

Abstract
Francisella tularensis is a Gram-negative bacterium and the causative agent of tularemia. Recent data indicate that F. tularensis replicates inside macrophages, but its fate in other cell types, including human neutrophils, is unclear. We now show that F. tularensis live vaccine strain (LVS), opsonized with normal human serum, was rapidly ingested by neutrophils but was not eliminated. Moreover, evasion of intracellular killing can be explained, in part, by disruption of the respiratory burst. As judged by luminol-enhanced chemiluminescence and nitroblue tetrazolium staining, neutrophils infected with live F. tularensis did not generate reactive oxygen species. Confocal microscopy demonstrated that NADPH oxidase assembly was disrupted, and LVS phagosomes did not acquire gp91/p22phox or p47/p67phox. At the same time, F. tularensis also impaired neutrophil activation by heterologous stimuli such as phorbol esters and opsonized zymosan particles. Later in infection, LVS escaped the phagosome, and live organisms persisted in the neutrophil cytosol for at least 12 h. To our knowledge, our data are the first demonstration of a facultative intracellular pathogen, which disrupts the oxidative burst and escapes the phagosome to evade elimination inside neutrophils, and as such, our data define a novel mechanism of virulence.
Keywords: NADPH oxidase, tularemia, superoxide, pathogenesis
INTRODUCTION
Human neutrophils [polymorphonuclear leukocytes (PMN)] are key players in innate defense that use toxic reactive oxygen species (ROS), degradative enzymes, and cationic peptides to kill ingested microbes. Oxidative host defense requires the NADPH oxidase, a multicomponent enzyme that catalyzes the conversion of molecular oxygen into superoxide anions [1]. As ROS are damaging to invading microorganisms and host tissues, NADPH oxidase activity is tightly regulated. In resting PMN, the inactive oxidase is unassembled, and components are segregated in the membrane and cytosol. Upon activation by particulate stimuli, specific granules and secretory vesicles carry the integral membrane subunits (gp91phox and p22phox) to forming phagosomes, and the cytosolic subunits (p47phox, p67phox, and p40phox) translocate en bloc to the phagosome as does Rac2. Only when all components have assembled is the oxidase active and able to generate ROS. The central role of the NAPDH oxidase in host defense is illustrated by the fact that persons with inherited defects in this enzyme have chronic granulomatous disease and suffer from repeated, severe bacterial and fungal infections [2].
Francisella tularensis is a Gram-negative, facultative intra-cellular bacterium and the causative agent of tularemia [3]. Interest in F. tularensis has increased recently because of its potential use as a biowarfare agent. Four subspecies of F. tularensis have been described, and two of these, F. tularensis subsp. tularensis and subsp. holarctica, cause disease in humans. Recent data indicate that these organisms escape the phagosome and replicate in the cytosol of human and murine macrophages [4]. In contrast, little is known about F. tularensis-PMN interactions. A few studies performed more than 20 years ago suggest that when opsonized with normal human serum, neither a clinical isolate of F. tularensis subsp. holarctica nor the live vaccine strain (LVS) induces a significant respiratory burst in PMN [5–7]. However, the basis for this host defense defect was not explored, and the fate of F. tularensis in PMN is unclear.
We now show that F. tularensis subsp. holarctica strain LVS opsonized with fresh serum is ingested rapidly and efficiently by human neutrophils. However, phagocytosis does not trigger a respiratory burst, and intracellular organisms are not eliminated. LVS phagosomes exclude NADPH oxidase subunits, and cell activation by heterologous stimuli is inhibited. Later in infection, LVS breaches the phagosome and resides in the cytosol. As relatively few pathogens evade killing by PMN, our data significantly advance our understanding of Francisella pathogenesis and at the same time, suggest that this organism uses a novel mechanism of virulence.
MATERIALS AND METHODS
Materials
Tryptic soy broth/agar, cysteine heart agar, and rabbit anti-F. tularensis LVS polyclonal antibodies (pAb) were from Becton Dickinson and Co. (Sparks, MD). Defibrinated sheep blood was from Colorado Serum Co. (Denver). HBSS was from Mediatech, Inc. (Herndon, VA). HEPES-RPMI 1640 was from BioWhittaker/Cambrex (Walkersville, MD). Murine anti-LVS mAb were from Biodesign International (Saco, ME). Rabbit pAb specific for p47phox and p67phox have been described [8, 9], as have murine mAb to gp91phox (54.1) and p22phox (44.1) [9, 10]. Affinity-purified FITC- or rhodamine-conjugated donkey anti-rabbit and goat anti-mouse IgG F(ab′)2 secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). Paraformaldehyde and glutaraldehyde were from Electron Microscopy Sciences (Hatfield, PA). Other reagents were obtained from Sigma-Aldrich (St. Louis, MO).
Neutrophil isolation and bacterial cultivation
Heparinized venous blood was obtained from healthy, adult volunteers in accordance with a protocol approved by the Institutional Review Board for Human Subjects at the University of Iowa (Iowa City). PMN were isolated using dextran sedimentation and density gradient separation on Ficoll-Hypaque followed by hypotonic lysis of erythrocytes [9]. PMN were resuspended in HBSS without divalent cations, counted, and then diluted into appropriate media as indicated.
Cultivation of bacteria
F. tularensis subspecies holarctica strain LVS (obtained from Dr. Michael Apicella, University of Iowa) was grown on 9% sheep blood/cysteine heart agar plates at 37°C in 5% CO2. Staphylococcus aureus (strain ALC1435) was cultured as we described previously [11]. Bacteria were washed three times in HBSS containing divalent cations, opsonized with 50% fresh autologous serum (AS), washed twice in HBSS without divalent cations, and quantified by measurement of absorbance at 600 nm. Where indicated, bacteria were killed by exposure to 1% periodate for 30 min at room temperature [12] or 5% formalin for 1 h at 37°C prior to washing and opsonization.
Phagocytosis and intracellular killing
Resting PMN or cells activated with 200 nM PMA for 5 min were infected with opsonized LVS [multiplicity of infection (MOI) 20:1] in HEPES-RPMI 1640 containing 10% AS. At the indicated times, live organisms were quantified by enumeration of CFU [13].
Respiratory burst
Generation of ROS by PMN was assessed using luminol-, isoluminol-, or lucigenin-ECL assays and NBT staining as described previously [11, 14].
Immunofluorescence and confocal microscopy
Our established methods were used to localize NADPH oxidase subunits in infected PMN [8, 9, 11]. LVS was detected using mAb from Biodesign International or pAb from Becton Dickinson and Co.. In all cases, phagocytosis was synchronized using centrifugation, and neutrophils infected for 5, 15, and 30 min were examined using confocal microscopy. All studies were performed in triplicate, and at least 50 cells in each sample were evaluated.
Transmission electron microscopy (TEM)
PMN (5×106/ml) in polypropylene tubes were infected with live or formalin-killed LVS (MOI 20:1) in RPMI 1640 containing 10% AS. After 5 min–12 h at 37°C, cells were processed for TEM as described [13]. Samples were examined using a Jeol JEM-1230 transmission electron microscope (Jeol Institute, Peabody, MA). Bacteria in sections from 60 independent cells were scored for each time-point and condition. Phagosome escape was defined as loss of >50% of the surrounding membrane.
Statistics
Statistical significance (P < 0.05) was assessed using the f-test, unpaired Student’s t-test, and one-way ANOVA.
RESULTS
Phagocytosis of live F. tularensis LVS does not trigger a respiratory burst
Luminol-ECL assays were used to assess production of ROS by PMN before and after infection. As shown in Figure 1, A and B, neutrophils infected with S. aureus (MOI = 10:1) underwent a strong respiratory burst, but cells infected with LVS did not. Indeed, responses of LVS-infected PMN barely exceeded the signal obtained from uninfected PMN, even when the MOI was increased to 100:1. Comparable data were obtained using lucigenin- or isoluminol-ECL to measure ROS (not shown). NBT staining confirmed the accumulation of superoxide inside 96 ± 1% of S. aureus phagosomes [11] and demonstrated that formazan deposits were present inside only 7 ± 2% of LVS compartments (Fig. 1C). Conversely, formalin- or periodate-killed LVS activated PMN in a dose-dependent manner (Fig. 1, A and B), and 23 ± 5% of phagosomes containing killed bacteria accumulated superoxide (Fig. 1C). As PMN ingested comparable numbers of live and killed LVS (Fig. 1A, inset), differential neutrophil activation was not a result of changes in infection efficiency. Collectively, these data demonstrate that LVS does not trigger a respiratory burst in human neutrophils.
Fig. 1.
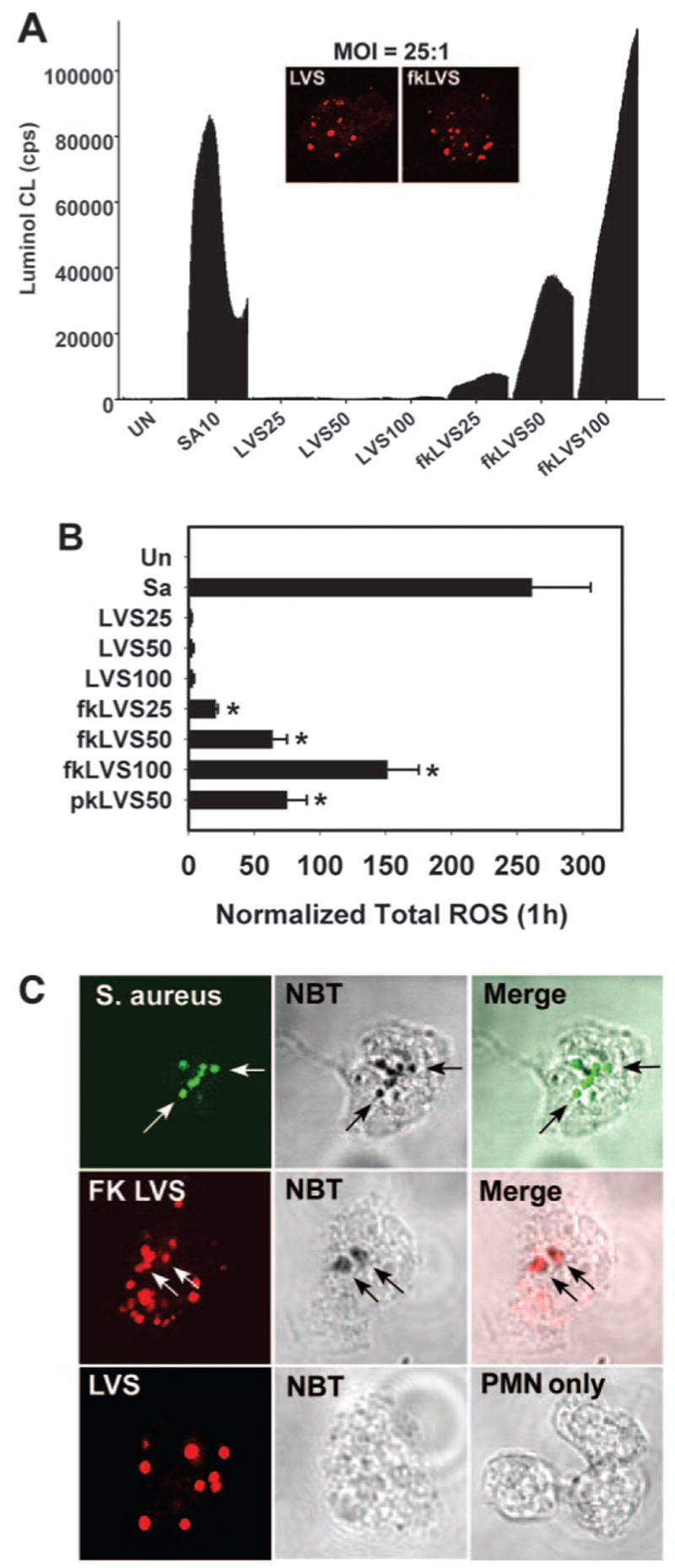
LVS does not trigger a respiratory burst in PMN. (A) Representative experiment depicting the total PMN oxidative burst over 1 h, as measured by luminol-ECL of uninfected cells (UN), PMN exposed to S. aureus MOI 10:1 (SA10), or live or formalin-killed (fk) LVS at MOI 25:1, 50:1, or 100:1. Data indicate CL as counts per second (cps) of triplicate samples from a representative experiment. (Inset) Confocal sections demonstrate equivalent phagocytosis of live and and fkLVS (both in red). (B) Total PMN ROS measured using luminol. Data are the mean ± SEM of three independent experiments performed in triplicate. pkLVS, Periodate-killed LVS. *, P < 0.05, versus LVS. (C) NBT staining of resting PMN or cells infected with S. aureus (green, MOI 5:1), LVS, or FK-LVS (red, both MOI 35:1) for 1 h. Arrows indicate formazan-positive phagosomes.
LVS inhibits NADPH oxidase assembly
Our published data indicate that the NADPH oxidase assembles rapidly and specifically on forming phagosomes containing opsonized zymosan (OpZ) and Neisseria meningitidis [8, 9] but is diverted to the plasma membrane in neutrophils infected with Helicobacter pylori [11]. Whether F. tularensis affects NADPH oxidase assembly is unknown. Therefore, we used confocal microscopy to assess recruitment of gp91phox/p22phox and p47phox/p67phox to LVS phagosomes. In good agreement with our biochemical data, LVS phagosomes rarely acquired NADPH oxidase components (5–13% positive) 5–30 min postinfection (Fig. 2). Conversely, a significantly larger fraction of phagosomes containing formalin-killed bacteria (24–25%) recruited gp91/p22phox and p47/p67phox within 15 min, and robust oxidase assembly was apparent on nearly all S. aureus compartments (80–86% positive). These data indicate that LVS inhibits the oxidative burst, at least in part, by preventing NADPH oxidase assembly at the phagosome.
Fig. 2.
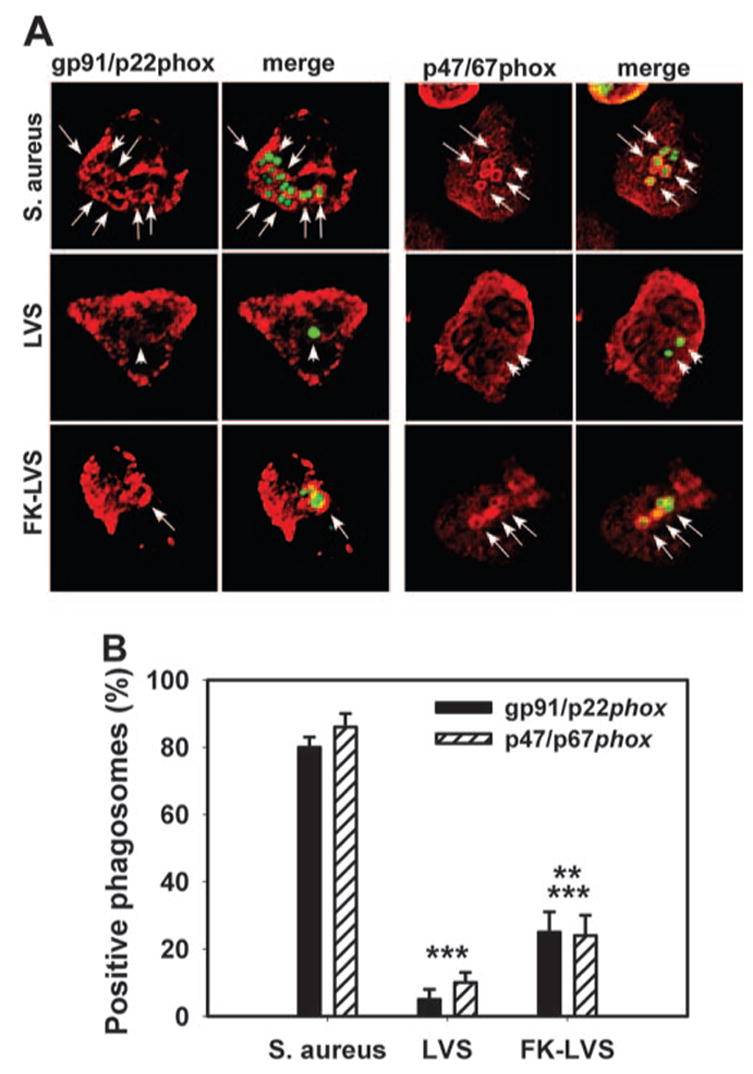
LVS phagosomes do not acquire NADPH oxidase subunits. (A) Confocal sections of PMN infected for 15 min show S. aureus, LVS, and formalin-killed (FK)-LVS in green and NAPDH oxidase components in red. Arrows and arrowheads indicate NADPH oxidase-positive and negative phagosomes, respectively. Comparable data were obtained at 5 min and 30 min. (B) Fraction of S. aureus, LVS, or FK-LVS phagosomes, which acquired gp91/p22phox or p47/67phox. Data are the average ± SEM from at least three independent experiments. **, P < 0.010, versus LVS; ***, P ≤ 0.001, versus S. aureus.
LVS evades killing by resting and PMA-activated neutrophils
Oxidants generated by S. aureus-infected PMN rapidly kill ingested organisms [15]. Therefore, we reasoned that inhibition of NADPH oxidase assembly and ROS generation in LVS-infected PMN may retard intracellular killing. To test this hypothesis, we quantified viable bacteria by enumeration of CFU. As shown in Figure 3, LVS did not survive in medium containing 10% AS for more than 3 h. In marked contrast, bacteria ingested by PMN remained viable for at least 8 h.
Fig. 3.
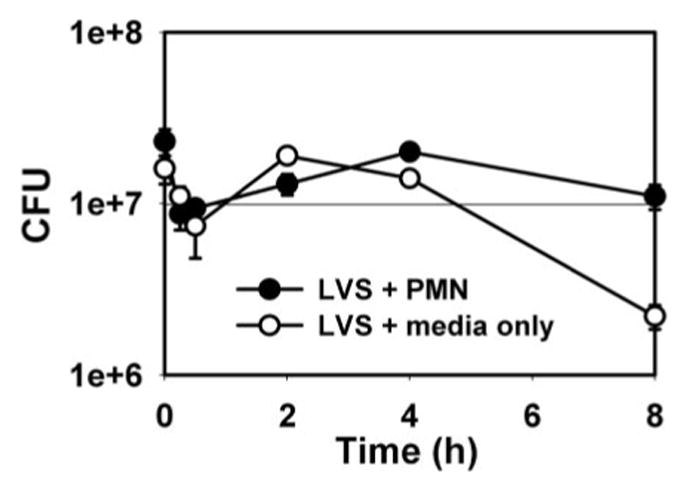
LVS evades killing inside PMN. Viability of LVS in media alone (RPMI 1640+10% AS) versus bacteria inside PMN (MOI 20:1). Data are the mean ± SEM for triplicate samples from one experiment representative of three independent determinations.
Neutrophils stimulated with PMA undergo a strong respiratory burst, and we have shown that active NADPH oxidase complexes accumulate throughout the plasma membrane of these cells [11]. The results of this study demonstrate that LVS phagosomes excluded NADPH oxidase components in resting PMN, but whether this is also true for activated cells is unknown. Therefore, we wished to assess whether PMA-stimulated neutrophils would exhibit enhanced killing of LVS. As shown in Figure 4A, we found that activated cells were no more able to inhibit LVS survival than resting neutrophils. Moreover, PMA pretreatment did not support accumulation of gp91/p22phox (not illustrated) or p47/p67phox (Fig. 4B) on LVS phagosomes. We conclude that LVS is not killed efficiently by resting or PMA-activated neutrophils.
Fig. 4.
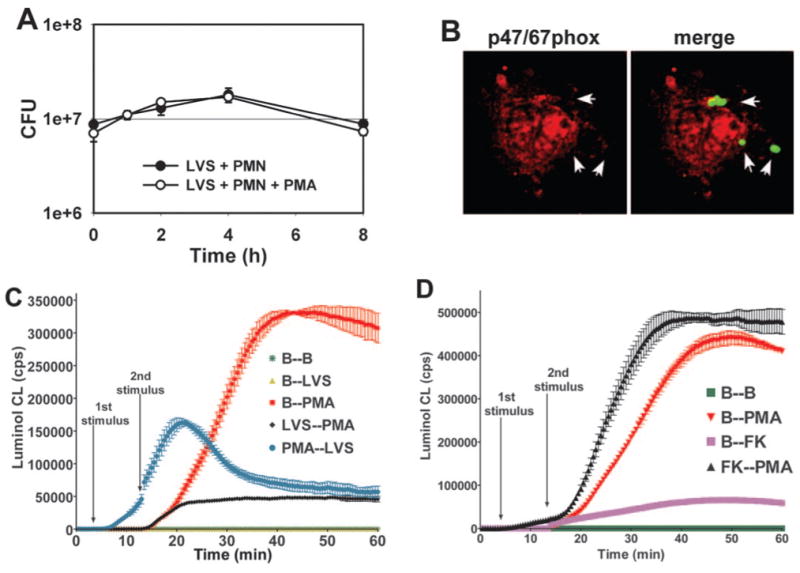
LVS survival is not impaired in PMA-activated PMN. (A) Resting and PMA-activated PMN (200 nM PMA, 5 min) were infected with LVS at MOI 20:1, and viable intracellular bacteria were quantified at the indicated time-points. Data are the mean ± SEM for triplicate samples from one experiment representative of three determinations. (B) LVS phagosomes (arrowheads) do not acquire p47/67phox in PMA-activated PMN. Data shown are representative of three independent experiments. (C) LVS impairs cell activation by PMA. PMN in luminol-containing buffer (B; 1st stimulus) were left untreated (B–B) or exposed to LVS (MOI 50:1; B–LVS) or 200 nM PMA (B–PMA) as indicated. Alternatively, cells were activated with PMA 10 min before (PMA–LVS) or 10 min after infection with LVS (LVS–PMA). Data are mean CL ± SD of triplicate samples from one experiment representative of four. (D) Same as C except formalin-killed (FK) LVS was used instead of live bacteria. Data are the mean ± SD of triplicate samples from one representative experiment.
LVS inhibits global NADPH oxidase activity
The ability of LVS to evade toxic ROS and survive in PMA-activated neutrophils suggested that this organism may have effects on PMN that extend beyond the bacterial phagosome. To test this hypothesis, we assessed the ability of LVS-infected cells to generate oxidants in response to heterologous stimuli. Luminol assays were performed using neutrophils, which were activated with PMA before or after infection with LVS (Fig. 4C). Concordant with the data shown in Figure 1, neither neutrophils maintained in buffer nor cells infected with LVS alone generated ROS. As expected, addition of 200 nM PMA to naïve PMN triggered a robust, respiratory burst, and CL increased linearly for approximately 20 min (Fig. 4C, red squares). In sharp contrast, neutrophils infected with LVS 10 min prior to (Fig. 4C, black diamonds) or 10 min after (Fig. 4C, blue circles) stimulation with PMA initiated, but were unable to sustain, oxidant production. Specifically, prior LVS infection reduced initial rates of ROS synthesis by approximately 20% and reduced peak CL by 85% compared with PMA controls. When LVS was added 10 min after PMA, the effect was less pronounced, and peak CL declined by 50% (Fig. 4C). Inhibition of the oxidative burst was specific for live Francisella, and exposure of neutrophils to formalin-killed bacteria and PMA generated more ROS than cells exposed to either agent alone (Fig. 4D). At the same time, TEM analyses indicated that PMN ingested nearly all LVS within 5 min (see below). Thus, inhibition is likely conferred by intracellular bacteria. Taken together, the data indicate that LVS inhibits neutrophil activation by PMA rapidly and efficiently.
As PMA activates the NADPH oxidase by a receptor-independent mechanism, we also asked if LVS could disrupt cell activation by a particulate stimulus such as yeast zymosan. Indeed, preinfection with LVS inhibited the oxidative burst triggered by phagocytosis of OpZ (MOI 4:1), as judged by a 40% decrease in the initial rate of ROS production and a 63% decrease in peak CL (Fig. 5A, red triangles vs. black diamonds). In contrast to data obtained with PMA, neither the initial rate of the OpZ-activated respiratory burst nor peak CL were altered significantly (≤7% change) when LVS was added 10 min after OpZ. However, peak oxidant production was not sustained relative to OpZ-treated controls, as indicated by a decline in CL after approximately 25 min (Fig. 5A, blue circles vs. red triangles). As seen with PMA, formalin-killed LVS and OpZ activated PMN in an additive manner (Fig. 5B). Collectively, these data suggest that LVS inhibits overall PMN function and as a result, responses to heterologous particulate and soluble stimuli are compromised, albeit to different extents.
Fig. 5.
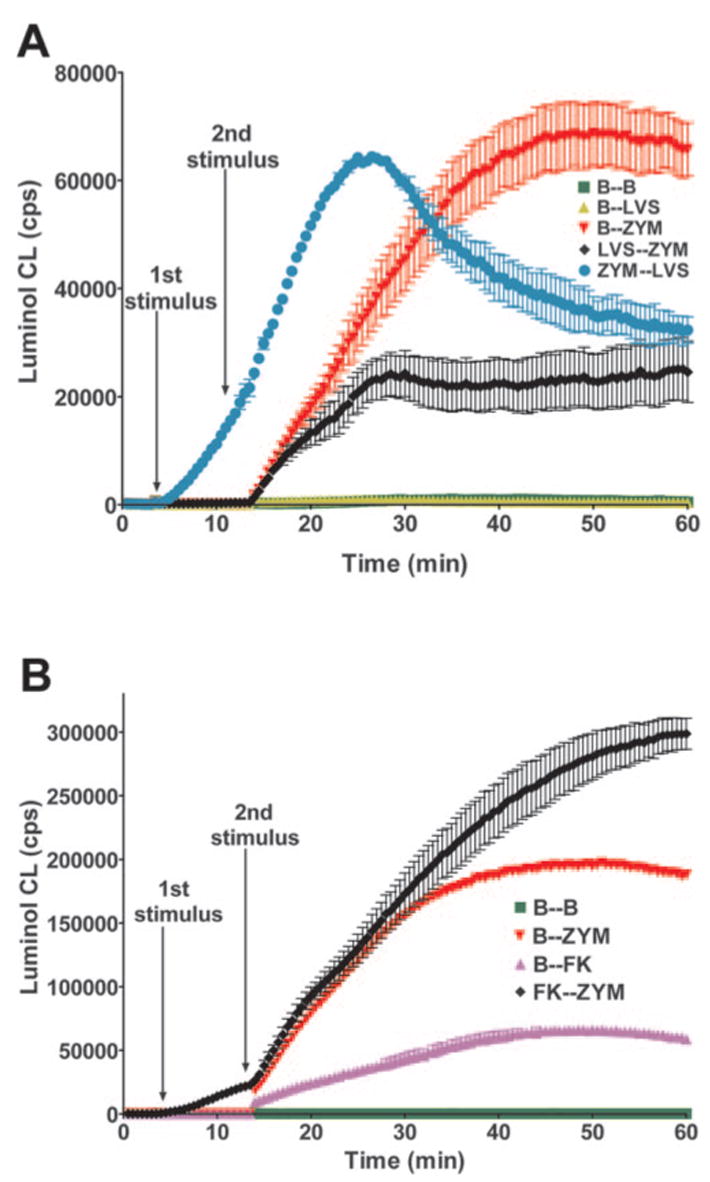
LVS inhibits PMN activation by OpZ. (A) PMN were left in buffer or infected with LVS (MOI 50:1) and/or OpZ (ZYM, MOI 4:1) as indicated. Data are the mean ± SD of triplicate samples from one experiment representative of three. (B) PMN were left in buffer or activated with formalin-killed LVS (FK) and/or OpZ (ZYM) as indicated. Data are the mean ± SD of triplicate samples from a representative experiment.
LVS escapes PMN phagosomes and enters the cytosol
It is now well established that LVS escapes from macrophage phagosomes and replicates in the cytosol after a lag of approximately 6 h [16, 17]. By contrast, the subcellular localization of this organism within PMN is unknown. Therefore, we performed ultrastructural analyses of neutrophils infected with LVS for up to 12 h (Fig. 6). Bacteria were rapidly ingested by PMN (Fig. 6A) and shortly thereafter (5–15 min), LVS resided in semispacious phagosomes (Fig. 6B). By 4 h hpi, disruptions were apparent in the phagosome membrane (Fig. 6C), and by 6 hpi, approximately half of all ingested LVS were no longer surrounded by a membrane and appeared free in the cytosol (Fig. 6, D, E, and J). In good agreement with the CFU data shown in Figure 3, bacteria that reached the cytosol remained intact for at least 12 h (Fig. 6, E, F, and H). Conversely, degradation of some intraphagosomal LVS was apparent at ≥9 hpi (Fig. 6F), and loss of organism integrity appeared to coincide with phagosome-granule fusion (Fig. 6G). Phagosome egress, like inhibition of the respiratory burst, was specific for live bacteria, and formalin-killed LVS did not reach the cytosol (not illustrated). Finally, over the time course of these studies, LVS-infected neutrophils remained viable and did not display morphological signs of apoptosis (Fig. 6I). Thus, our data demonstrate that LVS escapes the phagosome in human neutrophils and persists in the nutrient-rich cytosol.
Fig. 6.
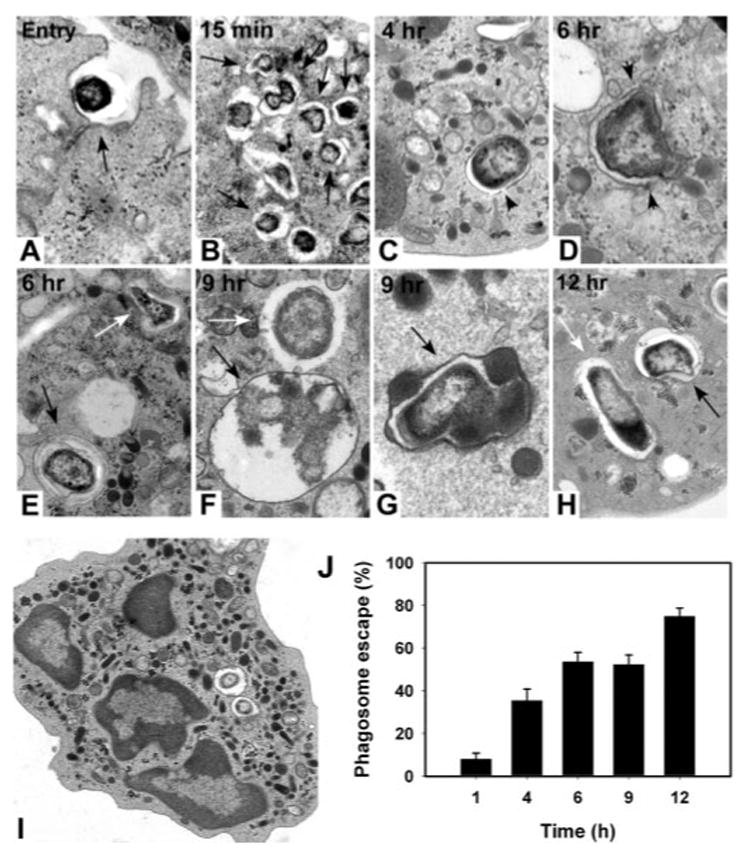
LVS escapes PMN phagosomes. TEM images depict LVS entry into PMN (A) as well as bacteria at 15 min (B) and 4 (C), 6 (D and E), 9 (F and G), and 12 h postinfection (hpi; H). Arrow in A indicates a forming phagosome. Other black arrows indicate intact phagosomes. Black arrowheads indicate disrupted phagosome membranes. White arrows indicate bacteria free in the cytosol. (I) Overall PMN morphology at 9 hpi. (J) Time course of phagosome escape. Data are the mean ± SEM (n=60) for each time-point.
DISCUSSION
PMNs are an essential first line of defense against invading microbes. Rapid assembly of the NADPH oxidase on forming phagosomes and subsequent ROS generation allow PMN to kill most ingested microbes within 30–60 min [15, 18]. Bacteria that evade PMN killing must avoid or resist toxic ROS. We show here that LVS prevents NADPH oxidase assembly in resting and activated PMN, and as a result, engulfment is not coupled to ROS synthesis as measured by luminol CL or NBT staining. Our data are in good agreement with previous studies, which found that neither LVS nor wild F. tularensis subsp. holarctica triggers oxidant production by PMN when bacteria are opsonized with normal serum [5–7]. Moreover, we show for the first time that LVS infection prevents cell activation by heterologous stimuli and that early evasion of ROS allows bacteria to escape the phagosome and reach the PMN cytosol.
How LVS prevents oxidase assembly is unknown. This bacterium secretes no known toxins and lacks Types III, IV, and V secretion systems common in other pathogens [19]. It has been suggested that F. tularensis AcpA may inhibit the respiratory burst, but the effects of this protein are controversial, and its function remains obscure [3]. A role for bacterial surface factors, particularly carbohydrates, is suggested by the inability of periodate-killed LVS to prevent ROS synthesis (Fig. 1B). At the same time, our data suggest that inhibition of the oxidative burst may be multifactorial, as formalin fixation completely abrogated the ability of F. tularensis to prevent cell activation by PMA and OpZ (Figs. 4D and 5B), yet only 25% of phagosomes containing killed LVS accumulated NADPH oxidase components and ROS (Figs. 1C and 2), and as a result, killed LVS was a less potent agonist than live S. aureus (Fig. 1, A and B). Regardless of mechanism, the ability of LVS to block NADPH oxidase activity triggered by PMA or yeast zymosan (Figs. 4C and 5A) suggests that this pathogen may affect a common signaling pathway or intermediate. In view of these data, it is tempting to predict that evasion of oxidative host defense mechanisms is an essential element of virulence important for Francisella survival in neutrophils.
Our ultrastructural data demonstrate for the first time that LVS escapes the PMN phagosome, and as such, the results of this study indicate that the effects of F. tularensis extend beyond blockade of the respiratory burst. LVS opsonized with complement factors in fresh serum was ingested rapidly by PMN (Fig. 6A) without elaboration of the pseudopod loops recently described on Francisella-infected macrophages [12]. Four to 6 h after uptake, LVS breached the phagosome and entered the cytosol, and bacteria remained viable in this locale for at least 12 h (Figs. 3 and 6 and our unpublished data). By contrast, organisms retained inside phagosomes were eventually degraded, coincident with phagosome-granule fusion (Fig. 6, F and G). These data suggest that phagosome egress may also be essential for LVS survival in PMN. In macrophages, virulence factors such as IglC and MglA are required for phagosome escape [3, 4]. However, their mechanism of action remains obscure, and the role of these proteins in LVS-PMN interactions is unknown.
The results of this study are significant, as relatively few pathogens evade elimination by PMN. In particular, it is noteworthy that Listeria monocytogenes and Shigella flexneri escape the phagosome in macrophages and/or epithelial cells, but neither of these organisms evades killing by neutrophils. Shigella virulence factors required for phagosome escape are degraded by PMN elastase [20, 21], and Listeria is rapidly killed by intraphagosomal ROS [15]. Conversely, Anaplasma phagocytophilum is an obligate intracellular pathogen of neutrophils. Like LVS, A. phagocytophilum subverts NADPH oxidase assembly at the bacterial phagosome [22, 23], and periodate-sensitive factors have been implicated in this process [24]. However, A. phagocytophilum differs from LVS in two key respects. Early in infection, A. phagocytophilum does not impair oxidant generation by heterologous stimuli [22], and this organism replicates inside vacuoles and does not reach the cytosol [24]. As the effects of LVS on PMN are distinct from other pathogens, we propose that this organism may exhibit a novel mechanism of virulence.
Unlike macrophages, PMN are short-lived cells, and recent data indicate that phagocytosis and ROS synthesis trigger an apoptosis differentiation program in PMN, which is essential for resolution of infection and control of inflammation [15, 25]. Over the time course examined here, LVS-infected neutrophils did not exhibit morphological signs of cell death (Fig. 6I). This finding is of considerable interest, as A. phagocytophilum and Chlamydia pneumoniae evade killing by PMN, and both of these pathogens prolong cell survival [25]. In future studies, it will be important to determine whether LVS alters the apoptosis differentiation program of PMN and to discern whether cell survival is prolonged as a direct consequence of NADPH oxidase inhibition or by some other mechanism.
In summary, the results of this study demonstrate that F. tularensis opsonized with AS survives inside human PMN. During entry, LVS prevents NADPH oxidase assembly at the phagosome and thereby evades early elimination by toxic ROS. Thereafter, LVS escapes the phagosome and enters the PMN cytosol, where it survives for at least 12 h. In this context, PMN may sustain infection by disseminating bacteria to distal sites without inducing a significant inflammatory response (a notion consistent with the low bioactivity of Francisella endotoxin [26, 27]). Alternatively, infected PMN may be ingested by macrophages and in this manner, propagate infection as has been reported recently for Leishmania [28]. Clearly, our understanding of the pathogenesis of tularemia is incomplete. In future studies, it will be important to determine at the molecular level how Francisella disrupts PMN function in general and NADPH oxidase activity in particular and as such, to define more precisely the role of PMN in tularemia.
Acknowledgments
This work was supported by funds from the National Institutes of Health (NIH; P01-AI44642) to L-A. H. A. R. L. M. is a postdoctoral fellow supported by NIH T32AI07343-17 awarded to the Division of Infectious Diseases, Department of Internal Medicine, University of Iowa. We thank Michael Apicella (Department of Microbiology, University of Iowa) for the generous gift of LVS and Randy Nessler of the University of Iowa Central Microscopy Research Facility for assistance with electron microscopy. We are indebted to Drs. Algirdas Jesaitis, Mark Quinn, and James Burritt (Montana State University, Bozeman) and Dr. William Nauseef (University of Iowa) for antibodies to NADPH oxidase subunits. In addition, we thank Lili Zhao and Kung-Sik Chan of the University of Iowa Department of Statistics and Actuarial Science for helpful discussions.
References
- 1.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 2.Curnutte JT. Chronic granulomatous disease: the solving of a clinical riddle at the molecular level. Clin Immunol Immunopathol. 1993;67:S2–S15. doi: 10.1006/clin.1993.1078. [DOI] [PubMed] [Google Scholar]
- 3.Oyston PCF, Sjostedt A, Titball RW. Tularemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 4.Santic M, Molmeret M, Klose KE, Abu Kwaik Y. Francisella tularensis travels a novel, twisted road within macrophages. Trends Microbiol. 2006;14:37–44. doi: 10.1016/j.tim.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 5.Proctor RA, White JD, Ayala E, Canonico PG. Phagocytosis of Francisella tularensis by rhesus monkey peripheral leukocytes. Infect Immun. 1975;11:146–152. doi: 10.1128/iai.11.1.146-151.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lofgren S, Tarnvik A, Carlsson J. Demonstration of opsonizing antibodies to Francisella tularensis by leukocyte chemiluminescence. Infect Immun. 1980;29:329–334. doi: 10.1128/iai.29.2.329-334.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lofgren S, Tarnvik A, Thore M, Carlsson J. A wild and an attenuated strain of Francisella tularensis differ in susceptibility to hypo-chlorous acid: a possible explanation of their different handling by polymorphonuclear leukocytes. Infect Immun. 1984;43:730–734. doi: 10.1128/iai.43.2.730-734.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen LAH, DeLeo FR, Gallois A, Toyoshima S, Suzuki K, Nauseef WM. Transient association of the nicotinamide adenine dinucleotide phosphate oxidase subunits p47phox and p67phox with phagosomes in neutrophils from patients with X-linked chronic granulomatous disease. Blood. 1999;93:3521–3530. [PubMed] [Google Scholar]
- 9.DeLeo FR, Allen LAH, Apicella M, Nauseef WM. NADPH oxidase activation and assembly during phagocytosis. J Immunol. 1999;163:6732–6740. [PubMed] [Google Scholar]
- 10.Burritt JB, Quinn MT, Jutila MA, Bond CW, Jesaitis AJ. Topological mapping of neutrophil cytochrome b epitopes with phage-display libraries. J Biol Chem. 1995;270:16974–16980. doi: 10.1074/jbc.270.28.16974. [DOI] [PubMed] [Google Scholar]
- 11.Allen LAH, Beecher BR, Lynch JT, Rohner OV, Wittine LM. Helicobacter pylori disrupts NADPH oxidase targeting in human neutrophils to induce extracellular superoxide release. J Immunol. 2005;174:3658–3667. doi: 10.4049/jimmunol.174.6.3658. [DOI] [PubMed] [Google Scholar]
- 12.Clemens DL, Lee BY, Horwitz MA. Francisella tularensis enters macrophages via a novel process involving pseudopod loops. Infect Immun. 2005;73:5892–5902. doi: 10.1128/IAI.73.9.5892-5902.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen LAH, Schlesinger LS, Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med. 2000;191:115–127. doi: 10.1084/jem.191.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J Immmunol Methods. 1999;232:3–14. doi: 10.1016/s0022-1759(99)00146-5. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi SD, Braughton KR, Whitney AR, Voyich JM, Schwan TG, Musser JM, DeLeo FR. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci USA. 2003;100:10948–10953. doi: 10.1073/pnas.1833375100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golovliov I, Baranov V, Krocova Z, Kovarova H, Sjostedt A. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect Immun. 2003;71:5940–5950. doi: 10.1128/IAI.71.10.5940-5950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clemens DL, Lee BY, Horwitz MA. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect Immun. 2004;72:3204–3217. doi: 10.1128/IAI.72.6.3204-3217.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rada BK, Geiszt M, Kaldi K, Timar C, Ligeti E. Dual role of phagocytic NADPH oxidase in bacterial killing. Blood. 2004;104:2947–2953. doi: 10.1182/blood-2004-03-1005. [DOI] [PubMed] [Google Scholar]
- 19.Larsson P, Oyston PC, Chain P, Chu MC, Duffield M, Fuxelius HH, Garcia E, Halltorp G, Johansson D, Isherwood KE, et al. The complete genome sequence of Francisella tularensis, the causative agent of tularemia. Nat Genet. 2005;37:153–159. doi: 10.1038/ng1499. [DOI] [PubMed] [Google Scholar]
- 20.Mandic-Mulec I, Weiss J, Zychlinsky A. Shigella flexneri is trapped in polymorphonuclear leukocyte vacuoles and efficiently killed. Infect Immun. 1997;65:110–115. doi: 10.1128/iai.65.1.110-115.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weinrauch Y, Drujan D, Shapiro SD, Weiss J, Zychlinsky A. Neutrophil elastase targets virulence factors of enterobacteria. Nature. 2002;417:91–94. doi: 10.1038/417091a. [DOI] [PubMed] [Google Scholar]
- 22.Ijdo JW, Mueller AC. Neutrophil NADPH oxidase is reduced at the Anaplasma phagocytophilum phagosome. Infect Immun. 2004;72:5392–5401. doi: 10.1128/IAI.72.9.5392-5401.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlyon JA, Abdel-Latif D, Pypaert M, Lacy P, Fikrig E. Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infect Immun. 2004;72:4772–4783. doi: 10.1128/IAI.72.8.4772-4783.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rikihisa Y. Ehrlichia subversion of host innate responses. Curr Opin Microbiol. 2006;9:95–101. doi: 10.1016/j.mib.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 25.DeLeo FR. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis. 2004;9:399–413. doi: 10.1023/B:APPT.0000031448.64969.fa. [DOI] [PubMed] [Google Scholar]
- 26.Ancuta P, Pedron T, Girard R, Sandstrom G, Chaby R. Inability of the Francisella tularensis lipopolysaccharide to mimic or to antagonize the induction of cell activation by endotoxins. Infect Immun. 1996;64:2041–2046. doi: 10.1128/iai.64.6.2041-2046.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandstrom G, Sjostedt A, Johannson T, Kuoppa K, Williams JC. Immunogenicity and toxicity of lipopolysaccharide from Francisella tularensis LVS. FEMS Microbiol Immunol. 1992;5:201–210. doi: 10.1111/j.1574-6968.1992.tb05902.x. [DOI] [PubMed] [Google Scholar]
- 28.Van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, Laskay T. Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J Immunol. 2004;173:6521–6525. doi: 10.4049/jimmunol.173.11.6521. [DOI] [PubMed] [Google Scholar]