

Abstract
Listeria monocytogenes is a bacterial pathogen that multiplies in the cytosol of host cells and spreads directly from cell to cell by using an actin-based mechanism of motility. The broad-range phospholipase C (PC-PLC) of L. monocytogenes contributes to bacterial escape from vacuoles formed upon cell-to-cell spread. PC-PLC is made as an inactive proenzyme whose activation requires cleavage of an N-terminal propeptide. During infection, PC-PLC is activated specifically in acidified vacuoles. To assess the importance of compartmentalizing PC-PLC activity during infection, we created a mutant that makes constitutively active PC-PLC (the plcBΔpro mutant). Results from intracellular growth and cell-to-cell spread assays showed that the plcBΔpro mutant was sensitive to gentamicin, suggesting that unregulated PC-PLC activity causes damage to host cell membranes. This was confirmed by the observation of a twofold increase in staining of live infected cells by a non-membrane-permeant DNA fluorescent dye. However, membrane damage was not sufficient to cause cell lysis and was dependent on bacterial cell-to-cell spread, suggesting that damage was localized to bacterium-containing filopodia. Using an in vivo competitive infection assay, we observed that the plcBΔpro mutant was outcompeted up to 200-fold by the wild-type strain in BALB/c mice. Virulence attenuation was greater when mice were infected orally than when they were infected intravenously, presumably because the plcBΔpro mutant was initially outcompeted in the intestines, reducing the number of mutant bacteria reaching the liver and spleen. Together, these results emphasize the importance for L. monocytogenes virulence of compartmentalizing the activity of PC-PLC during infection.
Listeria monocytogenes is an opportunistic bacterial pathogen that has the ability to remain sequestered within host cells during its entire life cycle (15, 26). Upon entry into host cells, bacteria escape from membrane-bound vacuoles and begin multiplying in the cytosol of their host. During intracellular growth, bacteria use an actin-based mechanism of motility to induce the formation of membrane filopodia, which are taken up by neighboring cells, enabling bacteria to spread directly from cell to cell. The virulence of L. monocytogenes is directly related to its efficacy at (i) escaping primary and secondary vacuoles, formed upon initial entry into a host cell and upon cell-to-cell spread, respectively, and (ii) remaining sequestered within host cells (4, 7, 8, 11, 23, 26).
Bacterial factors implicated in vacuolar lysis include a pore-forming hemolysin called listeriolysin O (LLO), a phosphatidylinositol-specific phospholipase C (PI-PLC), and a broad-range phospholipase C (PC-PLC). LLO is essential for bacterial escape from vacuoles of most cell lines (18), whereas the phospholipases are dispensable but enhance the efficacy of vacuolar escape (4, 23, 27). In the absence of PI-PLC, bacteria have a significant decrease in escape from primary vacuoles and a small defect in the ability to spread from cell to cell (4). The absence of PC-PLC does not affect escape from primary vacuoles but significantly reduces the bacterium's ability to escape secondary vacuoles (23, 27). The two bacterial phospholipases appear to have overlapping functions, as a double phospholipase mutant is more deficient in escape from secondary vacuoles than the sum of the defects observed with individual mutants (23). Exceptionally, LLO is dispensable for escape from vacuoles in cultured human cells, in which PC-PLC can substitute for LLO in mediating vacuolar escape (10, 12, 16, 18). In vivo, LLO-negative bacteria completely fail to grow in livers and spleens of infected mice (6), which is consistent with their inability to escape from primary vacuoles. On the other hand, PI-PLC and PC-PLC are not essential for growth of L. monocytogenes in vivo, although individual PI-PLC and PC-PLC deletion mutants are recovered in much lower numbers from livers and spleens of infected mice, supporting their role in virulence (4, 20).
Remarkably, the activities of these three membrane-targeting factors appear to be restricted to vacuolar membranes, as infected host cells show few signs of toxicity and intracellular bacteria are protected from gentamicin, an antibiotic that does not diffuse across membranes (4, 18, 23). In fact, LLO's pore-forming ability is optimal at pH 5.9, a pH value that would be reached only in vacuoles, and the protein has an intracellular half-life of <15 min (2, 22). Accordingly, mutations that affect compartmentalization of LLO activity are cytotoxic and compromise the ability of L. monocytogenes to remain sequestered within host cells, resulting in severe virulence attenuation (7, 8). The activity of PC-PLC is also tightly regulated during intracellular infection. PC-PLC is made as an inactive proenzyme whose activation requires proteolytic cleavage of a 24-residue propeptide (17, 19, 27). PC-PLC activation is assisted by a metalloprotease (Mpl) and is dependent on cell-to-cell spread and vacuolar acidification, indicating that it occurs specifically in vacuoles (13, 19). Bacteria multiplying in the cytosol of host cells secrete the inactive form of PC-PLC but also maintain a pool of inactive PC-PLC at the membrane-cell wall interface (14, 24). Similar to the case for LLO, PC-PLC secreted into the cytosol of infected cells is degraded by the proteasome, with a half-life of <15 min (13). In vacuoles, the pool of bacterium-associated PC-PLC is activated and translocated across the bacterial cell wall within minutes of a decrease in pH (14).
In this study, we investigated the biological relevance of compartmentalizing PC-PLC activity during intracellular infection. A mutant of L. monocytogenes making a constitutively active form of PC-PLC (the plcBΔpro mutant) was used. Previously, we showed that secretion and activation of this mutant form of PC-PLC are no longer regulated by pH and by Mpl (28). Here we report that the plcBΔpro mutant is not cytotoxic, although it compromises host cell membrane integrity during cell-to-cell spread. Remarkably, the plcBΔpro mutant shows strong attenuation in mice, indicating that similar to the case for LLO, correct compartmentalization of PC-PLC activity is imperative for the virulence of L. monocytogenes.
MATERIALS AND METHODS
Bacterial strains and cell cultures.
The L. monocytogenes 10403S wild-type strain (3) and the isogenic mutant HEL-335 were used in this study. HEL-335, which is identified as the plcBΔpro mutant, contains a deletion of the sequence coding for the PC-PLC propeptide, generating a constitutively active form of PC-PLC (28). In addition, strain DP-L3903, a fully virulent derivative of strain 10403S carrying Tn917 in its chromosome, was used for in vivo assays (1). DP-L3903 is erythromycin resistant and is identified as the Emr wild type in this paper. For tissue culture assays, the wild-type strain and the plcBΔpro mutant were grown statically overnight at 30°C in brain heart infusion medium. For in vivo assays, the Emr wild type and the plcBΔpro mutant were grown at 30°C (for oral infections) or 37°C (for intravenous [i.v.] infections) at 250 rpm in brain heart infusion broth supplemented with 200 μg/ml streptomycin to an optical density at 600 mm of 0.4 to 0.5. A regression formula was developed empirically to determine the number of CFU per ml. The mouse macrophage-like J774 cell line and mouse fibroblast L2 cell line were propagated in Dulbecco's modified Eagle's medium supplemented with 7.5% fetal bovine serum and 2 mM glutamine. Cell cultures were incubated at 37°C with 5% CO2.
Plaque assay.
A plaque assay was used as an indication of bacterial cell-to-cell spreading efficacy. The method described by Sun et al. (25) was adopted. L2 cells (7 × 105) were seeded in each well of a six-well plate 2 days prior to infection with 2 × 105 to 4 × 105 bacterial cells. At 60 min postinfection, cells were washed three times with phosphate-buffered saline (PBS) and overlaid with a semisolid medium containing defined concentrations of gentamicin. At 4 days postinfection, plaques were stained with neutral red. The mean plaque surface area formed by the mutant was compared with that of the wild type. The relative plaque size is reported as a percentage of the wild-type plaque size.
Membrane permeability assay.
To assess host cell membrane integrity, live-cell staining was performed on infected J774 cells with a fluorescent non-membrane-permeant SYTOX orange nucleic acid stain (Invitrogen). J774 cells (6 × 105) were seeded in glass-bottomed 35-mm dishes (MatTek Corporation) 1 day prior to infection with 5 × 106 to 10 × 106 bacterial cells. Cells were infected 20 to 60 min apart with the wild-type and plcBΔpro mutant strains. Infected cells were washed at 30 min postinfection, and gentamicin (50 μg/ml) was added at 1 h postinfection for a period of 2 h. At 6 h postinfection, infected cells were washed with PBS, stained for 20 min at room temperature with SYTOX diluted in TBS (25 mM Tris, pH 8.0, 150 mM NaCl, 1% bovine serum albumin), washed with TBS, and replenished with 1 ml of clear Dulbecco's modified Eagle's medium. Images were captured with a ×40 lens, using a confocal microscope with a krypton laser and transmitted light. For each experiment, images from 8 to 10 fields were captured, and 1 × 103 to 2 × 103 infected host cells were counted per sample. The fields were chosen at random. The experiment was repeated five times, and for each experiment the ratio of stained cells relative to the total number of cells was determined for cells infected with the wild type or the plcBΔpro mutant. Diff Quick staining was also performed on another set of coverslips to verify that every host cell was infected.
Intracellular growth kinetics.
To evaluate the bacterial ability to multiply inside host cells, J774 cells were infected, followed by enumeration of intracellular bacterial cells (18). J774 (1.5 × 106) cells were seeded in 60-mm petri dishes containing 14 12-mm alcohol-flamed round coverslips 1 day prior to infection. L. monocytogenes cells from overnight cultures were washed once with PBS, and 5 × 105 to 10 × 105 bacteria were used to infect host cells. At 30 min postinfection, cells were washed three times with PBS and replenished with fresh medium. Gentamicin was added at 1 h postinfection at a final concentration of 50 μg/ml to kill extracellular bacteria. To determine if gentamicin influences the growth kinetics of intracellular bacteria, cells were washed once more with PBS at 3 h postinfection and replenished with fresh medium, with or without gentamicin, as indicated in the figure legends. At defined time points (2, 5, 8, and 11 h postinfection), host cells on three coverslips were lysed by vortexing each coverslip in sterile H2O for 20 s. Appropriate dilutions were then plated on Luria-Bertani (LB) medium to determine the number of CFU per coverslip. To determine the intracellular growth of bacteria devoid of cell-to-cell movement, cytochalasin D, an inhibitor of actin polymerization, was added at a final concentration of 0.5 μg/ml at 3 h postinfection. Experiments were repeated a minimum of three times. The doubling time between 5 h and 8 h postinfection was calculated based on the following formula: ln 2/[ln (Nt/N0)/(tn − t0)], where Nt is the number of bacteria present at a given time t (in min), N0 is the number of bacteria present initially, and tn − t0 is the corresponding time interval between Nt and N0.
In vivo proliferation.
Six- to 8-week-old female BALB/c mice (Taconic, Hudson, NY) were infected with log-phase bacterial cells orally or i.v., using a competitive index (CI) approach (1). For oral infection, mice were fasted for 2 h prior to and after infection. Bacteria were washed once in PBS and suspended in an appropriate volume of PBS. The Emr wild type and the plcBΔpro mutant were mixed at equal concentrations, and each mouse received a 250-μl dose containing approximately 4 × 109 CFU with a gavage needle. For i.v. infection, 200-μl inocula containing approximately 2 × 104 bacteria in an equal mixture of the Emr wild type and the plcBΔpro mutant were injected into tail veins. At defined time points, livers, spleens, and mesenteric lymph nodes (MLN) were harvested from sacrificed mice and homogenized in 5 ml of buffer containing 0.5% NP-40 and 200 μg/ml streptomycin. Intestines were cut longitudinally, rid of fecal materials, rinsed in three changes of PBS (10 ml each), and homogenized in 5 ml of buffer containing 0.5% NP-40, 0.2 mM EDTA, and 200 μg/ml streptomycin. Suspensions were plated on LB agar supplemented with streptomycin to recover L. monocytogenes cells. Up to 200 colonies were streaked onto LB agar supplemented with 200 μg/ml streptomycin and 1 μg/ml erythromycin or onto activated charcoal (0.2%)-treated LB supplemented with 25 mM glucose-1-phosphate and 1.25% egg yolk (LB-G1P-EY). While the Emr wild-type strain was resistant to erythromycin, the plcBΔpro mutant was unable to grow in medium containing erythromycin. The plcBΔpro mutant also exhibited an elevated level of PC-PLC activity after 24 h of incubation at 37°C on LB-G1P-EY plates (28). The CI was calculated by the formula CIout/CIin, where CIout is the ratio of the plcBΔpro mutant to the Emr wild type postinfection and CIin is the ratio of the plcBΔpro mutant to the Emr wild type in the inoculum (i.e., preinfection). A CI of 1 indicates that the Emr wild type and the plcBΔpro mutant proliferated equally in the organ. In some events where no plcBΔpro mutant was recovered from >100 (but ≤200) colonies, we estimated the CIout by assuming that the next colony would have been from the mutant strain.
Statistical analyses.
Experimental data were analyzed statistically to evaluate the significance of differences between the wild-type and mutant strains. For each experiment from the plaque assay, the surface area of plaques formed by the plcBΔpro mutant was normalized relative to that of plaques formed by the wild type. A one-sample t test was conducted to test if the mutant plaque size was significantly different from 100%, which was defined as the wild-type plaque size. Likewise, for each experiment from the membrane permeability assay, the ratio obtained with plcBΔpro mutant-infected cells was normalized to that obtained with wild type-infected cells. The normalized value was tested, by a one-sample t test, with the null hypothesis of the mean value equaling 1. Data obtained from the intracellular growth kinetics assays were analyzed using a two-sample t test to determine the effect of cytochalasin D on the doubling time. The mouse CI assay was analyzed with a one-sample t test using log-transformed CI to determine if the CI were significantly less than 1.
RESULTS
Unregulated secretion of active PC-PLC by L. monocytogenes compromises host cell membrane integrity during intracellular infection.
During intracellular infection, Mpl-mediated PC-PLC activation occurs specifically in vacuoles and requires a decrease in vacuolar pH (13). However, the plcBΔpro mutant secretes active PC-PLC independently of Mpl and pH (28). Constitutive secretion of active PC-PLC is not cytotoxic to the producer bacteria. We hypothesized that the inability of L. monocytogenes to compartmentalize PC-PLC activity during intracellular infection would affect host cell membrane integrity. To test this hypothesis, we analyzed bacterial sensitivity to gentamicin during intracellular infection. Gentamicin was added to the extracellular milieu to specifically kill extracellular bacteria, as it cannot diffuse across intact membranes. However, gentamicin may access the intracellular milieu if host membrane integrity is compromised. Mouse fibroblast L2 cells were infected and then overlaid with medium-containing soft agar supplemented with three different concentrations of gentamicin. Four days after infection, the plaques were visualized by staining the monolayer with neutral red, a weak cationic dye that diffuses across intact membranes and cumulates intracellularly. Plaques are defined by the absence of staining, which is an indication of major cell damage, presumably as a result of overwhelming infection, and the size of plaques reflects the ability of bacteria to spread from cell to cell. If host membrane integrity was compromised early after infection, then bacterial intracellular growth and cell-to-cell spread would be reduced. Plaques formed by the plcBΔpro mutant were compared to those formed by the wild-type strain for the same gentamicin concentration. The surface areas of plaques formed by the plcBΔpro mutant were 93% (P = 0.05), 84% (P = 0.02), and 81% (P = 0.01) relative to that of wild-type plaques with 2, 5, and 10 μg/ml of gentamicin, respectively (Fig. 1). However, the most striking difference was the appearance of the plaques. Plaques formed by the wild-type strain were uniformly clear at all gentamicin concentrations. Similarly, plaques formed by the plcBΔpro mutant were uniformly clear at 2 μg/ml of gentamicin. At 5 μg/ml of gentamicin, plaques formed by the plcBΔpro mutant had a clear center surrounded by a zone of partial staining delimited by a thin clear zone. At 10 μg/ml of gentamicin, plaques formed by the plcBΔpro mutant showed a small clear central zone surrounded by a large zone of different staining intensity. The unusual appearance of plaques formed by the plcBΔpro mutant at 5 and 10 μg/ml of gentamicin suggested that the antibiotic had a protective effect on host cells, presumably by impairing the growth of intracellular bacteria.
FIG. 1.

Cell-to-cell spreading efficacy of L. monocytogenes wild-type strain (top row) and plcBΔpro mutant (bottom row) in infected mouse fibroblast L2 cells in the presence of gentamicin. Infected cells were washed at 60 min postinfection and overlaid with a semisolid medium containing 2, 5, or 10 μg/ml gentamicin. At 4 days postinfection, the monolayer was stained with neutral red, a weak cationic dye that diffuses across intact membranes and cumulates intracellularly. Plaques are defined by the absence of staining, which is indicative of major cell damage or cell lysis.
The plaque assay results indicated that unregulated secretion of active PC-PLC affects host membrane integrity, as the size and appearance of plaques formed by the plcBΔpro mutant differed from those of wild-type plaques in a gentamicin-dependent manner. To evaluate the extent of damage caused by infection, we quantified the levels of lactate dehydrogenase (LDH) released into the supernatants of infected murine macrophage-like J774 cells, using the Cytotox 96 nonradioactive cytotoxicity assay (Promega) according to the manufacturer's instructions. LDH is normally contained within the cytosol of eukaryotic cells but is released into the extracellular milieu upon cell lysis. Surprisingly, no differences were observed in LDH release from samples infected with the wild type or the plcBΔpro mutant strain, even when gentamicin was not used to allow intracellular bacterial growth in permeabilized host cells (data not shown). In agreement with this result, the appearances of J774 cells infected with either bacterial strain were indistinguishable, as determined by microscopic examination of fixed and stained cells (Fig. 2). The nuclei and cytosol of infected cells appeared to be normal, and bacterium-containing filopodia were observed whether or not gentamicin was present. These observations indicated that unregulated secretion of active PC-PLC during intracellular infection did not cause host cell lysis.
FIG. 2.

Micrographs of J774 cells infected with the wild-type strain or the plcBΔpro mutant. Infected host cells were washed at 30 min postinfection. Gentamicin (50 μg/ml) was added to the medium at 1 h postinfection and maintained until the end of the experiment (top row) or removed at 3 h postinfection (bottom row). Cells on coverslips were stained with Diff Quick at 8 h postinfection. Images were taken with a ×60 lens.
To evaluate host membrane integrity in a more sensitive manner, we stained live infected cells with a fluorescent, non-membrane-permeant nucleic acid stain. In this assay, infected J774 cells were maintained in a gentamicin-free medium from 3 h to 6 h postinfection to prevent inhibition of intracellular bacterial growth in damaged cells. The nucleic acid dye was added to live cells, and the ratio of stained to unstained cells was determined by microscopy. In this assay, twofold more stained cells were observed in samples infected with the plcBΔpro mutant than in samples infected with the wild-type strain (P = 0.018) (Fig. 3). Taken together, the results indicate that unregulated secretion of active PC-PLC by L. monocytogenes during intracellular infection compromises host cell membrane integrity without causing host cell lysis.
FIG. 3.
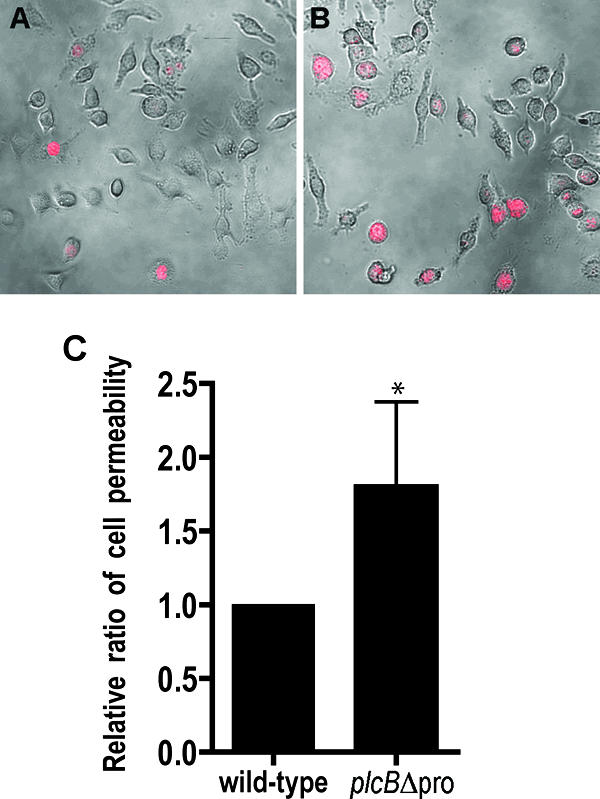
Staining of live infected J774 cells with a fluorescent, non-membrane-permeant nucleic acid dye. Host cells were infected with the wild type or the plcBΔpro mutant for 6 h, with the addition of 50 μg/ml gentamicin between 1 h and 3 h postinfection. Live infected cells were stained at 6 h postinfection. Images were captured with a ×40 lens, using a confocal microscope with a krypton laser and transmitted light. Host cells with compromised membranes are stained red. The images show overlays of phase-contrast and fluorescence images of host cells infected with the wild-type strain (A) or the plcBΔpro mutant (B). (C) Ratios of stained cells to the total number of infected host cells (1 × 103 to 2 × 103 per sample; n = 5 per sample). The wild type was set to have a ratio of 1. Statistical analysis of the data indicated that the P value was 0.018.
Host cell membrane damage occurs during cell-to-cell spread.
During intracellular infection, L. monocytogenes moves randomly in the host cytosol, bouncing on membranes and organelles (21). Sporadically, moving bacteria induce the formation of membrane filopodia, at which point bacteria become closely surrounded by host cell membranes. Based on the premise that constitutive secretion of active PC-PLC compromises host membrane integrity in the absence of major cell damage or cell lysis, we hypothesized that it acts specifically on filopodial membranes. To this end, we first determined the intracellular growth kinetics of bacteria. In the absence of gentamicin, the growth rate of the plcBΔpro mutant was the same as that of the wild-type strain at all time points (Fig. 4). However, the intracellular growth kinetics was influenced by the presence of extracellular gentamicin. While the growth rate of the plcBΔpro mutant was initially the same as that of the wild-type strain (between 2 and 5 h postinfection), it was lower than that of the wild-type strain after 5 h (Fig. 4 and Table 1). This change in kinetics is supported by our hypothesis that gentamicin acts on intracellular bacteria as they begin to spread from cell to cell. To test whether or not the damage was restricted to filopodial membranes, actin-based motility was restricted with cytochalasin D, and thus cell-to-cell movement was prohibited. Inhibition of cell-to-cell spread did not affect the intracellular kinetics of the wild-type strain or the plcBΔpro mutant between 2 and 5 h postinfection (data not shown). It also did not affect the intracellular kinetics of the wild-type strain between 5 and 8 h postinfection (Table 1). However, the growth rate of the mutant was nearly twofold higher between 5 and 8 h postinfection in the presence of cytochalasin D. The difference in growth rates between wild type- and plcBΔpro mutant-infected cells treated with cytochalasin D was not statistically significant (P = 0.36). These results indicate that the ability of the plcBΔpro mutant to damage host cell membranes is dependent on cell-to-cell spread.
FIG. 4.

Growth kinetics of wild-type (squares) and plcBΔpro mutant (inverted triangles) strains in J774 cells. Infected host cells were washed at 30 min postinfection, and gentamicin (50 μg/ml) was added at 1 h postinfection. At 3 h postinfection, the samples were washed, and cells were replenished with fresh medium alone (open symbols, dotted lines) or fresh medium containing gentamicin (closed symbols, solid lines). At 2, 5, 8, and 11 h postinfection, host cells on coverslips were lysed to release bacteria, which were subsequently enumerated by standard plate counts. Means ± standard deviations for one representative experiment are shown. The experiment was repeated at least three times.
TABLE 1.
Intracellular growth rates of L. monocytogenes in J774 cells between 5 and 8 h postinfectiona
| Strain | Presence of cytochalasin Db | Mean doubling time (min)c ± SD | n | Pd |
|---|---|---|---|---|
| Wild type | − | 81.3 ± 17.8 | 8 | 0.538 |
| + | 90.3 ± 34.1 | 5 | ||
| plcBΔpro mutant | − | 118.4 ± 33.7 | 7 | 0.032 |
| + | 74.3 ± 5.6 | 4 |
Gentamicin (50 μg/ml) was present in the extracellular medium from 1 h postinfection until the end of the experiment.
Cytochalasin D (0.5 μg/ml) was added to infected cells at 3 h postinfection.
Doubling times were calculated as indicated in Materials and Methods.
A two-sample t test was conducted to compare the intracellular growth rates with and without cytochalasin D for the wild-type strain (top value). The same test was conducted for the plcBΔpro mutant strain (bottom value).
Unregulated secretion of active PC-PLC compromises the virulence of L. monocytogenes.
Given that constitutive secretion of active PC-PLC induces host cell damage in tissue culture models, our next step was to evaluate the importance of compartmentalizing PC-PLC activity for the virulence of L. monocytogenes in an animal model. BALB/c mice were infected orally or intravenously with an equal mixture of two strains, i.e., a fully virulent erythromycin-resistant derivative of the wild-type strain (the Emr wild type) and the plcBΔpro mutant. The virulence of the plcBΔpro mutant was evaluated by calculating the ratio of the two strains recovered from various organs at specific times after infection. The results are reported as CI, which were calculated as described in Materials and Methods. A fully virulent strain would be expected to have a CI of ≥1, whereas an attenuated strain would have a CI below 1. For mice infected orally, CI were determined on days 2, 3, and 4 postinfection for intestines, MLN, livers, and spleens. CI were not evaluated on day 1 for orally infected mice because the numbers of bacteria per organ were often below detectable levels or too low. For mice infected intravenously, bacteria were recovered from livers and spleens as early as 7 h postinfection, and CI were calculated at 7 h and on days 1, 2, and 3.
In the intestines of orally infected mice, the plcBΔpro mutant was outcompeted ≈15- to 25-fold by the Emr wild type on days 2 and 3 postinfection and ≈100-fold by day 4 postinfection (Fig. 5). In livers of orally infected mice, the plcBΔpro mutant was outcompeted ≈50-fold by the Emr wild type on day 2 postinfection and ≈100-fold on days 3 and 4 postinfection. In spleens of orally infected mice, the plcBΔpro mutant was outcompeted ≈20-fold by the Emr wild type on day 2 postinfection and ≈200-fold on days 3 and 4 postinfection. In MLN, the plcBΔpro mutant was outcompeted by the Emr wild type to a lesser degree, and CI showed a large degree of variability between mice on days 2 and 4 postinfection. Statistical analysis of these results indicated that the plcBΔpro mutant was significantly outcompeted by the Emr wild type in intestines, livers, and spleens of infected mice at all times but not in MLN on days 2 and 4 postinfection. It is important that in some circumstances the mutant was not recovered and the median CI was calculated based on some estimated values (see Materials and Methods). The estimated CI, which are cross-marked on the figure, are potentially higher than the actual CI. Hence, the mutant is potentially attenuated to a greater extent than estimated.
FIG. 5.

CI for the plcBΔpro mutant relative to the Emr wild type after oral or intravenous infection of BALB/c mice. Six- to 8-week-old female BALB/c mice were initially infected with equal concentrations of the Emr wild type and the plcBΔpro mutant. At defined time points (2 to 4 days for oral infection; 7 h and 1 to 3 days for i.v. infection), mice were sacrificed, and bacteria residing in various organs were enumerated. Livers and spleens were harvested from mice infected intravenously. In addition to these two organs, whole intestines and MLN were harvested from mice infected orally. Each dot represents the CI for an individual mouse. Crossed dots represent estimated CI when the mutant was not recovered from the corresponding mice. The horizontal lines represent the median CI for each treatment group. A CI of <1 indicates that the plcBΔpro mutant's virulence was attenuated. Data were analyzed with a one-sample t test. *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
For mice infected intravenously, CI from livers and spleens were not significantly different from 1 at 7 h postinfection, indicating that the mutant was not defective in the first stage of infection. As with oral infection, CI dropped below 1 in the subsequent stages of infection, but to a lesser extent. In livers of mice infected intravenously, the plcBΔpro mutant was outcompeted nearly 10-fold by the Emr wild type on days 1 and 2 postinfection and ≈100-fold by day 3 postinfection. In spleens, it took 2 days for the Emr wild type to outcompete the plcBΔpro mutant. The Emr wild type outcompeted the plcBΔpro mutant 4- and 35-fold by days 2 and 3 postinfection, respectively.
The median of total CFU recovered from tissues was determined and compared to the median CI to evaluate the correlation between the two sets of data (Fig. 6). In most instances, the median CI within a tissue decreased, while the median CFU increased or remained constant, indicating that wild-type bacteria were proliferating as the mutant was being eliminated. Together, the results from in vivo infections indicate that the plcBΔpro mutant is significantly attenuated in the mouse.
FIG. 6.

Comparison of CI and total bacterial counts for tissues of infected mice. Each median CI reported in Fig. 5 is represented here in a bar graph, in parallel with the median CFU harvested from tissues of the same group of mice. Median CI are shown with black bars and the left y axis scale, and median CFU are shown with white bars and the right y axis scale. The left panel represents data for mice infected orally, and the right panel represents data for mice infected i.v.
DISCUSSION
L. monocytogenes has the remarkable ability to regulate the activities of two membrane-damaging virulence factors, namely, LLO and PC-PLC. During intracellular infection, bacteria are temporarily located in membrane-bound vacuoles, where LLO and PC-PLC operate (2, 13). Regulation of LLO and PC-PLC activities is controlled in part by pH, as inhibition of vacuolar acidification prohibits these factors from acting. L. monocytogenes mutants that have lost the ability to compartmentalize LLO activity are cytotoxic and strongly attenuated (7, 8). In this study, we show that an L. monocytogenes mutant that constitutively secretes active PC-PLC is not cytotoxic. However, this mutant compromises host cell membrane integrity in a manner that is dependent on cell-to-cell spread, suggesting that membrane damage is limited to filopodia formed by moving bacteria. Moreover, the lack of compartmentalization of PC-PLC activity during infection significantly decreases the virulence of L. monocytogenes.
PC-PLC is made as a proenzyme whose activation requires a decrease in pH (13). During intracellular infection, L. monocytogenes maintains a pool of PC-PLC in its proform at the membrane-cell-wall interface (24). Upon vacuolar acidification, the pool of bacterium-associated PC-PLC is rapidly activated and translocated across the cell wall in an Mpl-dependent manner (14, 28). To study the biological relevance of regulating PC-PLC activity during intracellular infection, we made a PC-PLC mutant whose activation and secretion are no longer regulated by pH and Mpl (28). The behavior of this mutant, the plcBΔpro mutant, was indistinguishable from that of the isogenic wild-type strain with regard to growth kinetics in broth and sensitivity to acid stress and bile salts (data not shown). The only remarkable phenotypic difference was the size of the zone of opacity generated on LB-G1P-EY plates, indicating increased levels of PC-PLC activity (28). The plcBΔpro mutant was used in this study to evaluate the importance of L. monocytogenes regulation of PC-PLC activity during infection.
Cells infected with the plcBΔpro mutant did not show signs of cytotoxicity. However, we have previously shown that pH-induced release and activation of bacterium-associated PC-PLC from intracellular bacteria are cytotoxic (14). In the previous study, the intracellular pH of infected cells was equilibrated to the desired pH by using a potassium-based buffer and a potassium ionophore, resulting in a synchronized and rapid release of active PC-PLC from every intracellular bacterium. This massive release of active PC-PLC in the cytosol of infected cells is cytotoxic. In contrast to that phenomenon, the plcBΔpro mutant does not cumulate a pool of PC-PLC, as the active form of PC-PLC appears to diffuse unrestrictively across the bacterial cell wall (28). In addition, the turnover rate of active PC-PLC in the cytosol of cells infected with the plcBΔpro mutant could be high, preventing accumulation of this enzyme to a concentration that is detrimental to host cells. Therefore, constant release and, perhaps, rapid turnover of active PC-PLC in the cytosol of infected cells prevented the enzyme from causing irreversible membrane damage.
The plcBΔpro mutant became sensitive to gentamicin during intracellular infection. This sensitivity to gentamicin was abolished by preventing bacteria from forming filopodia, indicating that it was not due to an intrinsic increased bacterial sensitivity to gentamicin but rather to leakage of the antibiotic within infected cells as a result of filopodium formation. Concomitantly, using a non-membrane-permeant nucleic acid stain, cells were shown to be twice as sensitive to permeabilization when infected with the plcBΔpro mutant as they were when infected with the wild-type strain. It is conceivable that membrane damage is more likely to occur during cell-to-cell spread when bacteria are tightly apposed to host cell membranes in filopodia. Release of active PC-PLC by plcBΔpro mutant bacteria in filopodia would result in localized and steady hydrolysis of membrane phospholipids, compromising membrane integrity and its ability to prevent diffusion of gentamicin. Consequently, intracellular growth of bacteria would begin to stall when moving bacteria initiate filopodium formation, which may explain the plaquing phenotype. Plaques formed by the wild-type strain in the presence of gentamicin at 10 μg/ml were relatively clear, whereas those formed by the plcBΔpro mutant were darker after staining of the monolayers with neutral red. Since neutral red diffuses across membranes and cumulates within live cells, the plaquing phenotype indicates that cells infected with the plcBΔpro mutant were not as damaged as cells infected with the wild-type strain, presumably as a result of a lower intracellular bacterial density.
The plcBΔpro mutant was outcompeted up to 200-fold by the Emr wild type in a mouse competitive infection model. It is not clear why the plcBΔpro mutant was attenuated in vivo. In tissue culture cells, the mutant strain behaved similarly to the wild-type strain with regard to invasion and escape from vacuoles (data not shown). Moreover, the plcBΔpro mutant did not show an intracellular growth defect or a cell-to-cell spreading defect in the absence of gentamicin. The only observed difference between the wild-type strain and the mutant was a twofold increase in host membrane permeability during intracellular growth. Conceivably, this increase in host membrane permeability carries greater consequences in vivo, as damaged cells may hasten and amplify the inflammatory response to infection. An amplified inflammatory response would have affected the wild-type strain as well as the mutant strain in a competitive assay. Absolute bacterial numbers recovered from internal organs did not decrease overtime, but CI did, indicating that Emr wild-type bacteria were proliferating. However, it remains a possibility that the rate of proliferation of the Emr wild type would have been greater in the absence of the plcBΔpro mutant.
The kinetics of L. monocytogenes in the initial phase of an intravenous infection in mice has been established (9). Within 10 min of infection, 60% and 5% of bacteria are found in the liver and the spleen, respectively, but 90% of the bacteria reaching the liver are killed by neutrophils within the first 6 h of infection (5, 9). It appears that the plcBΔpro mutant was not more susceptible than the Emr wild type to killing by neutrophils, as indicated by the CI obtained at 7 h postinfection. In addition, the plcBΔpro mutant was not more susceptible than the wild-type strain to killing by activated mouse bone marrow-derived macrophages (data not shown). However, these observations do not eliminate the possibility that damaged host cells are better targets for inflammatory cells and, consequently, that cells infected with the plcBΔpro mutant may be eliminated more readily than those infected with the Emr wild type at a time during infection when bacteria begin to spread from cell to cell.
Mesenteric lymph nodes from infected mice did not show the same CI pattern as that for other tissues. The data points were much more scattered on days 2 and 4 post-oral infection, and the day 4 median CI was 6- to 12-fold higher for MLN than for intestines, livers, and spleens. Perhaps MLN are not the main source of infection of the liver and spleen after oral infection with L. monocytogenes.
The plcBΔpro mutant was more attenuated in mice infected orally than in those infected intravenously. This difference is unlikely due to a decreased ability of the plcBΔpro mutant to invade cells of the intestinal tract, as it was not defective in invasion of different cell lines, such as Caco-2 human intestinal epithelial cells, L2 mouse fibroblasts, and J774 mouse macrophage-like cells (data not shown). In addition, the mutant was not more sensitive than the wild-type strain to acid stress and bile salts (data not shown). Conceivably, this difference only reflects the inability of the mutant to perform as well as the wild-type strain in any infected tissue. Invasion of the liver and spleen following an oral infection takes 1 to 2 days according to the absolute bacterial numbers obtained in the present study. In comparison, invasion of the liver and spleen was observed at 7 h postinfection and has been reported to occur as early as 10 min postinfection following an intravenous infection (9). The difference in attenuation between orally and intravenously infected mice may simply reflect a cumulative effect of the plcBΔpro mutant being outcompeted by the Emr wild type in the intestines before translocation to the liver and spleen.
In summary, this study emphasizes the importance for L. monocytogenes virulence of compartmentalizing the activity of PC-PLC, a membrane-acting factor that contributes to lysis of double-membrane vacuoles formed upon bacterial cell-to-cell spread. Constitutive secretion of active PC-PLC during intracellular infection causes host membrane damage in a cell-to-cell spread-dependent manner and results in virulence attenuation in the mouse. Future studies will aim at elucidating the mechanism contributing to plcBΔpro mutant attenuation in vivo.
Acknowledgments
We thank D. A. Portnoy for providing strain DP-L3903, which was used in the in vivo experiments. We are also grateful to Heather O'Neil and Avni Shah for their assistance on the competitive index and bile salts resistance assays, respectively.
This work was supported by U.S. Public Health Service grant AI52154 to H.M. from NIAID.
Editor: V. J. DiRita
Footnotes
Published ahead of print on 23 October 2006.
REFERENCES
- 1.Auerbuch, V., L. L. Lenz, and D. A. Portnoy. 2001. Development of a competitive index assay to evaluate the virulence of Listeria monocytogenes actA mutants during primary and secondary infection of mice. Infect. Immun. 69:5953-5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beauregard, K. E., K.-D. Lee, R. J. Collier, and J. A. Swanson. 1997. pH-dependent perforation of macrophage phagosomes by listeriolysin O from Listeria monocytogenes. J. Exp. Med. 186:1159-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bishop, D. K., and D. J. Hinrichs. 1987. Adoptive transfer of immunity to Listeria monocytogenes: the influence of in vitro stimulation on lymphocyte subset requirements. J. Immunol. 139:2005-2009. [PubMed] [Google Scholar]
- 4.Camilli, A., L. G. Tilney, and D. A. Portnoy. 1993. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol. Microbiol. 8:143-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conlan, J. W., and R. J. North. 1991. Neutrophil-mediated dissolution of infected host cells as a defense strategy against a facultative intracellular bacterium. J. Exp. Med. 174:741-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cossart, P., M. F. Vicente, J. Mengaud, F. Baquero, J. C. Perez-Diaz, and P. Berche. 1989. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect. Immun. 57:3629-3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Decatur, A. L., and D. A. Portnoy. 2000. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science 290:992-995. [DOI] [PubMed] [Google Scholar]
- 8.Glomski, I. J., A. L. Decatur, and D. A. Portnoy. 2003. Listeria monocytogenes mutants that fail to compartmentalize listeriolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect. Immun. 71:6754-6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregory, S. H., A. J. Sagnimeni, and E. J. Wing. 1996. Bacteria in the bloodstream are trapped in the liver and killed by immigrating neutrophils. J. Immunol. 157:2514-2520. [PubMed] [Google Scholar]
- 10.Grundling, A., M. D. Gonzalez, and D. E. Higgins. 2003. Requirement of the Listeria monocytogenes broad-range phospholipase PC-PLC during infection of human epithelial cells. J. Bacteriol. 185:6295-6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones, S., and D. A. Portnoy. 1994. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect. Immun. 62:5608-5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marquis, H., V. Doshi, and D. A. Portnoy. 1995. The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect. Immun. 63:4531-4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marquis, H., H. Goldfine, and D. A. Portnoy. 1997. Proteolytic pathways of activation and degradation of a bacterial phospholipase C during intracellular infection by Listeria monocytogenes. J. Cell Biol. 137:1381-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marquis, H., and E. J. Hager. 2000. pH-regulated activation and release of a bacteria-associated phospholipase C during intracellular infection by Listeria monocytogenes. Mol. Microbiol. 35:289-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mounier, J., A. Ryter, M. Coquis-Rondon, and P. J. Sansonetti. 1990. Intracellular and cell-to-cell spread of Listeria monocytogenes involves interaction with F-actin in the enterocytelike cell line Caco-2. Infect. Immun. 58:1048-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neumann, K., E. Eppler, L. Filgueira, P. Groscurth, E. Gasal, A. Schaffner, G. Schoedon, and M. Schneemann. 2003. Listeria species escape from the phagosomes of interleukin-4-deactivated human macrophages independent of listeriolysin. Immunol. Cell Biol. 81:431-439. [DOI] [PubMed] [Google Scholar]
- 17.Niebuhr, K., T. Chakraborty, P. Köllner, and J. Wehland. 1993. Production of monoclonal antibodies to the phosphatidylcholine-specific phospholipase C of Listeria monocytogenes, a virulence factor for this species. Med. Microbiol. Lett. 2:9-16. [Google Scholar]
- 18.Portnoy, D. A., P. S. Jacks, and D. J. Hinrichs. 1988. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 167:1459-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poyart, C., E. Abachin, I. Razafimanantsoa, and P. Berche. 1993. The zinc metalloprotease of Listeria monocytogenes is required for maturation of phosphatidylcholine phospholipase C: direct evidence obtained by gene complementation. Infect. Immun. 61:1576-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raveneau, J., C. Geoffroy, J.-L. Beretti, J.-L. Gaillard, J. E. Alouf, and P. Berche. 1992. Reduced virulence of a Listeria monocytogenes phospholipase-deficient mutant obtained by transposon insertion into the zinc metalloprotease gene. Infect. Immun. 60:916-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robbins, J. R., A. I. Barth, H. Marquis, E. L. de Hostos, W. J. Nelson, and J. A. Theriot. 1999. Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. J. Cell Biol. 146:1333-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schnupf, P., D. A. Portnoy, and A. L. Decatur. 2006. Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: role of the PEST-like sequence. Cell. Microbiol. 8:353-364. [DOI] [PubMed] [Google Scholar]
- 23.Smith, G. A., H. Marquis, S. Jones, N. C. Johnston, D. A. Portnoy, and H. Goldfine. 1995. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect. Immun. 63:4231-4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snyder, A., and H. Marquis. 2003. Restricted translocation across the cell wall regulates secretion of the broad-range phospholipase C of Listeria monocytogenes. J. Bacteriol. 185:5953-5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun, A. N., A. Camilli, and D. A. Portnoy. 1990. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect. Immun. 58:3770-3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tilney, L. G., and D. A. Portnoy. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109:1597-1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vazquez-Boland, J.-A., C. Kocks, S. Dramsi, H. Ohayon, C. Geoffroy, J. Mengaud, and P. Cossart. 1992. Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell-to-cell spread. Infect. Immun. 60:219-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yeung, P. S., N. Zagorski, and H. Marquis. 2005. The metalloprotease of Listeria monocytogenes controls cell wall translocation of the broad-range phospholipase C. J. Bacteriol. 187:2601-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]