

Abstract
Mycobacterium ulcerans is the etiologic agent of Buruli ulcer (BU), an emerging tropical skin disease. Virulent M. ulcerans secretes mycolactone, a cytotoxic exotoxin with a key pathogenic role. M. ulcerans in biopsy specimens has been described as an extracellular bacillus. In vitro assays have suggested a mycolactone-induced inhibition of M. ulcerans uptake by macrophages in which its proliferation has not been demonstrated. Therefore, and uniquely for a mycobacterium, M. ulcerans has been classified as an extracellular pathogen. In specimens from patients and in mouse footpad lesions, extracellular bacilli were concentrated in central necrotic acellular areas; however, we found bacilli within macrophages in surrounding inflammatory infiltrates. We demonstrated that mycolactone-producing M. ulcerans isolates are efficiently phagocytosed by murine macrophages, indicating that the extracellular location of M. ulcerans is not a result of inhibition of phagocytosis. Additionally, we found that M. ulcerans multiplies inside cultured mouse macrophages when low multiplicities of infection are used to prevent early mycolactone-associated cytotoxicity. Following the proliferation phase within macrophages, M. ulcerans induces the lysis of the infected host cells, becoming extracellular. Our data show that M. ulcerans, like M. tuberculosis, is an intracellular parasite with phases of intramacrophage and extracellular multiplication. The occurrence of an intramacrophage phase is in accordance with the development of cell-mediated and delayed-type hypersensitivity responses in BU patients.
Mycobacterium ulcerans is the etiologic agent of Buruli ulcer (BU), an emerging, devastating, difficult-to-treat skin disease reported in many countries, mostly in tropical areas (16), and BU has become the third most common mycobacterial infection in humans after tuberculosis and leprosy. BU assumes various nonulcerative clinical forms that can progress to ulcers.
M. ulcerans (26, 41, 47, 59), M. tuberculosis (24, 38, 42), M. marinum (25, 57), and M. haemophilum (23) are known to have cytotoxic activity, M. ulcerans being the most cytotoxic of all known mycobacteria. The M. ulcerans toxin is a mycolactone, a unique polyketide lipid exotoxin that has a potent destructive activity for cells that provokes the extensive necrotic lesions characteristic of BU (26). Mycolactone is considered to be the major virulence factor of this pathogen (1, 26, 41).
Intracellular parasites are defined on the basis of their lifestyles in the infected hosts and of the types of immune responses elicited (7, 37): they live and multiply predominantly within host cells, typically macrophages, and therefore are able to survive and grow within cultured macrophages. The host immune response against intramacrophage parasites involves mechanisms of cell-mediated immunity (CMI) accompanied by delayed-type hypersensitivity (DTH).
Genetic analysis places M. ulcerans very close to M. marinum and M. tuberculosis (71), two species of mycobacteria that are typical intracellular parasites that grow within macrophages in vivo (7, 12, 15, 31) and in vitro (25, 42, 45, 57, 58, 66) and elicit CMI and DTH responses (7, 9, 12, 14, 15, 18, 46, 72). Several reports indicate that M. ulcerans infections are also associated, at least at some stage of the disease, with CMI and DTH (18, 27, 28, 33, 39, 43, 44, 50, 54, 56, 61, 67, 68, 70, 74), strongly suggesting the existence of an intracellular phase in the life cycle of M. ulcerans inside macrophages. It is therefore intriguing that M. ulcerans has been described as an extracellular pathogen in humans and experimentally infected animals (1, 11, 12, 17, 26, 29, 33, 52), implying that this organism is an exception among pathogenic mycobacteria (12).
The interpretation that M. ulcerans is an extracellular pathogen stems mainly from reports that BU tissues show predominantly free bacilli in extensive necrotic acellular areas (8, 17, 20, 29, 35). Other arguments advanced in support of that interpretation are that (i) M. ulcerans and mycolactone, under some in vitro conditions, inhibit the phagocytic activity of macrophages (1, 13, 55, 58) and (ii) M. ulcerans fails to grow within macrophages in vitro (1, 58).
The observation that extracellular acid-fast bacilli (AFB) predominate in necrotic areas devoid of cells has also led to the concept that the total absence of, or only minimal, inflammation is typical of M. ulcerans infections (11, 17, 29, 33, 35). Although such a histopathological pattern is a diagnostic hallmark of BU, it may not reflect the actual host-parasite interactions at the active foci of infection in patients with BU, interactions that so far have not been analyzed in detail. However, a recent publication (32) reported inflammatory infiltrates with neutrophils and mononuclear cells in 92% of 78 BU specimens with AFB, but no description of the relative locations of bacilli and inflammatory cells was given. Additionally, we have recently described the consistent occurrence of inflammatory infiltrates with neutrophils and macrophages in the mouse model, with M. ulcerans bacilli colocalizing with the phagocytic cells and extracellular bacilli in necrotic acellular areas (51). Other, earlier investigators have reported bacilli within macrophages in experimental M. ulcerans infections (21, 41, 59). In this context, it is worth recalling that the initial description of BU noted large numbers of bacilli within phagocytes (43).
Because M. ulcerans infections are associated with CMI and DTH responses, and because of our previous observations with mice, we postulated that M. ulcerans would be an intracellular pathogen with a phase of extracellular existence.
The objective of the present work is to clarify the lifestyle of M. ulcerans by reevaluating M. ulcerans-phagocyte interactions in BU tissues, mouse tissues, and cultured macrophages. One advantage of the mouse footpad model is that it is possible to systematically observe the entire lesion and surrounding healthy tissues even in advanced infections. We complemented these observations with in vitro studies involving the interactions of M. ulcerans with mouse bone marrow-derived macrophages (BMDM) and with the macrophage cell line J774A.1. Our results show that, in a considerable percentage of BU biopsy specimens containing AFB and inflammatory infiltrates, intramacrophage mycobacteria are present in the infiltrates at the peripheries of the necrotic acellular areas. In this model of infection, we also found that virulent mycolactone-producing strains of M. ulcerans are efficiently phagocytosed by macrophages in vitro and in vivo, can be found within macrophages during the entire course of infection, and grow intracellularly within cultured macrophages. These data show that M. ulcerans, like M. tuberculosis, is an intracellular parasite with phases of extracellular and intramacrophage multiplication.
MATERIALS AND METHODS
BU biopsy specimens.
We studied 24 paraffin-embedded biopsy specimens from African patients with ulcerative or nonulcerative BU lesions, confirmed by histopathological features and the presence of AFB, from the pathology archives of the Armed Forces Institute of Pathology, Washington, DC. Serial sections of the entire specimens were viewed under the Olympus BX61 light microscope following staining with Ziehl-Neelsen stain (ZN; Merck, Darmstadt, Germany) and hematoxylin (Merck) or methylene blue.
For the isolation of M. ulcerans from BU samples, the tissues (±1 g) were homogenized in 1 ml saline, decontaminated, and inoculated onto Löwenstein-Jensen medium. The inoculated tubes were incubated for up to 12 months at 33°C and were observed weekly. For detection of the RD1 gene cluster, primers for esxA (Esat-6, 5′-GACAGAACAGCAGTGGAATTTCG-3′ and 5′-CTTCTGCTGCACACCCTGGTA-3′) and esxB (Cfp-10, 5′-TTTTGAAGAACGATGCCGCTAC-3′ and 5′-TGACGGATGTTCGTCGAAATC-3′) were used (48). “M. liflandii” ITM04-3050 and M. ulcerans ITM00-1240 were used as positive and negative controls, respectively.
Mycobacterial strains and preparation of inocula for experimental work.
M. ulcerans strains were selected based on different degrees of virulence in mice. M. ulcerans 98-912 and 97-1116 are highly virulent, and strain 5114 is avirulent (51). The origins and genetic and phenotypic characteristics of strains 98-912 and 5114 have been previously described in detail (51). Strain 5114 contains the RD1 gene cluster (48) and does not produce mycolactones (47, 69). Strain 97-1116 produces mycolactones A and B (47), and we found it to lack the RD1 gene cluster, as is typical of African strains (48). Strain 98-912 possesses the RD1 gene cluster (48) and produces mycolactones A and B slightly different from those in African strains (34). For the determination of the percentage of phagocytosis, M. tuberculosis H37Rv and M. bovis BCG Pasteur were used as controls. M. tuberculosis possesses RD1, and M. bovis BCG lacks this gene cluster (6).
The M. ulcerans strains were grown on Löwenstein-Jensen medium at 32°C for approximately 2 months, recovered from slants, diluted in phosphate-buffered saline to a final concentration of 1 mg/ml, and vortexed by using 2-mm-diameter glass beads. The number of AFB in the inocula was determined by the method of Shepard and McRae (62) using ZN. The final suspensions revealed more than 90% of the cells to be viable as assessed with the LIVE/DEAD BacLight kit (Molecular Probes, Leiden, The Netherlands).
Animals.
Eight-week-old female BALB/c mice were obtained from Charles River (Barcelona, Spain) and were housed under specific-pathogen-free conditions with food and water ad libitum.
Peritoneal model of infection.
Mice were infected in the peritoneal cavity with 0.1 ml of M. ulcerans suspensions containing 7 log10 AFB of M. ulcerans strain 98-912, 97-1116, or 5114. Phosphate-buffered saline was used as a control. At different time points after infection, four mice per group were sacrificed and peritoneal cells were recovered for cytospin preparations by peritoneal lavage. Cytospin preparations were fixed with 10% Formol in ethanol, stained with ZN, and counterstained with Hemacolor (Merck).
Footpad model of infection.
Mice were infected in the left hind footpad with 0.03 ml of suspensions containing 6 log10 AFB of M. ulcerans 98-912 or 97-1116. The right hind footpad was used as a control.
Culture of murine BMDM and J774A.1 macrophage cell line.
BMDM were prepared as previously described (51). For AFB counting and light microscopy analysis, BMDM were seeded in 24-well plates at a density of 5 × 105 cells/well; for electron microscopy, BMDM were seeded in 6-well plates at a density of 3.5 × 106 cells/well. The plates were kept at 37°C in a 5% CO2 atmosphere. Twelve hours before infection, macrophages were incubated at 32°C in a 5% CO2 atmosphere and maintained under these conditions thereafter.
Because BMDM survive in vitro for only a short time, the macrophage cell line J774A.1 was used for longer experimental infections. The J774A.1 mouse macrophage cell line was seeded in 12-well plates at a density of 2 × 105 cells/well and incubated at 37°C for 12 h to allow macrophage adherence. Cells were then washed with Dulbecco modified Eagle medium (DMEM) and incubated at 32°C in a 5% CO2 atmosphere and maintained in these conditions thereafter.
Macrophage infectivity assays.
Bacterial suspensions were further diluted in DMEM to obtain the selected multiplicity of infection (MOI; bacterium/macrophage ratio). For the assessment of phagocytosis and of intramacrophage growth of M. ulcerans, BMDM and J774A.1 cells were infected with the appropriate number of bacilli and incubated for 4 h at 32°C in a 5% CO2 atmosphere and then washed with warm Hank's balanced salt solution to remove noninternalized bacteria. For growth experiments, the macrophages were reincubated at 32°C in DMEM for maximum periods of 8 and 15 days, respectively. At 7 and 10 days after infection, J774A.1 cultures were supplemented with fresh medium by adding 1 ml of DMEM per well.
Since the M. ulcerans strains used in this study were found to grow in DMEM and to be susceptible to 20 μg/ml of amikacin (data not shown), this concentration of amikacin (Sigma, St. Louis, MO) was added to the culture wells after infection and kept during the whole period of the experiment to prevent extracellular M. ulcerans growth (45, 57).
To evaluate the intramacrophage growth of M. ulcerans, AFB were counted as follows: macrophages were lysed with 0.1% saponin (final concentration; Sigma), the suspensions were homogenized with 2-mm glass beads, and the AFB were counted (62). Monolayers were not washed previous to AFB counting to avoid losing nonadherent or loosely adherent cells.
Cytological analysis.
Adhesion of BMDM was carried out on 13-mm-diameter plastic coverslips (Nunc, Naperville, IL) in 24-well plates. For cytological analysis, plastic coverslips were removed and the cells were fixed with 10% Formol in ethanol, stained with ZN, and counterstained with hematoxylin. Cells were analyzed under a light microscope, and digital images were captured by using an Olympus DP70 camera (Hamburg, Germany).
To quantify dead macrophages, the trypan blue (Gibco, Paisley, United Kingdom) exclusion assay was employed. J774A.1 macrophages were carefully scraped off by using a cell scraper, stained, and counted.
For ultrastructural studies, BMDM cultures were processed for electron microscopy as previously described (63) and viewed and photographed under a Zeiss EM10 electron microscope (Hallbergmoos, Germany).
Histological studies.
Human macrophages were labeled with the antibody NCL-LN5 (Novocastra, Newcastle, United Kingdom) that specifically stains the cytoplasm of histocytes and macrophages (5) or with an isotype control antibody. Briefly, the tissue sections were deparaffinized with xylene, followed by sequential immersion in graded ethanol solutions and hydration. The sections were microwave incubated for 15 min with a commercial antigen retrieval solution, citrate buffer heat-induced epitope retrieval (Labvision Corporation, Fremont, CA). Endogenous peroxidase activity was blocked with 3% H2O2 for 10 min, and primary antibody was added and incubated for 1 h. Slides were developed with the Ultravision anti-polyvalent horseradish peroxidase ready-to-use detection system (Labvision Corporation). For diaminobenzidene detection, slides were incubated with the Ultravision large-volume diaminobenzidene substrate system (Labvision Corporation).
Excised tissues of mouse footpads were fixed in 10% phosphate-buffered formalin. Whole paws were decalcified (Thermo Shandon TBD-1; Runcorn, United Kingdom) and embedded in paraffin. Longitudinal sections of the entire paw were stained with ZN. For labeling of mouse macrophages, the specific marker antibody F4/80 or an isotype control antibody was used, according to the procedure referred to above for immunohistochemical staining. All digital images were captured by using an Olympus DP70 camera.
The present study was conducted under the guidelines and approval of the Research Ethics Committee of the Life and Health Sciences Research Institute.
RESULTS
BU biopsy specimens show intramacrophage M. ulcerans bacilli in inflammatory infiltrates.
M. ulcerans has been described as an extracellular parasite, an exception among pathogenic mycobacteria. On the other hand, reports of CMI and DTH responses to M. ulcerans infections (18, 27, 28, 33, 39, 43, 44, 50, 54, 56, 61, 67, 68, 70, 74) point to an intracellular residence of M. ulcerans. To reevaluate this issue, we started by performing histopathological analyses of BU tissues.
Nineteen specimens from the Armed Forces Institute of Pathology collection from active cases of African BU were selected based on the presence of AFB and the absence of secondary infections. Serial sections were searched for inflammatory infiltrates and for the localization of AFB in the extracellular and intracellular compartments. Among the 19 cases, inflammatory infiltrates were observed in 15 (Table 1). This result agrees with a recent report describing cellular inflammatory responses in a high percentage of BU cases (32). Among the 15 specimens containing inflammatory cells, 8 showed intracellular bacilli (Table 1), as shown for 2 representative specimens (Fig. 1A to F). In addition, NCL-LN5 antibody demonstrated that M. ulcerans bacilli were present within macrophages (Fig. 1C). The histopathological hallmark of BU, extracellular bacilli, was found in all cases and was particularly abundant in the necrotic acellular areas (Fig. 1B), while bacilli colocalizing with inflammatory cells were found in the infiltrates at the peripheries of the necrotic areas (Fig. 1A). Some intracellular bacilli appeared as globus-like structures (Fig. 1F) or within phagocytes undergoing apoptosis (Fig. 1E).
TABLE 1.
Clinical data and histopathologic features evaluated for human biopsy specimens from patients with active BUa
| Origin of specimen | Clinical feature | Extracellular AFB | Inflammation | Intracellular AFB | RD1 |
|---|---|---|---|---|---|
| Benin | Nonulcerative | + | − | − | ND |
| Togo | Nonulcerative | + | − | − | Negative |
| Benin | Ulcer | + | − | − | ND |
| Democratic Republic of Congo | Ulcer | + | − | − | Negative |
| Côte d'lvoire | Ulcer | + | + | − | ND |
| Benin | Nonulcerative | + | + | − | ND |
| Benin | Nonulcerative | + | + | − | Negative |
| Côte d'lvoire | Ulcer | + | + | − | ND |
| Democratic Republic of Congo | Ulcer | + | + | − | ND |
| Democratic Republic of Congo | Ulcer | + | + | − | ND |
| Democratic Republic of Congo | Ulcer | + | + | − | ND |
| Benin | Nonulcerative | + | + | + | ND |
| Benin | Nonulcerative | + | + | + | ND |
| Benin | Nonulcerative | + | + | + | Negative |
| Benin | Nonulcerative | + | + | + | Negative |
| Democratic Republic of Congo | Ulcer | + | + | + | ND |
| Democratic Republic of Congo | Ulcer | + | + | + | ND |
| Benin | Ulcer | + | + | + | ND |
| Benin | Ulcer | + | + | + | Negative |
| Totalb | 19 (100) | 15 (79) | 8 (42) |
+, present; −, absent; ND, not determined.
Number of specimens with the indicated characteristic; numbers in parentheses are percentages of the total number of specimens examined.
FIG. 1.

Intracellular M. ulcerans bacilli are found in specific areas of biopsy specimens from BU patients. The two cases shown (A to C and D to F) are representative of the human biopsy specimens showing inflammation and intracellular bacilli. (A) Extracellular bacilli are shown in the necrotic acellular area of the lesion, and bacilli colocalizing with inflammatory cells were found in the peripheral infiltrate. (B) Detail of the abundant extracellular bacilli in the central acellular area. (C) Specifically stained macrophages with internalized bacilli in the peripheral inflammatory area. (D to F) High magnifications of the areas containing inflammatory infiltrates colocalizing with AFB show intracellular bacilli associated with destruction of phagocytes (E) and intracellular, as well as extracellular, globus-like structures (F). Histological sections were stained with ZN and counterstained with hematoxylin or methylene blue. NCL-LN5 antibody was used to specifically label human macrophages. Magnifications, ×250 (A) and ×1,250 (B, C, D, E, and F).
Detection of the gene cluster RD1 carried out with M. ulcerans strains from six of the studied BU specimens, with and without detectable intracellular bacilli (Table 1), showed that all these strains were RD1 negative as was previously found with other M. ulcerans strains isolated from African patients (48). This result suggests that there is no correlation between the occurrence of intracellular bacilli and RD1.
Following experimental infections, M. ulcerans bacilli are phagocytosed by resident macrophages in vivo and by BMDM and J774A.1 macrophages in vitro.
Murine peritoneal leukocyte populations can be readily and precisely studied, and this model proved useful in the elucidation of host interactions with several species of mycobacteria (3, 53, 65). We have previously reported the recruitment of phagocytes to the peritoneal cavity in response to inoculated M. ulcerans (51). Therefore, we used this model to study the early interactions of phagocytes and M. ulcerans.
Microscopic analyses revealed that for all the strains of M. ulcerans tested (98-912, 97-1116, and 5114), bacilli were phagocytosed by resident macrophages and recruited neutrophils after the first few hours postinfection, as shown in Fig. 2A for strain 98-912. As previously described for experimental peritoneal infections caused by other mycobacteria (65), as well as the livers of mice infected with Listeria monocytogenes (30) or Salmonella enterica serovar Typhimurium (60), we found that neutrophils containing ingested bacilli were progressively phagocytosed by macrophages so that these bacteria were eventually taken up by macrophages.
FIG. 2.

M. ulcerans is phagocytosed by macrophages and neutrophils in vivo following experimental infections. (A) Cytospin preparations of mouse peritoneal leukocytes 6 h after inoculation with 7 log10 AFB of M. ulcerans 98-912, stained with ZN and counterstained with Hemacolor. (B to G) Mouse footpads infected subcutaneously with 6 log10 AFB of M. ulcerans 98-912, collected 24 h (B) and 15 (C to E) and 28 (F and G) days after infection. (A and B) Bacilli inside neutrophils and macrophages (arrows) early after infection. (C and D) Extracellular bacilli in the necrotic acellular center of the infection focus (shown in high magnification in panel D), surrounded by inflammatory cells with intracellular bacilli. (E) Higher magnification of the peripheral area with inflammatory infiltrates shows specific immunohistochemical staining of a macrophage with internalized bacilli. (F and G) Central acellular necrotic areas and peripheral inflammatory infiltrates in advanced lesions, with both intramacrophage and free bacilli, some packed in globus-like structures with intracellular (arrowheads) or extracellular (white asterisks) locations. Histological sections were stained by ZN and counterstained with methylene blue or hematoxylin. The F4/80 antibody was used to specifically label murine macrophages. Magnifications, ×1,250 (A, B, D, and E), ×550 (C and F), and ×1,000 (G).
As in our previous study (51), we consistently found intracellular M. ulcerans in the subcutaneous tissue of mouse footpads infected with M. ulcerans when the histological analysis of the entire footpad was performed. Mycolactone-producing M. ulcerans bacilli (strains 98-912 and 97-1116) were phagocytosed by mononuclear phagocytes and neutrophils a few hours postinoculation (Fig. 2B) and throughout the entire experimental period (Fig. 2B to G). The intramacrophage location of M. ulcerans was confirmed by specific immunohistochemical labeling (Fig. 2E). Some infected macrophages contained high numbers of packed clusters of bacilli, reminiscent of globi seen in lepromatous leprosy. These structures were seen both within distended macrophages (Fig. 2G) and free in the extracellular compartment (Fig. 2F and G), probably following the lysis of infected macrophages. Such clusters of AFB in BU have been previously described (10, 43) and were also found by histopathological analysis of BU specimens included in the present study (Fig. 1F). The occurrence of macrophages with high numbers of packed bacilli is suggestive of intracellular multiplication, as in the case of M. leprae infections. In the advanced lesions, the central necrotic acellular areas with high numbers of extracellular bacilli were prominent (Fig. 2F and G), but in the boundary between these areas and the surrounding inflammatory infiltrates, clumps of extracellular bacilli were found (Fig. 2F and G). The sizes of the clumps of free bacilli grew as the distance from the limit of the infiltrated areas increased (Fig. 2F and G), suggesting extracellular multiplication of M. ulcerans.
We then established an in vitro model of BMDM infection at 32°C to study the interactions of M. ulcerans with primary macrophages. Preliminary studies showed that macrophage viability, assessed by cell rounding, shrinkage, and detachment (26), as well as by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling staining (51), was compromised at high MOIs when mycolactone-producing, cytotoxic M. ulcerans strains were tested (not shown). An early cytotoxic effect on macrophages was also described for M. tuberculosis and M. marinum at high MOIs, being absent at an MOI of 1:1 or lower (25, 42, 57). We therefore tested lower MOIs and found that at an MOI of 1:1 to 1:3, the early cytotoxic effect was abolished. Microscopic analysis showed that M. ulcerans 98-912, 97-1116, and 5114 were internalized by macrophages as early as 4 h after infection at a 1:1 MOI, as shown in Fig. 3A for strain 98-912 (see also Fig. 4E for quantitative data). Ultrastructural analysis confirmed the presence of intramacrophage M. ulcerans, including the more-cytotoxic strain 98-912 (Fig. 3B and C). Bacilli were present within spacious (Fig. 4A) or tight (Fig. 4B and D) phagosomes, as described previously for other mycobacteria (4). Quantitative assessment of phagocytosis of M. ulcerans by BMDM showed that after 4 h at an MOI of 1:1, 26.3% ± 7.5% of the inoculum was internalized for the mycolactone-negative strain 5114; 22.1% ± 4.2% and 29.9% ± 8.7% of the bacteria were within macrophages for the mycolactone-producing strains 98-912 and 97-1116, respectively (Fig. 4E). The phagocytosis index of M. ulcerans at an MOI of 1:1 in the macrophage murine cell line J774A.1 was found to be similar to that in BMDM, as shown for strain 97-1116 (Fig. 4E). Similar values were found for M. tuberculosis H37Rv and M. bovis BCG (30.3% ± 11.4% and 24.9% ± 5.7%, respectively). These results suggest that RD1-positive and RD1-negative mycobacteria behave similarly regarding phagocytosis by BMDM at low MOIs, since M. tuberculosis H37Rv and M. ulcerans 5114 and 98-912 are RD1 positive (6, 48) and M. bovis BCG (6) and M. ulcerans 97-1116 (data not shown) are RD1 negative.
FIG. 3.
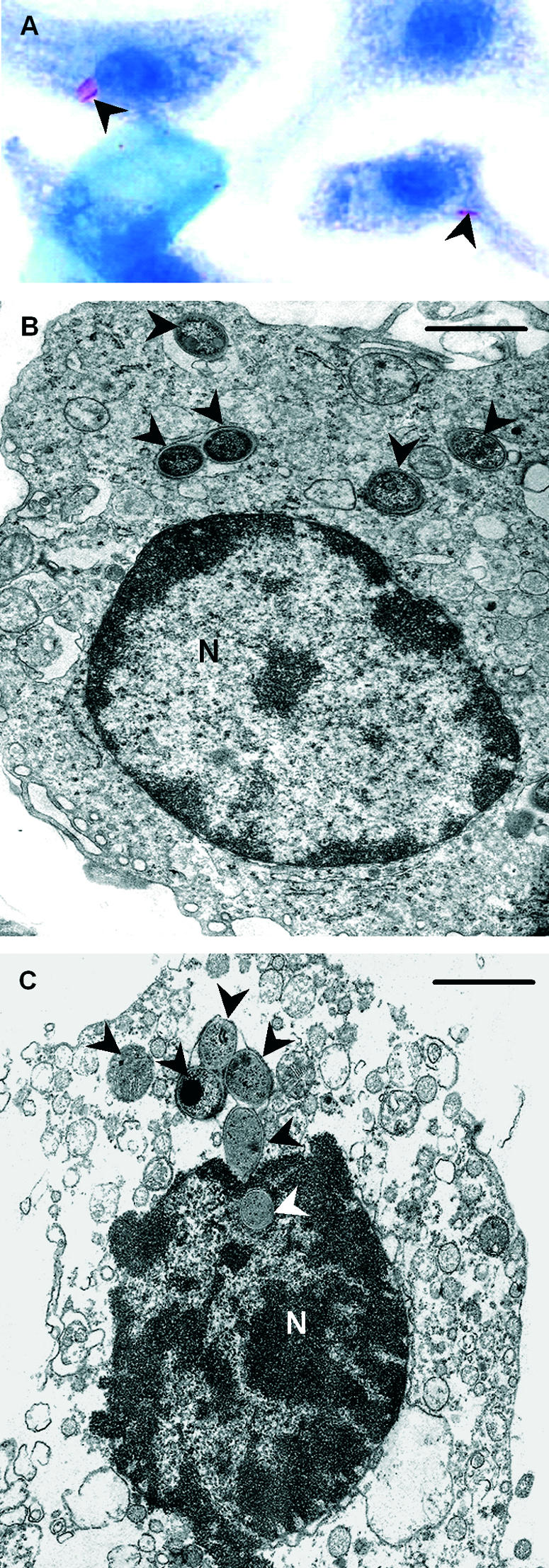
Intracellular M. ulcerans bacilli are found in mouse BMDM infected in vitro. BMDM were infected with M. ulcerans 98-912 (MOI, 1:1). (A) Coverslips were removed 4 h postinfection, and the cells were washed, stained with ZN, and counterstained with methylene blue. Microscopic analysis shows bacilli inside macrophages (arrowheads). Magnification, ×1,250. (B) Electron microscopy of BMDM infected for 4 h with M. ulcerans 98-912 and photographed 4 days later, showing bacilli inside an intact macrophage (arrowheads). Bar = 1 μm. (C) Six bacilli are seen within a lysing macrophage; one bacillus is inside the nucleus (N), which has ruptured nuclear membranes. This macrophage shows features typical of necrosis (75), namely, rupture of the cytoplasmic and nuclear membranes, condensation of chromatin, swelling and disruption of mitochondria, and loss of the normal density of the cytosol. Bar = 1 μm.
FIG. 4.

M. ulcerans is found within macrophage phagosomes, and all tested strains are phagocytosed by BMDM at an MOI of 1:1, irrespective of their cytotoxic activity. For electron microscope studies (A to D), BMDM were infected for 4 h with M. ulcerans 98-912 (MOI, 1:1) and sampled 4 days after infection. Intracellular bacilli can be found within spacious (A) or tight (B) phagosomes. Bars = 0.5 μm. PM, phagosomal membrane. (C) Detailed image of the cell envelope of extracellular bacilli. The typical layers of mycobacterial envelopes (64) are visible, namely, the cytoplasmic membrane (CM) covered by the cell wall with an innermost electron-dense peptidoglycan layer (PGL), an intermediate electron transparent layer (ETL), and an outer electron-dense layer (OL). Bar = 0.1 μm. (D) High magnification of the bacillary envelope at the zone labeled with an arrow in panel B, showing the same envelope layers shown in panel C plus a tightly apposed phagosomal membrane outside the OL. Bar = 0.1 μm. (E) BMDM (solid bars) were infected for 4 h with M. ulcerans 5114, 97-1116, or 98-912 (MOI, 1:1) or, for comparison, with M. tuberculosis H37Rv or M. bovis BCG (MOI, 1:1). To assess the phagocytosis of M. ulcerans by a different macrophagic cell, J774A.1 cultures were infected with M. ulcerans 97-1116 as described for BMDM (striped bar). Student's t test for the rates of phagocytosis gave nonsignificant P values. Results are from one representative experiment out of three independent experiments.
Taken together, these results show that soon after experimental infection with M. ulcerans, irrespective of the cytotoxicity of the clinical isolate, bacilli are phagocytosed by cultured macrophages as well as by resident macrophages and recruited neutrophils in vivo. Furthermore, intramacrophage bacilli are consistently seen in the inflammatory infiltrates at the peripheries of the necrotic acellular areas where extracellular bacilli accumulate.
M. ulcerans proliferates within cultured macrophages.
Following our observation of intramacrophage M. ulcerans both in vitro and in vivo, we evaluated the intracellular growth of M. ulcerans within BMDM by counting AFB. Amikacin was used to prevent extracellular bacterial multiplication (45, 57). At an MOI of 1:1, we found a 0.6 log10 increase in intracellular AFB counts at 8 days for the mycolactone-negative strain 5114 and the mycolactone-producing strain 97-1116 (Fig. 5A). However, for the most cytotoxic strain, 98-912 (51), bacterial counts did not increase when the same MOI was used (Fig. 5A). This is due to the macrophage damage induced by M. ulcerans 98-912, as shown by the necrotic alterations in most infected macrophages (Fig. 3C). Necrotic macrophages lose cytoplasmic membrane integrity (75), permitting access of amikacin to bacilli. However, at an MOI of 1:3, a significant growth of 0.6 log10 was observed over the 8-day experimental period (Fig. 5B), showing that this cytotoxic strain also proliferates inside macrophages when early damage to the macrophages is prevented.
FIG. 5.

M. ulcerans clinical isolates grow within mouse BMDM and J774A.1 macrophages before inducing cell lysis and becoming extracellular. All macrophage infections were performed in the presence of amikacin. (A and B) BMDM were infected for 4 h at an MOI of 1:1 with M. ulcerans 5114 (closed circles), 97-1116 (closed triangles), or 98-912 (closed squares) (A) or at an MOI of 1:3 (open squares) with M. ulcerans 98-912 (B). At an MOI of 1:1, M. ulcerans 5114 and 97-1116, but not M. ulcerans 98-912, proliferate inside macrophages for a period of 8 days. At a lower MOI, the early cytotoxic effect of M. ulcerans 98-912 is reduced and intracellular growth was detected. Statistically significant differences between results at day 0 and day 4 or 8, as evaluated by Student's t test, are labeled with double asterisks (P < 0.01) or triple asterisks (P < 0.001). (C and D) J774A.1 cells were infected for 4 h with the mycolactone-positive M. ulcerans strain 97-1116 (triangles) or the mycolactone-negative strain 5114 (circles) at an MOI of 1:1 (C) or 3:1 (D) for a period of 15 days. Panels C and D show the numbers of AFB/well, demonstrating that M. ulcerans 97-1116 grows inside macrophages before inducing their death and becoming extracellular as indicated by an arrest in bacterial growth (at day 3 for an MOI of 3:1 and day 10 for an MOI of 1:1). In contrast, strain 5114 at both MOIs grew continuously until the end of the experiment. Statistical significance was calculated by using Student's t test, comparing the results at each time point with those of the previous time point, and significant differences are labeled with a single asterisk (P < 0.05) or double asterisks (P < 0.01). Results are from one representative experiment out of two independent experiments.
M. ulcerans lyses infected macrophages after an initial phase of intracellular proliferation.
We then questioned whether or not the continuous intracellular growth of a cytotoxic M. ulcerans strain in macrophages would result in the lysis of the infected cells due to the activity of mycolactone, leading to the shedding of bacilli. To address this question, we infected J774A.1 macrophages, which can be cultured for 15 days, with the mycolactone-producing M. ulcerans strain 97-1116 or the mycolactone-negative strain 5114 at different MOIs in the presence of amikacin.
As with infected BMDM, we found that both the 97-1116 and 5114 M. ulcerans strains are phagocytosed and grow inside J774A.1 macrophages (Fig. 5C and D). With strain 97-1116 at an MOI of 1:1, the bacterial load increased, reaching 5.68 ± 0.11 log10 AFB by day 10 postinfection, when an arrest in bacterial proliferation was observed (Fig. 5C). At this time point, the number of dead macrophages had increased from 4.35 ± 0.16 log10 at time zero to 5.29 ± 0.15 log10 (an 8.7-fold increase). With strain 5114 at an MOI of 1:1, the bacterial load at 10 days was 5.61 ± 0.10 log10 AFB, with an increase in macrophage mortality of only 1.3-fold; bacterial growth continued, reaching 5.85 ± 0.17 log10 AFB by day 15, when macrophage mortality increased only 1.6-fold. When the higher MOI of 3:1 was tested, the cytotoxic load of strain 97-1116 was achieved at day 3, when the number of AFB reached 5.48 ± 0.17 log10 (Fig. 5D) and the number of dead macrophages was 5.26 ± 0.12 log10 (an 8.1-fold increase). At this MOI, strain 5114 reached 5.58 ± 0.13 log10 AFB at day 3, when macrophage mortality increased only 1.2-fold; growth continued, reaching 6.19 ± 0.09 log10 AFB by day 15. At this time point, the number of dead macrophages had increased only 3.5-fold. In contrast, with strain 97-1116 at the same MOI, the number of dead macrophages had increased 12.6-fold by day 15, with a lower bacterial load (5.54 ± 0.14 log10 AFB).
Altogether, these findings show that after an initial phase of intracellular proliferation, cytotoxic M. ulcerans becomes accessible to amikacin, suggesting that lysis of infected macrophages occurs when a certain bacterial load is reached. In vivo, intracellular growth of mycobacteria occurs by cycles of multiplication in individual macrophages followed by their lysis, egress of replicated bacilli, and entry of these bacilli into new macrophages where the growth cycle is repeated (25). The use in our in vitro experiments of amikacin to prevent extracellular proliferation of M. ulcerans (45, 57) blocks the spreading of the infection from lysed infected macrophages to healthy macrophages, resulting in an underestimation of the capacity of cytotoxic M. ulcerans to grow in cells.
DISCUSSION
The prevailing concept is that M. ulcerans is an extracellular pathogen in humans and experimentally infected animals (1, 11, 12, 17, 26, 29, 33, 52). This concept represents an exception among pathogenic mycobacteria (12) and is based mainly on the description of absent or minimal inflammation as a typical feature of M. ulcerans infections and on the fact that BU tissues show predominantly free bacilli in extensive necrotic acellular areas (8, 17, 20, 26, 29, 35). In our previous study, we demonstrated with the mouse model of BU that inflammatory infiltrates containing bacilli are consistently present at the peripheries of necrotic acellular areas. From this observation, we concluded that studies directed toward the evaluation of the interactions between phagocytes and M. ulcerans in human infections were justified (51). We have shown here that among tissue specimens from 19 BU patients, 8 of 15 specimens with inflammatory exudates and AFB revealed intramacrophage M. ulcerans in the inflammatory cells at the peripheries of the extensive necrotic acellular areas. This picture clearly parallels the scenario we described for footpads of mice with experimental BU. Intramacrophage M. ulcerans bacilli were readily found in the mouse model, because the entire lesion could be evaluated histopathologically. In specimens from BU patients, however, this phenomenon was not often appreciated because inflammatory exudates and bacilli were not always represented in the tissue fragments. BU lesions are usually extensive, and the chances that a fragment of tissue will fail to include areas of inflammatory infiltrates and bacilli are high. In many instances, intramacrophage M. ulcerans bacilli were found only after multiple consecutive sections had been evaluated.
Another argument has been put forward in favor of the concept that M. ulcerans is an extracellular pathogen: under some in vitro conditions, M. ulcerans and mycolactone inhibit the phagocytic activity of macrophages (1, 13, 55, 58). Extending previous data from our group (51), we now show quantitatively that mycolactone-producing M. ulcerans strains are efficiently ingested in vivo as well as by mouse macrophages in vitro at rates comparable to those for a mycolactone-negative M. ulcerans strain and for M. tuberculosis and M. bovis BCG. Our present results suggest that previous data showing inhibition of phagocytosis of M. ulcerans in vitro (1, 55, 58) are explained by the use of high MOIs resulting in early mycolactone-induced damage to the macrophages (26). The only previous study using low MOIs (13) concluded that M. ulcerans bacilli “were efficiently captured by RAW264.7 macrophages, bone marrow-derived macrophages, and the dendritic cell line FSDC after a 3 h contact,” showing only a partial, dose-dependent inhibition of the phagocytosis of wild-type M. ulcerans compared to that of a mycolactone-negative isogenic mutant. These findings and our results indicate that the presence of extracellular M. ulcerans in infectious foci is not due to the blockage of phagocytosis and explain the persistent occurrence of intramacrophage M. ulcerans in infected tissues. A recent publication on mouse intradermal M. ulcerans experimental infection (13) reports intracellular bacilli primarily within neutrophils and rarely in macrophages and only in the initial stages of the infection. Note, however, that a few early studies did report intramacrophage bacilli in experimental (21, 41, 59) and human (43) M. ulcerans infections.
The presence of M. ulcerans within macrophages in the focus of infection is not evidence that in vivo this mycobacterium is primarily an intracellular parasite. However, using in vitro models of macrophage infection, we show that M. ulcerans multiplies intracellularly in macrophages, as is the case for the M. ulcerans-related intracellular parasites M. marinum and M. tuberculosis (25, 42, 45, 57, 58, 66). Previous attempts to grow M. ulcerans within mammalian macrophages were unsuccessful (1, 58) due to the cytotoxicity of bacilli for macrophages at the high MOIs used (10:1 to 20:1). We avoided this early cytotoxic effect by using MOIs of 1:1 to 1:3, as previously done with M. tuberculosis and M. marinum (25, 42, 57). Our data show for the first time that virulent, mycolactone-producing strains of M. ulcerans grow inside mammalian macrophages at 32°C.
It would be intuitive to expect intracellular pathogens to have low direct cytotoxicity so that no damage would be imparted to the host cells. Indeed, it has been considered that one of the characteristics of intracellular pathogens is low cytotoxicity (37) and that cytokines produced locally by immune cells would be responsible for mycobacterium-associated cell destruction as seen in pulmonary tuberculosis (14, 22). However, it is becoming progressively evident that intracellular parasites can directly lyse macrophages to exit the cell efficiently after termination of the intracellular proliferation phase and proceed to infect other cells in the same host (extending the infection) or to pass to a new host (spreading the disease) and that direct microbial cytotoxicity is a mechanism that promotes cell lysis. In fact, it is known that many intracellular pathogens, including M. tuberculosis and M. marinum, have direct cytotoxicity toward professional and occasional phagocytes in vivo and macrophages in vitro (19, 24, 25, 38, 42, 57). In addition, it has been reported that several intracellular parasites, including not only M. ulcerans-related mycobacteria (14, 25, 31) but also Legionella pneumophila, Salmonella spp., and Shigella spp. (2, 40, 49, 60), also use cytotoxic mechanisms to lyse infected macrophages at the end of their life cycles within the host cell, thus becoming extracellular.
The consequences of the potentially conflicting features of an intracellular lifestyle and cytotoxicity would be minimized if M. ulcerans orchestrated a pattern of transcriptional control of gene expression to turn off the synthesis of mycolactone during intramacrophage growth. The genes involved in mycolactone synthesis would later be switched on, resulting in the lysis of the infected cells and egress of fully replicated bacilli. It has been reported for infections with Salmonella spp. (40) and Legionella pneumophila (2) that the pathogen machinery responsible for the lysis of host cells is turned off after the entry of the pathogen into the macrophage and expressed only at the end of an intramacrophage multiplication phase. Our results showing that multiplication of M. ulcerans at different MOIs within J774A.1 macrophages yields similar maximal bacterial loads and induces similar macrophage viability losses, although at different times, are compatible with the hypothesis of a temporal switching off of mycolactone synthesis within macrophages.
The above-described observations showing that several pathogens are able to reconcile cytotoxicity with intracellular parasitism are in accordance with the concept that low cytotoxicity is a conditional rather than an absolute characteristic of intracellular parasites (37).
In the mouse footpads and in BU tissues from humans, extracellular bacilli colocalized with inflammatory cells but were more abundant in the acellular necrotic areas. The presence of extracellular bacteria is compatible with intracellular parasitism. Accumulation of extracellular bacilli in necrotic areas, following the lysis of macrophages infected with intracellular mycobacteria, has been previously reported for human (31) and murine (36, 73) pulmonary tuberculosis, for human skin lesions due to M. haemophilum (17), and for animals infected with M. marinum (21, 25).
Our observation of progressive enlargement of the clumps of extracellular AFB away from the areas of colocalization with inflammatory cells suggests that there is extracellular growth of M. ulcerans following the shedding of the bacilli from lysed, infected macrophages. Taking into consideration this observation and the results showing that M. ulcerans bacilli (i) are efficiently phagocytosed by mouse macrophages in vivo and in vitro, (ii) are present inside macrophages in mouse footpad and peritoneal infections and in BU tissues, and (iii) grow within cultured macrophages, we propose that the growth of M. ulcerans would take place, in a manner similar to that of M. tuberculosis, by both intracellular and extracellular multiplication in distinct areas of the lesion (14, 31).
According to our model, some of the bacilli shed from lysing macrophages would be taken up by resident macrophages and by monocytes that are continuously attracted to the infectious foci (51) and resume intracellular growth. This continuous intracellular multiplication at the areas of inflammatory infiltrates in active, progressing infections depends on the permanent availability of resident macrophages and incoming monocytes as host cells (51). Our interpretation is that this focus of intramacrophage M. ulcerans residence and multiplication at the peripheries of the necrotic areas promotes the progression of the lesion by the invasion of healthy tissues and explains the occurrence of CMI, DTH, and granulomatous responses reported in murine and human M. ulcerans infections (18, 27, 28, 33, 39, 43, 44, 50, 54, 56, 61, 67, 68, 70, 74). In contrast, the extracellular bacilli abundant in the central necrotic acellular areas may be ultimately eliminated in the slough of necrotic tissue when the lesion becomes ulcerative, as is the case for extracellular M. tuberculosis in the necrotic caseous foci in human pulmonary tuberculosis (31).
In conclusion, our results showing the uptake of M. ulcerans by macrophages in experimentally infected mice and in vitro, the presence of intramacrophage bacilli in inflammatory infiltrates of M. ulcerans-infected tissues, and the multiplication of bacilli within cultured macrophages indicate that this mycobacterium fulfils the essential features for classification as an intracellular parasite like the other pathogenic mycobacteria. This interpretation is further supported by the reported protective CMI and DTH responses in mouse and human M. ulcerans infections and may have value for the design of novel prophylactic and therapeutic approaches for BU.
Acknowledgments
This work was supported by a grant from the Health Services of Fundação Calouste Gulbenkian and by FCT fellowships Praxis SFRH/BD/9757/2003 and SFRH/BD/15911/2005 to E. Torrado and A. G. Fraga, respectively.
We thank Goreti Pinto and Luis Martins for laboratory assistance.
Editor: J. L. Flynn
Footnotes
Published ahead of print on 4 December 2006.
REFERENCES
- 1.Adusumilli, S., A. Mve-Obiang, T. Sparer, W. Meyers, J. Hayman, and P. L. Small. 2005. Mycobacterium ulcerans toxic macrolide, mycolactone modulates the host immune response and cellular location of M. ulcerans in vitro and in vivo. Cell. Microbiol. 7:1295-1304. [DOI] [PubMed] [Google Scholar]
- 2.Alli, O. A., L. Y. Gao, L. L. Pedersen, S. Zink, M. Radulic, M. Doric, and K. Y. Abu. 2000. Temporal pore formation-mediated egress from macrophages and alveolar epithelial cells by Legionella pneumophila. Infect. Immun. 68:6431-6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Appelberg, R., J. M. Pedrosa, and M. T. Silva. 1991. Host and bacterial factors control the Mycobacterium avium-induced chronic peritoneal granulocytosis in mice. Clin. Exp. Immunol. 83:231-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Astarie-Dequeker, C., E. N. N′Diaye, V. Le Cabec, M. G. Rittig, J. Prandi, and I. Maridonneau-Parini. 1999. The mannose receptor mediates uptake of pathogenic and nonpathogenic mycobacteria and bypasses bactericidal responses in human macrophages. Infect. Immun. 67:469-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhoopat, L., R. R. Turner, E. Stathopoulos, P. R. Meyer, C. R. Taylor, R. J. Marder, and A. L. Epstein. 1988. Immunohistochemical characterization of two new monoclonal antibodies (LN-4, LN-5) reactive with human macrophage subsets and derived malignancies in B5-fixed, paraffin-embedded tissues. Blood 71:1079-1085. [PubMed] [Google Scholar]
- 6.Brodin, P., L. Majlessi, L. Marsollier, M. I. de Jonge, D. Bottai, C. Demangel, J. Hinds, O. Neyrolles, P. D. Butcher, C. Leclerc, S. T. Cole, and R. Brosch. 2006. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect. Immun. 74:88-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan, J., and S. H. E. Kaufmann. 1994. Immune mechanisms of protection, p. 389-415. In B. R. Bloom (ed.), Tuberculosis: pathogenesis, protection, and control. American Society for Microbiology, Washington, DC.
- 8.Clancey, J. K., O. G. Dodge, H. F. Lunn, and M. L. Oduori. 1961. Mycobacterial skin ulcers in Uganda. Lancet 278:951-954. [DOI] [PubMed] [Google Scholar]
- 9.Clark, R. B., H. Spector, D. M. Friedman, K. J. Oldrati, C. L. Young, and S. C. Nelson. 1990. Osteomyelitis and synovitis produced by Mycobacterium marinum in a fisherman. J. Clin. Microbiol. 28:2570-2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coloma, J. N., G. Navarrete-Franco, P. Iribe, and L. D. Lopez-Cepeda. 2005. Ulcerative cutaneous mycobacteriosis due to Mycobacterium ulcerans: report of two Mexican cases. Int. J. Lepr. Other Mycobact. Dis. 73:5-12. [DOI] [PubMed] [Google Scholar]
- 11.Connor, D. H., and H. F. Lunn. 1965. Mycobacterium ulcerans infection (with comments on pathogenesis). Int. J. Lepr. 33(Suppl.):698-709. [PubMed] [Google Scholar]
- 12.Cosma, C. L., D. R. Sherman, and L. Ramakrishnan. 2003. The secret lives of the pathogenic mycobacteria. Annu. Rev. Microbiol. 57:641-676. [DOI] [PubMed] [Google Scholar]
- 13.Coutanceau, E., L. Marsollier, R. Brosch, E. Perret, P. Goossens, M. Tanguy, S. T. Cole, P. L. Small, and C. Demangel. 2005. Modulation of the host immune response by a transient intracellular stage of Mycobacterium ulcerans: the contribution of endogenous mycolactone toxin. Cell. Microbiol. 7:1187-1196. [DOI] [PubMed] [Google Scholar]
- 14.Dannenberg, A. M., Jr. 1994. Rabbit model of tuberculosis, p. 149-156. In B. R. Bloom (ed.), Tuberculosis: pathogenesis, protection, and control. American Society for Microbiology, Washington, DC.
- 15.Dannenberg, A. M., Jr. 1994. Pathogenesis of pulmunary tuberculosis: an interplay of time-damaging and macrophage-activating immune responses. Dual mechanisms that control bacillary multiplication, p. 459-483. In B. R. Bloom (ed.), Tuberculosis: protection, pathogenesis, and control. American Society for Microbiology, Washington, DC.
- 16.Debacker, M. 2004. Mycobacterium ulcerans disease (Buruli ulcer) in rural hospital, southern Benin, 1997-2001. Emerg. Infect. Dis. 10:1391-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dobos, K. M., F. D. Quinn, D. A. Ashford, C. R. Horsburgh, and C. H. King. 1999. Emergence of a unique group of necrotizing mycobacterial diseases. Emerg. Infect. Dis. 5:367-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobos, K. M., E. A. Spotts, B. J. Marston, C. R. Horsburgh, Jr., and C. H. King. 2000. Serologic response to culture filtrate antigens of Mycobacterium ulcerans during Buruli ulcer disease. Emerg. Infect. Dis. 6:158-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dobos, K. M., E. A. Spotts, F. D. Quinn, and C. H. King. 2000. Necrosis of lung epithelial cells during infection with Mycobacterium tuberculosis is preceded by cell permeation. Infect. Immun. 68:6300-6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans, M. R., H. S. Thangaraj, and M. H. Wansbrough-Jones. 2000. Buruli ulcer. Curr. Opin. Infect. Dis. 13:109-112. [DOI] [PubMed] [Google Scholar]
- 21.Fenner, F. 1956. The pathogenic behavior of Mycobacterium ulcerans and Mycobacterium balnei in the mouse and the developing chick embryo. Am. Rev. Tuberc. 73:650-673. [DOI] [PubMed] [Google Scholar]
- 22.Filley, E. A., and G. A. Rook. 1991. Effect of mycobacteria on sensitivity to the cytotoxic effects of tumor necrosis factor. Infect. Immun. 59:2567-2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer, L. J., F. D. Quinn, E. H. White, and C. H. King. 1996. Intracellular growth and cytotoxicity of Mycobacterium haemophilum in a human epithelial cell line (Hec-1-B). Infect. Immun. 64:269-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gadea, I., J. Zapardiel, P. Ruiz, M. I. Gegundez, J. Esteban, and F. Soriano. 1993. Cytopathic effect mimicking virus culture due to Mycobacterium tuberculosis. J. Clin. Microbiol. 31:2517-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao, L. Y., S. Guo, B. McLaughlin, H. Morisaki, J. N. Engel, and E. J. Brown. 2004. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol. Microbiol. 53:1677-1693. [DOI] [PubMed] [Google Scholar]
- 26.George, K. M., D. Chatterjee, G. Gunawardana, D. Welty, J. Hayman, R. Lee, and P. L. Small. 1999. Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science 283:854-857. [DOI] [PubMed] [Google Scholar]
- 27.Gooding, T. M., P. D. Johnson, D. E. Campbell, J. A. Hayman, E. L. Hartland, A. S. Kemp, and R. M. Robins-Browne. 2001. Immune response to infection with Mycobacterium ulcerans. Infect. Immun. 69:1704-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gooding, T. M., P. D. Johnson, M. Smith, A. S. Kemp, and R. M. Robins-Browne. 2002. Cytokine profiles of patients infected with Mycobacterium ulcerans and unaffected household contacts. Infect. Immun. 70:5562-5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goutzamanis, J. J., and G. L. Gilbert. 1995. Mycobacterium ulcerans infection in Australian children: report of eight cases and review. Clin. Infect. Dis. 21:1186-1192. [DOI] [PubMed] [Google Scholar]
- 30.Gregory, S. H., and E. J. Wing. 2002. Neutrophil-Kupffer cell interaction: a critical component of host defenses to systemic bacterial infections. J. Leukoc. Biol. 72:239-248. [PubMed] [Google Scholar]
- 31.Grosset, J. 2003. Mycobacterium tuberculosis in the extracellular compartment: an underestimated adversary. Antimicrob. Agents Chemother. 47:833-836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guarner, J., J. Bartlett, E. A. Whitney, P. L. Raghunathan, Y. Stienstra, K. Asamoa, S. Etuaful, E. Klutse, E. Quarshie, T. S. Van Der Werf, W. T. Van Der Graaf, C. H. King, and D. A. Ashford. 2003. Histopathologic features of Mycobacterium ulcerans infection. Emerg. Infect. Dis. 9:651-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayman, J. 1993. Out of Africa: observations on the histopathology of Mycobacterium ulcerans infection. J. Clin. Pathol. 46:5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong, H., J. B. Spencer, J. L. Porter, P. F. Leadlay, and T. Stinear. 2005. A novel mycolactone from a clinical isolate of Mycobacterium ulcerans provides evidence for additional toxin heterogeneity as a result of specific changes in the modular polyketide synthase. Chembiochem 6:643-648. [DOI] [PubMed] [Google Scholar]
- 35.Johnson, P. D., T. P. Stinear, and J. A. Hayman. 1999. Mycobacterium ulcerans—a mini-review. J. Med. Microbiol. 48:511-513. [DOI] [PubMed] [Google Scholar]
- 36.Kaufmann, S. H., S. T. Cole, V. Mizrahi, E. Rubin, and C. Nathan. 2005. Mycobacterium tuberculosis and the host response. J. Exp. Med. 201:1693-1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaufmann, S. H. E. 1999. Immunity to intracellular bacteria, p. 1335-1371. In W. E. Paul (ed.), Fundamental immunology. Lippincott-Raven, New York, NY.
- 38.King, C. H., S. Mundayoor, J. T. Crawford, and T. M. Shinnick. 1993. Expression of contact-dependent cytolytic activity by Mycobacterium tuberculosis and isolation of the genomic locus that encodes the activity. Infect. Immun. 61:2708-2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiszewski, A. E., E. Becerril, L. D. Aguilar, I. T. Kader, W. Myers, F. Portaels, and P. R. Hernandez. 2006. The local immune response in ulcerative lesions of Buruli disease. Clin. Exp. Immunol. 143:445-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knodler, L. A., and B. B. Finlay. 2001. Salmonella and apoptosis: to live or let die? Microbes Infect. 3:1321-1326. [DOI] [PubMed] [Google Scholar]
- 41.Krieg, R. E., W. T. Hockmeyer, and D. H. Connor. 1974. Toxin of Mycobacterium ulcerans. Production and effects in guinea pig skin. Arch. Dermatol. 110:783-788. [DOI] [PubMed] [Google Scholar]
- 42.Lewis, K. N., R. Liao, K. M. Guinn, M. J. Hickey, S. Smith, M. A. Behr, and D. R. Sherman. 2003. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J. Infect. Dis. 187:117-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacCallum, P., J. C. Tolhurst, G. Buckle, and H. A. Sissons. 1948. A new mycobacterial infection in man. J. Pathol. Bacteriol. 60:93-122. [PubMed] [Google Scholar]
- 44.Marston, B. J., M. O. Diallo, C. R. Horsburgh, Jr., I. Diomande, M. Z. Saki, J. M. Kanga, G. Patrice, H. B. Lipman, S. M. Ostroff, and R. C. Good. 1995. Emergence of Buruli ulcer disease in the Daloa region of Cote d'Ivoire. Am. J. Trop. Med. Hyg. 52:219-224. [DOI] [PubMed] [Google Scholar]
- 45.Mehta, P. K., C. H. King, E. H. White, J. J. Murtagh, Jr., and F. D. Quinn. 1996. Comparison of in vitro models for the study of Mycobacterium tuberculosis invasion and intracellular replication. Infect. Immun. 64:2673-2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mor, N., I. Lutsky, and L. Levy. 1981. Response in the hindfoot pad and popliteal lymph node of C57BL mice to infection with Mycobacterium marinum. Isr. J. Med. Sci. 17:236-244. [PubMed] [Google Scholar]
- 47.Mve-Obiang, A., R. E. Lee, F. Portaels, and P. L. Small. 2003. Heterogeneity of mycolactones produced by clinical isolates of Mycobacterium ulcerans: implications for virulence. Infect. Immun. 71:774-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mve-Obiang, A., R. E. Lee, E. S. Umstot, K. A. Trott, T. C. Grammer, J. M. Parker, B. S. Ranger, R. Grainger, E. A. Mahrous, and P. L. Small. 2005. A newly discovered mycobacterial pathogen isolated from laboratory colonies of Xenopus species with lethal infections produces a novel form of mycolactone, the Mycobacterium ulcerans macrolide toxin. Infect. Immun. 73:3307-3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Navarre, W. W., and A. Zychlinsky. 2000. Pathogen-induced apoptosis of macrophages: a common end for different pathogenic strategies. Cell. Microbiol. 2:265-273. [DOI] [PubMed] [Google Scholar]
- 50.Noeske, J., C. Kuaban, S. Rondini, P. Sorlin, L. Ciaffi, J. Mbuagbaw, F. Portaels, and G. Pluschke. 2004. Buruli ulcer disease in Cameroon rediscovered. Am. J. Trop. Med. Hyg. 70:520-526. [PubMed] [Google Scholar]
- 51.Oliveira, M. S., A. G. Fraga, E. Torrado, A. G. Castro, J. P. Pereira, A. L. Filho, F. Milanezi, F. C. Schmitt, W. M. Meyers, F. Portaels, M. T. Silva, and J. Pedrosa. 2005. Infection with Mycobacterium ulcerans induces persistent inflammatory responses in mice. Infect. Immun. 73:6299-6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palenque, E. 2000. Skin disease and nontuberculous atypical mycobacteria. Int. J. Dermatol. 39:659-666. [DOI] [PubMed] [Google Scholar]
- 53.Pedrosa, J., B. M. Saunders, R. Appelberg, I. M. Orme, M. T. Silva, and A. M. Cooper. 2000. Neutrophils play a protective nonphagocytic role in systemic Mycobacterium tuberculosis infection of mice. Infect. Immun. 68:577-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phillips, R., C. Horsfield, S. Kuijper, S. F. Sarfo, J. Obeng-Baah, S. Etuaful, B. Nyamekye, P. Awuah, K. M. Nyarko, F. Osei-Sarpong, S. Lucas, A. H. Kolk, and M. Wansbrough-Jones. 2006. Cytokine response to antigen stimulation of whole blood from patients with Mycobacterium ulcerans disease compared to that from patients with tuberculosis. Clin. Vaccine Immunol. 13:253-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pimsler, M., T. A. Sponsler, and W. M. Meyers. 1988. Immunosuppressive properties of the soluble toxin from Mycobacterium ulcerans. J. Infect. Dis. 157:577-580. [DOI] [PubMed] [Google Scholar]
- 56.Prévot, G., E. Bourreau, H. Pascalis, R. Pradinaud, A. Tanghe, K. Huygen, and P. Launois. 2004. Differential production of systemic and intralesional gamma interferon and interleukin-10 in nodular and ulcerative forms of Buruli disease. Infect. Immun. 72:958-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramakrishnan, L., and S. Falkow. 1994. Mycobacterium marinum persists in cultured mammalian cells in a temperature-restricted fashion. Infect. Immun. 62:3222-3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rastogi, N., M. C. Blom-Potar, and H. L. David. 1989. Comparative intracellular growth of difficult-to-grow and other mycobacteria in a macrophage cell line. Acta Leprol. 7(Suppl. 1):156-159. [PubMed] [Google Scholar]
- 59.Read, J. K., C. M. Heggie, W. M. Meyers, and D. H. Connor. 1974. Cytotoxic activity of Mycobacterium ulcerans. Infect. Immun. 9:1114-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richter-Dahlfors, A., A. M. Buchan, and B. B. Finlay. 1997. Murine salmonellosis studied by confocal microscopy: Salmonella typhimurium resides intracellularly inside macrophages and exerts a cytotoxic effect on phagocytes in vivo. J. Exp. Med. 186:569-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rondini, S., C. Horsfield, E. Mensah-Quainoo, T. Junghanss, S. Lucas, and G. Pluschke. 2006. Contiguous spread of Mycobacterium ulcerans in Buruli ulcer lesions analysed by histopathology and real-time PCR quantification of mycobacterial DNA. J. Pathol. 208:119-128. [DOI] [PubMed] [Google Scholar]
- 62.Shepard, C. C., and D. H. McRae. 1968. A method for counting acid-fast bacteria. Int. J. Lepr. Other Mycobact. Dis. 36:78-82. [PubMed] [Google Scholar]
- 63.Silva, M. T., R. Appelberg, M. N. Silva, and P. M. Macedo. 1987. In vivo killing and degradation of Mycobacterium aurum within mouse peritoneal macrophages. Infect. Immun. 55:2006-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Silva, M. T., and P. M. Macedo. 1983. The interpretation of the ultrastructure of mycobacterial cells in transmission electron microscopy of ultrathin sections. Int. J. Lepr. Other Mycobact. Dis. 51:225-234. [PubMed] [Google Scholar]
- 65.Silva, M. T., M. N. Silva, and R. Appelberg. 1989. Neutrophil-macrophage cooperation in the host defence against mycobacterial infections. Microb. Pathog. 6:369-380. [DOI] [PubMed] [Google Scholar]
- 66.Silver, R. F., Q. Li, and J. J. Ellner. 1998. Expression of virulence of Mycobacterium tuberculosis within human monocytes: virulence correlates with intracellular growth and induction of tumor necrosis factor alpha but not with evasion of lymphocyte-dependent monocyte effector functions. Infect. Immun. 66:1190-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stanford, J. L., W. D. Revill, W. J. Gunthorpe, and J. M. Grange. 1975. The production and preliminary investigation of Burulin, a new skin test reagent for Mycobacterium ulcerans infection. J. Hyg. (London) 74:7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stienstra, Y., W. T. Van Der Graaf, G. J. te Meerman, T. H. The, L. F. de Leij, and T. S. van der Werf. 2001. Susceptibility to development of Mycobacterium ulcerans disease: review of possible risk factors. Trop. Med. Int. Health 6:554-562. [DOI] [PubMed] [Google Scholar]
- 69.Stinear, T. P., H. Hong, W. Frigui, M. J. Pryor, R. Brosch, T. Garnier, P. F. Leadlay, and S. T. Cole. 2005. Common evolutionary origin for the unstable virulence plasmid pMUM found in geographically diverse strains of Mycobacterium ulcerans. J. Bacteriol. 187:1668-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tanghe, A., J. Content, J. P. Van Vooren, F. Portaels, and K. Huygen. 2001. Protective efficacy of a DNA vaccine encoding antigen 85A from Mycobacterium bovis BCG against Buruli ulcer. Infect. Immun. 69:5403-5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tønjum, T., D. B. Welty, E. Jantzen, and P. L. Small. 1998. Differentiation of Mycobacterium ulcerans, M. marinum, and M. haemophilum: mapping of their relationships to M. tuberculosis by fatty acid profile analysis, DNA-DNA hybridization, and 16S rRNA gene sequence analysis. J. Clin. Microbiol. 36:918-925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Travis, W. D., L. B. Travis, G. D. Roberts, D. W. Su, and L. W. Weiland. 1985. The histopathologic spectrum in Mycobacterium marinum infection. Arch. Pathol. Lab. Med. 109:1109-1113. [PubMed] [Google Scholar]
- 73.Turner, O. C., R. G. Keefe, I. Sugawara, H. Yamada, and I. M. Orme. 2003. SWR mice are highly susceptible to pulmonary infection with Mycobacterium tuberculosis. Infect. Immun. 71:5266-5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Westenbrink, B. D., Y. Stienstra, M. G. Huitema, W. A. Thompson, E. O. Klutse, E. O. Ampadu, H. M. Boezen, P. C. Limburg, and T. S. Van Der Werf. 2005. Cytokine responses to stimulation of whole blood from patients with Buruli ulcer disease in Ghana. Clin. Diagn. Lab Immunol. 12:125-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wyllie, A. H., J. F. Kerr, and A. R. Currie. 1980. Cell death: the significance of apoptosis. Int. Rev. Cytol. 68:251-306. [DOI] [PubMed] [Google Scholar]