).
Masscode™ technology (BioServe Biotechnologies, Ltd., Laurel, MD).
2. Materials and methods
2.1. Study subjects
This study was conducted with healthy individuals who were enrolled in a hospital-based case-control study of pancreatic cancer at The University of Texas M. D. Anderson Cancer Center between October 2000 and November 2003. They were companions (non-blood relatives or friends) of patients who were diagnosed with various cancers. Individuals were considered eligible if they had no previous history of cancer (except for non-melanoma skin cancer) and donated a blood sample of at least 20 ml. There were no age and sex restrictions, and all study subjects were non-Hispanic whites. Each study participant provided written informed consent to an in-person interview and donation of a blood sample. The study was approved by the Institutional Review Board of M. D. Anderson Cancer Center.
During the study period, a total of 184 individuals met the eligibility criteria and adduct levels were measured. Nine subjects were excluded because of missing exposure information; four other subjects were excluded because of large variations (> 50%) in repeated measurements. Therefore, the final results were based on data from 171 study subjects consisting of 89 ever-smokers and 82 never-smokers. The distributions of age and sex were comparable between individuals who were included or not included in the study. None of the study subjects reported a history of occupational exposure to alkylating agents.
2.2. Data collection
A trained study coordinator administered a structured risk factor questionnaire to collect information on demographics, occupation, smoking and alcohol consumption. The definitions of exposure parameters (never, ever, former, and current smokers) have been published elsewhere [12]. Briefly, subjects were classified as ever-smokers if they had smoked more than 100 cigarettes in their lifetime. Former smokers were defined as those who had quit smoking for more than 1 year. The information on duration (the number of years smoked) and intensity of smoking (the number of cigarettes smoked per day) was also collected. Information on alcohol consumption was collected to evaluate its potential confounding effect on the association between polymorphism and adduct levels. The subject was asked whether he/she had consumed at least four alcoholic beverages a month, for six months or more. The answer of ‘no’, ‘yes, but quit’, or ‘yes’ to this question distinguishes the “never”, “former” and “current” drinker, respectively.
2.3. Isolation of lymphocytes and ENU treatment in cell culture
Blood samples were collected in heparinized Vacutainers (BD Biosciences, Franklin Lakes, NJ), and each specimen was labeled with a unique study identification number. Peripheral blood lymphocytes were isolated on Ficoll-Hypaque (Amersham Pharmacia Biotech, Piscataway, NJ) density gradients. All cell cultures were set up within 2 hours after blood collection. Cell concentrations were adjusted to 1–10 × 107/ml with 1 ml serum-free RPMI 1640 medium (GIBCO Invitrogen Corporation, Carlsbad, CA) supplemented with 2 mM L-glutamine, 100 unit/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA). ENU (Sigma, St. Louis, MO, CAS# 759-73-9, lot# 79H1011) was dissolved in H2O-free dimethyl sulfoxide (200 mM, stock solution) and stored at −80°C in the dark. That ENU stock solution was then added to 1 ml cell culture, with a final concentration of 0.64 mM (75 μg/ml). After incubation at 37°C with 5% CO2 for 1 hour, cells were harvested by centrifugation. Cell pellets were washed with serum-free RPMI 1640 medium twice to remove ENU and then resuspended in 400 μl Tris-EDTA buffer (pH = 8.0) and immediately stored at −80°C until DNA isolation.
2.4. Immunoslot blots, standard curves, and dose response
DNA was extracted by using a FlexiGene DNA kit according to the manufacturer’s instructions (Qiagen, Valencia, CA). DNA was dissolved in 10 mM Tris-HCl (pH = 8.0), quantified by UV spectrophotometry (Amersham Pharmacia Biotech), and stored at −80°C until assay. Semi-quantification of ENU-induced O6-EtGua was carried out by an immunoslot blot method [13]. Briefly, 5 μg DNA was sonicated and then denatured at 100°C for 10 minutes. The heat-denatured DNA was quickly chilled on ice and immediately slot-blotted onto Hybond-N+ Nylon membranes (Amersham Pharmacia Biotech) by using a Convertible Filtration Manifold System (GibcoBRL, Carlsbad, CA). All 48 slots were rinsed with 200 μl 1 M ammonium acetate and the membranes were baked for 2 hours at 80°C. After single-stranded DNA was immobilized onto the nitrocellular membranes, the membranes were blocked with 5% milk −1×TBS −0.05% Tween 20 and then hybridized with the (O6-EtGua)–specific mouse monoclonal antibody EM-2-3 (1:5000 diluted, kindly provided by Dr. Menfred F. Rajewsky’s laboratory in University of Essen, Germany) for 150 min at room temperature with shaking [14]. The membrane was then hybridized with the secondary antibody goat anti-mouse horseradish peroxidase–labeled IgG (1:1000 diluted) (Chemicon, Temecula, CA) by shaking for 1 hour at room temperature. Enzymatic activity was visualized by chemiluminescence reaction with an ECLTM western blotting detection kit (Amersham Pharmacia Biotech). The intensity of the bands was analyzed with Kodak 1D Image Analysis software (Kodak, New Haven, CT).
A standard for DNA measurement was generated by the in vitro reaction of calf thymus DNA with 3H-labeled N-methyl-N-nitrosourea (NCI Chemical Carcinogen Repository), and the DNA adduct levels were determined by scintillation counting. The labeled DNA was then diluted with naïve calf thymus DNA to a final concentration of 25, 50, 100, 250, 500, 1000, or 2000 fmol adduct per μg DNA. Standard curves were established from this series of DNA concentrations by using the immunoslot blot assay (Figure 1, A). The intensity of the band in each slot was compared with the standard curve to semiquantify the adduct levels. The arithmetic mean was computed from the parallel samples to represent the actual value of each sample.
Figure 1.
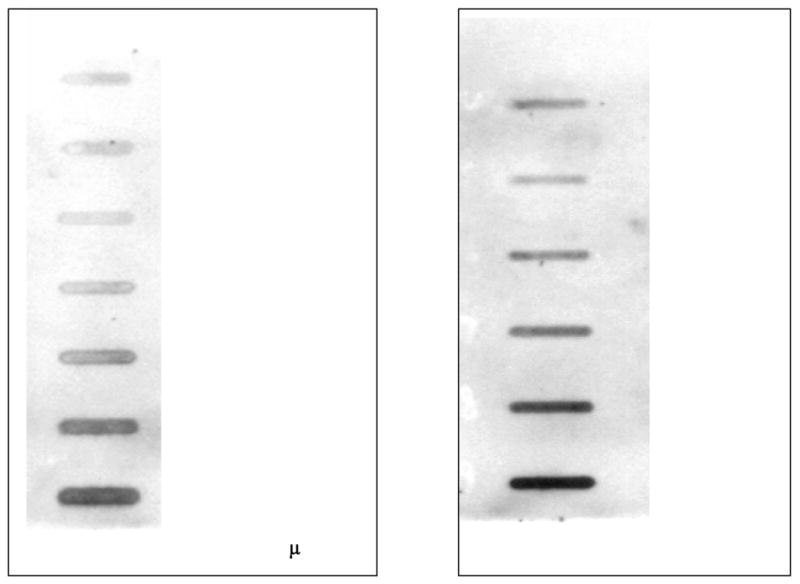
A. Immunoslot blot assay result for the standard curve. A standard for DNA measurement was generated by the in vitro reaction of calf thymus DNA with 3H-labeled N-methyl N-nitrosourea. The DNA adduct levels were determined by scintillation counting. The labeled DNA was then diluted with naïve calf thymus DNA to a final concentration of 25, 50, 100, 250, 500, 1000, or 2000 fmol adduct per μg DNA.
B. Immunoclot blot assay result for a dose-response experiment. The lymphocytes from 3 healthy volunteers were treated with ENU at doses of 0, 0.21, 0.43, 0.64, 0.85, and 1.60 mM.
At the beginning of the study, a dose-response experiment was performed with lymphocytes from 3 healthy volunteers at ENU doses of 0, 0.21, 0.43, 0.64, 0.85, and 1.60 mM; a dose-response relationship was observed (Figure 1, B). The 0.64 mM concentration was selected as the challenging dose because cellular viability was optimal (> 80%, tested by exclusion of 0.4% trypan blue) and the adduct levels were within the linear range of the standard curve. One hour was used as the treatment time because we were interested in the rapid mode of O6-EtGua repair.
2.5. Genotyping
Genomic DNA was extracted by using a FlexiGene DNA kit and the aliquots stored at 4°C. The genotyping methods are listed in Table 1 [15–17]. No data were missing for the GSTM1, GSTT1, MGMT I143V, and XPD K751Q polymorphisms. The rate of missing data varied from 4.3% to 7.4% for the remaining polymorphisms.
2.6. Quality control measures
The same batch of ENU and monoclonal antibody was used in all experiments. The reagents were aliquoted to a small volume and stored at −80°C. ENU was stored in light-protected vials, and the open stock vial was refreshed every 2 weeks and the pH value maintained below 5.0. A positive control (an adduct standard of 500 fmol/μg DNA) and a negative control (untreated calf thymus DNA) were included in every immunoslot blot assay. The same person conducted all the procedures, from the ENU treatment through the final quantification of adducts, according to a standard protocol. Samples were analyzed in parallel in the same experiment, and those with variation greater than 50% in duplicate experiments were eliminated from the final analysis. The two values from repeated measurements were significantly correlated (Pearson’s correlation coefficient ρ = 0.79, P < 0.001).
In genotyping assays, 10% of the specimens were randomly selected for duplication. For the polymerase chain reaction–restriction fragment length polymorphism assays, two investigators independently read the genotyping results. In the rare cases (0.03%) in which an inconsistency was noted, genotyping was repeated from the polymerase chain reaction step. For the MassCode assays, positive allele controls were run in every experiment. Only conservative calls were taken and any ambiguous call (owing to poor DNA quality and quantity) was excluded from the final report.
2.7. Statistical analyses
Data analyses were performed with STATA 7.0 (Stata Corporation, College Station, TX) and SPSS (SPSS Inc. Chicago, IL) software. All statistical tests were two-sided. P values ≤ 0.05 for any test indicated statistical significance. The distribution of original values of O6-EtGua was skewed to the left and was normalized by square-root transformation to ensure the normality assumption for parametric testing and to stabilize the group variance. The allele frequency for each polymorphism was estimated by gene counting. The Hardy-Weinberg equilibrium was tested by using the goodness-of-fit χ2 test.
Independent one-sample Student’s t test with equal variance or one way analysis of variance was used to compare induced adducts level according to demographic factors (age and sex) and exposure factors (smoking and alcohol consumption). An analysis of covariance (ANCOVA) was used to compare the difference in mean adduct levels between genotypes adjusted for age, sex, years of smoking, number of cigarettes smoked per day, and alcohol consumption (never, quit, and ever). Variable selection for the multiple linear regression models used the criterion of P value of less than 0.20 in the univariate analysis by smoking status. Generally, the heterozygous and homozygous variant genotype was combined when both showed the same effect on the level of adducts. In the multiple linear regression analyses, the percentage of the variance of dependent variable (adduct level) explained by each polymorphism and other variable was evaluated by subtracting the r2 value for the full model from the r2 for a model that excludes the test variable [18]. The full models contained covariates age (in years), sex (0, women; 1, men), alcohol consumption (0, never, 1, quit; 2, current), and all polymorphism terms selected from the univariate analyses. Separate models were run for the never-smokers and the ever-smokers. Multiplicity adjusted P-value was calculated [19]. Lastly, backward elimination regression analysis with exclusion criteria of P ≥ 0.10 was used to examine the contributions of the polymorphism to the prediction of the O6-EtGua level.
3. Results
3.1. DNA adduct phenotype according to study subject characteristics
O6-EtGua levels ranged from 3.2 to 508.1 fmol/μg DNA, with a mean of 94.6 and a median of 84.8 fmol/μg DNA. The distribution of O6-EtGua values was skewed to the left (P < 0.001 for joint skewness/Kurtosis test) and was approximately normalized by square-root transformation (P = 0.05 for joint skewness/Kurtosis test). The transformed values were expressed as mean ± standard error (SE). Table 2 shows mean adduct levels according to selected characteristics of the 171 study subjects. The O6-EtGua level did not differ significantly according to demographic variables. People who smoked more than 25 years had a significantly lower adduct level than did the never-smokers (8.20 versus 9.37, P = 0.03).
Table 2.
Characteristics of study subjects and levels of ENU-induced O6-EtGua
| Study Subjects | O6-EtGua levela | ||||
|---|---|---|---|---|---|
| N | Mean | S.E.b | |||
| Selected Variables | 171 | % | 9.21 | 0.25 | P valuec |
| Age (years) | |||||
| ≤ 50 | 37 | 21.6 | 9.32 | 0.44 | |
| 50–60 | 43 | 25.1 | 8.82 | 0.54 | |
| 61–70 | 61 | 35.7 | 9.54 | 0.46 | |
| > 70 | 30 | 17.5 | 8.76 | 0.45 | 0.60 |
| Sex | |||||
| Men | 87 | 50.9 | 8.84 | 0.35 | |
| Women | 84 | 49.1 | 9.52 | 0.35 | 0.17 |
| Smoking | |||||
| Never-smoker | 82 | 47.9 | 9.37 | 0.32 | |
| Ever-smoker | 89 | 52.0 | 9.00 | 0.37 | 0.62 |
| Former | 63 | 36.8 | 9.03 | 0.49 | 0.53d |
| Current | 26 | 15.2 | 8.92 | 0.46 | 0.48d |
| No. years smokede | |||||
| < 25 | 43 | 25.1 | 9.85 | 0.58 | 0.43d |
| ≥ 25 | 46 | 26.9 | 8.20 | 0.45 | 0.03d |
| No. cigarettes/daye | |||||
| < 20 | 27 | 15.8 | 9.36 | 0.90 | 0.99d |
| ≥ 20 | 62 | 36.2 | 8.84 | 0.37 | 0.28d |
| Alcohol | |||||
| Never | 76 | 44.5 | 8.91 | 0.40 | |
| Quit | 24 | 14.0 | 10.21 | 0.64 | |
| Current | 71 | 41.5 | 9.09 | 0.35 | 0.22 |
Square-root transformed value of fmol/μg DNA.
Standard error.
P value for the two-sample independent Student’s t test or one-way analysis of variance.
Compared with adduct levels of never-smokers.
Cutoff point was the median of the continuous variable.
3.2. Allele and genotype frequency and ANCOVA analysis of O6-EtGua level by genotypes
Table 3 describes the genotype or allelic frequency for each polymorphism. The frequencies of GSTM1, GSTT1, and GSTP1 A114V, MGMT L84F, and XPD D312N and K751Q were within the range of those reported by other studies [3,20,21]. The variant allele frequencies of the GSTP1 I105V and the MGMT I143V were 0.38 and 0.16, respectively, which were higher than those reported in other Caucasian populations [22,23]. However, this would not affect our study purpose in examining genotype–adduct associations. None of the genotype distributions differed significantly from those expected under Hardy-Weinberg equilibrium (data not shown). Table 3 presents the adjusted mean levels of O6-EtGua and shows the level of O6-EtGua did not significantly differ by any polymorphism. The GSTT1 null genotype carriers had borderline significantly lower adduct levels than did the wild-type carrier (8.20 versus 9.42, P = 0.05).
Table 3.
ANCOVA of single polymorphism and O6-EtGua adduct levels in 171 study subjects
| Genotype | N (%)a | Adduct levelb (adjusted mean ± SE) | % Relative increase | P valuec |
|---|---|---|---|---|
| GSTM1 | ||||
| Wild-type | 88 (51.5) | 9.13 ± 0.35 | Ref. | |
| Null | 83 (48.5) | 9.22 ± 0.36 | +0.98 | 0.88 |
| GSTT1 | ||||
| Wild-type | 137 (80.1) | 9.42 ± 0.28 | Ref. | |
| Null | 34 (19.9) | 8.20 ± 0.55 | −12.9 | 0.05 |
| GSTP1 I105V | ||||
| I/I | 59 (37.1) | 9.59 ± 0.43 | Ref. | |
| I/V | 78 (49.1) | 9.12 ± 0.37 | − 4.9 | |
| V/V | 22 (13.8) | 8.35 ± 0.71 | −12.9 | 0.32 |
| I/V + V/V | 100 (62.9) | 8.95 ± 0.34 | −6.7 | 0.24 |
| V allele frequency | 0.383 | |||
| GSTP1 A114V | ||||
| A/A | 133 (81.6) | 9.22 ± 0.28 | Ref. | |
| A/V | 30 (18.4) | 9.26 ± 0.59 | +0.43 | 0.95 |
| V/V | 0 | |||
| V allele frequency | 0.092 | |||
| MGMT I143V | ||||
| I/I | 119 (69.6) | 8.97 ± 0.30 | Ref. | . |
| I/V | 50 (29.2) | 9.70 ± 0.46 | +8.1 | |
| V/V | 2 (1.2) | 8.57 ± 0.31 | −4.4 | 0.40 |
| I/V + V/V | 52 (30.4) | 9.65 ± 0.45 | +7.6 | 0.21 |
| V allele frequency | 0.162 | |||
| MGMT L84F | ||||
| L/L | 131 (79.4) | 9.38 ± 0.28 | Ref. | |
| L/F | 34 (20.6) | 8.65 ± 0.56 | −7.8 | 0.25 |
| F/F | 0 | |||
| F allele frequency | 0.103 | |||
| XPD D312N | ||||
| D/D | 72 (45.3) | 9.05 ± 0.38 | Ref. | |
| D/N | 66 (41.5) | 9.21 ± 0.40 | +1.8 | |
| N/N | 21 (13.2) | 8.62 ± 0.71 | −4.7 | 0.76 |
| D/N + N/N | 87 (54.7) | 9.07 ± 0.35 | +0.2 | 0.95 |
| N allele frequency | 0.347 | |||
| XPD K751Q | ||||
| K/K | 70 (40.9) | 8.93 ± 0.39 | Ref. | |
| K/Q | 91 (53.2) | 9.50 ± 0.34 | +6.9 | |
| Q/Q | 10 (5.9) | 7.94 ± 1.02 | −11.1 | 0.26 |
| K/Q + Q/Q | 101 (59.1) | 9.34 ± 0.32 | +4.6 | 0.42 |
| Q allele frequency | 0.325 | |||
| XRCC3 T241M | ||||
| T/T | 66 (38.6) | 9.12 ± 0.40 | Ref. | |
| T/M | 75 (43.9) | 9.13 ± 0.38 | +0.1 | |
| M/M | 30 (17.5) | 9.39 ± 0.60 | +2.7 | 0.86 |
| T/M + M/M | 105 (61.4) | 9.21 ± 0.32 | +1.0 | 0.87 |
| M allele frequency | 0.394 | |||
Numbers do not add up to expected totals because of missing genotyping data.
Adduct levels were estimated by ANCOVA for all study subjects, adjusted for age (continuous), sex, smoking status (never-smokers and ever-smokers), and alcohol consumption (never, quit, current).
P value for ANCOVA F-test.
3.3. Univariate analyses of O6-EtGua level stratified by smoking status
Since the univariate analysis showed an association between O6-EtGua level and years of smoking, we further investigated the association between each polymorphism and O6-EtGua level according to smoking status (Figure 2). Among never-smokers, the GSTT1 null genotype carriers had significantly lower adduct levels than did the wild-type carriers (7.61 versus 9.76, P = 0.009) (Figure 2, A). The GSTP1 105V allele was associated with a lower adduct level compared with the GSTP1 I105I genotype (8.80 versus 10.29, P = 0.03) (Figure 2, B). Among ever-smokers, the MGMT 143V allele carriers had higher adduct level than did the I143I genotype carriers (9.80 versus 8.60, P = 0.13) (Figure 2, C). Compared with the adduct level of XRCC3 T241T wild-type carriers, the adduct level of the 241M allele carriers tended to be higher among ever-smokers (8.29 versus 9.50, P = 0.11), but lower among never-smokers (10.1 versus 8.97, P = 0.09) (Figure 2, D). The adduct level essentially did not differ by other polymorphisms (P for t test ≥ 0.20) (data not shown).
Figure 2.
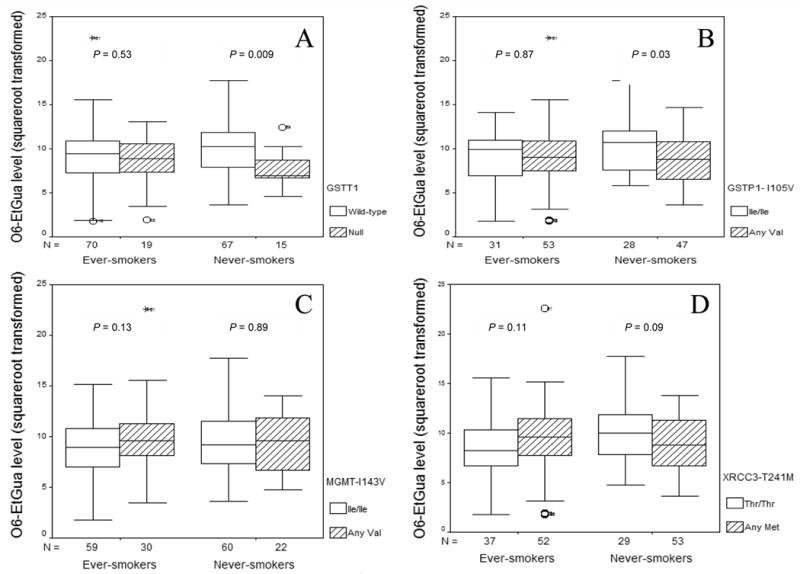
Box-plot of adduct levels by GSTT1 (panel A), GSTP1 I105V (panel B), MGMT I143V (panel C), and XRCC3 T241M (panel D) genotype in ever-smokers and never-smokers. P value was from Student’s t test. The solid line indicates median; the box extents mark the 25th and 75th percentiles of the observed values, and the capped bars indicate the 10th and 90th percentiles. Symbols indicate the outliers of adduct level.
3.4. Multiple linear regression analyses stratified by smoking status
Table 4 summarizes the relationship between O6-EtGua level and polymorphisms estimated by multiple linear regression models. Among never-smokers, a lower adduct level was related to the presence of the GSTT1 null genotype (β = −2.36, P = 0.009), the GSTP1 105V allele (β = −1.77, P = 0.02), the XPD 312N allele (β = −1.46, P = 0.05), and the XRCC3 241M allele (β = −1.75, P = 0.02). Among ever-smokers, a higher adduct level was related to the presence of the MGMT 143V allele (β = 1.64, P = 0.05) and the XRCC3 241M allele (β = 2.22, P = 0.007). These polymorphisms each can explain more than 2% but less than 10% of the variance of the dependent variable among never- or ever-smokers. These five polymorphisms, together with other covariates, can explain 27.3% and 25.3% of the variance of the dependent variable for never-smokers and ever-smokers, respectively. The number of years smoked was a significant determinant for the adduct level among ever-smokers (β = −0.08, P = 0.005). Assuming there were five independent tests in each smoking stratum, the Bonferroni adjusted P value remained ≤ 0.05 for the term GSTT1 in never smokers, the number of years smoked and XRCC3 T241M in ever smokers, with the adjusted P value of 0.045, 0.025, and 0.035, respectively. Notable was the differential effect of the XRCC3 T241M polymorphism on adduct levels for never-smokers and ever-smokers (Table 4).
Table 4.
Multiple linear regression analysis of predictor for O6-EtGua levela
| Never-smokers | Ever-smokers | |||||||
|---|---|---|---|---|---|---|---|---|
| Multivariate | r2 variance explainedf | Multivariate | ||||||
| Covariates | βc | Pd | Pe | βc | Pd | Pe | r2 variance explainedf | |
| GSTT1 (0, wild-type; 1, null) | −2.36 | 0.009 | 0.045 | 8.7% | −1.26 | 0.20 | 1.8% | |
| GSTP1 I105V (0, I/I; 1, any V) | −1.77 | 0.02 | 8.9% | −0.50 | 0.53 | 2.1% | ||
| MGMT I143V (0, I/I; 1, any V) | −0.69 | 0.37 | 1.0% | 1.64 | 0.05 | 4.3% | ||
| XPD D312N (0, D/D; 1, any N) | −1.46 | 0.05 | 2.9% | 0.91 | 0.25 | 12.3% | ||
| XRCC3 T241M (0, T/T; 1, any M) | −1.75 | 0.02 | 7.0% | 2.22 | 0.007 | 0.035 | 8.5% | |
| No. years of smoked | NAb | NAb | −0.08 | 0.005 | 0.025 | 9.4% | ||
| Age | −0.03 | 0.24 | 1.7% | −0.04 | 0.35 | 0.9% | ||
| Sex (0, women; 1, men) | 0.68 | 0.34 | 1.1% | 0.09 | 0.91 | 0.0% | ||
| Alcohol (0, never, 1, quit, 2 current) | 0.22 | 0.57 | 0.4% | −0.32 | 0.47 | 0.6% | ||
| r2 | 27.3% g | 25.3%g | ||||||
The square-root transformed value is the dependent variable. Age, sex, alcohol consumption are covariates.
For never-smoker stratum, the regression model did not include covariate ‘No. years of smoked’.
β coefficient: the unit change of dependent variable caused by 1 unit change of variable. For genotype covariates, β is the difference of the mean of each genotype coded as 0 or 1.
P value for the term in the multiple linear regression.
Bonferroni multiplicity adjusted P value = (listed P value x 5), assuming 5 independent hypothesis testing in each stratum.
Bolded indicates multiplicity adjust P value ≤ 0.05.
The difference of the r2 of the full model and the r2 of the reduced model without that variable.
r2, percentage of variation of the dependent variable that the model can explain.
Backward elimination regression with the exclusion criteria of P ≥ 0.10 was used to screen the importance of the explanatory variables. The GSTT1, GSTP1 I105V, and XRCC3 T241M polymorphism turned out to be crucial in determining the adduct level among never-smokers. The number of years smoked, the XRCC3 T241M and XPD D312N polymorphisms turned out to be the important determinants for the adduct level among ever-smokers (data not shown).
4. Discussion
By using an in vitro experiment system in cultured lymphocytes of non-cancer healthy study subjects, we have observed a significant association between GSTT1 null genotype in never smokers and the number of years smoked and XRCC3 T241M in ever-smokers with the level of ENU-induced O6-EtGua adducts. Inconclusive association (GSTP1 I105V, XPD D312N, MGMT I143V) or no association (GSTM1, GSTP1 A114V, MGMT L84F, XPD K751Q) was observed for the remaining SNPs. We also found that genetic polymorphisms only explain a small proportion of the variance of adduct level. These observations suggest a potential functional role of GSTT1 null genotype and XRCC3 T241M polymorphism in the formation and removal of the in vitro ENU-induced O6-EtGua. It also suggests that the effect of polymorphism may differ according to smoking status.
The GST genes encode phase II enzymes that catalyze the conjugation of electrophilic compounds in the detoxification process. Deletion or mutation of these genes is expected to be associated with higher levels of DNA damage [24]. However, previous studies provided inconsistent evidence on the effect of the GSTM1 and GSTT1 genotypes on various cytogenetic markers [25]. Polymorphisms of GSTP1 I105V and A114V are located in the electrophile-binding active site of the GSTP1, producing enzymes with different thermal stability and substrate affinity [26,27]. Decreased enzyme activity towards 1-chloro-2,4-dinitrobenzene [10] and increased binding activity towards benzo[a]pyrene diol epoxide [28] has been associated with the GSTP1 variant alleles. Very few phenotypic studies have investigated the association between GST enzyme and genotoxic effect of DNA alkylating agents. Salama et al [29] observed that the GSTM1, but not GSTT1 genotype might influence 4-(methylnitrosamino)-1- (3-pyridyl)-1-butanone (NNK)-induced genotoxicity. Lewis et al observed that, based on 44 DNA samples, N7-methylguanine level was higher in bronchial lavage cells of individuals with GSTM1 null, GSTT1 null or GSTP1 Ile/Ile genotypes [30]. Another study suggested that the biological consequences of GSTT1 genotype are difficult to predict because this enzyme has both detoxifying and activating properties in many environmental pollutants [31]. Although it is not known whether ENU is a substrate of glutathione conjugation catalyzed by any GSTs in human, the current study found a significantly lower level of ENU-induced O6-EtGua adducts in association with GSTT1 null and possibly GSTP1 105V variant allele among never-smokers, which suggests that these two GSTs may play a role in the metabolism of ENU. Because smoking was associated with the level of O6-EtGua, the genotype association with the level of adducts was perhaps masked by smoking therefore was not observed in ever-smokers.
O6-EtGua is mainly removed by the DNA repair enzyme MGMT. The MGMT codon 143 polymorphism is near the alkyl group acceptor pocket at codon 145. The replacement of Ile by Val has been postulated to affect the acceptance of an alkyl group. However, functional studies of the MGMT 143V variant protein have found either no difference from the wild-type or a higher activity [32,33]. The 84F polymorphism has been shown to have similar enzymatic and physicochemical properties to the wild type [34]. Our study showed that the MGMT 143V allele tended to be related to a higher adduct level among ever-smokers, but no effect was observed for the MGMT L84F polymorphism. The general lack of association between MGMT polymorphisms and O6-EtGua level may be partially explained by the inefficient capacity of the MGMT in repairing the bulky alkyl adduct. Pegg et al. showed that O6-EtGua was repaired more slowly than was O6-methylguanine by rat liver MGMT [35]. If this is the case in human beings, the effect of MGMT polymorphism may not be seen in our study since lymphocytes were treated with ENU for 1 hour. Moreover, the great variation in both expression and activity of MGMT related to other factors could mask any minor effect conferred by a SNP.
A human lymphoblastoid cell line lacking either MGMT or nucleotide excision repair (NER) capability had similar levels and types of mutation after ENU treatment, which suggested that MGMT and NER are of approximately equal quantitative importance for the repair of ENU-induced damage [36]. The XPD protein has been shown to correlate with resistance to alkylating agents in human tumor cell lines [37]. Previous studies have shown that the 312N allele was associated with an increased level of aromatic DNA adduct among never smoking lung cancer patients [38] and an increased frequency of chromosome aberrations in cells treated with NNK [39] or UV light [40] among healthy individuals. In contrast, Lunn et al. [41] showed that individuals with the XPD D321D genotype had insignificantly more X-ray induced chromosome aberrations than did 312N carriers. Similarly, one group showed that lymphoblastoid cell lines with the homozygous Asn genotype had a significantly higher apoptotic response to UV [42]. In our study, the influence of the XPD D312N polymorphism on adduct level was only revealed by multiple linear regression but not by univariate analysis, suggesting that this polymorphism may interact with other polymorphisms in determining the adduct level. Since the association was not stable, its effect is worthy of a further investigation. Many studies have investigated the association between the XPD K751Q polymorphism and the in vivo level of a variety of bulky DNA adducts in either cancer patients [38,43–45] or healthy individuals [17,46,47]. Most of the studies favored the association between the XPD 751Q allele and adduct formation, with a few exceptions [45,47]. However, we failed to observe any statistically significant association for this polymorphism.
The XRCC3 T241M polymorphism turned out to be one of the important factors in determining the variation of the adduct level in both never- and ever-smokers. Previous study has linked the XRCC3 T241M polymorphism to higher level of bulky DNA adducts among healthy individual [17]. Our study on XRCC3 is rather exploratory than hypothesis-driven because XRCC3 has not been directly associated with repair of alkylating DNA damage. Although homologous recombination is the major pathway of processing O6-methylguanine in replicating cells, the recombination induced by O6-methylguanine occurs only in the second postexposure replication cycle [48,49]. It is unlikely that this mechanism would occur within 1 hour of exposure to ENU in our experiment. The hamster cell line irs1SF (XRCC3 mutant) has been shown to be modestly sensitive to DNA alkylating agents [50]. Therefore, the mechanism of XRCC3 in repair of alkylating DNA adduct should be explored further. Alternatively, the XRCC3 T241M may be in linkage disequilibrium with other locus of gene involved in processing alkylating DNA damage.
In examining the relationship between O6-EtGua level and smoking indices, we observed that the number of years smoked was inversely related to the adduct level. It is postulated that higher expression of DNA repair genes among ever-smokers might be attributable to more efficient removal of adduct. We further found that polymorphisms, in particular XRCC3 T241M, had a different effect on adduct levels in never- or ever-smokers. Cigarette smokers may have altered DRC compared with never-smokers because of long-time exposure to tobacco carcinogens [51]. Wei et al. [52] have shown that the adaptation of DRC to smoking seems to be long-term rather than transient because former smokers and current smokers had similar DRC. This observation supports the argument that the mixture of ever-smokers and never-smokers would mask a true genotype-phenotype relationship when phenotype is related to smoking. Previous studies have suggested that in the examination of relationship between cancer risk, biomarker, and genotype, mixture of individuals with different exposures or different diseases biases the findings and compromises their interpretation [53]. Findings of the current study apparently support this notion.
In view of the complexity and the number of proteins involved in the repair of alkylating DNA damage, the phenotype is perhaps the aggregate of many low-penetrance gene effects. Our study suggests that none of the single polymorphisms can explain more than 10% of the variation of the adduct levels or play a dominant role in this sophisticated process. Furthermore, the effect of polymorphism on phenotypic trait is also likely to be different according to smoking exposure. This would support the gene-smoking interaction effect observed in risk association studies.
This exploratory study has many limitations. The immunoassay itself was plagued by issues of cross-reactivity of the antibody with other DNA alkylation adducts. However, measurement of the class of adducts as a whole, is still a useful marker [54]. The limited detection dynamics of the immunoassay made accurate adduct quantification impossible. The residual variance of the adduct level was large, suggesting that experimental random error was an important source of the total variance. The multiple comparisons of many factors in this study may increase the chance of false-positive results. Although the findings on GSTT1 among never-smokers and XRCC3 T241M among ever-smokers remained statistically significant after the P value was adjusted for multiple testing, our study findings still should be interpreted cautiously. The small sample size precluded us to further examine gene-gene interaction and allele-dosage effect in the current analysis.
In summary, the observed associations between GSTT1/XRCC3 genotypes and the level of ENU-induced DNA adducts should be confirmed in other experimental systems and future study on genotype and phenotype associations should consider the confounding effect of cigarette smoking and other exposure.
Acknowledgments
We thank the study participants. We are grateful for the laboratory support of Jijiang Zhu, Yanan Li, and Yingqiu Du. We also appreciate the fieldwork conducted by Manal Hassan, Rabia Khan, Kaustubh Mestry, Ajay Nooka, and Hui Liu. We thank Christine F. Wogan for scientific editing. This study was supported by National Institutes of Health grant CA98380, National Institute of Environmental Health Sciences grant P30 ES07784, National Institutes of Health Cancer Center Support (Core) Grant CA16672, and a fellowship from International Union Against Cancer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hecht SS, Hoffmann D. N-nitroso compounds and man: sources of exposure, endogenous formation and occurrence in body fluids. Eur J Cancer Prev. 1998;7:165–166. [PubMed] [Google Scholar]
- 2.Kyrtopoulos SA, Anderson LM, Chhabra SK, Souliotis VL, Pletsa V, Valavanis C, Georgiadis P. DNA adducts and the mechanism of carcinogenesis and cytotoxicity of methylating agents of environmental and clinical significance. Cancer Detect Prev. 1997;21:391–405. [PubMed] [Google Scholar]
- 3.Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002;11:1513–1530. [PubMed] [Google Scholar]
- 4.Albrecht T, Deng CZ, Abdel-Rahman SZ, Fons M, Cinciripini P, El-Zein RA. Differential mutagen sensitivity of peripheral blood lymphocytes from smokers and nonsmokers: effect of human cytomegalovirus infection. Environ Mol Mutagen. 2004;43:169–178. doi: 10.1002/em.20012. [DOI] [PubMed] [Google Scholar]
- 5.Li D, Wang M, Cheng L, Spitz MR, Hittelman WN, Wei Q. In vitro induction of benzo(a)pyrene diol epoxide-DNA adducts in peripheral lymphocytes as a susceptibility marker for human lung cancer. Cancer Res. 1996;56:3638–3641. [PubMed] [Google Scholar]
- 6.Li D, Firozi PF, Chang P, Wang LE, Xiong P, Sturgis EM, Eicher SA, Spitz MR, Hong WK, Wei Q. In vitro BPDE-induced DNA adducts in peripheral lymphocytes as a risk factor for squamous cell carcinoma of the head and neck. Int J Cancer. 2001;93:436–440. doi: 10.1002/ijc.1347. [DOI] [PubMed] [Google Scholar]
- 7.Stoica G, Koestner A. Diverse spectrum of tumors in male Sprague-Dawley rats following single high doses of N-ethyl-N-nitrosourea (ENU), Am J Pathol. 1984;116:319–326. [PMC free article] [PubMed] [Google Scholar]
- 8.Goth R, Rajewsky MF. Persistence of O6-ethylguanine in rat-brain DNA: correlation with nervous system-specific carcinogenesis by ethylnitrosourea. Proc Natl Acad Sci U S A. 1974;71:639–643. doi: 10.1073/pnas.71.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolt HMR. Thier Relevance of the deletion polymorphisms of the glutathione S-transferases GSTT1 and GSTM1 in pharmacology and toxicology. Curr Drug Metab. 2006;7:613–628. doi: 10.2174/138920006778017786. [DOI] [PubMed] [Google Scholar]
- 10.Watson MA, Stewart RK, Smith GB, Massey TE, Bell DA. Human glutathione S-transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19:275–280. doi: 10.1093/carcin/19.2.275. [DOI] [PubMed] [Google Scholar]
- 11.Strange RC, Fryer AA. The glutathione S-transferases: influence of polymorphism on cancer susceptibility. IARC Sci Publ. 1999:231–249. [PubMed] [Google Scholar]
- 12.Li D, Jiao L, Li Y, Doll MA, Hein DW, Bondy ML, Evans DB, Wolff RA, Lenzi R, Pisters PW, Abbruzzese JL, Hassan MM. Polymorphisms of cytochrome P4501A2 and N-acetyltransferase genes, smoking, and risk of pancreatic cancer. Carcinogenesis. 2006;27:103–111. doi: 10.1093/carcin/bgi171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nehls P, Adamkiewicz J, Rajewsky MF. Immunoslot-blot: a highly sensitive immunoassay for the quantitation of carcinogen-modified nucleosides in DNA. J Cancer Res Clin Oncol. 1984;108:23–29. doi: 10.1007/BF00390969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seiler F, Kamino K, Emura M, Mohr U, Thomale J. Formation and persistence of the miscoding DNA alkylation product O6-ethylguanine in male germ cells of the hamster. Mutat Res. 1997;385:205–211. doi: 10.1016/s0921-8777(97)00043-8. [DOI] [PubMed] [Google Scholar]
- 15.Xiong P, Bondy ML, Li D, Shen H, Wang LE, Singletary SE, Spitz MR, Wei Q. Sensitivity to benzo(a)pyrene diolepoxide associated with risk of breast cancer in young women and modulation by glutathione S-transferase polymorphisms: a case-control study. Cancer Res. 2001;61:8465–8469. [PubMed] [Google Scholar]
- 16.Deng C, Xie D, Capasso H, Zhao Y, Wang LD, Hong JY. Genetic polymorphism of human O6-alkylguanine-DNA alkyltransferase: identification of a missense variation in the active site region. Pharmacogenetics. 1999;9:81–87. doi: 10.1097/00008571-199902000-00011. [DOI] [PubMed] [Google Scholar]
- 17.Matullo G, Palli D, Peluso M, Guarrera S, Carturan S, Celentano E, Krogh V, Munnia A, Tumino R, Polidoro S, Piazza A, Vineis P. XRCC1, XRCC3, XPD gene polymorphisms, smoking and (32)P-DNA adducts in a sample of healthy subjects. Carcinogenesis. 2001;22:1437–1445. doi: 10.1093/carcin/22.9.1437. [DOI] [PubMed] [Google Scholar]
- 18.Dunning AM, Dowsett M, Healey CS, Tee L, Luben RN, Folkerd E, Novik KL, Kelemen L, Ogata S, Pharoah PD, Easton DF, Day NE, Ponder BA. Polymorphisms associated with circulating sex hormone levels in postmenopausal women. J Natl Cancer Inst. 2004;96:936–945. doi: 10.1093/jnci/djh167. [DOI] [PubMed] [Google Scholar]
- 19.Bland JM, Altman DG. Multiple significance tests: the Bonferroni method. Bmj. 1995;310:170. doi: 10.1136/bmj.310.6973.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garte S, Gaspari L, Alexandrie AK, Ambrosone C, Autrup H, Autrup JL, Baranova H, Bathum L, Benhamou S, Boffetta P, Bouchardy C, Breskvar K, Brockmoller J, Cascorbi I, Clapper ML, Coutelle C, Daly A, Dell'Omo M, Dolzan V, Dresler CM, Fryer A, Haugen A, Hein DW, Hildesheim A, Hirvonen A, Hsieh LL, Ingelman-Sundberg M, Kalina I, Kang D, Kihara M, Kiyohara C, Kremers P, Lazarus P, Le Marchand L, Lechner MC, van Lieshout EM, London S, Manni JJ, Maugard CM, Morita S, Nazar-Stewart V, Noda K, Oda Y, Parl FF, Pastorelli R, Persson I, Peters WH, Rannug A, Rebbeck T, Risch A, Roelandt L, Romkes M, Ryberg D, Salagovic J, Schoket B, Seidegard J, Shields PG, Sim E, Sinnet D, Strange RC, Stucker I, Sugimura H, To-Figueras J, Vineis P, Yu MC, Taioli E. Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiol Biomarkers Prev. 2001;10:1239–1248. [PubMed] [Google Scholar]
- 21.Benhamou S, Sarasin A. ERCC2 /XPD gene polymorphisms and lung cancer: a HuGE review. Am J Epidemiol. 2005;161:1–14. doi: 10.1093/aje/kwi018. [DOI] [PubMed] [Google Scholar]
- 22.Shepard TF, Platz EA, Kantoff PW, Nelson WG, Isaacs WB, Freije D, Febbo PG, Stampfer MJ, Giovannucci E. No association between the I105V polymorphism of the glutathione S-transferase P1 gene (GSTP1) and prostate cancer risk: a prospective study. Cancer Epidemiol Biomarkers Prev. 2000;9:1267–1268. [PubMed] [Google Scholar]
- 23.Kaur TB, Travaline JM, Gaughan JP, Richie JP, Jr, Stellman SD, Lazarus P. Role of polymorphisms in codons 143 and 160 of the O6-alkylguanine DNA alkyltransferase gene in lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2000;9:339–342. [PubMed] [Google Scholar]
- 24.Hayes JD, Strange RC. Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology. 2000;61:154–166. doi: 10.1159/000028396. [DOI] [PubMed] [Google Scholar]
- 25.Norppa H. Cytogenetic biomarkers and genetic polymorphisms. Toxicol Lett. 2004;149:309–334. doi: 10.1016/j.toxlet.2003.12.042. [DOI] [PubMed] [Google Scholar]
- 26.Johansson AS, Stenberg G, Widersten M, Mannervik B. Structure-activity relationships and thermal stability of human glutathione transferase P1-1 governed by the H-site residue 105. J Mol Biol. 1998;278:687–698. doi: 10.1006/jmbi.1998.1708. [DOI] [PubMed] [Google Scholar]
- 27.Ali-Osman F, Akande O, Antoun G, Mao JX, Buolamwini J. Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase Pi gene variants. Evidence for differential catalytic activity of the encoded proteins. J Biol Chem. 1997;272:10004–10012. doi: 10.1074/jbc.272.15.10004. [DOI] [PubMed] [Google Scholar]
- 28.Hu X, Herzog C, Zimniak P, Singh SV. Differential protection against benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide-induced DNA damage in HepG2 cells stably transfected with allelic variants of pi class human glutathione S-transferase. Cancer Res. 1999;59:2358–2362. [PubMed] [Google Scholar]
- 29.Salama SA, Abdel-Rahman SZ, Sierra-Torres CH, Hamada FA, Au WW. Role of polymorphic GSTM1 and GSTT1 genotypes on NNK-induced genotoxicity. Pharmacogenetics. 1999;9:735–743. [PubMed] [Google Scholar]
- 30.Lewis SJ, Cherry NM, Niven RM, Barber PV, Povey AC. Associations between smoking, GST genotypes and N7-methylguanine levels in DNA extracted from bronchial lavage cells. Mutat Res. 2004;559:11–18. doi: 10.1016/j.mrgentox.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 31.Pavanello S, Clonfero E. [Biomarkers of gentotoxic risk and metabolic polymorphism] Med Lav. 2000;91:431–469. [PubMed] [Google Scholar]
- 32.Mijal RS, Thomson NM, Fleischer NL, Pauly GT, Moschel RC, Kanugula S, Fang Q, Pegg AE, Peterson LA. The repair of the tobacco specific nitrosamine derived adduct O6-[4-Oxo-4-(3-pyridyl)butyl]guanine by O6-alkylguanine-DNA alkyltransferase variants. Chem Res Toxicol. 2004;17:424–434. doi: 10.1021/tx0342417. [DOI] [PubMed] [Google Scholar]
- 33.Ma S, Egyhazi S, Ueno T, Lindholm C, Kreklau EL, Stierner U, Ringborg U, Hansson J. O6-methylguanine-DNA-methyltransferase expression and gene polymorphisms in relation to chemotherapeutic response in metastatic melanoma. Br J Cancer. 2003;89:1517–1523. doi: 10.1038/sj.bjc.6601270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoue R, Abe M, Nakabeppu Y, Sekiguchi M, Mori T, Suzuki T. Characterization of human polymorphic DNA repair methyltransferase. Pharmacogenetics. 2000;10:59–66. doi: 10.1097/00008571-200002000-00008. [DOI] [PubMed] [Google Scholar]
- 35.Pegg AE, Scicchitano D, Dolan ME. Comparison of the rates of repair of O6-alkylguanines in DNA by rat liver and bacterial O6-alkylguanine-DNA alkyltransferase. Cancer Res. 1984;44:3806–3811. [PubMed] [Google Scholar]
- 36.Bronstein SM, Cochrane JE, Craft TR, Swenberg JA, Skopek TR. Toxicity, mutagenicity, and mutational spectra of N-ethyl-N-nitrosourea in human cell lines with different DNA repair phenotypes. Cancer Res. 1991;51:5188–5197. [PubMed] [Google Scholar]
- 37.Xu Z, Chen ZP, Malapetsa A, Alaoui-Jamali M, Bergeron J, Monks A, Myers TG, Mohr G, Sausville EA, Scudiero DA, Aloyz R, Panasci LC. DNA repair protein levels vis-a-vis anticancer drug resistance in the human tumor cell lines of the National Cancer Institute drug screening program. Anticancer Drugs. 2002;13:511–519. doi: 10.1097/00001813-200206000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Hou SM, Falt S, Angelini S, Yang K, Nyberg F, Lambert B, Hemminki K. The XPD variant alleles are associated with increased aromatic DNA adduct level and lung cancer risk. Carcinogenesis. 2002;23:599–603. doi: 10.1093/carcin/23.4.599. [DOI] [PubMed] [Google Scholar]
- 39.Affatato AA, Wolfe KJ, Lopez MS, Hallberg C, Ammenheuser MM, Abdel-Rahman SZ. Effect of XPD/ERCC2 polymorphisms on chromosome aberration frequencies in smokers and on sensitivity to the mutagenic tobacco-specific nitrosamine NNK. Environ Mol Mutagen. 2004;44:65–73. doi: 10.1002/em.20032. [DOI] [PubMed] [Google Scholar]
- 40.Au WW, Salama SA, Sierra-Torres CH. Functional characterization of polymorphisms in DNA repair genes using cytogenetic challenge assays. Environ Health Perspect. 2003;111:1843–1850. doi: 10.1289/ehp.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lunn RM, Helzlsouer KJ, Parshad R, Umbach DM, Harris EL, Sanford KK, Bell DA. XPD polymorphisms: effects on DNA repair proficiency. Carcinogenesis. 2000;21:551–555. doi: 10.1093/carcin/21.4.551. [DOI] [PubMed] [Google Scholar]
- 42.Seker H, Butkiewicz D, Bowman ED, Rusin M, Hedayati M, Grossman L, Harris CC. Functional significance of XPD polymorphic variants: attenuated apoptosis in human lymphoblastoid cells with the XPD 312 Asp/Asp genotype. Cancer Res. 2001;61:7430–7434. [PubMed] [Google Scholar]
- 43.Tang D, Cho S, Rundle A, Chen S, Phillips D, Zhou J, Hsu Y, Schnabel F, Estabrook A, Perera FP. Polymorphisms in the DNA repair enzyme XPD are associated with increased levels of PAH-DNA adducts in a case-control study of breast cancer. Breast Cancer Res Treat. 2002;75:159–166. doi: 10.1023/a:1019693504183. [DOI] [PubMed] [Google Scholar]
- 44.Terry MB, Gammon MD, Zhang FF, Eng SM, Sagiv SK, Paykin AB, Wang Q, Hayes S, Teitelbaum SL, Neugut AI, Santella RM. Polymorphism in the DNA repair gene XPD, polycyclic aromatic hydrocarbon-DNA adducts, cigarette smoking, and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2004;13:2053–2058. [PubMed] [Google Scholar]
- 45.Pastorelli R, Cerri A, Mezzetti M, Consonni E, Airoldi L. Effect of DNA repair gene polymorphisms on BPDE-DNA adducts in human lymphocytes. Int J Cancer. 2002;100:9–13. doi: 10.1002/ijc.10463. [DOI] [PubMed] [Google Scholar]
- 46.Palli D, Russo A, Masala G, Saieva C, Guarrera S, Carturan S, Munnia A, Matullo G, Peluso M. DNA adduct levels and DNA repair polymorphisms in traffic-exposed workers and a general population sample. Int J Cancer. 2001;94:121–127. doi: 10.1002/ijc.1433. [DOI] [PubMed] [Google Scholar]
- 47.Pavanello S, Pulliero A, Siwinska E, Mielzynska D, Clonfero E. Reduced nucleotide excision repair and GSTM1-null genotypes influence anti-B[a]PDE-DNA adduct levels in mononuclear white blood cells of highly PAH-exposed coke oven workers. Carcinogenesis. 2005;26:169–175. doi: 10.1093/carcin/bgh303. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H, Tsujimura T, Bhattacharyya NP, Maher VM, McCormick JJ. O6-methylguanine induces intrachromosomal homologous recombination in human cells. Carcinogenesis. 1996;17:2229–2235. doi: 10.1093/carcin/17.10.2229. [DOI] [PubMed] [Google Scholar]
- 49.Kaina B, Haas S, Kappes H. A general role for c-Fos in cellular protection against DNA-damaging carcinogens and cytostatic drugs. Cancer Res. 1997;57:2721–2731. [PubMed] [Google Scholar]
- 50.Wang ZM, Chen ZP, Xu ZY, Christodoulopoulos G, Bello V, Mohr G, Aloyz R, Panasci LC. In vitro evidence for homologous recombinational repair in resistance to melphalan. J Natl Cancer Inst. 2001;93:1473–1478. doi: 10.1093/jnci/93.19.1473. [DOI] [PubMed] [Google Scholar]
- 51.Shen H, Spitz MR, Qiao Y, Guo Z, Wang LE, Bosken CH, Amos CI, Wei Smoking Q. DNA repair capacity and risk of nonsmall cell lung cancer. Int J Cancer. 2003;107:84–88. doi: 10.1002/ijc.11346. [DOI] [PubMed] [Google Scholar]
- 52.Wei Q, Cheng L, Amos CI, Wang LE, Guo Z, Hong WK, Spitz MR. Repair of tobacco carcinogen-induced DNA adducts and lung cancer risk: a molecular epidemiologic study. J Natl Cancer Inst. 2000;92:1764–1772. doi: 10.1093/jnci/92.21.1764. [DOI] [PubMed] [Google Scholar]
- 53.Harms C, Salama SA, Sierra-Torres CH, Cajas-Salazar N, Au WW. Polymorphisms in DNA repair genes, chromosome aberrations, and lung cancer. Environ Mol Mutagen. 2004;44:74–82. doi: 10.1002/em.20031. [DOI] [PubMed] [Google Scholar]
- 54.Santella RM. Immunological methods for detection of carcinogen-DNA damage in humans. Cancer Epidemiol Biomarkers Prev. 1999;8:733–739. [PubMed] [Google Scholar]