

Abstract
Fulminant type 1 diabetes is a new subtype of type 1 diabetes. The term was established in 2000. It is a syndrome characterized by a markedly rapid and almost complete destruction of pancreatic β-cells. Several lines of evidence suggest that both genetic factors, such as human leukocyte antigen (HLA), and environmental factors, such as viral infection, contribute to the development of this disease. It is also suggested that autoimmune processes contribute less critically to fulminant type 1 diabetes than to classic type 1A diabetes. Based on the findings made to date, both viral infection and the subsequent immune reaction in genetically susceptible individuals cause β-cell destruction and lead to fulminant type 1 diabetes.
Keywords: diabetes, autoimmunity, idiopathic, HLA, enterovirus
Introduction
Type 1 diabetes is now defined as "diabetes characterized by β-cell destruction, usually leading to absolute insulin deficiency" by the American Diabetes Association, the World Health Organization and the Japan Diabetes Society [1-3]. In this context, fulminant type 1 diabetes is a typical type 1 diabetes characterized by its clinical features of abrupt onset with ketosis or ketoacidosis and virtually no C-peptide secretion [4-6]. This disease, which accounts for 20% of acute onset type 1 diabetes in Japan, is also characterized by high plasma glucose levels accompanied with almost normal glycosylated hemoglobin levels. This means that affected individuals experience sudden increases in blood glucose together with elevated serum pancreatic enzyme levels at the onset of overt diabetes and have a negative status of islet-associated autoantibodies, such as islet cell antibodies (ICA), anti-glutamic acid decarboxyalse antibody (GADAb), insulin autoantibody (IAA) or anti-insulinoma-associated antigen 2 antibody (IA-2Ab). On this account fulminant type 1 diabetes is classified not as autoimmune (type 1A) but as idiopathic (type 1B) diabetes. Type 1B diabetes exhibits permanent insulinopenia, ketoacidosis and no evidence of autoimmunity, but its etiology is unknown. Fulminant type 1 diabetes is a first phenotype described according to definite criteria within type 1B diabetes. The clinical characteristics of fulminant type 1 diabetes are summarized in Table 1.
Table 1. Clinical characteristics of fulminant type 1 diabetes.

To date most of the patients with fulminant type 1 diabetes have been reported within Japan, and a few outside Japan. Two Korean and one Filipino patient were confirmed to have fulminant type 1 diabetes by Hahm et al. [7-8] and Taniyama et al. [9], respectively. Two Chinese patients were also reported [10-11]. In Caucasians, Pozzilli et al. did not identify any patients with fulminant type 1 diabetes among 82 consecutive cases newly diagnosed in Italy [12], but a suspected case was reported from the Netherlands [13].
Sekine et al. reported on a patient whose plasma glucose and serum C-peptide levels were monitored for a couple of weeks at approximately the onset of fulminant type 1 diabetes. Blood glucose levels in this patient were within normal limits a day before onset but suddenly increased to more than 1000 mg/dl on the day of onset. This event was accompanied by a drop in serum C-peptide levels to almost zero [14]. This raises the questions of why hyperglycemia and insulinopenia appear so suddenly and why pancreatic β-cells are destroyed so suddenly in fulminant type 1 diabetes. In this review, we want to discuss these questions and provide explanations for this phenomenon.
Lessons from "ground zero"
Initially we need to perform histological investigations of the pancreatic β-cells, which produce insulin. We have reported that the pancreatic β-cell area in patients with fulminant type 1 diabetes was 0.4% of that in normal subjects [15]. As shown in Figure 1, pancreatic β-cells were almost completely absent from the sections of patients obtained at 1-5 months after the onset of overt diabetes. The β-cell area in type 1A diabetic patients was 14.5% of that of normal subjects at 1-9 months after onset. In an animal model, rodents developed diabetes when 80-90 % of native pancreas was surgically removed [16], which is consistent with human type 1A diabetes. Compared with type 1A diabetes, fulminant type 1 diabetes is characterized by the more aggressive and almost complete destruction of β-cells. Clinically, the rapid loss of pancreatic β-cells is confirmed by low levels of glycosylated hemoglobin in spite of high serum glucose concentrations. HbA1c levels reflect average plasma glucose concentrations during the preceding 1 to 2 months, so very high plasma glucose concentrations for only a few days, as seen in fulminant type 1 diabetes, is too short a time for HbA1c to increase.
Figure 1.
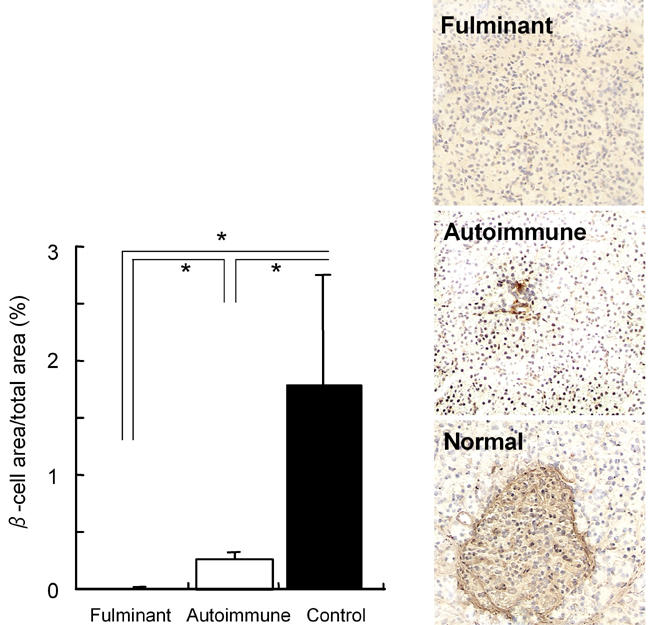
Pancreatic β-cells are markedly reduced in fulminant type 1 diabetes. β-cells are almost completely destroyed in fulminant dia-betes, but low numbers remain in autoimmune type 1 diabetes (right panel). In fulminant patients the β-cell area is reduced to 1/37 of that in autoimmune patients and 1/255 of that in normal controls (left panel). * p < 0.05.
We shall now consider the cells around the β-cells. The second important histological finding is that pancreatic α-cells are also damaged in fulminant type 1 diabetes [15]. The α-cell area is decreased to 33.1% of that seen in normal controls, although this is not the case in autoimmune type 1 diabetic patients.
We now turn to the cells outside the islet. The other portion of the pancreas, the exocrine portion, keeps its structure for at least 1-5 months after the onset of fulminant type 1 diabetes. Neither edematous change, necrosis, hemorrhage, suppuration, cyst formation, fatty degeneration nor the apparent atrophy of the exocrine pancreas was observed in any patients. However, CD3+ T cell-predominant infiltration to the exocrine pancreas was observed at the same time [4]. Elevations in serum pancreatic amylase, lipase, elastase-1 or phospholipase level are consistent with the histological findings in fulminant type 1 diabetes [4]. Pancreatic swelling is rarely reported in computed tomography or ultrasonography [17]. Some patients had disorders similar to pancreatitis, before the onset of overt diabetes and others after onset [18]. In both cases, symptoms resulting from exocrine dysfunction, such as diarrhea or fatty stool, were not long-standing. Increased serum pancreatic enzyme levels and pancreatic swelling disappear during the treatment of diabetic ketoacidosis alone, without drug treatment for pancreatitis, suggesting that the disorders in question are different from those seen in pancreatitis.
We now return to the β-cells. Hypoglycemia is sometimes observed just before the onset of fulminant type 1 diabetes [4]. This is probably because β-cell destruction is so rapid that insulin in the destroyed β-cells may enter the blood stream within a short period of time. A similar phenomenon is frequently observed when streptozotocin (STZ) or alloxan is injected into rodents to provide models of type 1 diabetes. In these models, pancreatic β-cells are completely destroyed in the same way as is evident in fulminant type 1 diabetes [19]. Do these chemical agents then contribute to β-cell destruction in fulminant type 1 diabetes?
Chemical agent and Reye's syndrome
Many drugs, chemical agents and toxins are known to induce diabetes through impaired insulin secretion or impaired insulin action or both. Diuretics (thiazide or loop diuretics), diphenylhydantoin, pentamidine, pyriminil (vacor), β-adrenergic antagonists, diazoxide, cyclosporine and opiates induce diabetes through impaired insulin secretion [20]. However, most fulminant type 1 diabetic patients have not received any of those drugs [4-6]. Antibiotics and/or non-steroidal or steroidal anti-inflammatory agents are sometimes used to treat the symptoms preceding fulminant type 1 diabetes such as fever, sore throat or cough. A case of fulminant type 1 diabetes associated with brain edema and fatty degeneration in the liver, both of which are hallmarks of Reye's syndrome, was reported [21]. Aspirin, a representative non-steroidal anti-inflammatory agent, is known to trigger Reye’s syndrome [22]. Mitochondrial dysfunction causes Reye's syndrome through fatty acid and other metabolic disorders. These findings suggest that anti-inflammatory drugs may kill β-cells, indirectly rather than directly, in some patients with fulminant type 1 diabetes. The question remains, however, of how the β-cells are destroyed in the remaining patients with fulminant type 1 diabetes, who do not take those agents.
Virus-primary candidate?
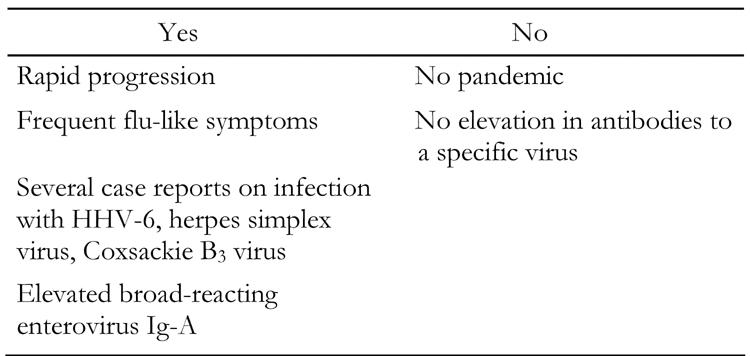
Viral involvement has been proposed in the pathogenesis of fulminant type 1 diabetes because of its markedly acute onset (Table 2) [4-6]. As shown in the report by Sekine et al., pancreatic β-cells are destroyed within a few days in fulminant type 1 diabetes [14]. It appears that targeted epithelial cells are destroyed by a virus within a few days in acute pharyngitis or tonsillitis. A nationwide survey revealed that flu-like symptoms were observed in 71.2 % of fulminant type 1 diabetic patients [6], also suggesting that viral infection was critical in the development of fulminant type 1 diabetes. Of those symptoms, fever was the most frequent and was observed in 60.0% of patients, followed in descending order of frequency by sore throat (25.2%), cough (12.0%), headache (11.5%) and nasal discharge (7.9%). Symptoms associated with gastrointestinal infection, such as diarrhea (5.5%) or lower abdominal pain (11.0%), were also reported. Indeed, some cases of fulminant type 1 diabetes were reported, in which the onset of diabetes was accompanied by the reactivation of human herpes virus-6 [11, 14, 23-26] and infection with herpes simplex virus [27] or coxsackie B3 virus [28-29].
Table 2. Viral infection in the pathogenesis of fulminant type 1 diabetes.
In addition, IgA antibody titers to enterovirus were significantly higher in patients with recent-onset fulminant type 1 diabetes than in those with typical type 1A diabetes and controls [30]. In this study, the assay system was able to detect IgA antibodies reacting with several different serotypes of enterovirus such as coxsackie A, coxsackie B, and echoviruses. Titers of IgA antibodies would be increased if different serotypes of enteroviruses repeatedly infected a single patient. Thus, these results suggest that fulminant type 1 diabetic patients are more susceptible to enterovirus infections than autoimmune diabetic patients and controls.
Shimada and Maruyama reported that encephalomyocarditis (EMC)-induced diabetes in a mouse model resembled fulminant type 1 diabetes in humans [31]. Intraperitoneal injection of male DBA/2 mice with a diabetic strain of EMC virus can induce very acute onset diabetes [32]. The EMC-induced diabetes model resembles fulminant type 1 diabetes, not only in its pattern of very rapid onset but also in the involvement of exocrine tissue damage, as indicated by histological findings and high serum amylase levels.
These lines of evidence prompted us to suppose that virus is a likely candidate for the pathogenesis of fulminant type 1 diabetes. However, an objection to this hypothesis has been raised. IgA antibody titers to enterovirus were significantly higher in fulminant type 1 diabetes, but there was no elevation in anti-coxsackie A2-10 or coxsackie B1-6 virus serotype-specific neutralizing antibodies in paired sera of 20 recent-onset fulminant type 1 diabetic patients in a nationwide survey (unpublished observation). This was also the case in human herpes virus (HHV)-6, although 6 patients were reported to suffer from fulminant type 1 diabetes after infection with or reactivation of this virus [11, 14, 23-26]. Titers of other viruses, HHV-7, cytomegalovirus, Epstein-Barr virus and rotavirus, were not elevated either in paired sera of 20 recent-onset fulminant type 1 diabetic patients (unpublished observation). This result suggests that viral infection is a candidate for fulminant β-cell killing, but no definite evidence is available to date. In addition, the nationwide survey revealed that no pandemic of fulminant type 1 diabetes has ever occurred. It is factors within the host, therefore, which are more significant for the development of fulminant diabetes than the virus itself. This was also demonstrated in the EMC-induced diabetes model. In this model, the same variant of EMC virus exerts effects in some inbred strains of mice such as DBA/2, SJL, or SWR, but not in C57BL/6 or BALB/C [32].
Recently, we took care of a 64-year-old patient who suffered from fulminant type 1 diabetes after influenza B infection (Sano et al., manuscript submitted). This virus is very common in Japan, so it was unlikely that it was the first time he had been infected. A new variant of influenza B virus may be involved, but it is likely that not only the influenza B virus itself but additional factors affect the development of fulminant type 1 diabetes.
In the next section, we discuss the host-defence mechanism, as part of the immune reaction, in the development of fulminant type 1 diabetes.
Immune mechanism
Islet autoantibodies seldom appear in fulminant type 1 diabetes. In the 11 patients with fulminant type 1 diabetes, who were initially reported, all these antibodies were negative in all patients [4]. Also in a nationwide survey, GAD antibody was positive only in 4.7% of cases and the titer was low. Neither IA-2Ab, IAA, nor ICA was positive in fulminant diabetic patients at all [6]. Positive GAD antibodies were detected in some cases, but the titer was low and positivity was transient, suggesting that those antibodies were produced as a result rather than a cause of β-cell destruction, as is the case in subacute thyroiditis. In this disease, thyroid autoantibody is sometimes transiently detected in patients' sera [33-34]. In addition, thyroid autoantibodies or thyroid disorders and other autoimmune diseases such as Hashimoto's thyroiditis or Graves’ disease, are less frequent in fulminant diabetes than in autoimmune type 1 diabetes [6].
Insulitis, the mononuclear cell infiltration of islets, and hyperexpression of MHC class I antigens in islets, are hallmarks of immune disorder and were observed in autoimmune type 1 diabetes [35], but were not found in the patients with fulminant type 1 diabetes in our first report. Their pancreases were studied at 1-5 months after onset [4]. Yamazaki and Hayashi also reported no insulitis in the biopsy specimens taken on Day 33 after the onset of fulminant diabetes [36]. In patients with autoimmune type 1 diabetes of similar duration, CD8+ T-cell predominant insulitis was observed in the pancreas biopsy tissue. Fas was expressed in islet cells and Fas ligand was expressed in islet-infiltrating T cells. Pancreatic β-cells, therefore, are supposed to be destroyed through Fas-Fas ligand interaction in type 1A diabetes [37]. We had hypothesized that the absence of both islet-related autoantibodies in patients’ sera and insulitis in patients' pancreas suggest that an autoimmune mechanism does not contribute to the development of fulminant type 1 diabetes [4-5].
On the other hand, Tanaka et al. reported an autopsy case of fulminant type 1 diabetes where lymphocytes had infiltrated into both the endocrine and exocrine pancreas a few days after the onset of overt diabetes [38]. We also confirmed cellular infiltration of the pancreatic islets in patients with fulminant type 1 diabetes who died 3 days after onset (unpublished observation). On the basis of these findings, we now think that cellular infiltration of islets exists at approximately the time of onset of fulminant type 1 diabetes but disappears soon after β-cell destruction (Table 3).
Table 3. Immune reaction in the pathogenesis of fulminant type 1 diabetes.

GAD-reactive Th1 cells and insulin B9-23-reactive Th1 cells were identified in peripheral blood mononuclear cells in 9 out of 13 and 3 out of 12 fulminant type 1 diabetic patients by using ELISPOT assay [39]. Shimada et al. reported a fulminant diabetic patient with high serum levels of CXCL10, a chemokine inducing migration of activated T-cells to local lesions and GAD-reactive CD4+ cells in the periphery [40]. These findings indicate that cellular immunoreactivity to β-cell antigens may be unregulated in fulminant type 1 diabetes, at least after the onset of overt diabetes.
However, the presence of insulitis does not necessarily mean that β-cell death is caused by autoimmunity. Immune mechanisms include not only autoimmunity but also other mechanisms such as antivirus immunity following viral infection of β-cells, as was demonstrated in the EMC-infected mouse model [32]. In addition, the presence of T cell immunoreactivity after onset would not be the cause but the result of β-cell death.
The histological findings obtained shortly after onset and peripheral blood data suggest that the immune reaction to pancreatic β-cells is important in the development of fulminant type 1 diabetes. The human leukocyte antigen (HLA) plays a significant part in immune reaction. So, our next theme is the role of HLA subtypes in fulminant type 1 diabetes.
Genetic factors - the role of HLA
It has been reported that HLA confers susceptibility and resistance to the development of type 1 diabetes [41]. HLA DR4-DQ4 (usually encoded by DRB1*0405-DQB1*0401) and DR9-DQ3 (encoded by DRB1*0901-DQB1*0303) have been reported to confer susceptibility, and DR2-DQ1 (encoded by DRB1*1501/*1502-DQB1*0602/*0601) resistance, to type 1 diabetes in Japanese subjects. On the other hand, HLA DR4-DQ1 (encoded by DRB1*0401-DQB1*0302) and DR3-DQ2 (encoded by DRB1*0301-DQB1*0201), but not DR4-DQ4, confer susceptibility, and DR2-DQ1 (encoded by DRB1*1501-DQB1*0602) also confers resistance, to type 1 diabetes in Caucasians [41-42].
As a part of the nationwide survey, HLA-A, -DR and -DQ serotypes were investigated in 91 fulminant and 81 autoimmune (type 1A) diabetic patients together with 190 normal controls [43]. The distribution of HLA-A, class I HLA, did not differ among the 3 groups. As regards class II HLA, DR4-DQ4 was observed in 41.8% and was thus significantly more frequent, whereas both DR2-DQ1 and DR8-DQ1 were less frequent in fulminant diabetes. In type 1A diabetes, DR2-DQ1 was extremely rare, while DR9-DQ3 was significantly more frequent. In the combined analysis, the homozygote of DR4-DQ4 in fulminant type 1 diabetes and DR9-DQ3 in typical type 1A diabetes showed high odds ratios (13.3 and 13.3 respectively). These results suggest that class II HLA also contributes to the development of fulminant type 1 diabetes, but that the susceptibility and resistance of the HLA subtype to type 1 diabetes are different in fulminant and typical autoimmune type 1 diabetes. The HLA-DR4-DQ4 haplotype is common in Japanese subjects but rare in the Caucasian population. It might contribute to the different incidence of fulminant type 1 diabetes between Japanese and Caucasians. The role of class II HLA has been emphasized in the context of the antigen-presenting process in typical autoimmune type 1 diabetes [42], but it remains to be elucidated how a certain class II HLA can contribute towards the molecular mechanisms of β-cell destruction in fulminant type 1 diabetes. One possibility is that the HLA molecule is associated with an immune reaction in fulminant diabetes-like type 1A diabetes. It is also possible that it may interact with some kind of virus as shown in mice [44]. In addition, small pilot studies suggest that DR4-DQ4, which is susceptible to fulminant diabetes, is encoded by DRB1*0405-DQB1*0401 [45-46].
Other genetic factors contributing to the development of fulminant type 1 diabetes are largely unknown. The preliminary study of CTLA-4 gene polymorphism in exon 1 showed no susceptibility to the development of this disease (Kawasaki E, Imagawa A, et al., unpublished observation).
Association with pregnancy
Fulminant type 1 diabetes is not predominant in females, but pregnancy is sometimes associated with this disease [47-49]. Almost all patients who suffered from type 1 diabetes during pregnancy or just after delivery showed characteristics similar to the fulminant type. Shimizu et al. reported on the clinical characteristics of 22 patients who developed fulminant diabetes associated with pregnancy [49]. Out of those 22 patients, 18 patients developed diabetes during pregnancy and 4 patients developed diabetes within 2 weeks after delivery. Onset in 13 patients took place in the third trimester and fetal demise occurred in 12 out of 18 patients who developed fulminant diabetes during pregnancy. It is well known that autoimmune thyroid disease is ameliorated during pregnancy because of a shift in a Th1- to a Th2-type response, but is aggravated after delivery. This phenomenon is well known as a postpartum autoimmune disease, especially postpartum thyroid disease [50]. Because postpartum aggravation of Hashimoto's disease usually occurs 1-4 months after delivery, a postpartum rebound in cellular immunity is assumed to occur around this period. However, the onset of fulminant type 1 diabetes associated with pregnancy occurred either during pregnancy or shortly after delivery. Therefore, it may be caused by a mechanism other than that of postpartum autoimmune disease.
Tentative hypotheses for the destruction of β-cells
Figure 2 illustrates our tentative hypothesis of β-cell destruction in fulminant type 1 diabetes. Both genetic and environmental factors contribute to the development of fulminant type 1 diabetes. The results of HLA analyses and antibodies to enterovirus suggest that they are risk factors contributing to the susceptibility of fulminant type 1 diabetes development. Viral infection triggers the destruction of β-cells in susceptible individuals. The first pathway to β-cell death is via viral infection of, β-cells and the self-replication of the infected cells. Viral infection also activates an innate immune response to delete viruses and infected cells, predominantly through macrophage-derived agents, for example, cytokines and nitric oxide. This would be the second and main pathway and would play an important role in the destruction of β-cells in fulminant diabetes. It is noteworthy that the damage to both α- and β-cells suggests a less specific mechanism to β-cells in fulminant diabetes than that in typical type 1A diabetes. We can speculate that some kind of bystander effect on the part of cytokines or nitric oxide might play a role in the destruction of islet cells. In the final phase, the adaptive immune system would be activated and the remaining viruses and their host, the β-cells, would be destroyed by T cells. This is the third pathway, although the detailed mechanism remains to be clarified.
Figure 2.
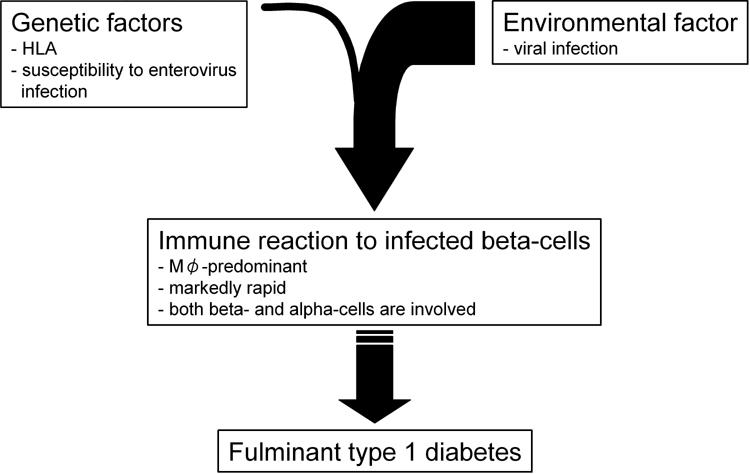
Tentative hypothesis for the development of fulminant type 1 diabetes.
Is this hypothesis different from that of type 1A diabetes or not? Is fulminant type 1 diabetes a subtype of type 1A diabetes, but one that does not have enough time to develop islet autoantibodies? Do viruses, macrophages and T cells also play a part of some kind in the destruction of β-cells in type 1A diabetes? These questions are difficult to answer because the molecular mechanism of type 1A diabetes is not yet fully understood [51]. However, the bimodal distribution of glycosylated hemoglobin at the onset of overt diabetes suggests a discontinuous etiology between fulminant and classic type 1A diabetes.
In addition, insulin resistance, which is another operator of glucose intolerance and which plays a critical part in type 2 diabetes, might also play a significant role in fulminant type 1 diabetes. Viral infection, which is commonly detected at the onset of fulminant diabetes, induces insulin resistance. A higher insulin dose needs to be injected in fulminant type 1 diabetes than in type 1A diabetes [6]. However, no detailed findings are available to date about insulin resistance in patients with fulminant type 1 diabetes.
For the better understanding of the pathogenesis of fulminant type 1 diabetes, the identification of the patients with this disease is essential. For this purpose, the committee of the Japan Diabetes Society on the Research of Fulminant Type 1 Diabetes Mellitus determined the criteria for the definite diagnosis of fulminant type 1 diabetes mellitus in 2004 (Table 4) [52].
Table 4. Criteria for the diagnosis of fulminant type 1 diabetes mellitus.

Conclusions
Fulminant type 1 diabetes is a recently discovered subtype of type 1 diabetes. The evidence accumulated to date suggests that both the viral infection itself and the subsequent immune reaction are likely to cause β-cell destruction and finally fulminant type 1 diabetes. The next step in studying this novel clinical entity would be to clarify the molecular mechanism of β-cell destruction in detail.
Acknowledgments
We are indebted to Drs. Naoto Itoh, Jun-ichiro Miyagawa, David M Harlan, Hiroshi Ikegami and Yuji Matsuzawa for their contribution to the discussion and also to the doctors of the Osaka IDDM study group (shown in [4]) and the Japan Diabetes Society Committee on Fulminant Type 1 Diabetes Mellitus Research (shown in [6]) for their collaboration. This study was supported by a grant-in-aid for scientific research from the Japan Society for the Promotion of Science. The authors are also indebted to the Japan Diabetes Society for its continuous support for this study.
References
- 1.Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20(7):1183–1197. doi: 10.2337/diacare.20.7.1183. [DOI] [PubMed] [Google Scholar]
- 2.Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998;15:539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 3.Kuzuya T, Nakagawa S, Satoh J, Kanazawa Y, Iwamoto Y, Kobayashi M, Nanjo K, Sasaki A, Seino Y, Ito C, Shima K, Nonaka K, Kadowaki T. Report of the Committee on the classification and diagnostic criteria of diabetes mellitus. Committee of the Japan Diabetes Society on the diagnostic criteria of diabetes mellitus. Diabetes Res Clin Pract. 2002;55(1):65–85. doi: 10.1016/s0168-8227(01)00365-5. [DOI] [PubMed] [Google Scholar]
- 4.Imagawa A, Hanafusa T, Miyagawa J, Matsuzawa Y. A novel subtype of type 1 diabetes mellitus characterized by a rapid onset and an absence of diabetes-related antibodies. Osaka IDDM study group. N Engl J Med. 2000;342:301–307. doi: 10.1056/NEJM200002033420501. [DOI] [PubMed] [Google Scholar]
- 5.Imagawa A, Hanafusa T, Miyagawa J, Matsuzawa Y. A proposal of three distinct subtypes of type 1 diabetes mellitus based on clinical and pathological evidence. Ann Med. 2000;32:539–543. doi: 10.3109/07853890008998833. [DOI] [PubMed] [Google Scholar]
- 6.Imagawa A, Hanafusa T, Uchigata Y, Kanatsuka A, Kawasaki E, Kobayashi T, Shimada A, Shimizu I, Toyoda T, Maruyama T, Makino H. Fulminant type 1 diabetes: a nationwide survey in Japan. Diabetes Care. 2003;26:2345–2352. doi: 10.2337/diacare.26.8.2345. [DOI] [PubMed] [Google Scholar]
- 7.Jung TS, Chung SI, Kim MA, Kim SJ, Park MH, Kim DR, Kang MY, Hahm JR. A Korean patient with fulminant autoantibody-negative type 1 diabetes. Diabetes Care. 2004;27:3023–3024. doi: 10.2337/diacare.27.12.3023. [DOI] [PubMed] [Google Scholar]
- 8.Jung JH, Hahm JR, Kim MA, Park MH, Kim DR, Jung TS, Chung SI. Fulminant autoantibody-negative and type 1A diabetes phenotypes in a Korean HLA identical dizygotic twin. Diabetes Care. 2005;28:2330–2331. doi: 10.2337/diacare.28.9.2330. [DOI] [PubMed] [Google Scholar]
- 9.Taniyama M, Katsumata R, Aoki K, Suzuki S. A Filipino patient with fulminant type 1 diabetes. Diabetes Care. 2004;27:842–843. doi: 10.2337/diacare.27.3.842. [DOI] [PubMed] [Google Scholar]
- 10.Katsumata K, Katsumata K. A Chinese patient presenting with clinical signs of fulminant type 1 diabetes mellitus. Intern Med. 2005;44:967–969. doi: 10.2169/internalmedicine.44.967. [DOI] [PubMed] [Google Scholar]
- 11.Chiou CC, Chung WH, Hung SI, Yang LC, Hong HS. Fulminant type 1 diabetes mellitus caused by drug hypersensitivity syndrome with human herpesvirus 6 infection. J Am Acad Dermatol. 2006;54(2 Suppl):S14–S17. doi: 10.1016/j.jaad.2005.03.057. [DOI] [PubMed] [Google Scholar]
- 12.Pozzilli P, Visalli N, Leslie D. No evidence of rapid onset (Japanese) type I diabetes in Caucasian patients. IMDIAB Group. Diabetologia. 2000;43(10):1332. doi: 10.1007/pl00022647. [DOI] [PubMed] [Google Scholar]
- 13.Vreugdenhil GR, Schloot NC, Hoorens A, Rongen C, Pipeleers DG, Melchers WJ, Roep BO, Galama JM. Acute onset of type I diabetes mellitus after severe echovirus 9 infection: putative pathogenic pathways. Clin Infect Dis. 2000;31(10):1025–1031. doi: 10.1086/318159. [DOI] [PubMed] [Google Scholar]
- 14.Sekine N, Motokura T, Oki T, Umeda Y, Sasaki N, Hayashi M, Sato H, Fujita T, Kaneko T, Asano Y, Kikuchi K. Rapid loss of insulin secretion in a patient with fulminant type 1 diabetes mellitus and carbamazepine hypersensitivity syndrome. JAMA. 2001;285(9):1153–1154. doi: 10.1001/jama.285.9.1153. [DOI] [PubMed] [Google Scholar]
- 15.Sayama K, Imagawa A, Okita K, Uno S, Moriwaki M, Kozawa J, Iwahashi H, Yamagata K, Tamura S, Matsuzawa Y, Hanafusa T, Miyagawa J, Shimomura I. Pancreatic beta and alpha cells are both decreased in patients with fulminant type 1 diabetes: a morphometrical assessment. Diabetologia. 2005;48:1560–1564. doi: 10.1007/s00125-005-1829-9. [DOI] [PubMed] [Google Scholar]
- 16.Yonemura Y, Takashima T, Miwa K, Miyazaki I, Yamamoto H, Okamoto H. Amelioration of diabetes mellitus in partially depancreatized rats by poly(ADP-ribose) synthetase inhibitors. Evidence of islet B-cell regeneration. Diabetes. 1984. 33(4):401–404. doi: 10.2337/diab.33.4.401. [DOI] [PubMed] [Google Scholar]
- 17.Kahara T, Takamura T, Sakurai M, Misu H, Usuda R, Hayakawa T, Nishimura Y, Bando Y, Nagaoka T, Nagai Y, Kaneko S. Pancreatic exocrine and endocrine events occur concomitantly but independently during the course of fulminant type 1 diabetes. Diabetes Res Clin Pract. 2006;71:241–246. doi: 10.1016/j.diabres.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Fukui K, Imagawa A, Iwahashi H, Kashine S, Moriwaki M, Nanmo T, Misugi S, Sekiguchi K, Yamagata K, Miyagawa JI, Matsuzawa Y. A case of diabetes mellitus after acute pancreatitis with histological findings compatible to non-autoimmune fulminant type 1 diabetes. J Japan Diab Soc. 2004;45:759–765. [Google Scholar]
- 19.Waguri M, Yamamoto K, Miyagawa JI, Tochino Y, Yamamori K, Kajimoto Y, Nakajima H, Watada H, Yoshiuchi I, Itoh N, Imagawa A, Namba M, Kuwajima M, Yamasaki Y, Hanafusa T, Matsuzawa Y. Demonstration of two different processes of beta-cell regeneration in a new diabetic mouse model induced by selective perfusion of alloxan. Diabetes. 1997. 46:1281–1290. doi: 10.2337/diab.46.8.1281. [DOI] [PubMed] [Google Scholar]
- 20.Ganda OP. In: Kahn CR, et al. (eds) Joslin's Diabetes Mellitus 14. ed. Lippincott Williams and Wilkins; Philadelphia: 2005. Secondary forms of diabetes; pp. 477–504. [Google Scholar]
- 21.Hasegawa G, Ohashi R, Naito M, Takagi A. An autopsy case of fulminant type 1 diabetes accompanying Reye's syndrome. Endocrine J. 2005;52(suppl 1):159. [Google Scholar]
- 22.Glasgow JF, Middleton B. Reye syndrome - insights on causation and prognosis. Arch Dis Child. 2001;85:351–353. doi: 10.1136/adc.85.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sommers LM, Schoene RB. Allopurinol hypersensitivity syndrome associated with pancreatic exocrine abnormalities and new-onset diabetes mellitus. Arch Intern Med. 2002;27:1190–1192. doi: 10.1001/archinte.162.10.1190. [DOI] [PubMed] [Google Scholar]
- 24.Seino Y, Yamauchi M, Hirai C, Okumura A, Kondo K, Yamamoto M, Okazaki Y. A case of fulminant type 1 diabetes associated with mexiletine hypersensitivity syndrome. Diabet Med. 2004;21:1156–1157. doi: 10.1111/j.1464-5491.2004.01307.x. [DOI] [PubMed] [Google Scholar]
- 25.Ohta K, Takamoto T, Izumi H, Yoshida T, Ishihara Y, Hirata Y. A case of fulminant type 1 diabetes mellitus during steroid therapy for hypersensitivity syndrome. J Japan Diab Soc. 2001;44:907–912. [Google Scholar]
- 26.Yamamoto S, Fujimoto H, Hirakawa N, Morimoto I. A case of fulminant type 1 diabetes mellitus accompanied by hypersensitivity syndrome. J Japan Diab Soc. 2003;46:949–954. [Google Scholar]
- 27.Nagaoka T, Terada M, Miyakoshi H. Insulin-dependent diabetes mellitus following acute pancreatitis caused by herpes simplex virus; a case report. J Japan Diab Soc. 2001;44:335–340. [Google Scholar]
- 28.Hashimoto N, Makino H, Kanatsuka A, Fujiwara T, Sakurada M, Iwaoka H, Taira M, Osawa K, Yamaguchi T, Yoshida S. A case of insulin-dependent diabetes mellitus with coxsackie B3 virus infection following diaminodiphenyl sulfone (DDS) syndrome. J Japan Diab Soc. 1987;30:761–765. [Google Scholar]
- 29.Nishida W, Hasebe S, Kawamura R, Hashiramoto M, Onuma H, Osawa H, Makino H. A case of fulminant type 1 diabetes associated with high titer of coxsackie B3 virus antibody. J Japan Diab Soc. 2005;48(suppl 1):A23–A27. [Google Scholar]
- 30.Imagawa A, Hanafusa T, Makino H, Miyagawa JI, Juto P. High titres of IgA antibodies to enterovirus in fulminant type-1 diabetes. Diabetologia. 2005;48:290–293. doi: 10.1007/s00125-004-1624-z. [DOI] [PubMed] [Google Scholar]
- 31.Shimada A, Maruyama T. Encephalomyocarditis-virus-induced diabetes model resembles "fulminant" type 1 diabetes in humans. Diabetologia. 2004;47:1854–1855. doi: 10.1007/s00125-004-1538-9. [DOI] [PubMed] [Google Scholar]
- 32.Jun HS, Yoon JW. The role of viruses in type I diabetes: two distinct cellular and molecular pathogenic mechanisms of virus-induced diabetes in animals. Diabetologia. 2001;44:271–285. doi: 10.1007/s001250051614. [DOI] [PubMed] [Google Scholar]
- 33.Strakosch CR, Joyner D, Wall JR. Thyroid stimulating antibodies in patients with subacute thyroiditis. J Clin Endocrinol Metab. 1978;46:345–348. doi: 10.1210/jcem-46-2-345. [DOI] [PubMed] [Google Scholar]
- 34.Wagar G, Makinen T. Thyrotrophin-binding inhibiting immunoglobulins (TBII) in subacute thyroiditis. Acta Endocrinol Suppl (Copenh) 1983;251:53–57. [PubMed] [Google Scholar]
- 35.Itoh N, Hanafusa T, Miyazaki A, Miyagawa J, Yamagata K, Yamamoto K, Waguri M, Imagawa A, Tamura S, Inada M, Tarui S, Kono N, Matsuzawa Y. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92:2313–2322. doi: 10.1172/JCI116835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamazaki M, Hayashi T. Rapid-onset type 1 diabetes mellitus without pancreatic exocrine dysfunction. Ann Intern Med. 2002;137:145–146. doi: 10.7326/0003-4819-137-2-200207160-00024. [DOI] [PubMed] [Google Scholar]
- 37.Moriwaki M, Itoh N, Miyagawa J, Yamamoto K, Imagawa A, Yamagata K, Iwahashi H, Nakajima H, Namba M, Nagata S, Hanafusa T, Matsuzawa Y. Fas and Fas ligand expression in inflamed islets in pancreas sections of patients with recent-onset type I diabetes mellitus. Diabetologia. 1999;42:1332–1340. doi: 10.1007/s001250051446. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka S, Kobayashi T, Momotsu T. A novel subtype of type 1 diabetes mellitus. N Engl J Med. 2000;342:1835–1838. [PubMed] [Google Scholar]
- 39.Kotani R, Nagata M, Imagawa A, Moriyama H, Yasuda H, Miyagawa J, Hanafusa T, Yokono K. T lymphocyte response against pancreatic beta cell antigens in fulminant type 1 diabetes. Diabetologia. 2004;47:1285–1291. doi: 10.1007/s00125-004-1441-4. [DOI] [PubMed] [Google Scholar]
- 40.Shimada A, Morimoto J, Kodama K, Oikawa Y, Irie J, Nakagawa Y, Narumi S, Saruta T. T-cell-mediated autoimmunity may be involved in fulminant type 1 diabetes. Diabetes Care. 2002;25:635–636. doi: 10.2337/diacare.25.3.635. [DOI] [PubMed] [Google Scholar]
- 41.Ikegami H, Ogihara T. Genetics of insulin-dependent diabetes mellitus. Endocr J. 1996;43:605–613. doi: 10.1507/endocrj.43.605. [DOI] [PubMed] [Google Scholar]
- 42.Eisenbarth GS, Polonsky KS, Buse JB. In: P Reed Larsen et al. (eds). Williams Textbook of Endocrinology 10th ed. Saunders; Philadelphia: 2003. Type 1 diabetes mellitus; pp. 1485–1504. [Google Scholar]
- 43.Imagawa A, Hanafusa T, Uchigata Y, Kanatsuka A, Kawasaki E, Kobayashi T, Shimada A, Shimizu I, Maruyama T, Makino H. Different contribution of class II HLA in fulminant and typical autoimmune type 1 diabetes mellitus. Diabetologia. 2005;48:294–300. doi: 10.1007/s00125-004-1626-x. [DOI] [PubMed] [Google Scholar]
- 44.Wykes MN, Shellam GR, McCluskey J, Kast WM, Dallas PB, Price P. Murine cytomegalovirus interacts with major histocompatibility complex class I molecules to establish cellular infection. J Virol. 1993;67:4182–4189. doi: 10.1128/jvi.67.7.4182-4189.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka S, Kobayashi T, Nakanishi K, Koyama R, Okubo M, Murase T, Odawara M, Inoko H. Association of HLA-DQ genotype in autoantibody-negative and rapid-onset type 1 diabetes. Diabetes Care. 2002;25:2302–2307. doi: 10.2337/diacare.25.12.2302. [DOI] [PubMed] [Google Scholar]
- 46.Nakamura T, Nagasaka S, Kusaka I, Yatagai T, Yang J, Ishibashi S. HLA-DR-DQ haplotype in rapid-onset type 1 diabetes in Japanese. Diabetes Care. 2003;26:1640–1641. doi: 10.2337/diacare.26.5.1640. [DOI] [PubMed] [Google Scholar]
- 47.Otsubo M, Shiozawa T, Kimura K, Konishi I. Nonimmune "fulminant" type 1 diabetes presenting with diabetic ketoacidosis during pregnancy. Obstet Gynecol. 2002;99:877–879. doi: 10.1016/s0029-7844(01)01628-3. [DOI] [PubMed] [Google Scholar]
- 48.Shimizu I, Makino H, Osawa H, Kounoue E, Imagawa A, Hanafusa T, Kawasaki E, Fujii Y. Association of fulminant type 1 diabetes with pregnancy. Diabetes Res Clin Pract. 2003;62:33–38. doi: 10.1016/s0168-8227(03)00147-5. [DOI] [PubMed] [Google Scholar]
- 49.Shimizu I, Makino H, Imagawa A, Iwahashi H, Uchigata Y, Kanatsuka A, Kawasaki E, Kobayashi T, Shimada A, Maruyama T, Hanafusa T. Clinical and immunogenetic characteristics of fulminant type 1 diabetes associated with pregnancy. J Clin Endocrinol Metab. 2006;91:471–476. doi: 10.1210/jc.2005-1943. [DOI] [PubMed] [Google Scholar]
- 50.Amino N, Mori H, Iwatani Y, Tanizawa O, Kawashima M, Tsuge I, Ibaragi K, Kumahara Y, Miyai K. High prevalence of transient post-partum thyrotoxicosis and hypothyroidism. N Engl J Med. 1982;306:849–852. doi: 10.1056/NEJM198204083061405. [DOI] [PubMed] [Google Scholar]
- 51.Gale EA. Declassifying diabetes. Diabetologia. 2006;49:1989–1995. doi: 10.1007/s00125-006-0348-7. [DOI] [PubMed] [Google Scholar]
- 52.Hanafusa T, Imagawa A, Iwahashi H, Uchigata Y, Kanatsuka A, Kawasaki E, Kobayashi T, Shimada A, Shimizu I, Maruyama T, Makino H. Epidemiological and clinical analysis and proposal of diagnostic criteria. Report of Japan Diabetes Society Committee on Fulminant Type 1 Diabetes Mellitus Research. J Japan Diab Soc. 2005;48(suppl 1):A1–A13. [Google Scholar]