

Summary
Axin1 and its homolog Axin2/conductin/Axil are negative regulators of the canonical Wnt pathway that suppress signal transduction by promoting degradation of β -catenin. Mice with deletion of Axin1 exhibit defects in axis determination and brain patterning during early embryonic development. We show that Axin2 is expressed in the osteogenic fronts and periosteum of developing sutures during skull morphogenesis. Targeted disruption of Axin2 in mice induces malformations of skull structures, a phenotype resembling craniosynostosis in humans. In the mutants, premature fusion of cranial sutures occurs at early postnatal stages. To elucidate the mechanism of craniosynostosis, we studied intramembranous ossification in Axin2-null mice. The calvarial osteoblast development is significantly affected by the Axin2 mutation. The Axin2 mutant displays enhanced expansion of osteoprogenitors, accelerated ossification, stimulated expression of osteogenic markers and increases in mineralization. Inactivation of Axin2 promotes osteoblast proliferation and differentiation in vivo and in vitro. Furthermore, as the mammalian skull is formed from cranial skeletogenic mesenchyme, which is derived from mesoderm and neural crest, our data argue for a region-specific effect of Axin2 on neural crest dependent skeletogenesis. The craniofacial anomalies caused by the Axin2 mutation are mediated through activation of β -catenin signaling, suggesting a novel role for the Wnt pathway in skull morphogenesis.
Keywords: Axin, Axin2, Wnt, Neural crest, Craniosynostosis
Introduction
Craniofacial morphogenesis is regulated by complex interactions between the surface and neural ectoderms, endoderm, paraxial mesoderm and cranial neural crest (Francis-West et al., 1998). This morphogenetic process is highly dependent on the patterning information of emigrant cranial neural crest (CNC) cells (Couly et al., 1996; Kontges and Lumsden, 1996). CNC cells arise along the lateral margins of the neural fold at the boundary between the surface and neural ectoderms in the brain region (Anderson, 2000; Dorsky et al., 1998; LaBonne and Bronner-Fraser, 1999; Trainor and Krumlauf, 2002). Upon closure of the neural fold, CNC cells migrate ventrolaterally to populate the head and neck regions and give rise to a wide variety of tissues, including nerves, ganglia, connective tissues, cartilages and bones (Le Douarin and Kalcheim, 1999). As a result, the majority of craniofacial malformations are caused by defects in CNC (Wilkie and Morriss-Kay, 2001). Therefore, understanding the mechanisms that control craniofacial development, particularly the CNC and its contribution to various facial tissues and structures, might provide new insights into the molecular basis of these defects in humans (Le Douarin and Kalcheim, 1999).
The cranial skull consists of the neurocranium and the viscerocranium that are formed from cranial skeletogenic mesenchyme derived from both mesoderm and neural crest (Jiang et al., 2002). The cranial skeletogenic mesenchyme undergoes intramembranous ossification to form the skull bones during calvarial morphogenesis (Hall, 1990). Expansion of the brain is accommodated by calvarial growth. This developmental process is regulated by cranial sutures, which serve as growth centers for osteogenesis. Individuals with craniosynostosis develop abnormal skull shapes due to premature fusion of the cranial sutures. Craniosynostosis is one of the most common human congenital craniofacial deformities affecting one in approximately 2,500 individuals (Cohen and MacLean, 2000). Premature suture closure results in cranial dysmorphism, which can be familial or sporadic in origin (Cohen and MacLean, 2000). Although linkage analyses have shown that the Fgfr (Burke et al., 1998), Msx2 (Jabs et al., 1993) and Twist (el Ghouzzi et al., 1997; Howard et al., 1997) genes are associated with craniosynostosis-related syndromes, the mechanisms underlying suture development remains largely unknown. Therefore, identification of genes and signaling pathways that mediate calvarial morphogenesis is critical for deciphering the pathogenesis of craniosynostosis.
Axin1, which regulates embryonic axis determination by modulating the canonical Wnt pathway, was first identified in a mouse mutant strain (Zeng et al., 1997). Substantial evidence has established that Axin1 and its homolog Axin2/conductin/Axil plays a central role in regulating the stability of β -catenin, which is a crucial event in cellular response to Wnt signaling (Kikuchi, 2000; Miller et al., 1999; Moon et al., 2002; Peifer and Polakis, 2000). Axins serve as scaffold proteins directly associating with several Wnt signaling molecules, including disheveled, the serine/threonine kinase GSK-3, β -catenin, adenomatous polypopsis coli (APC) and the serine/threonine protein phosphatase 2A (PP2A) (Behrens et al., 1998; Fagotto et al., 1999; Hedgepeth et al., 1999; Hsu et al., 1999; Itoh et al., 1998; Julius et al., 2000; Kishida et al., 1998; Sakanaka et al., 1998). In the absence of a Wnt signal, the Axin-dependent complex mediates β -catenin degradation, while Wnt signals perturb formation of this complex (Farr et al., 2000; Li et al., 1999; Smalley et al., 1999; Yanagawa et al., 1995). Therefore, β -catenin is accumulated and binds to LEF/TCF family proteins to activate target genes (Behrens et al., 1996; Brannon et al., 1997; Molenaar et al., 1996).
Wnt signaling controls early craniofacial morphogenesis (Parr et al., 1993). Wnt1 and Wnt3a are both expressed in the dorsolateral region of the neural tube that gives rise to CNC (McMahon et al., 1992). Although inactivation of either Wnt1 or Wnt3a gene did not cause defects in craniofacial development (McMahon and Bradley, 1990; Takada et al., 1994), mice in which both the Wnt1 and Wnt3a genes are inactivated showed a marked deficiency in CNC derivatives (Ikeya et al., 1997). Furthermore, downstream components of the Wnt signaling pathway, including Lrp6, APC and β -catenin, have also been implicated in craniofacial development (Brault et al., 2001; Hasegawa et al., 2002; Mitchell et al., 2001). Nevertheless, the importance of the Wnt pathway in intramembranous ossification during mammalian skull formation remains unclear.
In this study, we have investigated the involvement of Axin2 in cranial skeletogenesis. Targeted disruption of Axin2 did not cause obvious embryonic abnormalities, although Axin2 is highly expressed in CNC. However, our data demonstrate that Axin2 is required for skull development at early postnatal stages. The inactivation of Axin2 in mice induces craniosynostosis, a common human congenital defect. The premature fusion of cranial sutures is mediated by alterations in intramembranous ossification in the mutants. The neural crest dependent skeletogenesis is particularly sensitive to the loss of Axin2 that stimulates β -catenin signaling in the developing calvarium. These findings demonstrate not only the importance of Axin2, but also a novel role of the canonical Wnt pathway, in calvarial morphogenesis and craniosynostosis.
Materials and methods
Mouse strains
Specific targeting strategy to generate the Axin2-deficient mice will be reported elsewhere (B.J. and W.B., unpublished). PCR genotyping was performed using primers 5′ -agtccatcttcattccgcctagc-3′ and 5′ -tggtaatgctgcagtggcttg-3′ for the wild type, and primers 5′ -agtccatcttcattccgcctagc-3′ and 5′ -aagctgcgtcggatacttgcga-3′ for the Axin2 mutant. TOPGAL (DasGupta and Fuchs, 1999) mice were obtained form the Jackson Laboratory.
Histology, skeletal preparation and β -gal staining
Skulls were fixed in formaldehyde/formic acid (Cal-Rite, Richard-Allan Scientific) and paraffin embedded. Samples were sectioned, and stained with Hematoxylin/Eosin/Orange G for histological evaluation. Staining for β -galactosidase activity in cranial skulls (Whiting et al., 1991) and mouse skeletal preparation (Selby, 1987) was performed as described. The stained skulls were photographed for whole-mount analyses and then processed for analyses in sections.
Primary osteoblast isolation, culture and differentiation
Primary osteoblast precursors were isolated from P1 mouse calvaria as described (Mansukhani et al., 2000). Isolated osteoblasts were cultured in α MEM media containing 10% fetal calf serum. Only the first passage cells were used. Primary osteoblasts (2 × 104) were seeded in 12 well plates for 48 hours. To induce maturation of the osteoblasts, cells were maintained in differentiation media containing 50μg/ml ascorbic acid and 4 mM β -glycerophosphate.
Alkaline phosphatase and mineralized bone matrix formation assays
For histochemical staining in situ, cells were fixed in 10% formalin. Alkaline phosphatase staining was performed according to manufacturer’s protocol (Pierce). For liquid assays, cell lysates were prepared by M-PER (Pierce). Protein concentrations were determined (BioRad) and alkaline phosphatase activities were analyzed with solutions containing 0.5 mg/ml of p-nitrophenylphosphate (Sigma-Aldrich) in AMP buffer (0.5 M 2-methyl-1, 2-aminopropanol and 2 mM magnesium chloride, pH 10.3) at 37°C for 20–30 minutes. The reaction was stopped by a solution containing 300 mM sodium phosphate (pH 12.3). The differentiated culture was monitored by mineralized nodule formation with the standard von Kossa staining method.
Immunoblot and immunohistochemistry
Tissue sections were subject to immunological staining with avidin:biotinlylated enzyme complex as described (Hsu et al., 2001). Protein extracts were subject to immunoblotting as described (Hsu et al., 1999). Bound primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies, followed by ECL mediated visualization (Amersham) and autoradiography. Mouse monoclonal antibodies α -ABC (van Noort et al., 2002), α -Actin (Lab Vision) and α -BrdU (Lab Vision); rabbit monoclonal antibody α -Ki67 (Lab Vision); or rabbit polyclonal antibodies α -Cyclin D1 (Lab Vision), α -β -catenin (Lab Vision) and α -FGFR1 (Santa Cruz) were used as primary antibodies.
Isolation of RNA and real time RT-PCR
RNA was isolated using TRIzol (Invitrogen). Total RNA concentration was determined by ultraviolet spectroscopy at 260 and 280 nm. cDNA was synthesized with RNase H-free reverse transcriptase (Invitrogen) using oligoT primers (0.03 A260 units/reaction, Sigma) in 20 μ l for 1 hour at 42°C. The cDNA was then amplified by PCR (45 cycles, 94°C for 15 seconds, 55°C for 20 seconds and 72°C for 20 seconds) with SYBR Green Master Mix (Applied Biosystems) in 20 μ l buffered solution containing 1 μ l of the diluted (1:5) reverse transcription product in the presence of 20 pmoles each of the sense and antisense primers specific for the various target sequences. The primers were: osteopontin (5′ -tcccggtgaaagtgactgattct-3′ and 5′ -catcatcgtcatcatcgtcgtcca-3′ ), osteo-calcin (5′ -cttgaagaccgcctacaaac-3′ and 5′ -gctgctgtgacatccatac-3′ ), fgf4 (5′ -tactgcaacgtgggcatcggatt-3′ and 5′ -aggcttcgtaggcgttgtagttgt-3′ ), or fgf18 (5′ -tggtactagcaaggagtgcgtgtt-3′ and 5′ -tcgcagtt-tcctcgttcaagtcct-3′ ). In the same PCR reaction, expression of β -actin (5′ -agatgtggatcagcaagcag-3′ and 5′ -gcgcaagttaggttttgtca-3′ ) was analyzed as an internal control.
Results
Inactivation of Axin2 causes cranial skull defects in mice
We generated a lacZ knock-in allele (Axin2lacZ), which inactivates Axin2, to investigate a potential role of Axin2 in mammalian development (Lustig et al., 2002). The Axin2 homozygous (hereafter referred to as Axin2−/−) mice appeared to be viable and fertile. However, closer investigation revealed developmental defects of the cranial skull at postnatal stages (Fig. 1). The Axin2−/− mice were born initially without any noticeable distinctions. A reduced growth of the head, which became obvious within the first 3 weeks after birth, permitted an easy identification of the Axin2−/− mice among their wild-type and heterozygous littermates (Fig. 1A–C). Although the mutant mice were in a mixed 129 and C57BL/6 background, the genetic background seemed to cause little phenotypic variation and the head abnormality was present in all Axin2−/−mice examined (n>50), with only slight variation in severity among individuals. At 4 weeks, the average length from the nose tip to the medial point of the coronal suture was 15.95 mm (s.d. 0.94 mm; n=8) in control mice (Axin2+/+ or +/−), and was significantly reduced to 13.77 mm (s.d. 0.42 mm; n=5, P=0.00017) in the Axin2−/− mice. This phenotypic defect prompted us to examine the cranial sutures, which are critical for calvarial osteogenesis. The Jugum Limitans, which separates the frontal and nasal bones, were often missing in the Axin2−/− mice (Fig. 1D–I). The metopic sutures, which lie in the midline between two nasal bones, were fused in the Axin2−/− mutants (Fig. 1F–I).
Fig. 1.

Cranial skull defects in the Axin2−/− mice. Lateral (A) and dorsal (B,C) views of 27-day-old control (+/+) and Axin2 mutant (–/–) mice show that the skull is shortened in the Axin2−/− mice. Whole-mount (D,E) and skeletal staining (F–I) analyses of the cranial skull reveal abnormalities of the Axin2−/−suture at 4 weeks. The arrowheads indicate the sutures (black, metopic suture; white, jugum limitans) that lie between the nasal (N) and frontal (F) bones. The jugum limitans has disappeared and there is unilateral fusion of the metopic suture (arrows). Scale bars: 4 mm in A–C; 2 mm in D,E; 1 mm in F–I.
The metopic suture remains patent in early postnatal stages. The fusion process is initiated around 4 weeks, and completed by 7 weeks after birth. Histological analyses showed that inactivation of Axin2 induced premature closure of the metopic suture (Fig. 2). In Axin2−/− mutants, the suture remained patent at birth (Fig. 2A,B) but had been fused by postnatal day 8 (P8) (Fig. 2C–E). Unilateral or bilateral fusion of the anterior metopic suture was observed. The overlaying of cranial bones and endocranial bridging normally does not occur before the onset of suture closure, but was evident on the Axin2−/− suture at P17 (Fig. 2F,G). These data suggest that skeletogenesis was accelerated in the Axin2 mutants. This could be characterized by increases in the presence of the FGFR1-expressing osteoblast precursors in the Axin2−/− suture at P17 (Fig. 2H–K). A patent suture, which serves as an osteogenic center, is necessary for growth of the calvarial bones to accommodate postnatal expansion of the brain. As a result of the premature suture closure, the adult Axin2−/− skulls became abnormally structured and shaped. Thus, the Axin2-null mice exhibit a phenotypic defect resembling craniosynostosis in humans.
Fig. 2.
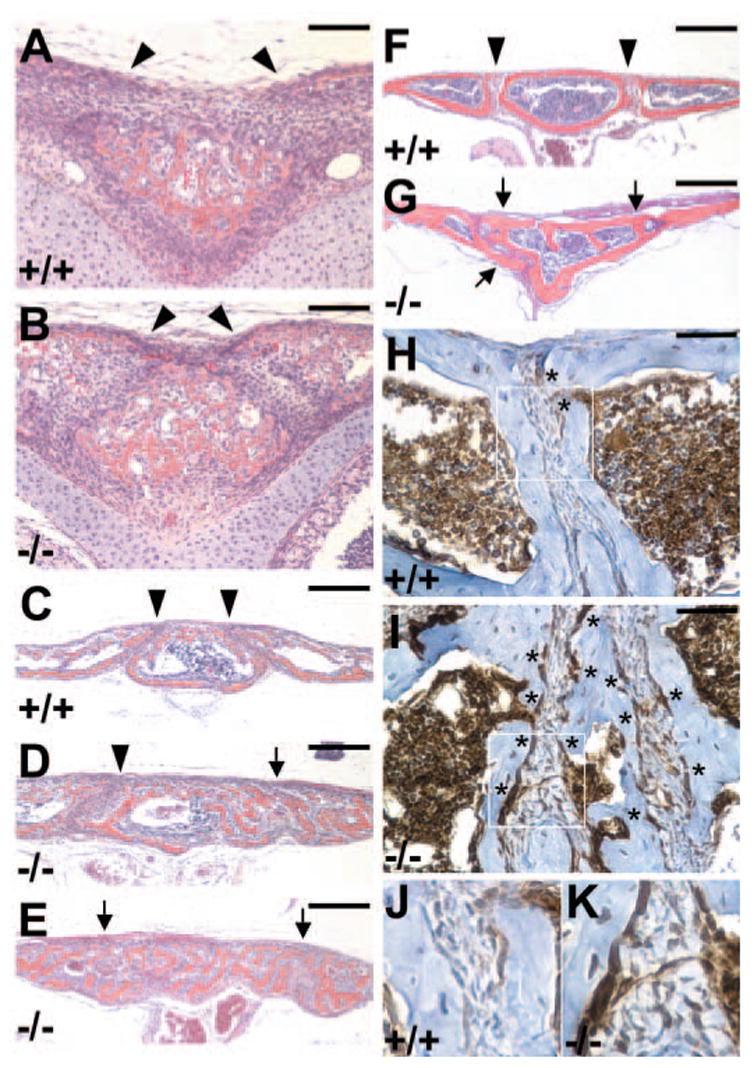
Targeted disruption of Axin2 in mice induces craniosynostosis. Histological sections show that cranial sutures are prematurely fused in the Axin2−/− mice. The sutures of control (A,C,F) and Axin2-null (B,D,E,G) mice at postnatal day 0 (A,B), 8 (C–E) and 17 (F,G) were analyzed by histology. In the controls (A,C,F), the metopic suture remains patent (indicated by arrowheads) in the first 4 weeks of postnatal development. The Axin2−/− suture did not show fusion at birth (B). However, unilateral (D) or bilateral (E) fusion of the Axin2−/− suture (arrows) occurred at day 8. The overlaying of cranial bones and endocranial bridging (arrows) is evident by day 17 (G). Compared with controls, the Axin2−/− sutures are morphologically more advanced with increased ossification (orange). The skeletogenesis stimulated in the Axin2 mutant was characterized by expression of FGFR1, a marker for osteoblast precursor (H–K). Immunohistochemical staining reveals an increase in FGFR1-expressing osteoblast precursors (asterisks) in the Axin2−/−suture at P17 (I,K). Sections were immunostained with α -FGFR1 antibody (brown) and counterstained with Hematoxylin (blue). Enlargements of the insets (H,I) are shown in J and K. Scale bars: 100 μ m in A,B; 200 μ m in C–G; 50 μ m in H,I.
Axin2 deficiency interferes with expansion of osteoprogenitors
The craniosynostosis phenotype induced by the Axin2 mutation suggests that Axin2 regulates intramembranous ossification during calvarial morphogenesis. The intramembranous ossification is regulated by cell proliferation, differentiation and apoptosis that have been implicated in the development of craniosynostosis (Ornitz and Marie, 2002). We investigated the involvement of apoptosis by TUNEL staining and did not detect any significant differences in the Axin2+/+ and Axin2−/− mice at various developmental stages or in primary cultured osteoblasts. To investigate if expansion of osteoblast precursors is affected by the Axin2 inactivation, a mitotic marker Ki67 was used to identify the proliferating cells in Axin2+/+ and Axin2−/− skulls. The lack of Axin2 drastically enhanced proliferation of osteoblast precursors in the developing metopic suture at late embryonic and early postnatal stages (Fig. 3A–C). The average percentage of proliferating cells were increased by ~1.9-fold at early postnatal stages. However, no significant changes were detected in the sagittal suture (data not shown), suggesting that Axin2 deficiency affects anterior skull development.
Fig. 3.

Axin2 deficiency stimulates expansion of osteoprogenitors. The osteoblast precursors undergoing active divisions in the Axin2+/+ (A) and Axin2−/− suture (B) were identified by the expression of Ki67. Immunohistochemical staining reveals increases in the presence of the mitotic osteoprogenitors in the Axin2−/− suture at P8. In the defined suture region (broken lines), osteoprogenitors positive (brown) and negative (blue) for Ki67 were counted to measure proliferation abnormalities. At least six different regions from the same developmental stage were analyzed to obtain the average percentage of Ki67 positive (Ki67+) cells at E16.5, P8 and P17 (C). Primary osteoblast precursors isolated from nasal and frontal bones of the Axin2+/+ (D) and Axin2−/− (E) were grown in culture media for in vitro BrdU incorporation analysis. Cells were immunostained with α -BrdU antibody (brown), and counterstained with Hematoxylin (blue). The stained images were taken randomly to determine the percentage of proliferating cells by counting the BrdU-positive cells in a total of 1000 cells. The graph shows the average percentage of Axin2+/+ and Axin2−/− in three independent experiments (F). Scale bars: 50 μ m in A,B; 200 μ m in D,E.
The emigrant CNC cells that give rise to a variety of facial tissues and structures regulate craniofacial morphogenesis. It has been shown that CNC is involved in formation of the mammalian skull (Jiang et al., 2002). The skull is formed from cranial skeletogenic mesenchyme derived from both mesoderm and neural crest, as was demonstrated by lineage tracing analysis using a genetic labeling system (Chai et al., 2000; Jiang et al., 2000). In mice carrying both the Wnt1-Cre transgene (Danielian et al., 1998) and the Cre-dependent ROSA26RlacZ reporter allele (Soriano, 1999), the lacZ reporter gene is expressed in CNC derivatives in addition to the region where Wnt1 is normally expressed. CNC contributes to the formation of the anterior skull, with the exception of the interparietal bones, which are located at the posterior skull (Jiang et al., 2002). Because anterior regions of the skull are most affected by the Axin2 mutation, the CNC-dependent skeletogenesis might be particularly sensitive to the loss of Axin2. We therefore isolated primary osteoblasts from nasal/frontal bones (CNC-derived) or parietal bones (mesoderm-derived) of the 1-day-old Axin2+/+ and Axin2−/− mouse calvaria for in vitro proliferation analyses (Fig. 4C). Owing to a contribution of CNC to the interparietal bone, it was excluded in these experiments. Indeed, only cells isolated from the nasal/frontal bones exhibited a ~2.1-fold increase in the BrdU proliferation assay (Fig. 3D–F). These data suggest that hyper proliferation of the CNC-derived osteoblast precursors contributes to the developmental defects in the Axin2−/− suture.
Fig. 4.
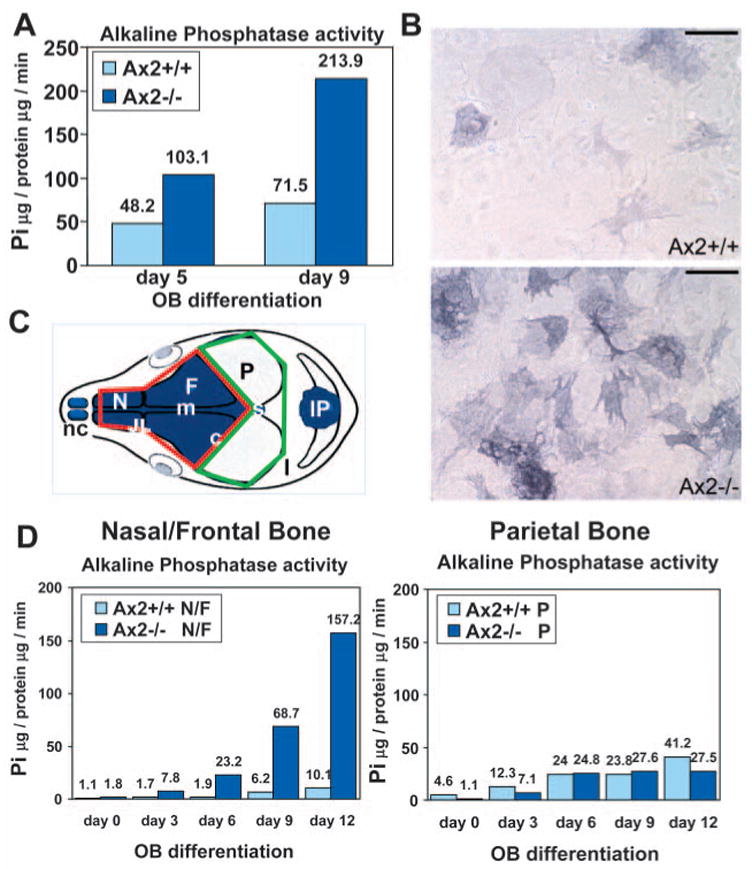
Defects of calvarial osteoblast differentiation caused by the Axin2 mutation. Primary calvarial osteoblast precursors isolated from the Axin2+/+ and Axin2−/− littermates were cultured in differentiation media for up to 9 days. Liquid (A) and histochemical (B) assays for alkaline phosphatase were performed at different time points of osteoblast (OB) differentiation as indicated. The enzyme activity is expressed as micrograms of p-nitrophenol (Pi) released per microgram of protein per minute. A diagram summarizes neural crest contribution (blue) to the skeletal elements and sutures of the mouse skull vault [diagram modified from Jiang et al. (Jiang et al., 2002)] (C). The neural crest or mesoderm-derived osteoblasts were isolated from nasal/frontal (highlighted by a red line) or parietal (highlighted by a green line) bones, respectively. c, coronal suture; F, frontal bone; IP, interparietal bone; JL, jugum limitans; l, lambdoid suture; m, metopic suture; N, nasal bone; nc, nasal cartilage; P, parietal bone; s, sagittal suture. (D) The neural crest or mesoderm-derived primary osteoblasts of Axin2+/+ and Axin2−/−were cultured in differentiation media for up to 12 days. Liquid assays for alkaline phosphatase were performed at different time points as indicated. A and D are representative of three independent experiments. Scale bar: 200 μ m in B.
The effects of Axin2 mutation on intramembranous ossification
To study osteoblast differentiation during intramembranous bone development of the cranial skull, we established an in vitro differentiation system. Primary osteoblast precursors were cultured and protein lysates of the Axin2+/+ and Axin2−/−cells were extracted at various time points during osteoblast differentiation. The activity of alkaline phosphatase, a marker for mature osteoblasts, was measured by histochemical staining and liquid assays. The Axin2 deletion had an obvious affect on osteoblast differentiation in vitro. Inactivation of Axin2 promotes osteoblast differentiation, as alkaline phosphatase activity was elevated by two- to threefold in the Axin2−/− cells (Fig. 4A). Increased numbers of cells were stained by the alkaline phosphatase histochemical analysis by day 9 of osteoblast differentiation (Fig. 4B). These findings reveal that disruption of Axin2 enhances osteoblast differentiation, which can lead to an accelerated intramembranous ossification and premature suture closure.
We next examined if the CNC-dependent osteogenesis is particularly defective in the Axin2−/− mice (Fig. 4C). The activity of alkaline phosphatase was determined in liquid assays during the course of osteoblast differentiation (days 0, 3, 6, 9 and 12) in vitro. In three independent experiments, our results demonstrated that the stimulation of osteoblast differentiation caused by the Axin2 mutation occurs predominantly in the anterior calvarium (Fig. 4D). Cells isolated from the Axin2−/− nasal/frontal bones showed an average 13-fold increase in alkaline phosphatase activity on differentiation days 6, 9 and 12, compared with controls. However, Axin2+/+ and Axin2−/− parietal osteoblasts, which originate from the mesoderm, showed similar alkaline phosphatase activities. These data suggest that osteoblasts derived from different calvarial regions possess distinct cellular and developmental properties.
To further examine the effects of the Axin2 mutation on developmental stages of the differentiated cell, the temporal expression patterns of two late-stage osteoblast specific markers, osteopontin and osteocalcin, were determined quantitatively by real time RT-PCR analyses (Fig. 5A,B). In Axin2−/− cells, the expression of osteopontin was prematurely elevated between 6 and 7.5 fold during the course of differentiation (day 3 to day 9). The level of osteocalcin was unaffected by the mutation in the early stages of the differentiation, but on differentiation day 9, it was abruptly activated in the Axin2−/− cells (~3-fold induction). The results, which agree with the data on alkaline phosphatase activity, suggest that inactivation of Axin2 promotes maturation of the CNC-derived osteoblasts. Furthermore, the developmental properties of mesoderm-derived osteoblasts are not affected significantly by the Axin2 ablation (Fig. 4D; data not shown).
Fig. 5.
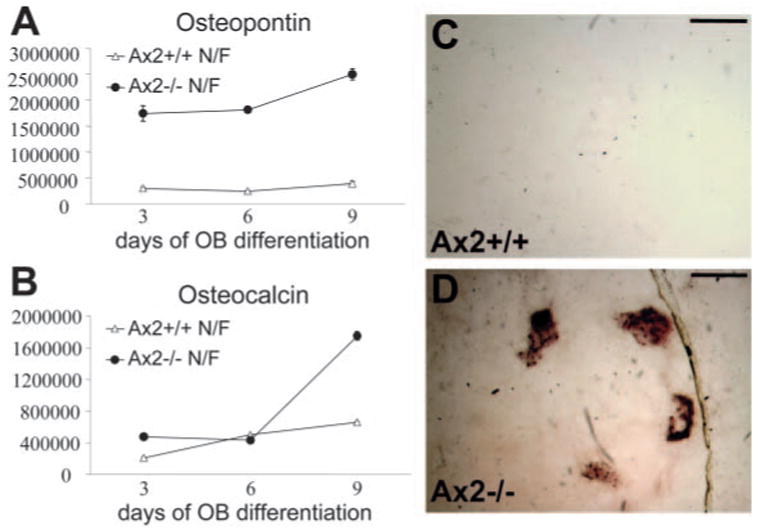
Alteration of osteoblast specific gene expression and increased mineralization by the Axin2 deletion. Primary calvarial osteoblast precursors isolated from nasal/frontal bones of the Axin2+/+ and Axin2−/− littermates were cultured in differentiation media for up to 9 days. Quantitative real time RT-PCR analyses were performed to examine expression of osteopontin and osteocalcin. The graphs represent the expression levels (in arbitrary units) of osteopontin (A) and osteocalcin (B) during the course of osteoblast differentiation. Primary calvarial osteoblasts from nasal/frontal bones of the Axin2+/+ (C) and Axin2−/− (D) littermates were maintained in differentiation media for 3 weeks. Von Kossa staining analyses show increased mineralization in Axin2−/− osteoblast cultures compared with wild-type cultures (C,D). Scale bars: 0.1 mm in C,D.
Long-term differentiation of osteoblasts in vitro induces matrix production, followed by the mineralization of the extracellular matrix. To investigate the effect of Axin2 inactivation on mineralization, primary calvarial osteoblasts isolated from nasal/frontal bones of the Axin2+/+ and Axin2−/−littermates were induced to differentiate in vitro. After 3 weeks, the cells were stained by the Von Kossa method to visualize mineralized nodules. In three independent experiments, no mineralized nodules were observed in the Axin2+/+ osteoblast cultures (Fig. 5C). However, an average of 14 nodules was formed in the Axin2−/− culture (Fig. 5D), suggesting that the formation of mineralized nodules was markedly enhanced by the loss of Axin2. The above data demonstrate the importance of Axin2 in calvarial osteoblast differentiation and strongly argue for a region-specific effect of Axin2 on cranial skeletogenesis. The ablation of Axin2 promotes differentiation of the anterior osteoblasts that are CNC derivatives.
Expression of Axin2 in cranial sutures
To investigate the role of Axin2 in cranial skull development, we then examined the expression of Axin2 using the Axin2lacZ allele (Fig. 6). β -Gal staining revealed that Axin2 is expressed in the cranial suture and bone during calvarial morphogenesis. At late embryonic and early postnatal stages, Axin2 was expressed strongly in nasal cartilages, nasal bones and cranial sutures (Fig. 6A,B,E,F). In the suture regions, Axin2 expression was mostly restricted to the osteogenic fronts and periosteum, which are enriched with pre-osteoblasts and osteoblasts by E16.5 (Fig. 6I) and P4 (Fig. 6J). The expression of Axin2 was maintained at elevated levels in the nasal bone at these stages. On day 14, Axin2 was highly expressed in the developing nasal and frontal bones, as well as the osteogenic fronts (Fig. 6C,G,K,L). By day 31, very little Axin2 expression could be detected after the metopic suture had initiated the fusion process (Fig. 6D,H). This spatial and temporal expression pattern of Axin2 correlates with the onset of premature suture closure in the Axin2−/− mice (Fig. 2), suggesting an inhibitory role of Axin2 in osteoblast development. These results demonstrate an important role of Axin2 in skull morphogenesis.
Fig. 6.
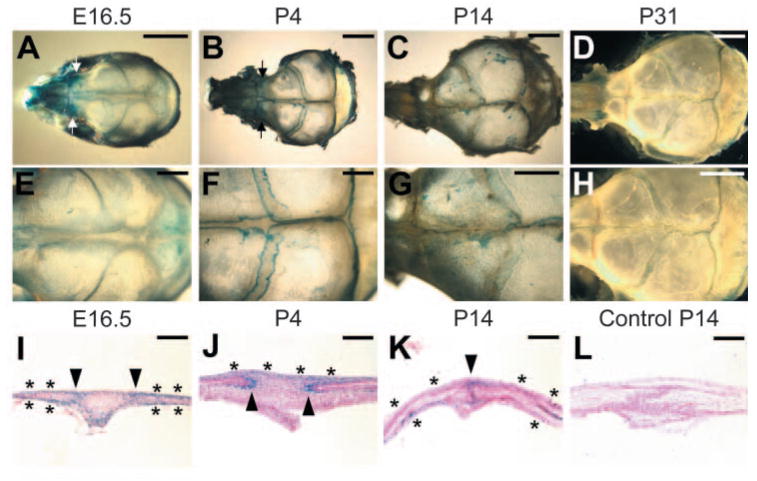
Expression of Axin2 in developing skull. The Axin2 expression pattern was visualized by β -gal staining in whole mounts (A–H) or sections (I–L). Axin2 is present in metopic and coronal sutures at E16.5 (A,E,I). Axin2 is detected at high levels in the osteogenic fronts (arrowheads), periosteum (asterisks) and mesenchyme of the sutures, as well as in the nasal bone (arrows) and nasal cartilage. At P4, Axin2 is highly expressed in the osteogenic fronts (arrowheads), periosteum (asterisks), nasal bone (arrows) and nasal cartilage (B,F,J). At P14, high levels of Axin2 are observed in nasal and frontal bones (C,G) in addition to the osteogenic fronts (arrowhead) and periosteum (asterisks; K,L). By P31, expression of Axin2 appears to be diminished to very low levels (D,H). First and second rows, the Axin2lacZ allele shows Axin2 expression. Third row, coronal sections of the β -gal stained metopic sutures, counterstained with nuclear Fast Red. Scale bars: 3 mm in A,H; 5 mm in B–D; 1 mm in E,F; 2 mm in G; 100 μ m in I–L.
β -Catenin signaling in skull development
The involvement of Axin family genes in the regulation of β -catenin stability (Behrens et al., 1998; Hsu et al., 2001; Zeng et al., 1997) implies that the Wnt pathway plays a role in calvarial morphogenesis. The inactivation of Axin2 might stimulate β -catenin signaling during cranial suture development. Therefore, we first examined whether the canonical Wnt pathway is activated during mammalian skull formation. Using transgenic mice harboring TOPGAL (DasGupta and Fuchs, 1999), a lacZ reporter under the control of a LEF/TCF and β -catenin inducible promoter, we monitored the stimulation of β -catenin and LEF/TCF signaling by the expression of lacZ in early skull development (Fig. 7). The β -catenin and LEF/TCF signaling was markedly activated in nasal cartilage and craniofacial bones at E16.5 (Fig. 7C,E), but diminished at P1 (Fig. 7D,F). In the nasal bone, this signaling pathway was activated in the medial region (Fig. 7C,D), which appeared to compliment the expression pattern of Axin2 in the adjacent lateral region (Fig. 6A,B). Both Axin2 expression and β -catenin and LEF/TCF signaling were present at moderate levels in the coronal suture at E16.5 (Fig. 6A,E and Fig. 7C,E). However, as the Axin2 expression gradually increased at P1 (data not shown) and P4 (Fig. 6B,F), the β -catenin and LEF/TCF signaling was no longer active (Fig. 7D,F). Similarly, Axin2 was expressed strongly in the metopic suture (Fig. 6A,B,E,F), where the β -catenin and LEF/TCF signaling showed no activation at these stages (Fig. 7C,D). In late osteogenesis, β -catenin signaling was stimulated in the mature osteoblasts and osteocytes where Axin2 was no longer present (data not shown). These results support the inhibitory effect of Axin2 on the canonical Wnt pathway during cranial suture and bone development.
Fig. 7.

Activation of β -catenin and LEF/TCF signaling in skull development. Mice carrying the TOPGAL transgene, capable of responding to β -catenin and LEF/TCF signaling, were used to monitor activation of this pathway by β -gal staining analyses in cranial skulls at E16.5 (C,E) and P1 (D,F). Whole-mount staining shows activation of the canonical Wnt pathway during early skull formation (C–F). Arrowheads indicate the coronal sutures and arrows indicate the activation of β -catenin and LEF/TCF signaling in medial parts of the nasal bones. The non-transgenic mice, with no background staining, indicate the specificity of the assay (A,B). Scale bars: 2 mm in A–D; 500 μ m in E,F.
The Wnt-Axin regulatory network in calvarial morphogenesis
To investigate whether the Wnt/β -catenin pathway is inappropriately activated in the Axin2−/− suture, immunostaining analyses were performed with a monoclonal antibody α -ABC. This antibody recognizes only the non-phosphorylated form of β -catenin at (Ser37 and Thr41 residues), which activates downstream signaling events (van Noort et al., 2002). β -Catenin signaling was highly activated in the skeletogenic mesenchyme and periosteum of the Axin2−/−sutures, which exhibit premature closure, but not in the Axin2+/+ patent sutures (Fig. 8A–D). Activation of β -catenin signaling is accompanied by stimulated expression of its downstream target cyclin D1 that is likely to mediate the effects of Axin2 on expansion of osteoprogenitors (Fig. 8E,F).
Fig. 8.
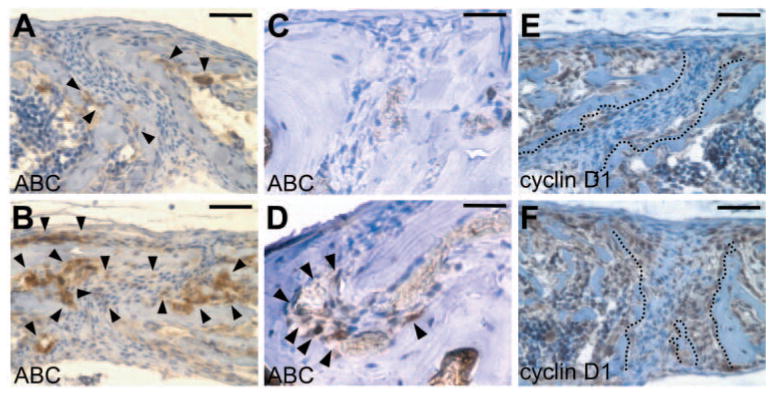
Alteration of the canonical Wnt pathway in the Axin2 mutants during skull morphogenesis. Sections of the Axin2+/+ (A,C,E) and Axin2−/− (B,D,F) sutures were immunostained with the primary antibody (brown) and counterstained with Hematoxylin (blue). Immunohistochemical staining with an antibody that recognizes only the activated form of β -catenin (α -ABC antibody) reveals stimulation of β -catenin signaling (arrowheads) in the Axin2−/− suture at P8 (B) and P28 (D). Expression of cyclin D1, a bona fide target of β -catenin, is also elevated in the Axin2−/− suture (broken lines) at P8 (F). Scale bars: 50 μ m in A,B,E,F; 25μ m in C,D.
We next examined whether cellular levels of the activated form of β -catenin were affected by the Axin2 mutation in calvarial osteoblasts during the course of differentiation in vitro. β -Catenin signaling was apparently activated at a low, but steady level, in the wild-type cells (Fig. 9A). Inactivation of Axin2 caused an abrupt activation of the Wnt/β -catenin pathway in the mutant osteoblasts at day 5 (Fig. 9A). Furthermore, this was primarily due to an effect of Axin2 on the CNC-derived osteoblasts as β -catenin signaling is drastically elevated (Fig. 9A).
Fig. 9.
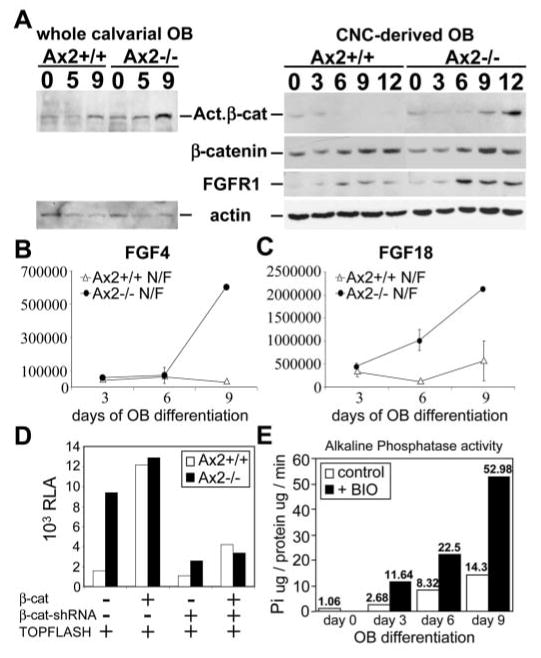
Stimulation of β -catenin signaling promotes osteoblast differentiation. Whole calvarial (A, left panel) or CNC-derived (A, right panel) osteoblast precursors, isolated from the Axin2+/+ and Axin2−/− littermates, were cultured in differentiation media for up to 12 days. Lysates, isolated at different differentiation days as indicated, were first analyzed by the alkaline phosphatase liquid assay (shown in Fig. 4A,D). The same lysates were then used for immunoblotting (A). Immunoblot analyses with the α -ABC (activated) and α -β -catenin (total) antibodies reveal that β -catenin signaling is significantly stimulated by inactivation of Axin2 during osteoblast differentiation. The expression of FGFR1, which is increased upon differentiation, is elevated in the Axin2−/− cells. The level of actin was also analyzed as a control for protein content of the lysates. (B,C) Quantitative real-time RT-PCR analyses were performed to examine expression of two Wnt targets FGF4 and FGF18 in the CNC-derived osteoblasts. The graphs represent the expression levels (in arbitrary units) of FGF4 (B) and FGF18 (C) during the course of differentiation in vitro. (D) The CNC-derived osteoblasts of Axin2+/+ and Axin2−/− were cultured in differentiation media for 7 days. The TOPFLASH reporter plasmid was then transfected by lipofectamine with different combinations of the β -catenin expression (β -cat) (Korinek et al., 1997), β -catenin RNA interference (β -cat-shRNA) (Cellogenetics) and pUC19 carrier plasmids, as indicated (0.5 μ g for each plasmid plus the carrier to a total of 1.5 μ g DNA). Relative luciferase activity (RLA) for each sample was determined after 48 hours. (E) Activation of β -catenin signaling by BIO stimulates osteoblast differentiation. Primary osteoblast precursors isolated from nasal and frontal bones were cultured in differentiation media with or without 2 μ M BIO for up to 9 days. Liquid assays for alkaline phosphatase were performed at different time points as indicated. The diagram is representative of three independent experiments.
To determine if activation of β -catenin in the Axin2−/− osteoblasts induced transcriptional activation of the canonical Wnt pathway, the TOPFLASH reporter plasmid was transfected into the differentiating primary osteoblasts. The β -catenin and LEF/TCF-dependent transcription was highly stimulated in the Axin2-null mutants (Fig. 9D). Additional expression of β -catenin enhanced the transcriptional activity only in the control cells, but had minimal effects on the mutants. Transfection of a β -catenin RNA interference plasmid to knockdown its expression diminished the TOPFLASH activity in the Axin2-null cells, suggesting that β -catenin is responsible for the transcriptional activation. Using quantitative real-time RT-PCR, we then tested for direct targets of β -catenin and LEF/TCF affected by the Axin2 mutation during osteoblast differentiation. Among the genes we examined, expression of FGF18 and FGF4 was drastically elevated in the Axin2−/− cells by differentiation day 6 and day 9, respectively (Fig. 9B,C). Together with stimulation of FGFR1 in the Axin2 mutants, these data imply a role of FGF signaling in the Wnt-Axin regulatory network during calvarial morphogenesis. Our studies demonstrated that the anterior skull defects caused by the Axin2 mutation are mediated through its effects on the canonical Wnt pathway. The ablation of Axin2 promotes differentiation of the CNC-derived osteoblasts that results in premature suture fusion in the Axin2−/− mice.
Finally, we investigated if stimulation of β -catenin signaling is sufficient to promote osteoblast differentiation. A pharmacological compound BIO (Meijer et al., 2003) was added to the calvarial osteoblast cultures in vitro. BIO is a potent GSK-3 specific inhibitor, which has been shown to induce axis formation in frogs (Meijer et al., 2003) and maintain pluripotency in mouse and human embryonic stem cells (Sato et al., 2004) through activation of Wnt signaling. The control culture without BIO showed a gradual increase in alkaline phosphatase activity during the course of differentiation (Fig. 9E). However, the addition of BIO greatly accelerated osteoblast differentiation, as alkaline phosphatase activity was elevated at ~three to fourfold on days 3, 6 and 9 (Fig. 9E). Therefore, stimulation of β -catenin signaling is sufficient to promote osteoblast differentiation. Our studies strongly support a significant role of the Wnt-Axin regulatory network in cranial skeletogenesis and craniosynostosis.
Discussion
Although human genetic analyses have linked craniosynostosis to mutations in the FGFR family (Burke et al., 1998), MSX2 (Jabs et al., 1993) and TWIST genes (el Ghouzzi et al., 1997; Howard et al., 1997), they only account for ~20% of clinical cases (Wilkie and Morriss-Kay, 2001). The molecular mechanisms underlying suture development remain largely unknown. Here, we show that disruption of Axin2 in mice induces premature suture closure, a phenotype resembling craniosynostosis in humans. Craniosynostosis is the major developmental disorder of the skull vault and is often associated with difficulties in vision, hearing and breathing (Wilkie and Morriss-Kay, 2001). In the adult Axin2 mutants, we have frequently noticed eye abnormalities that might be caused by the skull deformity with increased intracranial pressure. The spatial and temporal expression pattern of Axin2 in skull development correlates with craniofacial abnormalities caused by its ablation. Axin2 is expressed in osteogenic fronts and periosteum of the developing suture during skull formation. However, its expression gradually diminishes before the metopic suture has initiated its normal fusion processes. This might permit the suture fusion to occur slowly at early postnatal stages. By contrast, the lack of Axin2 greatly accelerates the fusion processes. A recent study, which linked mutations in AXIN2 to familial tooth agenesis/oligodontia in humans, also suggests that modulating the intensity of cellular signaling is necessary during specific stages of organ development (Lammi et al., 2004). Aulehla et al. (Aulehla et al., 2003) have proposed that the role of Axin2 as a negative feedback-regulator of Wnt/β -catenin signaling is crucial for segmentation during vertebrate development. However, our results show that the ablation of Axin2 has no discernible event on segmentation.
The expression of Axin2 in the pre-osteoblasts and osteoblasts is consistent with its role in intramembranous bone development. Targeted disruption of Axin2 enhances expansion of osteoprogenitors and facilitates osteoblast differentiation in vivo and in vitro, suggesting a mechanism of craniosynostosis. Based on the expression and the loss-of-function analyses in vivo and in vitro, we demonstrate that Axin2 is required for proper development of the calvarial osteoblast. Furthermore, stimulation of β -catenin signaling is not only necessary but also sufficient to promote osteoblast maturation. As a negative regulator in the osteoblasts, Axin2 prevents their development and premature suture closure, thereby controlling the timing of these developmental processes. This raises the possibility that therapeutic Axin2 could be exploited to correct defects in postnatal skull development.
The enhanced β -catenin signaling induced by the Axin2 mutation during cranial suture and bone development suggests a novel role of the canonical Wnt pathway in skull morphogenesis. Axin2 has been suggested to function as a negative-feedback regulator for Wnt (Jho et al., 2002; Lustig et al., 2002). This raises the issue of whether Axin2 is regulating the response to a Wnt signal, or the stability of β -catenin in the absence of Wnts. Based on the analyses in TOPGAL mice, β -catenin signaling is active during calvarial morphogenesis, even though there is a lack of information on the expression of Wnts. Axin2 is constantly present between E16.5 and P4 in both metopic and coronal sutures. However, TOPGAL transgene was transiently active in the coronal suture, and never showed activity in the metopic suture. These differences might imply suture specificity in skull development. Therefore, it is possible that Axin2 is modulating the response to a Wnt signal in the coronal suture, whereas this mechanism seems unlikely to occur in the metopic suture.
The neural crest dependent skeletogenesis is particularly sensitive to the loss of Axin2. Inactivation of Axin2 apparently stimulates development of the CNC-derived osteoblasts. By contrast, mesoderm-derived osteoblasts of the parietal bone are not significant affected by the Axin2 mutation. This region-specific effect on calvarial osteoblast development suggests that Axin2 is crucial for the CNC dependent skeletogenesis. Even though Axin2 is highly activated in migratory neural crest cells in the cranial and trunk regions (W.H., unpublished), it seems to be dispensable for most of the CNC development. This could be due to a redundant function of Axin1 and Axin2. When the Axin2−/− mice were crossed into the AxinTg1 (a Axin1-null) background, a genetic interaction between these two genes in early craniofacial morphogenesis was revealed (B.J., W.H., F.C. and W.B., unpublished). Axin2−/− embryos in the Axin1+/− background exhibited severe abnormalities in craniofacial regions. Together with the present study, our results reveal the importance of Axin1 and Axin2 in CNC development. Craniofacial development, especially the neural crest derived tissues and structures, is particularly sensitive to the loss of the Axin family genes.
The canonical Wnt pathway is intimately involved in the CNC and craniofacial development during early embryogenesis, as demonstrated by mutations affecting β -catenin signaling (Brault et al., 2001; Hasegawa et al., 2002; Ikeya et al., 1997; Mitchell et al., 2001). Using a genetic labeling system, CNC was further shown to derive from the Wnt1-expressing neural progenitor (Chai et al., 2000; Jiang et al., 2000). However, the role of Wnt/β -catenin signaling in craniofacial bone development remained elusive. Our present study shows that stimulation of β -catenin signaling occurs not only in the Axin2−/− suture displaying premature fusion, but also in the Axin2−/− cells undergoing intramembranous ossification. These data suggest that the inhibition of suture closure and osteoblast development by the Axin family genes is mediated through the regulation of β -catenin signaling, implying a novel role for this signaling pathway in cranial skeletogenesis. Indeed, the β -catenin and LEF/TCF mediated transcription is highly elevated during normal skull formation. Interestingly, activation of this signaling pathway seems to occur in a temporally and spatially restricted pattern that is the reverse of the Axin2 expression pattern. In the anterior cranium, Axin2 is expressed in the area immediately adjacent to where β -catenin signaling is stimulated during late embryogenesis. As the expression of Axin2 is enhanced at early postnatal stages, β -catenin signaling becomes inactivated. Furthermore, activation of β -catenin signaling is necessary and sufficient to induce intramembranous ossification. Although it remains to be determined whether stimulation of β -catenin signaling leads to craniosynostosis in mice, these results strongly support the hypothesis that the presence of Axin2 antagonizes β -catenin signaling to inhibit intramembranous ossification and prevent suture closure.
In addition to the origin of calvarial osteoblasts, the region-specific effect may be attributed to differences in fundamental properties between anterior and posterior parts of the cranium. The metopic suture fuses in the first 45 days of life, whereas the sagittal suture remains patent. It has been suggested that differential activation of FGF2, which inhibits the bone morphogenetic protein (BMP) antagonist Noggin, might be responsible for normal closure of the metopic suture (Warren et al., 2003). BMP belongs to the transforming growth factor β (TGFβ) superfamily, which plays an important role in bone morphogenesis (McCarthy et al., 2000; Serra and Chang, 2003). Targeted disruption of Axin2 apparently interferes with cellular signaling of the TGFβ superfamily (H.-M.Y. and W.H., unpublished). This raises the possibility that Axin2 interacts with the TGF-β /BMP pathways. It has been suggested that Axin1/Axin2 binds directly to Smad2/3 to stimulates TGFβ signaling (Furuhashi et al., 2001). Wnt signaling also has been shown to coordinately regulate expression of the BMP target gene Msx2 (Hussein et al., 2003). As activation of Msx2 has been associated with craniosynostosis, inactivation of Axin2 might induce this synergistic effect of Wnt and BMP. Finally, as direct targets of Wnt, FGF4 (Kratochwil et al., 2002) and FGF18 (Shimokawa et al., 2003) are stimulated by the Axin2 inactivation. The former is required for the downstream events mediated by Wnt in odontogenesis (Kratochwil et al., 2002), whereas the latter is important for osteogenesis and chondrogenesis (Liu et al., 2002; Ohbayashi et al., 2002). It remains to be elucidated whether FGF singling mediates the effect of Axin2 during calvarial morphogenesis. Future studies focused on delineating the interplay of these cellular signaling pathways promise new insights into the calvarial morphogenetic regulatory mechanism, and the molecular basis of craniosynostosis.
Footnotes
We thank Hiromu Ito for technical advice, Krystal Benyamein for technical assistance, and Hans Clevers and Ali H. Brivanlou for reagents. This work was supported in part by a grant (CA106308) from the National Institutes of Health to W.H.
References
- Anderson DJ. Genes, lineages and the neural crest: a speculative review. Philos Trans R Soc Lond B Biol Sci. 2000;355:953–964. doi: 10.1098/rstb.2000.0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulehla A, Wehrle C, Brand-Saberi B, Kemler R, Gossler A, Kanzler B, Herrmann BG. Wnt3a plays a major role in the segmentation clock controlling somitogenesis. Dev Cell. 2003;4:395–406. doi: 10.1016/s1534-5807(03)00055-8. [DOI] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280:596–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- Brannon M, Gomperts M, Sumoy L, Moon RT, Kimelman D. A beta-catenin/XTcf-3 complex binds to the siamois promoter to regulate dorsal axis specification in Xenopus. Genes Dev. 1997;11:2359–2370. doi: 10.1101/gad.11.18.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Burke D, Wilkes D, Blundell TL, Malcolm S. Fibroblast growth factor receptors: lessons from the genes. Trends Biochem Sci. 1998;23:59–62. doi: 10.1016/s0968-0004(97)01170-5. [DOI] [PubMed] [Google Scholar]
- Chai Y, Jiang X, Ito Y, Bringas P, Jr, Han J, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127:1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- Cohen MM, MacLean RE. Craniosynostosis: Diagnosis, Evaluation, and Management. New York: Oxford University Press; 2000. [Google Scholar]
- Couly G, Grapin-Botton A, Coltey P, le Douarin NM. The regeneration of the cephalic neural crest, a problem revisited: the regenerating cells originate from the contralateral or from the anterior and posterior neural fold. Development. 1996;122:3393–3407. doi: 10.1242/dev.122.11.3393. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Moon RT, Raible DW. Control of neural crest cell fate by the Wnt signalling pathway. Nature. 1998;396:370–373. doi: 10.1038/24620. [DOI] [PubMed] [Google Scholar]
- el Ghouzzi V, le Merrer M, Perrin-Schmitt F, Lajeunie E, Benit P, Renier D, Bourgeois P, Bolcato-Bellemin AL, Munnich A, Bonaventure J. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997;15:42–46. doi: 10.1038/ng0197-42. [DOI] [PubMed] [Google Scholar]
- Fagotto F, Jho E, Zeng L, Kurth T, Joos T, Kaufmann C, Costantini F. Domains of axin involved in protein-protein interactions, Wnt pathway inhibition, and intracellular localization. J Cell Biol. 1999;145:741–756. doi: 10.1083/jcb.145.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr GH, 3rd, Ferkey DM, Yost C, Pierce SB, Weaver C, Kimelman D. Interaction among GSK-3, GBP, axin, and APC in Xenopus axis specification. J Cell Biol. 2000;148:691–702. doi: 10.1083/jcb.148.4.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis-West P, Ladher R, Barlow A, Graveson A. Signalling interactions during facial development. Mech Dev. 1998;75:3–28. doi: 10.1016/s0925-4773(98)00082-3. [DOI] [PubMed] [Google Scholar]
- Furuhashi M, Yagi K, Yamamoto H, Furukawa Y, Shimada S, Nakamura Y, Kikuchi A, Miyazono K, Kato M. Axin facilitates Smad3 activation in the transforming growth factor beta signaling pathway. Mol Cell Biol. 2001;21:5132–5141. doi: 10.1128/MCB.21.15.5132-5141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BK. Bone. Caldwell, NJ: Telford Press; 1990. [Google Scholar]
- Hasegawa S, Sato T, Akazawa H, Okada H, Maeno A, Ito M, Sugitani Y, Shibata H, Miyazaki Ji J, Katsuki M, et al. Apoptosis in neural crest cells by functional loss of APC tumor suppressor gene. Proc Natl Acad Sci USA. 2002;99:297–302. doi: 10.1073/pnas.012264999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgepeth CM, Deardorff MA, Rankin K, Klein PS. Regulation of glycogen synthase kinase 3beta and downstream Wnt signaling by axin. Mol Cell Biol. 1999;19:7147–7157. doi: 10.1128/mcb.19.10.7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI, Garcia Delgado C, Gonzalez-Ramos M, Kline AD, Jabs EW. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15:36–41. doi: 10.1038/ng0197-36. [DOI] [PubMed] [Google Scholar]
- Hsu W, Zeng L, Costantini F. Identification of a domain of Axin that binds to the serine/threonine protein phosphatase 2A and a self-binding domain. J Biol Chem. 1999;274:3439–3445. doi: 10.1074/jbc.274.6.3439. [DOI] [PubMed] [Google Scholar]
- Hsu W, Shakya R, Costantini F. Impaired mammary gland and lymphoid development caused by inducible expression of Axin in transgenic mice. J Cell Biol. 2001;155:1055–1064. doi: 10.1083/jcb.200107066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein SM, Duff EK, Sirard C. Smad4 and beta-catenin co-activators functionally interact with lymphoid-enhancing factor to regulate graded expression of Msx2. J Biol Chem. 2003;278:48805–48814. doi: 10.1074/jbc.M305472200. [DOI] [PubMed] [Google Scholar]
- Ikeya M, Lee SM, Johnson JE, McMahon AP, Takada S. Wnt signalling required for expansion of neural crest and CNS progenitors. Nature. 1997;389:966–970. doi: 10.1038/40146. [DOI] [PubMed] [Google Scholar]
- Itoh K, Krupnik VE, Sokol SY. Axis determination in Xenopus involves biochemical interactions of axin, glycogen synthase kinase 3 and beta-catenin. Curr Biol. 1998;8:591–594. doi: 10.1016/s0960-9822(98)70229-5. [DOI] [PubMed] [Google Scholar]
- Jabs EW, Muller U, Li X, Ma L, Luo W, Haworth IS, Klisak I, Sparkes R, Warman ML, Mulliken JB, et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell. 1993;75:443–450. doi: 10.1016/0092-8674(93)90379-5. [DOI] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241:106–116. doi: 10.1006/dbio.2001.0487. [DOI] [PubMed] [Google Scholar]
- Julius MA, Schelbert B, Hsu W, Fitzpatrick E, Jho E, Fagotto F, Costantini F, Kitajewski J. Domains of axin and disheveled required for interaction and function in wnt signaling [In Process Citation] Biochem Biophys Res Commun. 2000;276:1162–1169. doi: 10.1006/bbrc.2000.3607. [DOI] [PubMed] [Google Scholar]
- Kikuchi A. Regulation of beta-catenin signaling in the Wnt pathway. Biochem Biophys Res Commun. 2000;268:243–248. doi: 10.1006/bbrc.1999.1860. [DOI] [PubMed] [Google Scholar]
- Kishida S, Yamamoto H, Ikeda S, Kishida M, Sakamoto I, Koyama S, Kikuchi A. Axin, a negative regulator of the wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of beta-catenin. J Biol Chem. 1998;273:10823–10826. doi: 10.1074/jbc.273.18.10823. [DOI] [PubMed] [Google Scholar]
- Kontges G, Lumsden A. Rhombencephalic neural crest segmentation is preserved throughout craniofacial ontogeny. Development. 1996;122:3229–3242. doi: 10.1242/dev.122.10.3229. [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Kratochwil K, Galceran J, Tontsch S, Roth W, Grosschedl R. FGF4, a direct target of LEF1 and Wnt signaling, can rescue the arrest of tooth organogenesis in Lef1(−/−) mice. Genes Dev. 2002;16:3173–3185. doi: 10.1101/gad.1035602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBonne C, Bronner-Fraser M. Molecular mechanisms of neural crest formation. Annu Rev Cell Dev Biol. 1999;15:81–112. doi: 10.1146/annurev.cellbio.15.1.81. [DOI] [PubMed] [Google Scholar]
- Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, Pirinen S, Nieminen P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74:1043–1050. doi: 10.1086/386293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin N, Kalcheim C. The Neural Crest. Cambridge, UK: Cambridge University Press; 1999. [Google Scholar]
- Li L, Yuan H, Weaver CD, Mao J, Farr GH, 3rd, Sussman DJ, Jonkers J, Kimelman D, Wu D. Axin and Frat1 interact with dvl and GSK, bridging Dvl to GSK in Wnt-mediated regulation of LEF- 1. EMBO J. 1999;18:4233–4240. doi: 10.1093/emboj/18.15.4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002;16:859–869. doi: 10.1101/gad.965602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, van de Wetering M, Clevers H, Schlag PM, Birchmeier W, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22:1184–1193. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansukhani A, Bellosta P, Sahni M, Basilico C. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J Cell Biol. 2000;149:1297–1308. doi: 10.1083/jcb.149.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy TL, Ji C, Centrella M. Links among growth factors, hormones, and nuclear factors with essential roles in bone formation. Crit Rev Oral Biol Med. 2000;11:409–422. doi: 10.1177/10454411000110040201. [DOI] [PubMed] [Google Scholar]
- McMahon AP, Bradley A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62:1073–1085. doi: 10.1016/0092-8674(90)90385-r. [DOI] [PubMed] [Google Scholar]
- McMahon AP, Joyner AL, Bradley A, McMahon JA. The midbrain-hindbrain phenotype of Wnt-1-/Wnt-1- mice results from stepwise deletion of engrailed-expressing cells by 9.5 days postcoitum. Cell. 1992;69:581–595. doi: 10.1016/0092-8674(92)90222-x. [DOI] [PubMed] [Google Scholar]
- Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Miller JR, Hocking AM, Brown JD, Moon RT. Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene. 1999;18:7860–7872. doi: 10.1038/sj.onc.1203245. [DOI] [PubMed] [Google Scholar]
- Mitchell KJ, Pinson KI, Kelly OG, Brennan J, Zupicich J, Scherz P, Leighton PA, Goodrich LV, Lu X, Avery BJ, et al. Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat Genet. 2001;28:241–249. doi: 10.1038/90074. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science. 2002;296:1644–1646. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- Ohbayashi N, Shibayama M, Kurotaki Y, Imanishi M, Fujimori T, Itoh N, Takada S. FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 2002;16:870–879. doi: 10.1101/gad.965702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- Parr BA, Shea MJ, Vassileva G, McMahon AP. Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development. 1993;119:247–261. doi: 10.1242/dev.119.1.247. [DOI] [PubMed] [Google Scholar]
- Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis–a look outside the nucleus. Science. 2000;287:1606–1609. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- Sakanaka C, Weiss JB, Williams LT. Bridging of beta-catenin and glycogen synthase kinase-3beta by axin and inhibition of beta-catenin-mediated transcription. Proc Natl Acad Sci USA. 1998;95:3020–3023. doi: 10.1073/pnas.95.6.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- Selby PB. A rapid method for preparing high quality alizarin stained skeletons of adult mice. Stain Technol. 1987;62:143–146. doi: 10.3109/10520298709107984. [DOI] [PubMed] [Google Scholar]
- Serra R, Chang C. TGF-beta signaling in human skeletal and patterning disorders. Birth Defects Res Part C Embryo Today. 2003;69:333–351. doi: 10.1002/bdrc.10023. [DOI] [PubMed] [Google Scholar]
- Shimokawa T, Furukawa Y, Sakai M, Li M, Miwa N, Lin YM, Nakamura Y. Involvement of the FGF18 gene in colorectal carcinogenesis, as a novel downstream target of the beta-catenin/T-cell factor complex. Cancer Res. 2003;63:6116–6120. [PubMed] [Google Scholar]
- Smalley MJ, Sara E, Paterson H, Naylor S, Cook D, Jayatilake H, Fryer LG, Hutchinson L, Fry MJ, Dale TC. Interaction of axin and Dvl-2 proteins regulates Dvl-2-stimulated TCF-dependent transcription. EMBO J. 1999;18:2823–2835. doi: 10.1093/emboj/18.10.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Takada S, Stark KL, Shea MJ, Vassileva G, McMahon JA, McMahon AP. Wnt-3a regulates somite and tailbud formation in the mouse embryo. Genes Dev. 1994;8:174–189. doi: 10.1101/gad.8.2.174. [DOI] [PubMed] [Google Scholar]
- Trainor P, Krumlauf R. Development. Riding the crest of the Wnt signaling wave. Science. 2002;297:781–783. doi: 10.1126/science.1075454. [DOI] [PubMed] [Google Scholar]
- van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H. Wnt signaling controls the phosphorylation status of beta-catenin. J Biol Chem. 2002;277:17901–17905. doi: 10.1074/jbc.M111635200. [DOI] [PubMed] [Google Scholar]
- Warren SM, Brunet LJ, Harland RM, Economides AN, Longaker MT. The BMP antagonist noggin regulates cranial suture fusion. Nature. 2003;422:625–629. doi: 10.1038/nature01545. [DOI] [PubMed] [Google Scholar]
- Whiting J, Marshall H, Cook M, Krumlauf R, Rigby PW, Stott D, Allemann RK. Multiple spatially specific enhancers are required to reconstruct the pattern of Hox-2.6 gene expression. Genes Dev. 1991;5:2048–2059. doi: 10.1101/gad.5.11.2048. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Morriss-Kay GM. Genetics of craniofacial development and malformation. Nat Rev Genet. 2001;2:458–468. doi: 10.1038/35076601. [DOI] [PubMed] [Google Scholar]
- Yanagawa S, van Leeuwen F, Wodarz A, Klingensmith J, Nusse R. The dishevelled protein is modified by wingless signaling in Drosophila. Genes Dev. 1995;9:1087–1097. doi: 10.1101/gad.9.9.1087. [DOI] [PubMed] [Google Scholar]
- Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL, 3rd, Lee JJ, Tilghman SM, Gumbiner BM, Costantini F. The mouse Fused locus encodes Axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell. 1997;90:181–192. doi: 10.1016/s0092-8674(00)80324-4. [DOI] [PubMed] [Google Scholar]