

Summary
Although each of the five mammalian long-chain acyl-CoA synthetases (ACSL) can bind saturated and unsaturated fatty acids ranging from 12 to 22 carbons, ACSL4 prefers longer chain polyunsaturated fatty acids. In order to gain a better understanding of ACSL4 fatty acid binding, we based a mutagenesis approach on sequence alignments related to ttLC-FACS crystallized from Thermus thermophilus HB8. Four residues selected for mutagenesis corresponded to residues in ttLC-FACS that comprise the fatty acid binding pocket; the fifth residue aligned with a region thought to be involved in fatty acid selectivity of the Escherichia coli acyl-CoA synthetase, FadD. Changing an amino acid at the entry of the putative fatty acid binding pocket, G401L, resulted in an inactive enzyme. Mutating a residue near the pocket entry, L399M, did not significantly alter enzyme activity, but mutating a residue at the hydrophobic terminus of the pocket, S291Y, altered ACSL4’s preference for 20:5 and 22:6 and increased its apparent Km for ATP. Mutating a site in a region previously identified as important for fatty acid binding also altered activation of 20:4 and 20:5. These studies suggested that the preference of ACSL4 for long-chain polyunsaturated fatty acids can be modified by altering specific amino acid residues.
Keywords: acyl-CoA synthetase, polyunsaturated fatty acid, mutagenesis, fatty acid binding, kinetics, arachidonic acid
Introduction
For virtually every metabolic pathway except that of eicosanoid synthesis, the use of long-chain fatty acids requires their activation to fatty acyl-CoAs. Long-chain acyl-CoAs are substrates for the synthesis of complex lipids, including triacylglycerol, phospholipids, ceramide, sphingolipids, and cholesterol esters, and for the degradation of fatty acids via mitochondrial and peroxisomal β-oxidation and microsomal ω-oxidation [1, 2]. In addition, long-chain acyl-CoAs alter a number of intracellular processes, including the modulation of insulin secretion [3], and are ligands for the transcription factor HNF4α [4, 5]. All acyl-CoA synthetases use the energy from ATP hydrolysis to catalyze a similar two step reaction. In the first step fatty acid and ATP yield a fatty acyl-AMP with the release of pyrophosphate. In the second step the fatty acyl-AMP is converted to fatty acyl-CoA with the release of AMP. The reaction is made essentially irreversible by the hydrolysis of the pyrophosphate.
Acyl-CoA synthetases have been classified by their preferences for short, medium, long, and very long chain fatty acids, although there is considerable overlap [2, 6]. Of the long chain acyl-CoA synthetases, which use fatty acids of 12 to 22 carbons, five rat, mouse and human isoforms have been identified [7]. The biochemical function of these isoforms can be distinguished by the chain length of their fatty acid substrate preference, their CoA and ATP affinities, their relative specific activities, and their tissue and organelle locations [2]. Although the amino acid sequence of each long-chain acyl-CoA synthetase isoform is unique, all contain considerable homology and several highly conserved regions [6].
Rat ACSL4 is a membrane-associated long chain acyl-CoA synthetase [8] that can activate both saturated and unsaturated fatty acids from 14 to 26 carbons [9]. In human brain a longer splice variant is present and mutations are associated with a rare form of X-linked mental retardation [10, 11]. The specific activity of rat ACSL4 is twice as high with arachidonate as with palmitate [9], and the apparent IC50 of ACSL4 is ≤ 5 μM for 18:2, 18:3, 20:4, 22:5 and 22:6 with 16:0 as the labeled substrate [12].
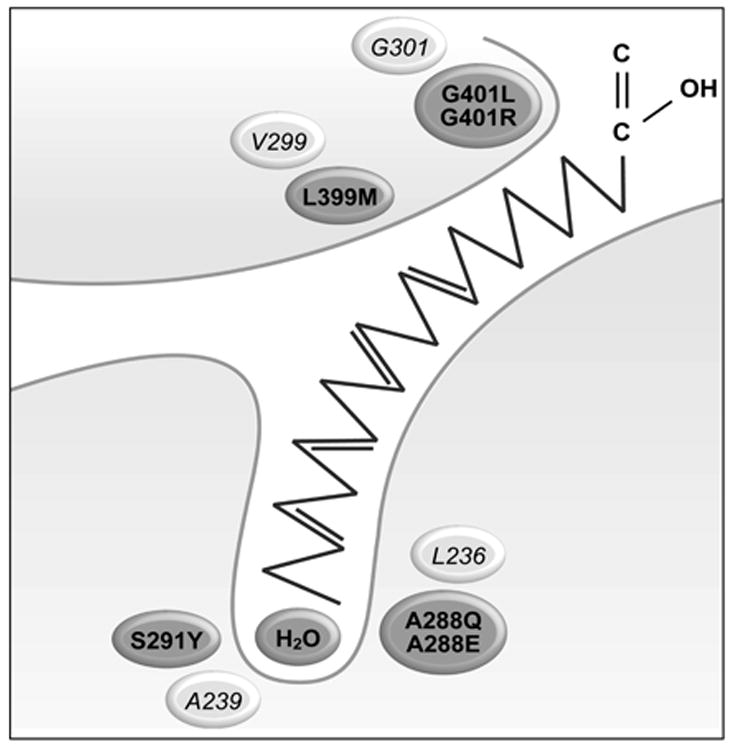
No NMR or X-ray crystal studies are available for mammalian ACSLs. In fact, the only structural information available is from studies of ttLC-FACS, a long chain acyl-CoA synthetase isolated from the thermophilic bacterium Thermus thermophilus HB8 [13]. The ttLC-FACS functions as a homodimer with interactions at the N-terminus. Specific residues in this region from different organisms are conserved in long-chain ACSs but not in other adenylate-forming families, suggesting that all long-chain ACSs may function as homodimers. Based on studies of ttLC-FACS crystal structure, Hisanaga, et al. identified a gated fatty acid binding tunnel with a unidirectional movement of fatty acid [13]. In addition, these authors suggested that long-chain acyl-CoA synthetases contain several structural motifs and conserved regions, including a 6-amino acid linker region that connects the C- and N-terminal domains, an adenine binding motif that has also been designated an ATP/AMP binding domain, a gate motif, and a phosphate-binding P-loop (Fig. 1A) [13]. Based on site-directed mutagenesis studies of FadD, the Escherichia coli long chain acyl-CoA synthetase, it was suggested that a conserved group of 25 amino acids, termed the fatty acid signature motif, is required for fatty acid recognition [14]. The crystal structure data from ttLC-FACS, however, suggests that additional amino acid residues located in a region that is distant from the fatty acid signature motif are also involved in recognizing fatty acid substrates. These studies suggest the presence of a “tunnel” that forms a pocket in which the fatty acid binds (Fig. 1B).
Figure 1A.


Fig 1B.
We wondered whether ACSL4 also formed a tunnel that accommodates the binding of a wide range of fatty acid substrates including the long chain polyunsaturated fatty acids. To address the question of substrate specificity, we used a site-directed mutagenesis approach based on structural information from ttLC-FACS. We generated mutations at four unique amino acid positions, each of which would correspond to a residue in the fatty acid binding pocket based on ttLC-FACS sequence alignments, as well as a fifth position corresponding to a fatty acid binding region of E. coli FadD. These studies provide novel structure-function information on the mammalian ACSL4.
Materials and Methods
Site directed mutagenesis
PCR based site directed mutagenesis was performed essentially as described in Stratagene’s Quickchange site directed mutagenesis kit using recombinant ACSL4 containing a C-terminal M2-flag-tag (DYKDDDDK) as a template. The primers used in the mutagenesis study (shown below) were synthesized at the UNC-Chapel Hill Nucleic Acid Facility. (The reverse complementary primer for each mutation is not shown.) The base changed to generate the amino acid change is underlined. For easy screening of two of the mutations, an additional base was changed to either remove or add a restriction enzyme site. This change is indicated in bold. S291Y, 5′-acagcagagatatactgctttac-3′; A288E, 5′-tgctggaactgacagaagagatatcctgctttacctacgg-3′; G401R, 5′-tgatgctgtcccgcggggccccactgtc-3′; L399M, 5′-gcatgatgatgtccggcggggccccact-3′; G401L, 5′-catgatgctgtccctcggcgccccactgtc-3′; A288Q, 5′-gtgctggaactgacacaagcgatgtggtctttacctac-3′; Q525K, 5′-gcttaaagattatcgaccgtaagaaag-3′. Mutations were confirmed by automated sequencing at the UNC-Chapel Hill Genome Analysis Facility.
Protein expression and partial purification
Proteins were expressed and partially purified essentially as described [15]. Briefly, cells containing plasmids that expressed ACSL4 or mutants of ACSL4 were grown in Terrific Broth. Protein expression was induced for 10–14 hours at 30°C with 1mM IPTG. Cells were pelleted and resuspended in TE buffer (10 mM Tris-HCl, 1mM EDTA). (For some samples, a protease inhibitor cocktail for bacterial lysates [Sigma P2714] was added as well.) Cells were lysed using 100 μg/ml lysozyme followed by sonication on ice (10 second pulse followed by 10 second break repeated 6 times). Membranes were isolated with an SW41 swinging bucket rotor on a sucrose cushion prepared with 2 ml of 55% (w/w) sucrose overlaid with 0.5 ml of 5% (w/w) sucrose via ultracentrifugation at 35,000 rpm for 3 hours with no brake. Membranes were removed from the cushion with a needle. An equal volume of 2% Triton X-100 in TBS buffer was added to the membranes. Partial purification was achieved by loading the membrane fraction onto an M2-anti-FLAG agarose column (Sigma). The column was washed with TBS buffer and the protein was eluted with 5 ml of 100 μg/ml FLAG peptide (Sigma). Samples were analyzed by SDS-PAGE. Protein purity was assessed by staining with Coomassie dye. The concentration of protein in the eluted fractions was determined using the Coomassie Plus assay (Pierce).
ACS kinetics
Acyl-CoA synthetase activity for ACSL4 and the ACSL4 mutants was determined using either an isotopic method (8) or a coupled assay (9). The assay mix for the isotopic method included 175 mM Tris-HCl, pH 7.4, 8 mM MgCl2, 5 mM dithiothreitol, 10 mM ATP, 0.25 mM CoA, and 50 μM [14C]palmitic acid (New England Nuclear) in 0.5 mM Triton X-100, 0.01 mM EDTA in a total volume of 200 μl. The reaction was started by adding enzyme (0.75–1 μg protein) and was measured for 5 min at 37°C. Reactions were terminated with 1 ml of Dole’s solution, isopropanol: heptane: 1M H2 SO4 (80/20/2; v/v). Water (0.5 ml) was added and unreacted palmitate was removed in the organic phase by extracting 2–3 times with 2 ml heptane. [14C]Palmitoyl-CoA formed during the reaction was measured by scintillation counting. Km values for palmitate, CoA and ATP were determined using subsaturating and saturating concentrations of the ligand being monitored while holding all other ligands at published saturating concentrations (15). Km and Vmax for each substrate were determined by double reciprocal analysis using a substrate range between 0.3 and 2.0 times the Km as reported previously [16]
A coupled spectrophotometric assay was used to determine activity using substrates of different chain lengths. The spectrophotometric assay contained 100 mM Tris-HCl pH 7.4, 150 mM KCl, 8 mM MgCl2, 5 mM DTT, 250 mM CoA, 10 mM ATP, 250 mM phosphoenylpyruvate, 1.5 mM NADH, 80 units myokinase, 17 units lactate dehydrogenase, 11 units pyruvate kinase (Sigma), and 50 μM fatty acid in 0.5 mM Triton X-100, 0.01 mM EDTA. Reactions were started by adding 0.75–1 μg of enzyme and NADH depletion was measured at 340 nm for 5 min at room temperature using a Hitachi U-2000 spectrophotometer.
Results and Discussion
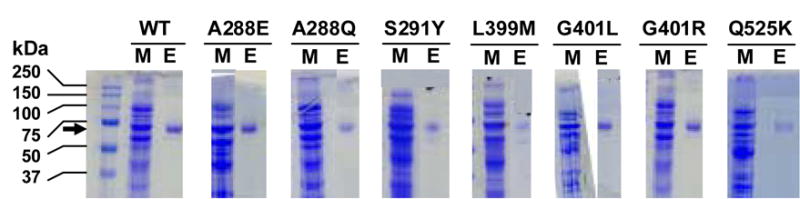
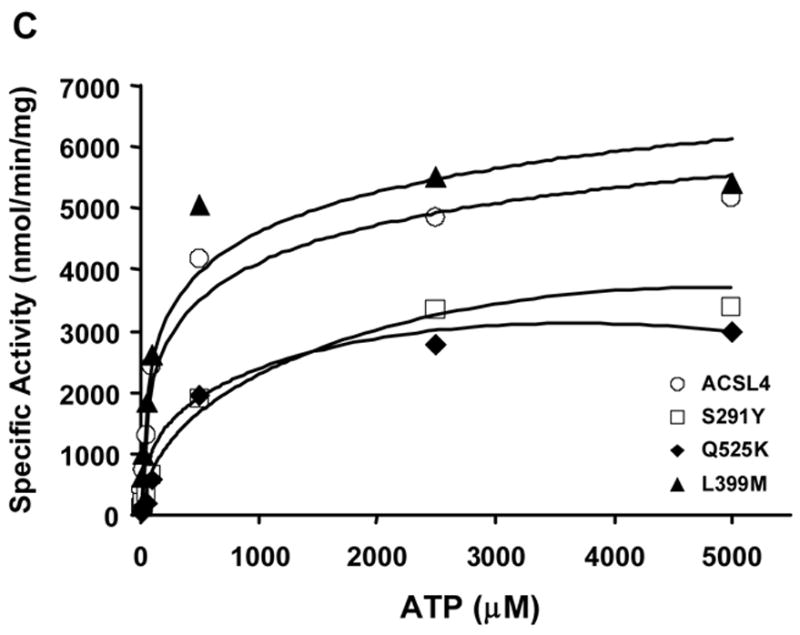
Although the long-chain ACSLs have considerable homology and overlapping use of fatty acids from 12 to 22 carbons, ACSL4 has a distinct preference for the longer chain polyunsaturated fatty acids 20:4 and 22:5 [12]. ACSL4 also has a splice variant that is not an intrinsic membrane protein [8], so its properties would not be altered by mutations that affected a transmembrane domain. In order to gain a better understanding of the features that contribute to FA selectivity in rat ACSL4, we used a mutagenesis approach. Based on sequence alignments, four of the residues selected for mutagenesis corresponded to residues in ttLC-FACS that comprise the fatty acid binding pocket [13]. L399 and G401 were predicted to lie at or near the entrance of the fatty acid binding pocket, and A288 and S291 were predicted to lie at the hydrophobic end of the pocket (Fig. 1B) [13]. We hypothesized that these residues, although distant from the putative fatty acid binding site previously identified for E. coli FadD (Fig. 1A) [14], would play a role in ACSL4 fatty acid preference, and that altering them would change the marked preference that ACSL4 has for long-chain polyunsaturated fatty acids [12]. The fifth residue selected, Q525, aligns with the R449 in FadD, which has also been proposed to be involved in fatty acid selectivity [14]. Each mutant protein, purified on an M2-anti-Flag column, shows similar expression in E. coli membranes and a similar degree of purification (Fig. 2)
Figure 2.
Why do mutations at the putative entry to the fatty acid pocket result in an inactive enzyme?
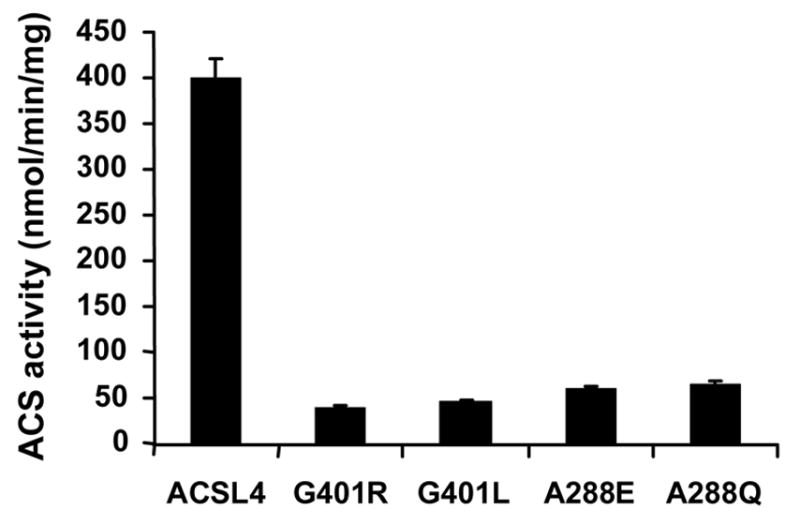
In ttLC-FACS, G301 is located at the entrance to the fatty acid binding pocket and V299 forms part of the initial tunnel for the fatty acid [13]. The T-Coffee alignment (Figure 1A) suggests that G401 in ACSL4 corresponds to G301 in ttLC-FACS and that L399 in ACSL4 corresponds to V299. L399 mutated to methionine was a modest change and the mutated enzyme retained the ability to form radiolabeled palmitoyl-CoA with similar activity as compared to wildtype ACSL4 (Table I). The L399M mutation had little effect on CoA, ATP, or palmitate binding as evidenced by Km values comparable to wild type ACSL4 (Table I). When G401 of ACSL4, predicted to lie at the entry to the fatty acid binding site (Fig. 1B), was mutated to an arginine residue, the resulting protein retained only 10% of wildtype activity with palmitate as its substrate. We then reasoned that this charged residue, if located in a hydrophobic pocket and not counterbalanced with a corresponding opposite charge, might destabilize the pocket/protein resulting in an inactive enzyme. Hence, G401 was mutated to leucine (Fig. 3). However, despite substitution with a neutral amino acid, the mutated enzyme was also inactive with respect to palmitate, and no activity was detected when saturated fatty acids from 2 to 16 carbon atoms were used as substrates. It is likely that G401 can accommodate a much greater number of phi/psi angles than can leucine, possibly resulting in a global structural change. If leucine constrains the conformations allowed by that region of the polypeptide chain, fatty acid binding could be affected. Another possibility is that a mutation in this region might disturb ATP and/or CoA binding. This interpretation is supported by performing a T-Coffee realignment that omits comparison with the luciferase sequence; when luciferase is omitted, T-Coffee predicts that G401 of ACSL4 aligns with G302 of ttLC-FACS, a residue that has been shown to be involved in ATP binding [13].
Table I.
Kinetic constants for ACSL4, S291Y, L399M, and Q525K
| ATP | CoA | Palmitate | ||
|---|---|---|---|---|
| ACSL4 | Km (μM) | 87 ± 52 | 6.7 ± 1.5 | 28.1 ± 6.7 |
| Vmax (nmol/min/mg) | 3828 ± 1153 | 3444 ± 1503 | 10159 ± 4747 | |
| S291Y | Km (μM) | 502 ± 134.6* | 6.4 ± 2.7 | 21.1 ± 6.3 |
| Vmax (nmol/min/mg) | 1471 ± 1052* | 1667 ± 0.1 | 5417 ± 3155 | |
| L399M | Km (μM) | 125 ± 41 | 8.8 ± 1.1 | 23.8 ± 8.8 |
| Vmax (nmol/min/mg) | 3393 ± 2677 | 3333 ± 543 | 8056 ± 4321 | |
| Q525K | Km (μM) | 242 ± 29* | 6.8 | 20.0 |
| Vmax (nmol/min/mg) | 2579 ± 229 | 2000 | 3000 |
Km and Vmax for ATP, CoA and palmitate were determined by double reciprocal analysis. Regression coefficients were all greater than 0.97. The range of each substrate was between 0.3 and 2.0 times the Km in order to accurately determine Vmax and Km, as described previously [16]. n ≥ 3 for each of 2 independent protein preparations, accounting for the large SD observed in some measurements. n=2 for Q525K with CoA and palmitate, therefore no SD is indicated.
indicates P ≤ 0.05.
Figure 3.
In contrast to the severe inhibition resulting from a G401 mutation, mutating L399 to methionine did not significantly alter enzyme specific activity (Table I), suggesting that substituting with another neutral amino acid residue in this position had relatively little effect on the ability of the protein to interact with the fatty acid within the putative pocket.
Mutations at the end of the fatty acid binding pocket
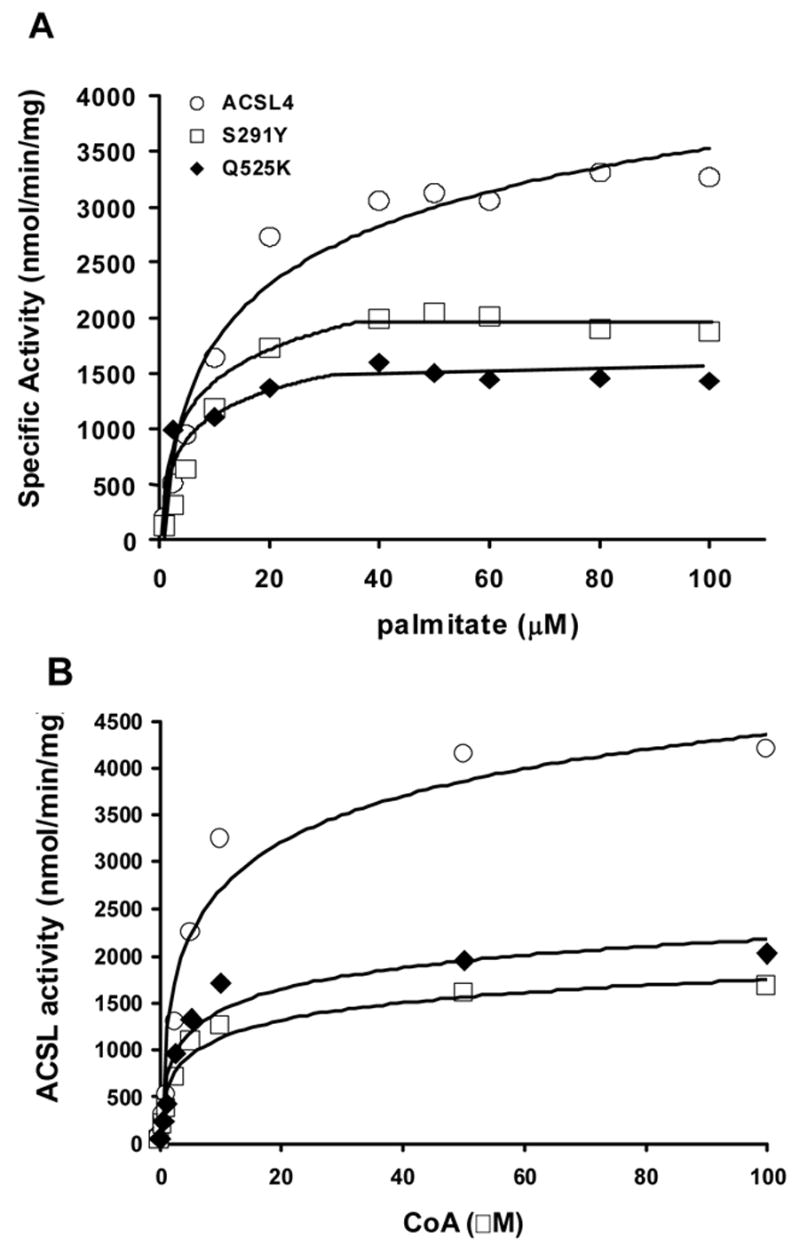
We reasoned that altering amino acids at the terminus of the putative fatty acid binding pocket might block access of the longer chain polyunsaturated fatty acids preferred by ACSL4. L236 which comprises the terminus of the fatty acid binding pocket in ttLC-FACS [13], aligns with A288 in ACSL4 (Fig. 1B). When this residue was mutated to glutamic acid in order to both increase bulk and place a negatively charged amino acid in the region that would normally accommodate the methyl end of the fatty acid, the resulting protein was virtually inactive with palmitate as substrate. We then reasoned that these charged residues, if located in a hydrophobic pocket and not counterbalanced with a corresponding opposite charge, could destabilize the pocket or the protein and result in an inactive enzyme. However when A288 was mutated to glutamine, which is uncharged at neutral pH, this protein only retained 13–15% of activity with [14C]palmitate as a substrate, and A288Q was not able to use shorter fatty acids (2:0, 4:0, 8:0, 12:0) as substrates (data not shown). A288E was not tested with other fatty acids. In contrast, the specific activity of the S291Y mutation was diminished and the binding of palmitate or of CoA was not altered, but the Km for ATP was 5.8-fold higher than wildtype ACSL4 (Fig. 4, Table I). It likely that the glutamine residue, being bulkier, resulted in local conformational changes that affected ligand binding and/or catalysis.
Figure 4.
In analyzing our data it must be considered that, since rat ACLS4 has not been crystallized and since the mutations chosen were based on the crystal structure for a bacterial long chain acyl-CoA synthetase, the mutagenized residues might not correspond to residues in ACSL4 that comprise the fatty acid binding pocket, but instead, distort the protein such that it is completely inactive. In this light, it is of interest that in ttLC-FACS, the amino acids L236 and A239 (corresponding to ACSL4 A288 and S291) that lie at the termination of the putative fatty acid binding pocket correspond to rat and human ACSL6 amino acids 306 to 331. This amino acid region is formed by two exons that are alternatively used to encode ACSL6 splice variants [12]. Amino acid changes arising from the two splice variants cause the apparent Km for ATP for ACSL6_v1 to be 8-fold higher than that for ACSL6_v2 [12]. Thus, this region seems clearly to interact with or alter the interaction of ACSL6 with ATP; these changes in ATP binding suggest that total correspondence may be lacking between ttLC-FACS and the mammalian ACSLs.
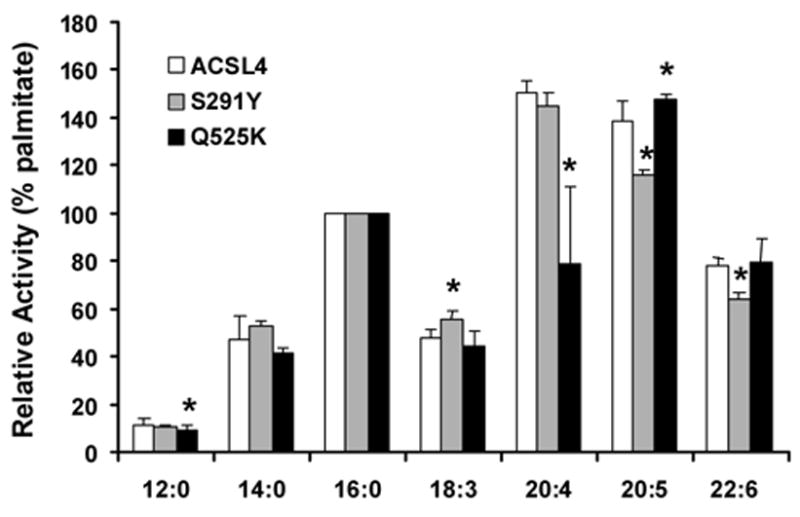
On the other hand, the use of information from the crystal structure of ttLC-FACS appears to be justified by the results from mutating ACSL4 S291. This amino acid corresponds to A239 in ttLC_FACS which, like L236, would lie at the termination of the putative fatty acid binding pocket. ACSL4 S291 was mutated to tyrosine in order to place a bulky amino acid in this region and block access by the longer chain fatty acids that ACSL4 prefers. In order to determine whether S291Y altered the ability of ACSL4 to recognize substrates other than palmitate, kinetics were performed using a spectrophotometric coupled assay which monitors depletion of NADH at 340 nm over time. We tested fatty acid substrates with chain lengths varying from C12:0 to C22:6. The results suggested that S291 does play a role in substrate selectivity (Fig. 5). Compared to the wild-type enzyme, mutating S291 to tyrosine diminished the conversion of long chain fatty acids to long chain fatty acyl-CoAs when 20:5 and 22:6 were provided as substrates, but not with other shorter chain fatty acids. The S291Y mutation decreased the activity of ACSL4 17% towards 20:5 and 18% towards 22:6 relative to palmitate, and minimally increased activity with 18:3. Since both serine and tyrosine contain hydroxyl groups, it is likely that the bulky benzyl portion of tyrosine, rather than the hydroxyl group, contributed to the decrease in activity for S291Y compared to wildtype ACSL4. In addition, the altered Km value for ATP suggests that S291 may, in fact, play a dual role in interactions with both fatty acid binding and with ATP like the splice variants of ACSL6 [12]. Alternatively, the S291Y mutation may still allow the fatty acid to bind, but in an altered conformation that interferes with ATP binding.
Figure 5.
What is the role of Q525?
In addition to the four residues predicted to lie within the fatty acid binding pocket by the ttLC-FACS crystal study, Q525 was chosen for mutagenesis because it had been previously demonstrated that the corresponding residue (R449) in the E. coli acyl-CoA synthetase FadD lies within a region involved in fatty acid selectivity [14]. In addition, unlike the positively charged R449 of FadD and the lysines at the comparable position in other mammalian ACSLs which do not exhibit a strong preference for long-chain polyunsaturated fatty acids, ACSL4 contains a glutamine at this position (Fig. 1A) [6]. To determine whether Q525 is important for the preference of ACSL4 for polyunsaturated fatty acids, we mutated Q525 to lysine, which is present in the other four rat ACSL isoforms. The Q525K mutation did not alter binding of palmitate or CoA, but the Km for ATP was 2.8-fold higher than wildtype ACSL4 (Fig. 4, Table I).
Our results suggest that Q525 also is important for substrate selectivity. Mutating Q525 to lysine decreased the activity of ACSL4 48% towards 20:4, and minimally increased activity with 20:5. (Fig. 5). This result is interesting since this residue in long chain acyl-CoA synthetases from rat, mouse, and yeast is usually a positively charged lysine [6]. That a mutation of Q525 to lysine decreased activity only with arachidonate, a preferred substrate for ACSL4, suggests that Q525 is, indeed, involved in arachidonic acid selectivity. Further, the altered Km values for ATP suggest that, like S291, Q525 may be involved in both interactions with ATP and with fatty acid binding.
In their study of the crystal structure of ttLC-FACS, Hisanaga et al. identified a fatty acid binding tunnel to convey the fatty acid unidirectionally to the ATP-binding site [13]. Basing our mutagenesis targets on ttLC-FACS, we modified selected residues calculated to lie at or near the entry of the putative fatty acid binding pocket and at the pocket terminus. The major findings of our study were 1) that mutations of a residue at the terminus of the putative fatty acid binding pocket and at a site lying outside this region, but previously identified as important for fatty acid binding, both altered the preference of ACSL4 for long-chain polyunsaturated fatty acids, and 2) that the structure of ttLC-FACS can serve as a guide for structure-function studies of fatty acid binding by the rat ACSL4.
Acknowledgments
This work was supported by grants from the National Institutes of Health DK59935 (RAC) and DK059931 (TML) and from the American Heart Association 023032N (TML).
Abbreviations
- ttLC-FACS
Thermus thermophilus long chain-fatty acyl-CoA synthetase
- FadD
Escherichia coli long chain acyl-CoA synthetase
- ACSL
long chain acyl-CoA synthetase
- AA
arachidonic acid
- EPA
eicosapentaenoic acid
- DHA
docosahexaenoic acid
- DTT
dithiothreitol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coleman RA, Lewin TM, Van Horn CG, Gonzalez-Baró MR. Do acyl-CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? J Nutr. 2002;132:2123–2126. doi: 10.1093/jn/132.8.2123. [DOI] [PubMed] [Google Scholar]
- 2.Coleman RA, Van Horn CG, Gonzalez-Baró MR, Lewin TM. In: Lipids: Glycerolipid Metabolizing Enzymes. Haldar D, Das SK, editors. Research Signpost; Trivandrum: 2002. pp. 1–15. [Google Scholar]
- 3.Corkey BE, Deeney JT, Yaney GC, Tornheim K, Prentki M. The role of long-chain fatty acyl-CoA esters in beta-cell signal transduction. J Nutr. 2000;130:299S–304S. doi: 10.1093/jn/130.2.299S. [DOI] [PubMed] [Google Scholar]
- 4.Hertz R, Magenheim J, Berman I, Bar-Tana J. Fatty acyl-CoA thioesters are ligands of hepatic nuclear factor-4alpha. Nature. 1998;392:512–516. doi: 10.1038/33185. [DOI] [PubMed] [Google Scholar]
- 5.Hertz R, Kalderon B, Byk T, Berman I, Za’tara G, Mayer R, Bar-Tana J. Thioesterase activity and acyl-CoA/fatty acid cross-talk of hepatocyte nuclear factor-4α. J Biol Chem. 2005;280:24451–24461. doi: 10.1074/jbc.M500732200. [DOI] [PubMed] [Google Scholar]
- 6.Steinberg SJ, Morgenthaler J, Heinzer AK, Smith KD, Watkins PA. Very long-chain acyl-CoA synthetases: Human “bubblegum” represents a new family of proteins capable of activating very long-chain fatty acids. J Biol Chem. 2000;275:35162–35169. doi: 10.1074/jbc.M006403200. [DOI] [PubMed] [Google Scholar]
- 7.Mashek DG, Bornfeldt KE, Coleman RA, Berger J, Bernlohr DA, Black P, DiRusso CC, Farber SA, Guo W, Hashimoto N, Khodiyar VK, Kuypers FA, Maltais LJ, Nebert DW, Renieri A, Schaffer JE, Stahl A, Watkins PA, Vasiliou V, Yamamoto TT. Revised nomenclature for the mammalian long chain acyl-CoA synthetase gene family. J Lipid Res. 2004;45:1958–1961. doi: 10.1194/jlr.E400002-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Lewin TM, Van Horn CG, Krisans SK, Coleman RA. Rat liver acyl-CoA synthetase 4 is a peripheral-membrane protein located in two distinct subcellular organelles, peroxisomes and mitochondrial associated membrane. Arch Biochem Biophys. 2002;404:263–270. doi: 10.1016/s0003-9861(02)00247-3. [DOI] [PubMed] [Google Scholar]
- 9.Kang MJ, Fujino T, Sasano H, Minekura H, Yabuki N, Nagura H, Iijima H, Yamamoto TT. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc Natl Acad Sci. 1997;94:2880–2884. doi: 10.1073/pnas.94.7.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Longo I, Frints SG, Fryns JP, Meloni I, Pescucci C, Ariani F, Borghgraef M, Raynaud M, Marynen P, Schwartz C, Renieri A, Froyen G. A third MRX family (MRX68) is the result of mutation in the long chain fatty acid-CoA ligase 4 (FACL4) gene: proposal of a rapid enzymatic assay for screening mentally retarded patients. J Med Genet. 2003;40:11–17. doi: 10.1136/jmg.40.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meloni I, Muscettola M, Raynaud M, Longo I, Bruttini M, Moizard MP, Gomot M, Chelly J, des Portes V, Fryns JP, Ropers HH, Magi B, Bellan C, Volpi N, Yntema HG, Lewis SE, Schaffer JE, Renieri A. FACL4, encoding fatty acid-CoA ligase 4, is mutated in nonspecific X-linked mental retardation. Nat Genet. 2002;30:436–440. doi: 10.1038/ng857. [DOI] [PubMed] [Google Scholar]
- 12.Van Horn CG, Caviglia JM, Li LO, Wang S, Granger DA, Coleman RA. Characterization of recombinant long-chain rat acyl-CoA synthetase isoforms 3 and 6: Identification of a novel variant of isoform 6. Biochemistry. 2005;44:1635–1642. doi: 10.1021/bi047721l. [DOI] [PubMed] [Google Scholar]
- 13.Hisanaga Y, Ago H, Nakagawa N, Hamada K, Ida K, Yamamoto M, Hori T, Arii Y, Sugahara M, Kuramitsu S, Yokoyama S, Miyano M. Structural basis of the substrate-specific two-step catalysis of long chain fatty acyl-CoA synthetase dimer. J Biol Chem. 2004;279:31717–31726. doi: 10.1074/jbc.M400100200. [DOI] [PubMed] [Google Scholar]
- 14.Black PN, Zhang Q, Weimar JD, DiRusso CC. Mutational analysis of a fatty acyl-coenzyme A synthetase signature motif identifies seven amino acid residues that modulate fatty acid substrate specificity. J Biol Chem. 1997;272:4896–4903. doi: 10.1074/jbc.272.8.4896. [DOI] [PubMed] [Google Scholar]
- 15.Kim JH, Lewin TM, Coleman RA. Expression and characterization of recombinant rat acyl-CoA synthetases 1, 4, and 5: Selective inhibition by triacsin C and thiazolidinediones. J Biol Chem. 2001;276:24667–24673. doi: 10.1074/jbc.M010793200. [DOI] [PubMed] [Google Scholar]
- 16.Segel IH. Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. John Wiley & Sons, Inc.; New York: 1975. Enzyme Kinetics. [Google Scholar]
- 17.Notredame C, Higgins D, Heringa J. T-Coffee: A novel method for multiple sequence alignments. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]