

Abstract
Although respiratory sinus arrhythmia (RSA) is a commonly quantified physiological variable, the methods for quantification are not consistent. This manuscript questions the assumption that respiration frequency needs to be manipulated or monitored to generate an accurate measure of RSA amplitude. A review of recent papers is presented that contrast RSA amplitude with measures that use respiratory parameters to adjust RSA amplitude. In addition, data from two studies are presented to evaluate empirically both the relation between RSA amplitude and respiration frequency and the covariation between RSA frequency and respiration frequency. The literature review demonstrates similar findings between both classes of measures. The first study demonstrates, during spontaneous breathing without task demands, that there is no relation between respiration frequency and RSA amplitude and that respiration frequency can be accurately derived from the heart period spectrum (i.e., frequency of RSA). The second study demonstrates that respiration frequency is unaffected by atropine dose, a manipulation that systematically mediates the amplitude of RSA, and that the tight linkage between the RSA frequency and respiration frequency is unaffected by atropine. The research shows that the amplitude of RSA is not affected by respiration frequency under either baseline conditions or vagal manipulation via atropine injection. Respiration frequency is therefore unlikely to be a concern under these conditions. Research examining conditions that produce (causal) deviations from the intrinsic relation between respiratory parameters and the amplitude of RSA combined with appropriate statistical procedures for understanding these deviations are necessary.
Keywords: Cardiac vagal control, Cardiac vagal tone, Respiratory sinus arrhythmia, Heart rate variability, Methodology, Vagus, Respiration
Methodological Issues in the Quantification of Respiratory Sinus Arrhythmia
Respiratory sinus arrhythmia (RSA) is a frequently quantified and naturally occurring rhythm in the beat-to-beat heart rate pattern. RSA is characterized by periodic increases and decreases in heart rate that occur at a frequency similar to respiration. Although respiratory sinus arrhythmia is a commonly used variable in psychophysiological research, the methods for quantification are not standardized. Currently, several strategies are used to quantify RSA (see Berntson et al, 1997). Implicit in these strategies are different assumptions regarding the operational definition of RSA and the dependence of RSA on respiration. For example, if one accepts the literal definition of RSA as a sinus rhythm coincident with breathing (i.e., acceleration during inhalation, deceleration during exhalation), then RSA can only occur in the presence of breathing. Historically, before knowledge of the neuroanatomical structures and neurophysiological mechanisms involved in regulating RSA was available, RSA was an observed phenomenon described by the measurement of heart rate and respiration. However, as knowledge of the mechanisms involved in regulating RSA became known, alternative operational definitions of RSA and measurement strategies were developed.
Based on our current understanding of the neurophysiology of RSA, this manuscript questions the need to manipulate or monitor respiration in order to generate an accurate measure of RSA. Using information obtained from both current literature and data presented from two studies, this manuscript is structured to answer four empirical questions. First, is the frequency of spontaneous breathing embedded in the beat-to-beat heart period as the frequency of RSA? Second, is the amplitude of RSA related to the frequency of spontaneous breathing? Third, does manipulating the amplitude of RSA (i.e., atropine) result in a convergent adjustment in respiration frequency? Fourth, is a time domain method of quantifying RSA, which extracts only the heart rate variability within a defined band of breathing frequencies, equivalent to values derived with spectral analyses?
Methods used to quantify RSA can be organized along a continuum of the assumed causal influence that breathing has on RSA. Table 1 provides an example of how these methods could be categorized. For example, a causal model might consist of pacing breathing at different frequencies and measuring the amplitude of RSA at each breathing frequency. A parallel output model might assume a common brainstem modulator of a respiratory rhythm that outputs independently to both the pulmonary system and the sinoatrial node (e.g., Eckberg, 2003). Consistent with the parallel model, the peak of the spectrum within the frequencies of spontaneous breathing would be used to quantify RSA. Other parallel approaches may be solely descriptive and quantify the variance in the heart rate time series (e.g., range, standard deviation, log variance, mean successive difference, or mean square successive difference).
Table 1.
Implicit neurophysiological models.
| Assumed relation between breathing and RSA | Method used to quantify RSA |
|---|---|
| Causal | Pace respiratory parameters |
| Causal | Statistically adjust using respiratory parameters as covariates |
| Causal | On a breath-by-breath basis apply respiratory parameters to identify features in heart rate pattern (i.e., peak-to-valley) |
| Parallel | Use respiratory frequency to identify frequency component in the heart rate spectrum |
| Parallel | Use global frequency bands associated with breathing rates to identify frequency band in the heart rate spectrum |
| Parallel | Global measures of heart rate variability without concern for respiration frequency |
A model focusing on the neural genesis of RSA, suggests alternative quantification strategies. This model assumes that RSA and respiratory frequency are parallel outputs of a “common cardiopulmonary oscillator” (see Eckberg, 2003; Richter & Spyer, 1990). Accordingly, the frequency for this “common cardiopulmonary oscillatory” is an emergent property of a complex neural circuit that includes brainstem structures, parallel vagal efferents to the sinoatrial node and the bronchi, and afferent feedback from the myocardium, chemoreceptors, baroreceptors, and stretch receptors (e.g., Porges, 1995). This neural model is consistent with several studies (e.g., Harper, Woo, & Alger, 2000; McAllen & Spyer, 1978; Richter & Spyer, 1990) that have described the neural mechanisms of a brainstem rhythm that regulates both respiration frequency and RSA. Empirically this may be observed as vagal efferent pathways conveying a respiratory rhythm to both the bronchi (Haselton et al., 1992) and the sinoatrial node (Jordan et al., 1982). Data from electrophysiological studies have been so consistent that functional properties including a respiratory rhythm have been used to determine when a neuron is vagal cardioinhibitory (Gilbey et al., 1983; Jordan et al., 1982).
Based on this neural model, the frequency of RSA would be the same as the frequency of respiration. Thus, respiration frequency information could be gained from quantifying the periodic processes observed in the heart rate data (i.e., the periodicity of RSA would be at the same frequency as respiration). The neural model would also argue that paced breathing might create confounds in the interpretation of both the frequency and amplitude components of RSA. The argument rests primarily on the atypical manner in which paced breathing would challenge the normal feedback from the periphery. Paced breathing would drive the feedback system via the potent stretch receptors. It might compromise the normal feedback regulating the frequency of respiration and the vagal efferent tone to the heart by changing inspiration/expiration ratios, respiratory resistance, and blood gases. In addition, a paced breathing task would place an additional mental processing load on the subject and limit the conditions when RSA could be effectively monitored. Thus, based on the neural model, adjustments would not be necessary even if the amplitude of RSA were correlated with respiratory parameters.
Neurophysiological arguments have been made for using respiration frequency as a covariate to correct the amplitude of RSA. One argument is based on the transfer function of the SA node, which attenuates the amplitude of fast frequencies (Saul, Berger, Chen, and Cohen, 1989). This argument contrasts the true vagal outflow being conveyed through the vagal fibers with the functional impact of the vagus on the heart. The model argues that to estimate the true vagal outflow, the amplitude of RSA needs to be corrected, because at faster breathing frequencies the amplitude of RSA will be underestimated. Thus, consistent with this model, there is an inconsistency between a hypothetical “true” vagal tone and the functional impact of vagal activity on the heart. However, since neural regulation of the sinoatrial node is feedback driven, the functional impact of the vagal efferents would be driven by the afferent feedback to the brainstem structures regulating the vagal outflow. Thus, if our interests were to measure accurately the functional impact of the vagal efferents on the heart, then correction would not be necessary.
Equally important is the fact that the use of covariate analysis as a correction procedure may be statistically unsound. Analysis of covariance (ANCOVA) assumes a linear relation between the covariate and the dependent variable, a relation not supported in the literature relating the amplitude of RSA to respiration frequency. While covariate analysis may be used to address (e.g., to rule out) whether respiration frequency has a causal effect on the amplitude of RSA, adjusting means based on the covariate (i.e. respiration frequency) and comparing them is entirely inappropriate. Miller and Chapman (2001) have provided an informative reminder to researchers of this fact. Their evaluation of covariate analysis and its use (or misuse) provides pertinent information for researchers and proposes that “...in many cases, there is no means of achieving the superficially appealing goal of ‘correcting’ or ‘controlling for’ real group differences on a potential covariate (pg. 40)”. The implication in the literature proposing correction procedures for accurately quantifying the amplitude of RSA is that the relationship between respiration frequency and task (or group in the case of between subjects resting data) is statistically independent. If the relation is not independent, ANCOVA will not yield interpretable results and may in fact eliminate valid effects. This is pertinent to the argument against correcting RSA using respiration frequency as a covariate. Furthermore, it is suggested that the arguments proposed by Miller and Chapman (2001) be given just consideration in evaluating research using respiration frequency as a covariate for quantifying the amplitude of RSA. Here it is proposed that differences reported between corrected and uncorrected measures of the amplitude of RSA are at best questionable and may in fact be a function of either methodology or inappropriate statistical procedures.
Reported relations between the amplitude of RSA and respiration frequency might be method dependent
RSA has been operationally defined in terms of its co-occurrence with features of the respiratory pattern (e.g., Grossman, Karemaker, & Wieling, 1991; Grossman, VanBeek, & Wientjes, 1990; Ritz, Thons, & Dahme, 2001). Algorithms have been described that seek both the maximum heart rate following the onset of inhalation and the minimum heart rate following the onset of exhalation (Grossman, VanBeek, & Wientjes, 1990; Fouad, Tarazi, Ferrario, Fighaly, & Alicandri, 1984; Katona, & Jih, 1975). Studies have manipulated breathing frequency without controlling for other parameters of breathing (e.g., inhalation-exhalation ratio, respiratory resistance, or deviation from spontaneous breathing). In some studies the range of breathing extended well beyond the range of spontaneous breathing (e.g., Hirsch & Bishop, 1981).
The studies supporting a relation between the amplitude of RSA and respiration frequency tend to apply descriptive statistics such as range (i.e., peak-valley or peak-to-trough) and other measures of variability (e.g., standard deviation, variance, mean successive difference, mean square successive difference) as indices of the amplitude of RSA (i.e. Grossman & Kollai, 1993; Grossman, Karemaker & Weiling, 1991). These studies, by employing global measures of variability, may be more vulnerable to respiratory influences than the methods designed to decompose the complex time series into constituent periodic components (see Byrne & Porges, 1993; Porges & Bohrer, 1990; Kettunen & Keltikangas-Jarvinen, 2001).
Underlying the arguments to quantify respiratory parameters or to control respiratory influences via paced breathing is an assumption that the amplitude of RSA, as a variable, is confounded by “lawful” relations with respiration frequency and other ventilatory parameters (Grossman & Kollai, 1993, Hirsch & Bishop, 1981). Hatfield et. al. (1998) investigated the relation between RSA and both respiration frequency and tidal volume during exercise. Hatfield et al. reported that the between subject correlations of the amplitude of RSA and both respiration frequency and tidal volume within each stage of exercise were weak and inconsistent. However, the within subject correlations between the amplitude of RSA and respiration frequency across the various stages of exercise exhibited great variability, with some subjects exhibiting robust negative correlations (i.e., faster breathing associated with lower amplitude RSA), while for others the correlations were weak and nonsignificant. Interestingly, contrary to the assumption that tidal volume is positively correlated with the amplitude of RSA, due to the severe exercise demands in this study the within subject correlations between the amplitude of RSA and tidal volume were negatively correlated.
Statistical Adjustments for RSA: Does it make a difference?
Researchers who have investigated the impact of respiration on the amplitude of RSA report similar statistical findings whether they have statistically adjusted or not (Burleson et al., 2003; Gianaros, Quigley, Mordkoff & Stern, 2001; Thayer, Friedman & Borkovec, 1996; Houtveen, Rietveld & DeGeus, 2002). Similarly, nonsignificant differences have been reported in studies investigating the effects of respiration rate and tidal volume on the amplitude of RSA (i.e., Berntson, Cacciopo & Fieldstone, 1996; Gianaros & Quigley, 2001; Rottenberg, Wilhelm, Gross & Gotlib, 2003). Ritz and colleagues (Ritz, Altupa, Thons & Dahme, 2002; Ritz, George & Dahme, 2000; Ritz, Thons and Dahme, 2000), using a ventilatory-adjustment procedure that corrected the amplitude of RSA by tidal volume during paced-breathing, reported that unadjusted and adjusted RSA amplitude produced similar findings.
In general, the literature does not provide evidence that findings change when the amplitude of RSA is adjusted for respiration parameters. Table 2 provides a brief summary of research published in Biological Psychology and the journal Psychophysiology since the year 2000. The table includes only manuscripts that provide comparisons between adjusted and unadjusted measures of the amplitude of RSA. As shown in Table 2, no study contrasting corrected and uncorrected measures of RSA, using respiration frequency as a covariate, provides support for correction procedures. Furthermore, neither do studies evaluating tidal volume or respiration frequency and tidal volume as covariates. Ritz et. al. (2000) concluded that their “…ventilation-adjustment procedure for RSA did not seem to be a critical factor, as analysis using unadjusted RSA did not improve the results… (pg. 131).” Houtveen et. al. (2002) provide evidence that during mental stress under normal breathing conditions, correction procedures using respiratory frequency did not in fact “…produce a better estimate than uncorrected RSA…” and stated that “…uncorrected within-subject changes in RSA are acceptable to index within-subject changes in tonic vagal modulation of heart rate for most clinical stress studies (pg. 434).” Houtveen et. al. (2002) suggested evaluating both corrected and uncorrected measures of the amplitude of RSA in studies where stressors may influence central respiratory drive (e.g., stressors involving in hypo- or hyperventilation or altered metabolic demands).
Table 2.
Summary of research reporting comparisons between RSA measures unadjusted and adjusted for respiratory parameters conducted since 2000 published in Biological Psychology and Psychophysiology. A more detailed description of the studies presented here, and related studies from additional journals is available upon request.
| Covariate | Spontaneous breathing | Paced breathing | Findings/Conclusions |
|---|---|---|---|
| Respiration frequency | Burleson et. al. 2003
Gianaros et. al. 2001 Gianaros et. al. 2003 Hawkley et. al. 2001 Althaus et. al. 2004 |
No difference found
No difference found No difference found No difference found No difference found |
|
| Tidal volume | Wilhelm et. al.2001 |
Ritz et. al. 2000
Ritz, George et. 2000 Ritz et. al. 2001 Ritz et. al. 2002 |
No difference found
No difference found Changes in regression intercept No difference found No difference found |
| Respiration frequency + Tidal volume |
Gianaros/Quigley 2001
Rottenberg et. al 2003 Houtveen et. al. 2002 |
No difference found
No difference found No difference found |
It should be noted that these papers use of variety of designs (i.e., within, between, and mixed designs) during varying task demands (i.e., baseline, physical tasks, cognitive, emotional, visual and auditory stimuli) with different populations (i.e., healthy controls, depressed, socially anxious). The fact that there were no significant differences between corrected and uncorrected amplitude of RSA argues against the use of correction or control procedures in quantifying the amplitude of RSA. Based on this literature, the causal assumption relating the amplitude of RSA to breathing finds no empirical support.
Evidence from basic research
Two studies provide data to evaluate the argument for using respiration-based correction or control procedures when quantifying RSA. The studies address two basic questions related to the mandate frequently voiced to either control respiration by paced breathing or to use respiration frequency as a covariate to adjust the amplitude of RSA. The first question evaluates whether an accurate estimate of respiration frequency can be extracted from the heart period time series by identifying the frequency of RSA. The second question evaluates the assumed linear relation between respiration frequency and the amplitude of RSA. The first study also contrasts a time domain procedure (see Porges, 1985), designed to quantify the amplitude of RSA with the frequency specific measures of RSA generated by spectral analysis.
Study 1
Methods
Participants
Forty-four healthy adults (23 female, 21 male) between the ages of 17–41 (mean 21.39, SD 5.08) were tested. Participants were given full disclosure of the research protocol and signed consent forms prior to their participation.
Apparatus
A three-lead configuration was used for collecting ECG data. Disposable electrodes were attached to the upper forearms with a reference electrode placed on the left wrist. The ECG leads were connected to a pre-amplifier (Scope Inc., Urbana, Il.) and the output of the amplifier was input to a Vagal Tone Monitor (VTM-III) (Delta-Biometrics, Bethesda, Md.). The VTM-III detected the peak of the R-wave from the ECG to the nearest msec. and timed sequential R-R intervals (i.e., heart periods) to the nearest msec. Respiration was monitored with a strain gauge placed around the thorax using a Resp-1 Pneumograph System (UFI, Morro Bay, Ca.). The analog output from the Resp-1 was input to the VTM-III. The VTM-III sampled the analog respiration signal every 500 msec. Sequential heart periods and measurements of respiration were stored by the VTM-III in ASCII format for off-line analyses.
Procedure
Participants were seated in a comfortable chair in a quiet room. After the electrodes and respiration strain gauge were placed on the subject, the signals were monitored on the VTM-III to ensure that the ECG presented a detectable R-wave and that the respiration signal was clean. Participants were asked to relax and to sit quietly. The subjects were instructed to breathe naturally during data collection. Once the subjects were relaxed and the VTM-III reliably detected the peaks of the R-waves and the positioning of the strain gauge ensured a clean respiration signal, data collection started and continued for approximately 10 minutes. Following the data collection, the electrodes and strain guage were removed. Prior to leaving the laboratory, the participants were debriefed.
Data Reduction
The heart period files were visually scanned for artifacts on a PC using MXedit software (Delta-Biometrics, Bethesda, Md.). To remove any possibility of a confound from editing outliers, a two-minute segment of sequential heart periods without false or missed R-wave detections, synchronized with rhythmic respiration, was extracted for each participant. The analyses described below were conducted on these two-minute segments.
Design and Analyses
Spectral analyses were calculated to identify the frequency components of the respiration and heart period time-series. The analyses were computed using Statistica software (StatSoft, Tulsa, Ok.). Statistica displays the spectral density for each frequency within the spectrum. Prior to computing the spectral densities, the heart period data were re-sampled every 500 msec. to generate a new time-series of data sampled at equal intervals. Low frequency oscillations and slow trends were removed from the data set using a 3rd-order, 21-point moving polynomial filter (MPF; Porges, 1985; Porges & Bohrer, 1982). The MPF functions as a high pass filter with a cutoff frequency of approximately .09Hz. Spectral analyses were conducted using a Hamming window (9 weights). The peak frequency for the respiration data was obtained by locating the frequency at which the maximum spectral density occurred. The peak of the heart period spectrum, within the frequency band of spontaneous respiration for this sample (.12 – .40 Hz.) defined the frequency of RSA. In addition, a time domain method incorporated into MXedit (Porges, 1985) used to calculate the amplitude of RSA was contrasted with the values derived from the spectral methods.
Results
Question 1: Is the frequency of spontaneous breathing correlated with the frequency of RSA?
Peak frequencies from the spectral decomposition of heart period and respiration were identified for each participant. These pairs of frequencies were correlated to assess the relation between the respiration frequency and the frequency of RSA. The correlation between these frequencies approached unity (r2 = .993, N = 44). Figure 1 illustrates that the relation between respiration frequency and frequency of RSA is virtually an identity function with an intercept passing close to the origin (0,0).
Figure 1.
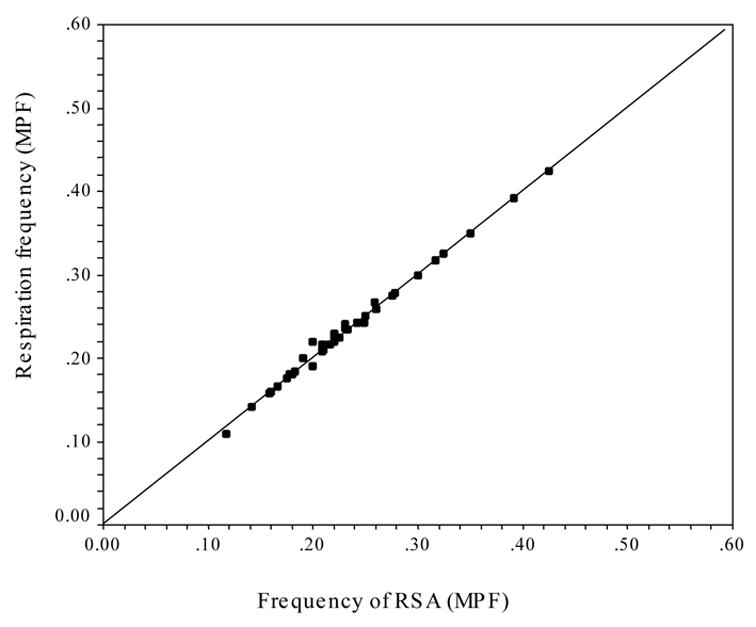
Scatterplot of the peak frequencies of RSA (MPF) and respiration (MPF).
Potential Confound I. The statistical characteristics of the MPF may distort the identification of the dominant frequency of respiration
The strong finding between the frequency of respiration and the frequency of RSA, does not preclude a possible confound by the filtering methods. The respiration and heart period data files were transformed using the MPF filter. Since the MPF removes virtually all the variance from frequencies slower than the spontaneous breathing frequencies, it may have contributed to the strength of the relation by statistically limiting the range of frequencies available to describe the periodicities of both heart rate and respiration. To assess this possibility, additional analyses were performed on the respiration data to determine if the MPF distorted the extraction of the dominant respiration frequency. An analysis was conducted in which the respiration data were detrended with a linear function instead of the MPF. The relation between the peak frequencies of the linear detrended (LIN) respiration data and the peak frequencies of heart period (i.e., the frequency of RSA) derived using the MPF were correlated. The relation between the two frequencies remains near unity (r2 = .988, N = 44) with the intercept passing close to the origin (0,0). Thus, the frequency of RSA is equivalent to the frequency of spontaneous breathing when the respiration data are transformed with either the MPF or a linear function.
Potential Confound II. Variations in within-subject depth of breathing may distort the identification of the dominant frequency of respiration
The nature of how spectral analysis decomposes the variance of a time-series might potentially confound the identification of the dominant “breathing” frequency in either heart period or respiration data. Spectral analysis decomposes the variance of a time series into constituent frequencies with the peak of the spectrum representing the frequency accounting for the most variance. Because spectral analysis decomposes variance and assumes that the time series is statistically stationary, an infrequent high-amplitude slow oscillation (e.g., deep breath) in a background of low-amplitude fast oscillations (e.g., shallow breaths) at a different frequency would be detected as the dominant frequency. To assess the possibility that spurious changes in the depth of breathing may have affected the frequency of respiration reported in the previous analyses, the amplitude component of respiration was removed. To remove the influence of depth of breathing, the respiration data (i.e., sequential values sampled every 500 msec.) were transformed into a binary time series (i.e., 1, 0, −1). The analog measures of respiration sampled 2 Hz were successive differenced. The difference scores were assigned a +1 if positive, a −1 if negative, and a 0 if no change. These binary data (BIN) were analyzed with spectral analysis and the peak of the spectrum was contrasted with the frequency of RSA derived via the MPF. Consistent with the previous analyses, the relation approached unity (r2 = .984, N=44) with the intercept passing close to the origin (0,0). Table 3 provides the correlation matrix evaluating the relation between the frequency of RSA derived via the MPF, linear detrend and binary transformation. Note that all correlations are above .99.
Table 3.
Correlations between RSA and respiration peak spectral frequencies derived by different transformation methods. MPF – moving polynomial filter, LIN – linear transformation, BIN – binary transformation.
| RESP Frequency (MPF) | RESP Frequency (LIN) | RESP Frequency (BIN) | |
|---|---|---|---|
| RSA Frequency (MPF) | .997* | .994* | .992* |
| RSA Frequency (LIN) | .993* | .994* | .993* |
p< .01 (N = 44, 2-tailed).
Potential Confound III. The statistical characteristics of the MPF may distort the identification of the frequency of RSA
To deal with the possible influence of the MPF filter on the frequency of RSA, the heart period files were detrended with a linear function. The frequency of RSA derived via the linear detrend procedures (LIN) was correlated with the frequency of respiration. Only peaks in the heart period spectrum within the range of spontaneous breathing (i.e., above .10 Hz) were selected. Similar to the contrasts reported above, the relation between the frequency of RSA derived from the linear detrending procedure and the respiration frequencies derived via the MPF filter approached unity (r2 = .986, N = 44). A similar relation was obtained when both respiration and heart period were detrended with a linear function (r2 = .988, N = 44).
Question 2: Is the amplitude of RSA correlated to the frequency of spontaneous breathing?
To evaluate this question, the frequency of respiration derived from spectral analysis (using the MPF filter) was compared with the spectral density associated with the peak of the heart period spectrum (using the MPF filter) within the band of spontaneous breathing (i.e., .12 – .40). Figure 2 illustrates that there was no relation between the amplitude of RSA and respiration frequency (r2 = .015, N = 44). Similar comparisons were made using respiration frequency derived using both the linear detrend (r2 = .018, N = 44) and binary transformation (r2 = .019, N = 44) procedures. Regardless of the method used to detrend or transform the respiration data, the amplitude of RSA was not related to respiration frequency.
Figure 2.
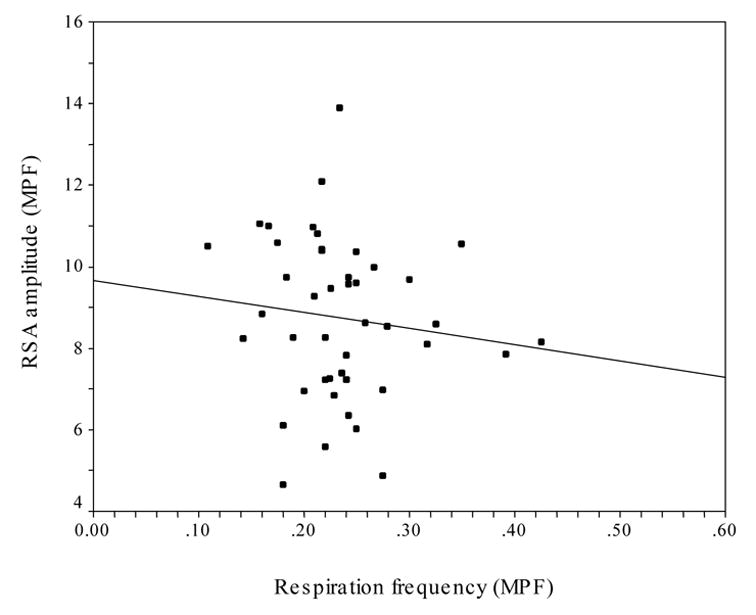
Scatterplot of the frequency of respiration (MPF) and the amplitude of RSA (MPF).
Because our findings deviate from the assumed deterministic relation between respiration frequency and the amplitude of RSA, we applied a bootstrapping methodology to our data (see Manly, 1997; Riniolo & Porges, 2000; Rosenfeld, Soskins, Bosh & Ryan; 2003). Bootstrapping is sampling with replacement. By sampling with replacement 10,000 times we generated a distribution of correlation coefficients that are assumed to be representative of the underlying population. We conducted this procedure to deal with the possibility that the lack of relation between respiration frequency and the amplitude of RSA was due to a sampling anomaly or bias caused by only a few subjects. The distribution of the bootstrapped correlations between respiration frequency (derived with the MPF) and the amplitude of RSA (derived with the MPF) are illustrated in Figure 3. The mean correlation of these analyses was −.122 (the same as we report) with a standard deviation of .11. Additionally, 95% of the correlations were between −.34 and −.10 with r2 values falling between .11 and .01. When we bootstrapped the data derived from the other detrending methods (i.e., LIN), the central tendency and standard deviation of the bootstrapped correlations consistently approximated these findings. Based on these data, the relation between respiration frequency and the amplitude of RSA is far from “lawful” (see Grossman, VanBeek, & Wientjes, 1990). The shared variance (i.e., r2) centered at approximately 1%. Furthermore, 95% of the bootstrapped analyses generated a shared variance between 1% and 11%. Thus, reports that respiratory frequency contributes to the amplitude of RSA with normal adults during a baseline condition while breathing spontaneously, must be due to the methodology of quantification or the context of the experiment.
Figure 3.
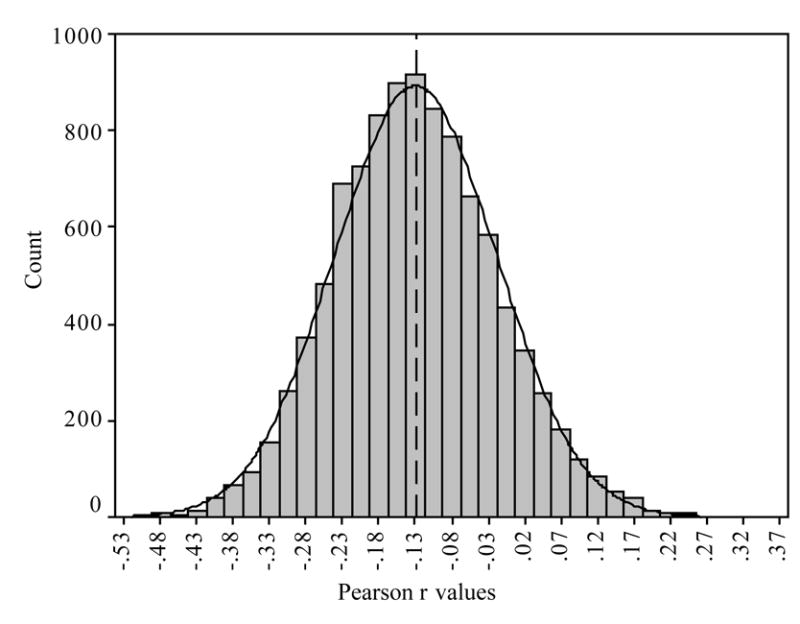
Histogram of bootstrapped correlations of frequency of respiration and amplitude of RSA derived using the MPF.
QUESTION 3: Is the time domain method of calculating RSA used in MXedit correlated with the spectral methods?
Because several laboratories use the time domain methods incorporated in MXedit to quantify RSA, analyses were performed to assess the relation between the amplitude of RSA derived via spectral analysis and the MXedit procedure. MXedit performs the MPF filtering process and outputs a measure of the amplitude of RSA defined as the variance within the frequency band associated with spontaneous breathing (i.e., .12 – .40). The amplitude of RSA derived from MXedit uses a time domain equivalent of spectral analyses. As illustrated in Figure 4, the MXedit estimate of the amplitude of RSA was highly correlated with the amplitude of RSA derived from spectral analysis (r2 = .988, N = 44). Consistent with the above findings demonstrating a statistical independence between the amplitude of RSA and respiration frequency, there was no relation between the amplitude of RSA derived from MXedit and respiration frequency. This was true when respiration was detrended with either: the MPF (r2 = .014, N = 44), the linear function (r2 = .017, N = 44), or the binary transformation (r2 = .019, N = 44).
Figure 4.
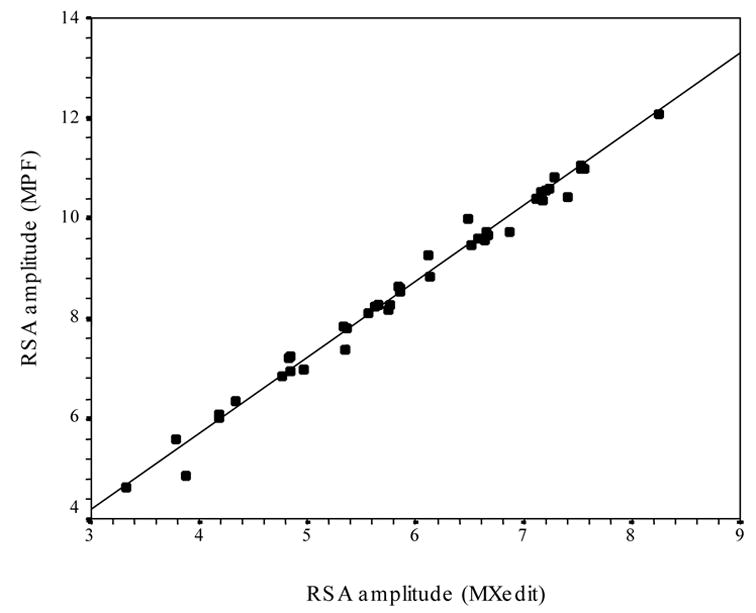
Scatterplot of the amplitude of RSA derived by both spectral analysis (MPF) and MXedit.
Study 2
Methods
Heart period and respiration data were re-analyzed from an earlier study evaluating the effect of atropine on aviator performance and autonomic activity in a flight simulator, while heart period and respiration were monitored (see Dellinger, Taylor & Porges, 1987). These data provide an opportunity to evaluate the effects of modulating the amplitude of RSA via atropine dose on respiration frequency. Since the brainstem areas regulating the cardiopulmonary system are constantly receiving afferent information via the afferent vagus, a causal model of RSA would hypothesize that respiration frequency would increase as the atropine reduced the amplitude of RSA. Data were analyzed from baseline conditions (i.e., prior to flight simulator trials) during each of five randomly ordered doses of atropine (0, .5, 1, 2 and 4 mg/75kg−1 of individual weight). Subjects with complete respiration and heart period data for each dose of atropine were included in the analyses. Data from13 male participants between the ages of 19 and 30 are reported.
Data reduction
Heart period data were derived from ECG digitized at 1 KHz. (see Dellinger et. al., 1987). For the current analyses, the heart period data were visually scanned for artifacts. For one subject the first few seconds of data were removed from the file due to artifacts from equipment noise. No other artifact editing was required. An MPF filter was applied to the heart period time series as described in Study 1.
Design and analysis
Similar to the procedures described in Study 1, spectral analyses of the heart period and respiration data were conducted to identify peak frequencies and the corresponding spectral density representing the peak amplitude. As in the first study, the peak of the heart period spectrum, within the frequency band of spontaneous respiration (.12 – .40 Hz.), defined the frequency of RSA. The amplitude of RSA was defined as the natural logarithm of the spectral density at the peak frequency within this range. Repeated measures analysis of variance analyzed respiration frequency and the frequency of RSA as a function of atropine dose. For the repeated measures analyses the Greenhouse–Geisser corrected p-values are reported, along with the uncorrected degrees of freedom. Correlations evaluated the covariation between respiration frequency and RSA.
Results
Consistent with the findings in Study 1, the correlation between Frequency of RSA and respiration frequency approached unity (see Table 4) regardless of the dose of atropine. However, independent of the profound and predicted effects of atropine on heart period and RSA previously reported (see Dellinger et. al., 1987), the effect of atropine dose did not influence respiration frequency (F [4, 48] = 1.47, p = .245, eta2 = .109) or frequency of RSA (F [4, 48] = 1.58, p = .219, eta2 = .116). As expected, significant differences in the amplitude of RSA were found in across atropine doses (F [4,48] = 56.03, p < .001, eta2 = .824).
Table 4.
Correlations between the frequency of RSA and respiration frequency, peak respiration and RSA frequencies, and amplitude of RSA as a function of atropine dose (in mg/kg−1 of individual’s weight).
| Atropine Dose | |||||
|---|---|---|---|---|---|
| Variables | 0 | .5 | 1 | 2 | 4 |
| RSA frequency/Respiration frequency (correlation) | .998 | .977 | .963 | .973 | .986 |
| Peak respiration frequency | .258 Hz. | .274 Hz. | .251 Hz. | .251 Hz. | .243 Hz. |
| Peak RSA frequency | .258 Hz | .273 Hz. | .248 Hz. | .250 Hz. | .242 Hz. |
| Amplitude of RSA (ln peak spectral density) | 10.6a | 10.3a | 9.6 | 7.2 | 5.3 |
Not significantly different.
Discussion
Results from the two studies clarify several points related to the quantification of RSA. First, during both baseline conditions (i.e., spontaneous breathing without task demands) and vagal manipulation via atropine, it is possible to accurately extract respiration frequency from the heart period time series. Second, under neither condition was there a relation between respiration frequency and the amplitude of RSA. In Study 1 this was examined by correlations between the amplitude of RSA and respiration frequency during a baseline condition. In Study 2 this was examined by manipulating the amplitude of RSA with atropine and evaluating whether the respiration frequency adjusted with the systematic changes in RSA.
The data from both studies confirm that the relation between respiration frequency and the frequency of RSA was virtually an identity function with a regression line that passed close to the origin (0,0). In the first study it was shown that this strong relation was observed regardless of the method used to transform the respiration time series (i.e., MPF, LIN, BIN). In the second study, the robustness of this relation was demonstrated by manipulating the amplitude of RSA with atropine.
In contrast to the strong relation between the respiration frequency and the frequency of RSA, the relation between respiration frequency and the amplitude of RSA was far from causal. In Study 1 respiration frequency accounted for less than 2% of the variance of the amplitude of RSA. In Study 2 across the atropine conditions respiration frequency accounted for less than 10% of the variance of the amplitude of RSA. Statistical corrections based on linear models, should be used if and only if there is a strong linear relation between the amplitude of RSA and respiration frequency.
Miller and Chapman’s (2001) provide a strong argument against the inappropriate application of covariance methods, and have emphasized that it is not possible that analysis of covariance (ANCOVA) procedures can overcome the confounding of the covariate and condition. Although ANCOVA can address whether accounting for a covariate (i.e., respiration frequency) leaves an effect intact, statistically adjusting metrics based on covariate analysis can eliminate valid effects. For example, using respiration frequency as a covariate when looking at task effects (or group differences) on RSA may provide information suggesting the valid inference that the covariate is not of concern as a possible cause for the effect of interest (e.g., that respiration frequency is not the reason that two conditions differ, or that two groups differ). However, it would be statistically inappropriate to use the covariate to “correct for” effects when there is an inherent difference (e.g., differential respiration frequencies as a function of condition or group membership).
Thus, based on the data presented from our laboratory, there is no statistical justification to use respiration frequency as a covariate to accurately quantify RSA amplitude during baseline conditions. Interestingly, the formalizing of respiration manipulations, such as paced breathing, as a necessary component in the quantification of the amplitude of RSA was based on an assumed linear dependence of the amplitude of RSA on spontaneous respiration frequency. The recent literature (e.g., see Table 2) supports our conclusion that the amplitude of RSA is not dependent on respiration frequency. This holds true under both baseline conditions and during vagal manipulation via atropine.
These conclusions do not preclude either the importance of monitoring respiration parameters as physiological variables, or the possible relation between respiration parameters (e.g., PaCo2) and the amplitude of RSA during psychological and physical tasks. While the literature does not provide strong evidence for the usefulness of applying respiratory parameters to correct RSA during baseline conditions (see Table 2), this does not diminish the importance of conducting research designed to evaluate the mechanisms that influence the functional impact of the vagus on the heart (i.e., amplitude of RSA). From a neural perspective, increased mobilization behaviors require physiological adjustments that include both increasing heart rate (i.e., a reduction of cardiac vagal tone) and adjusting breathing parameters (i.e., rate, inhalation/exhalation ratio, gas exchange parameters) to support the increased cardiac output. It is proposed that instead of arguing for respiratory adjustments to RSA, researchers should focus on the contextual conditions and neurobiological mechanisms that may result in a deviation from the intrinsic relation between the amplitude of RSA and specific respiratory parameters (i.e., PaCo2). Understanding these mechanisms would provide important information regarding the neural feedback system responsible for the generation, maintenance and regulation of vagal efferent influences on the sinoatrial node as measured by quantifying the amplitude of respiratory sinus arrhythmia.
Acknowledgments
This manuscript was supported in part by grant MH 60625 from the National Institute of Mental Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The preparation of this manuscript was supported in part by grant MH 60625 from the National Institute of Mental Health.
References
- Althaus M, Van Roon AM, Mulder LJM, Mulder G, AArnoudse CC, Minderaa RB. Autonomic response patterns observed during the performance of an attention-demanding task in two groups of children with autistic-type difficulties in social adjustment. Psychophysiology. 2004;41:893–904. doi: 10.1111/j.1469-8986.2004.00252.x. [DOI] [PubMed] [Google Scholar]
- Berntson GG, Bigger JT, Jr, Eckberg DL, Grossman P, Kaufmann PG, Malik M, Nagaraja HN, Porges SW, Saul JP, Stone PH, van der Molen MW. Heart rate variability: origins, methods, and interpretive caveats. Psychophysiology. 1997;34:623–48. doi: 10.1111/j.1469-8986.1997.tb02140.x. [DOI] [PubMed] [Google Scholar]
- Byrne EA, Porges SW. Data-dependent filter characteristics of peak-valley respiratory sinus arrhythmia estimation: A cautionary note. Psychophysiology. 1993;30:397–404. doi: 10.1111/j.1469-8986.1993.tb02061.x. [DOI] [PubMed] [Google Scholar]
- Chambers AS, Allen JB. Vagal tone as an indicator of treatment response in major depression. Psychophysiology. 2002;39:861–864. doi: 10.1111/1469-8986.3960861. [DOI] [PubMed] [Google Scholar]
- Dellinger JA, Taylor HL, Porges SW. Atropine sulfate effects on aviator performance and on respiratory-heart period interactions. Aviation and Space Environment Medicine. 1987;54:1242–1253. [PubMed] [Google Scholar]
- Ekberg DL. The human respiratory gate. Journal of Physiology. 2003;548:339–352. doi: 10.1113/jphysiol.2003.037192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouad FM, Tarazi RC, Ferrario CM, Fighaly S, Alicandri C. Assessment of parasympathetic control of heart rate by a noninvasive method. American Journal of Physiology. 1984;246:H838–H842. doi: 10.1152/ajpheart.1984.246.6.H838. [DOI] [PubMed] [Google Scholar]
- Frazier TW, Strauss ME, Steinhauer SR. Respiratory sinus arrhythmia as an index of emotional response in young adults. Psychophysiology. 2004;41:75–83. doi: 10.1046/j.1469-8986.2003.00131.x. [DOI] [PubMed] [Google Scholar]
- Gianaros PJ, Quigley KS, Mordkoff JT, Stern RM. Gastric myoelectrical and autonomic cardiac reactivity to laboratory stressors. Psychophysiology. 2001;38:642–652. [PMC free article] [PubMed] [Google Scholar]
- Gilbey MP, Coote JH, Fleetwood-Walker S, Peterson DF. The influence of the paraventriculo-spinal pathway, and oxytocin and vasopressin on sympathetic preganglionic neurons. Brain Research. 1983;251:283–90. doi: 10.1016/0006-8993(82)90745-4. [DOI] [PubMed] [Google Scholar]
- Grossman P, Karemaker J, Wieling W. Prediction of tonic parasympathetic cardiac control using respiratory sinus arrhythmia: The need for respiratory control. Psychophysiology. 1991;28:201–216. doi: 10.1111/j.1469-8986.1991.tb00412.x. [DOI] [PubMed] [Google Scholar]
- Grossman P, Kollai M. Respiratory sinus arrhythmia, cardiac vagal tone, and respiration: Within and between individual relations. Psychophysiology. 1993;30:486–495. doi: 10.1111/j.1469-8986.1993.tb02072.x. [DOI] [PubMed] [Google Scholar]
- Grossman P, VanBeek J, Wientjes C. A comparison of three quantification methods for estimation of respiratory sinus arrhythmia. Psychophysiology. 1990;27:702–714. doi: 10.1111/j.1469-8986.1990.tb03198.x. [DOI] [PubMed] [Google Scholar]
- Grossman P, Wilhelm FH, Kawachi I, Sparrow D. Gender differences in psychophysiological responses to speech stress among older social phobics: Congruence and incongruence between self-evaluative and cardiovascular reactions. Psychosomatic Medicine. 2001;63:765–777. doi: 10.1097/00006842-200109000-00010. [DOI] [PubMed] [Google Scholar]
- Harper RM, Woo MA, Alger JR. Visualization of sleep influences on cerebellar and brainstem cardiac and respiratory control mechanisms. Brain Research Bulletin. 2000;53:125–131. doi: 10.1016/s0361-9230(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Haselton JR, Solomon IC, Motekaitis AM, Kaufman MP. Bronchomotor vagal preganglionic cell bodies in the dog: An anatomic and functional study. Journal of Applied Physiology. 1992;73:1122–1179. doi: 10.1152/jappl.1992.73.3.1122. [DOI] [PubMed] [Google Scholar]
- Hatfield BD, Santa Maria DL, Porges SW, Potts JT, Spalding T, Byrne EA. Respiratory sinus arrhythmia during exercise in aerobically trained and untrained men. Medical and Science in Sports and Exercise. 1998;30:206–214. doi: 10.1097/00005768-199802000-00006. [DOI] [PubMed] [Google Scholar]
- Hirsch JA, Bishop B. Respiratory sinus arrhythmia in humans: How breathing pattern modulates heart rate. American Journal of Physiology. 1981;241:H206–H629. doi: 10.1152/ajpheart.1981.241.4.H620. [DOI] [PubMed] [Google Scholar]
- Houtveen JH, Rietveld S, De Geus EJ. Contributions of tonic vagal modulation of heart rate, central respiratory drive, respiratory depth and respiratory frequency to respiratory sinus arrhythmia during mental stress and physical exercise. Psychophysiology. 2002;39:427–436. doi: 10.1017.S0048577202394022. [DOI] [PubMed] [Google Scholar]
- Jones JFX, Wang Y, Jordan D. Heart rate responses to selective stimulation of cardiac vagal C fibers in anesthetized cats, rats and rabbits. Journal of Physiology (London) 1995;489:203–214. doi: 10.1113/jphysiol.1995.sp021042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson P, Sonnby-Borgstrom M. The effects of pictures of emotional faces on tonic and phasic autonomic cardiac control in women and men. Biological Psychology. 2003;62:157–173. doi: 10.1016/s0301-0511(02)00114-x. [DOI] [PubMed] [Google Scholar]
- Jordan D, Khalid MEM, Schneiderman N, Spyer KM. The location and properties of preganglionic vagal cardio-motor neurons in the rabbit. Pflugers Archive. 1982;395:244–250. doi: 10.1007/BF00584817. [DOI] [PubMed] [Google Scholar]
- Katona PG, Jih R. Respiratory sinus arrhythmia: A noninvasive measure of parasympathetic cardiac control. Journal of Applied Physiology. 1975;39:801–805. doi: 10.1152/jappl.1975.39.5.801. [DOI] [PubMed] [Google Scholar]
- Keltikangas-Jarvinen L, Heponiemi T. Vital exhaustion, temperament, and cardiac reactivity in task-induced stress. Biological Psychology. 2004;65:121–135. doi: 10.1016/s0301-0511(03)00112-1. [DOI] [PubMed] [Google Scholar]
- Kettunen J, Keltikangas-Jarvinen L. Intraindividual analysis of instantaneous heart rate variability. Psychophysiology. 2001;38:659–668. [PubMed] [Google Scholar]
- Kettunen J, Ravaja N, Naatanen P, Keltikangas-Jarvinen L. The relationship of respiratory sinus arrhythmia to the co-activation of autonomic and facial responses during the Rorschach test. Psychophysiology. 2000;37:242–250. [PubMed] [Google Scholar]
- Manly B. Randomization, Bootstrap, and Monte Carlo Methods in Biology. 2. London: Chapman & Hall; 1997. [Google Scholar]
- McAllen RM, Spyer KM. Two types of vagal preganglionic motorneurons projecting to the heart and lungs. Journal of Physiology (London) 1978;282:353–364. doi: 10.1113/jphysiol.1978.sp012468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor MF, Allen JB, Kasniak AW. Emotional disclosure for whom? A study of vagal tone in bereavement. Biological Psychology. 2005;68:135–146. doi: 10.1016/j.biopsycho.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Porges SW. US patent 4,510,944 Method and apparatus for evaluating rhythmic oscillations in aperiodic physiological response systems. 1985
- Porges SW. Orienting in a defensive world: Mammalian modifications of our evolutionary heritage. A Polyvagal Theory. Psychophysiology. 1995;32:301–318. doi: 10.1111/j.1469-8986.1995.tb01213.x. [DOI] [PubMed] [Google Scholar]
- Porges SW, Bohrer RE. Analyses of periodic processes in psychophysiological research. In: Caccioppo TJ, Tassinary LG, editors. Principles of Psychophysiology: Physical, Social, and Inferential Elements. New York: Cambridge University Press; 1990. pp. 708–753. [Google Scholar]
- Ravaja N. Effects of a small talking facial image on autonomic activity: The moderating influence of dispositional BIS and BAS sensitivities and emotions. Biological Psychology. 2004;65:163–183. doi: 10.1016/s0301-0511(03)00078-4. [DOI] [PubMed] [Google Scholar]
- Riniolo TC, Porges SW. Evaluating group distributional characteristics: Why psychophysiologists should be interested in qualitative departures from the normal distribution. Psychophysiology. 2000;37:21–28. [PubMed] [Google Scholar]
- Richter DW, Spyer KM. Cardiorespiratory control. In: Loewy AD, Spyer KM, editors. Central Regulation of Autonomic Function. New York: Oxford University Press; 1990. pp. 189–207. [Google Scholar]
- Ritz T, Altupa S, Thons M, Dahme B. Effects of affective picture viewing and imagery on respiratory resistance in nonasthmatic individuals. Psychophysiology. 2002;39:86–94. [PubMed] [Google Scholar]
- Ritz T, Thons M, Dahme B. Increases in total respiratory resistance during forhead temperature stimulation. Biological Psychology. 2000;50:119–135. doi: 10.1016/s0301-0511(00)00075-2. [DOI] [PubMed] [Google Scholar]
- Ritz T, Thons M, Dahme B. Modulation of respiratory sinus arrhythmia by respiration rate and volume: Stability across posture and volume variations. Psychophysiology. 2001;38:858–862. [PubMed] [Google Scholar]
- Rosenfeld JP, Soskins M, Bosh G, Ryan A. Simple, Effective Countermeasures to P300-based Tests of Detection of Concealed Information. Psychophysiology. 2003;41:205–219. doi: 10.1111/j.1469-8986.2004.00158.x. [DOI] [PubMed] [Google Scholar]
- Saul JP, Berger RD, Chen MH, Cohen RJ. Transfer function analysis of autonomic regulation. II. Respiratory sinus arrhythmia. American Journal of Physiology. 1989;256:H153–H161. doi: 10.1152/ajpheart.1989.256.1.H153. [DOI] [PubMed] [Google Scholar]
- Rottenberg J, Wilhelm FH, Gross JJ, Gotlib IH. Vagal rebound during resolution of tearful crying among depressed and nondepressed individuals. Psychophysiology. 2003;40:1–6. doi: 10.1111/1469-8986.00001. [DOI] [PubMed] [Google Scholar]
- Thayer JF, Friedman BH, Borkovec TD. Autonomic characteristics of generalized anxiety disorders and worry. Biological Psychiatry. 1996;39:255–266. doi: 10.1016/0006-3223(95)00136-0. [DOI] [PubMed] [Google Scholar]
- Thayer JF, Sollers JJ, 3rd, Ruiz-Padial E, Vila J. Estimating respiratory frequency from autoregressive spectral analysis of heart period. IEEE Engineering in Medicine and Biology. 2002;21(4):41–45. doi: 10.1109/memb.2002.1032638. [DOI] [PubMed] [Google Scholar]
- Umhau JC, George DT, Reed S, Petrulis SG, Rawlings RR, Porges SW. Psychophysiology. 2002;39:117–123. doi: 10.1017/S0048577202990669. [DOI] [PubMed] [Google Scholar]
- Watkins LL, Grossman P, Krishnan R, Sherwood A. Anxiety and vagal control of heart rate. Psychosomatic Medicine. 1998;60 (4):498–502. doi: 10.1097/00006842-199807000-00018. [DOI] [PubMed] [Google Scholar]
- Wilhelm FH, Kochar AS, Roth WT, Gross JJ. Social anxiety and response to touch: Incongruence between self-evaluative and physiological reactions. Biological Psychology. 2001;58:181–202. doi: 10.1016/s0301-0511(01)00113-2. [DOI] [PubMed] [Google Scholar]