

Abstract
An important phase of cardiac outflow tract (OFT) formation is the remodeling of the distal region of the common outlet in which the myocardial sleeve is replaced by with smooth muscle. Here we demonstrate that expression of the proteoglycan versican is reduced before the loss of myocardium from the distal cardiac outlet concomitant with an increase in production of the N-terminal cleavage fragment of versican. To test whether versican proteolysis plays a role in OFT remodeling, we determined the effects of adenoviral-mediated expression of a versican isoform devoid of known matrix metalloproteinase cleavage sites (V3) and an N-terminal fragment of versican (G1). V3 expression promoted an increase in thickness of the proximal OFT myocardial layer independent of proliferation. In contrast, the G1 domain caused thinning and interruptions of the OFT myocardium. These in vivo findings were consistent with findings using cultured primary cardiomyocytes showing that the V3 promoted myocardial cell–cell association while the G1 domain caused a loss of myocardial cell–cell association. Taken together, we conclude that intact versican and G1-containing versican cleavage products have opposing effects on myocardial cells and that versican proteolysis may facilitate the loss of distal myocardium during OFT remodeling.
Keywords: versican, matrix metalloproteinase, ADAMTS, outflow tract, fibulin-1, hyaluronan, myocardium, cardiac neural crest, hdf
INTRODUCTION
Versican is an extracellular matrix (ECM) chondroitin sulfate proteoglycan with abundant expression in several regions of the developing heart (Henderson and Copp, 1998; Mjaatvedt et al., 1998; Zanin et al., 1999). All four versican variants V0, V1, V2, and V3 are present throughout cardiac development (Ito et al., 1995; Zako et al., 1995; Kern et al., 2006). Although these variants contain a combination of functional domains, all contain both an N-terminal G1 domain, which binds hyaluronan (HA; Margolis and Margolis, 1994), and a C-terminal G3 domain, which interacts with other extracellular matrix molecules (Zhang et al., 1998; Day et al., 2004; Wu et al., 2005).
Previous studies have identified versican as critical for the formation of the cardiac outlet, which becomes the future aortic and pulmonary arteries of the mature heart (Yamamura et al., 1997; Mjaatvedt et al., 1998). At the initial stages of outflow tract (OFT) development, versican is found between the outer myocardial cell layer and the inner endothelial layer. As OFT development proceeds, versican is required for the recruitment of new mesoderm from the anterior heart field (AHF), which contributes to the myocardium and lengthens the OFT (Yamamura et al., 1997; Mjaatvedt et al., 1998). At approximately 11.5 days post coitum (dpc), the cardiac outlet begins a remodeling process, where it is shortened relative to the rest of the heart and the myocardial wall is replaced with smooth muscle and becomes arterial tissue. Because the versican insertional mutant (hdf) dies before this stage of development, the role of versican in OFT “remodeling” is unclear.
Several processes have been proposed to account for the apparent shortening of the cardiac outlet: retraction of the distal myocardial rim toward the ventricle, perhaps including absorption of conal myocardium predominantly into the right ventricle (Anderson et al., 1974; Thompson and Fitzharris, 1979; Thompson et al., 1987); the loss of myocardium through apoptosis, prominent along the conal (proximal) myocardium (Watanabe et al., 1998; van den Hoff et al., 2000; Watanabe et al., 2001; Cheng et al., 2002; Kubalak et al., 2002; Sugishita et al., 2004), myocardialization of the proximal cushions (van den Hoff et al., 1999) and nonapoptotic loss of myocardium from the distal end (truncus) of the outlet through reorganization as arterial muscle (Arguello et al., 1978; Ya et al., 1998b; Yang et al., 2004). However, the underlying mechanisms for the replacement of the myocardial wall with smooth muscle are largely unknown and a topic of controversy.
Turnover of OFT ECM constituents represents a potentially important aspect of OFT remodeling that has not been extensively examined. Given the critical importance of versican to the process of OFT formation (Yamamura et al., 1997; Mjaatvedt et al., 1998) and evidence showing that it is a substrate of extracellular metalloproteinases belonging to the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs; Sandy et al., 2001; Gao et al., 2002; Somerville et al., 2003) and matrix metalloproteinase (MMP) families (Perides et al., 1995; Passi et al., 1999), we speculate that the proteolytic breakdown of versican may be a key component of the mechanism by which the truncus myocardium is remodeled (i.e., retraction, loss of myocardium, and replacement of myocardium with smooth muscle; Anderson et al., 1974; Thompson and Fitzharris, 1979; Thompson et al., 1987; van den Hoff et al., 1999). In this study, we examine versican proteolysis during the process of OFT morphogenesis as well as test the possibility that products of versican breakdown have functions distinct from intact versican.
RESULTS
Versican Expression Correlated With Myocardial Recruitment Into the Developing Cardiac Outlet
The hdf mouse phenotype suggests that both the proximal and distal regions of the outlet segment (conus and truncus) do not develop under conditions of versican deficiency. We compared the expression of versican from early (9.5 dpc) through later stages (12.5 dpc) of OFT development. In these studies, we detected versican using an antibody recognizing the GAGβ domain within versican V0 and V1 variants (Kern et al., 2006). We observed versican and HA immunolocalization in the developing OFT at 9.5 and 10.5 dpc (Fig. 1A–C,G–I). In general, versican expression correlates very closely with recruitment of α -sarcomeric actin–positive cells within the myocardium (Fig. 1A–C). At 9.5 dpc and 10.5 dpc, expression of versican extended beyond the myocardial sleeve of the forming outlet and into the aortic sac (AS; open arrowheads Fig. 1A–C). We also assessed versican degradation in the OFT using an antibody that recognizes the N-terminal neoepitope (α -DPEAAE) released upon cleavage of versican by MMPs (Sandy, 2001; Kern et al., 2006). Faint anti-DPEAAE immunostaining was detected in the distal OFT, AS, and endocardium during the phase of myocardial recruitment (Fig. 1D–F). In these studies, immunolocalization of the intact variants of versican V0 and V1 was detected using the α -GAGβ antibody. This GAG domain is not present in the N-terminal proteolytically cleaved fragment of versican, recognized by the neoepitope antibody DPEAAE. Therefore, we could distinguish between the expression of versican’s intact variants V0 and V1 using α-GAGβ and its proteolytically cleaved fragment using the antibody against DPEAAE (Sandy, 2001; Kern et al., 2006).
Fig. 1.
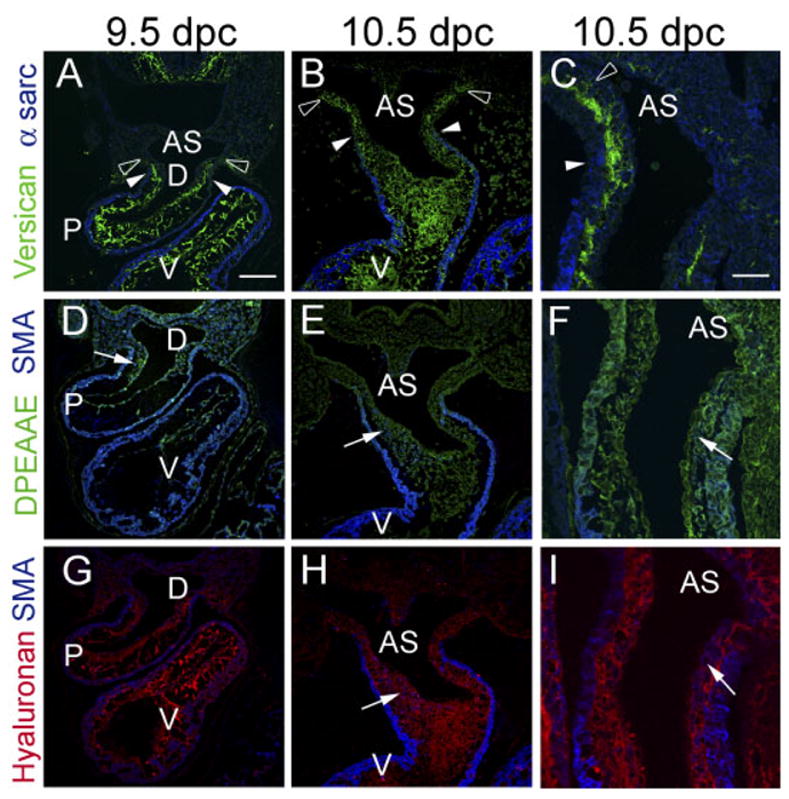
Versican expression in the distal outflow tract correlates with myocardial recruitment and emigration of neural crest cells. A,D,G: Immunohistological analysis of the developing outflow tract (OFT) of the mouse heart at 9.5 days post coitum (dpc). B,C,E,F,H,I:, Immunohistological analysis of the developing OFT of the mouse heart at 10.5 dpc. C,F,I: High magnifications of additional sections of the truncus region of the OFT at 10.5 dpc. A–C: Mouse embryos immunolabeled with antibodies to “intact” versican (anti-GAGβ, green), while the myocardium was immunolabeled using α-sarcomeric actin. Note that versican staining at 9.5 and 10.5 dpc extends beyond the boundary of the myocardial wall (closed arrowheads) of the distal OFT. D–F: Anti-DPEAAE antibody immunolabeling (green). Anti-DPEAAE is reactive with neoepitope DPEAAE generated by N-terminal proteolytically cleavage of versican. Anti-DPEAAE staining is present at relatively low levels at 9.5 and 10.5 dpc. G–I: Hyaluronan (HA) labeling using biotinylated hyaluronan binding protein. Within the mesenchyme of the OFT at 9.5 and 10.5 dpc, α-smooth muscle actin–positive neural crest cells can be seen within the mesenchyme (D–I, blue cells, arrows). At this stage, α-smooth muscle actin antibodies also immunolabel the myocardium (D–I). AS, aortic sac; D, distal aspect of the cardiac outlet; P, proximal region of the OFT. Scale bar = 150 μm in A (applies to B,D,E,G,H), 50 μm in C (applies to F,I).
Intact Versican Was Reduced While Anti-DPEAAE Reactive Versican Was Increased Before the Loss of Myocardium From the Distal Cardiac Outlet
Between 10.5 and 11.5 dpc, there was a loss of intact (α-GAGβ ) versican immunostaining in the distal truncus (Fig. 2A, open arrowheads, asterisks), while expression α-sarcomeric actin (Fig. 2A, filled arrowheads) in the myocardial sleeve of the cardiac outlet persisted. Concomitant with the loss of versican expression, the levels of anti-DPEAAE–reactive immunostaining increased, indicating that the proteolytically cleaved form of versican V0/V1 variants, was more apparent in regions of the distal cardiac outlet at 11.5 dpc (Fig. 2C) than at earlier stages of myocardial recruitment (Fig. 1C,D). The pattern of increased anti-DPEAAE immunolocalization corresponded to, but was not limited to, areas of anti-α-smooth muscle actin–positive cells (Fig. 2C arrows blue cells) within the OFT and AS mesenchyme. Such cells have been identified by others to be neural crest–derived extra cardiac cells (Epstein et al., 2000; Waller et al., 2000). Loss of intact versican during cardiac outlet remodeling preceded loss of fibulin-1 and HA, which were still detected beyond or at the myocardial boundary of the distal truncus at 11.5 dpc (Fig. 2E,G). This finding suggested that proteolytic processing of versican preceded temporally, and spatially, the loss of myocardium from the developing cardiac outlet.
Fig. 2.
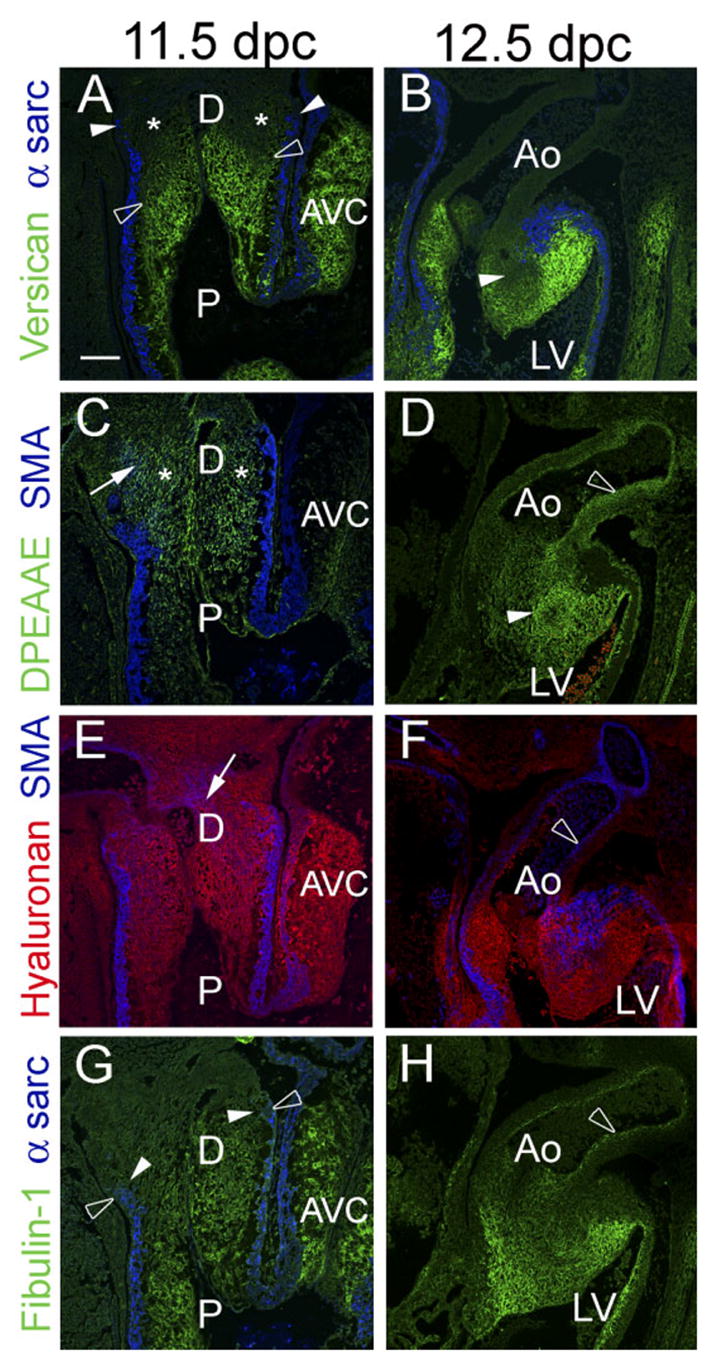
Intact versican levels are reduced before loss of myocardium in the distal outflow tract (OFT). A,C,E,G: Immunohistological analysis of embryonic mouse hearts at 11.5 days post coitum (dpc). B,D,F,H: Immunohistological analysis of embryonic mouse hearts at 12.5 dpc. A,B: Immunolabeling with antibodies to versican (green) and α-sarcomeric actin (blue). In A, versican immunolabeling (open arrowheads) appears to have regressed proximal to the myocardial boundary (filled arrowheads). Versican expression is not detected at 12.5 dpc in the aortic wall (B). C,D: Immunodetection of the N-terminal neoepitope cleavage product of versican using DPEAAE antibody. The distal portion of the outlet displayed relatively high levels of anti-DPEAAE immunolabeling and low levels of intact versican immunolabeling (A,C). E: Hyaluronan (HA) labeling using biotinylated HA-binding protein. HA was present throughout the entire OFT at 11.5 dpc. F: HA labeling at 12.5 dpc. HA was not detected in the aortic wall at this stage. G: Fibulin-1 immunolabeling at 11.5 dpc. In contrast to intact versican, fibulin-1 expression remained at the level of the myocardial sleeve at 11.5 dpc. H: Fibulin-1 immunolabeling at 12.5 dpc. Fibulin-1 expression is overlapping with that of α-smooth muscle actin and DPEAAE in the developing aortic medial layer at 12.5 dpc. A, B, and G, show the myocardium is immunolabeled (blue) with anti–α-sarcomeric actin. C, E, and F, shows anti–α-smooth muscle actin immunolabeling (blue) of the OFT myocardium and a subpopulation of CNCs in the OFT at 11.5 dpc and smooth muscle formation in the aortic medial layer at 12.5 dpc. AVC, atrioventricular cushion; P, proximal OFT; AS, aortic sac; Ao, aorta; LV, left. Scale bar = 150 μm in A (applies to B–H).
After septation of the cardiac outlet was completed at 12.5 dpc, there was no versican detected in the aorta (Fig. 2B). However, anti-DPEAAE–reactive versican was apparent in the developing medial layer of the aortic artery (Fig. 2D). By comparison with early stages, there was a marked reduction of α-sarcomeric actin in the wall of the developing aorta (Fig. 2B) coincident with an increase in α-smooth muscle actin–positive cells (Fig. 2F, open arrow). Colocalization of the cleaved fragment of versican, DPEAAE, and α-smooth muscle actin was evident in the medial layers of the aorta subjacent to the aortic lumen (Fig. 2D,F). Furthermore, fibulin-1 was also colocalized in the forming aortic medial layer with DPEAAE and α-smooth muscle actin (Fig. 2H).
There was a complementary distribution of intact versican vs. anti-DPEAAE reactivity, indicative of the cleaved form of versican, within the forming aorticopulmonary septum (Fig. 2B,D, solid arrows). This region of condensed mesenchyme, identified by others as neural crest derived, was largely devoid of intact versican (Fig. 2B) but demonstrated significant anti-DPEAAE immunolocalization (Fig. 2D). This finding is consistent with the spatial and temporal correlation of DPEAAE reactivity with the migration of cardiac neural crest (CNC) cells in the distal outlet at earlier stages of development (Fig. 2C).
Adenovirus Engineered to Express the G1 Domain of Versican and the V3 Variant
An adenovirus was developed to express the versican amino terminal G1 domain, which approximates the amino terminal fragment generated by ADAMTS cleavage of versican variants V0 and V1 (Sandy et al., 2001; Wu et al., 2002; Russell et al., 2003). We also developed an adenovirus that expresses V3, a variant of versican that lacks the ADAMTS cleavage region (Fig. 3A). Both of these adenoviruses were engineered with an “HA-tag” at the C-terminus of the versican proteins to verify viral expression of the recombinant proteins. Using an antibody that recognizes the HA-tag of the virally expressed versican proteins, recombinant polypeptides of the expected size were detected in extracts of infected HEK-293 cells (Fig. 3B).
Fig. 3.
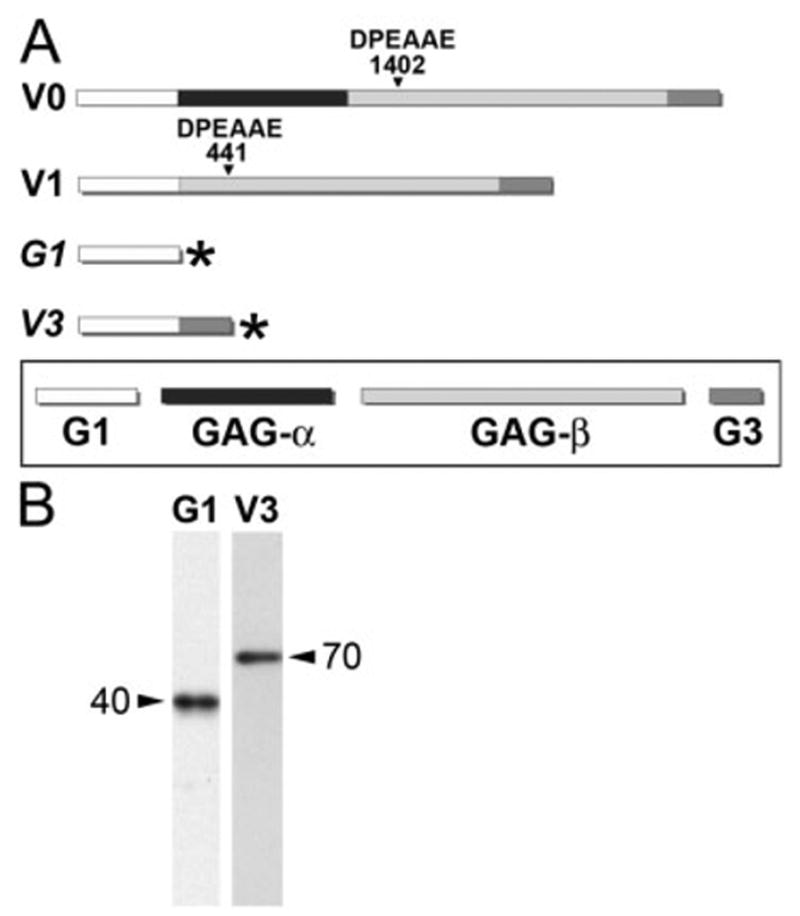
Adenoviral constructs of the versican G1 domain and versican variant V3. A: A schematic diagram of the versican variants. V0 and V1 variants include the G1 domain, which contains hyaluronan (HA) -binding sites, and the G3 that contains binding sites for extracellular matrix (ECM) components fibulin-1 and tenacin. The V0 variant contains both GAGα and GAGβ domains, while V1 contains only the GAGβ domain. V3 lacks both GAG attachment domains. ADAMTS metallo-proteinases cleavage site DPEAAE is denoted at amino acid number 1402 for V0 and 441 for V1. The G1 domain is similar to the ADAMTS, N-terminal proteolytically cleaved fragment of versican. Adenoviral constructs are depicted, italicized G1 and V3. The asterisk denotes the hemagglutinin “tag” at the C-terminus. B: Immunoblot of G1- and V3-infected HEK cells using a HA-tag antibody. Molecular weights of the virally produced proteins are indicated next to arrows in B.
Infection of Cardiac Myocytes With the G1 Domain of Versican Resulted in a Loss of Myocardial Cell Clustering
Primary cardiomyocyte cultures were infected with the G1 versican domain. We observed a loss of myocardial cell clustering (Fig. 4B) compared with control, LacZ-infected cultures (Fig. 4A). Myocardial cells (α-sarcomeric actin–positive, green) infected with G1 detected by the anti–HA-tag antibody (Fig. 4B, blue) appeared to integrate with other non–α-sarcomeric actin–stained cells. Although these cultures are composed primarily of myocardial cells, there are also non–α-sarcomeric actin–stained cells, presumably fibroblasts and mesenchymal cells, within the primary cardiac cell population. When primary cardiomyocytes were infected with LacZ, myocardial cells exhibited greater cell–cell association than G1-infected cultures. In the LacZ-expressing cultures, myocardial cells did not appear to interdigitate with other non–α-sarcomeric actin– expressing cells compared with G1-infected cultures (Fig. 4A). This finding resulted in a greater number of myocardial cell aggregates in LacZ- vs. G1-expressing primary cardiac myocyte cultures (Fig. 5).
Fig. 4.
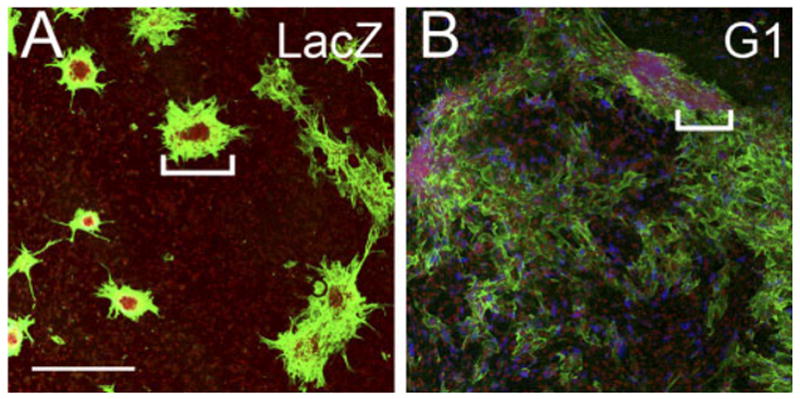
Versican G1 expression inhibits myocyte aggregate formation. A: Primary cardiomyocyte cultures infected with the recombinant adenovirus expressing LacZ. B: Primary cardiomyocyte cultures infected with the G1 domain of versican. White brackets in A and B indicate aggregates of myocardial cells. Cardiomyocytes were detected using antibodies against α-sarcomeric actin (green). The adenovirally expressed versican G1 domain was detected using an antibody that recognized the hyaluronan (HA) tag at the C-terminus of virally expressed G1 (blue). Propidium iodide was used to stain the nuclei of cells (red). Scale bar = 600 μm in A (applies to B).
Fig. 5.
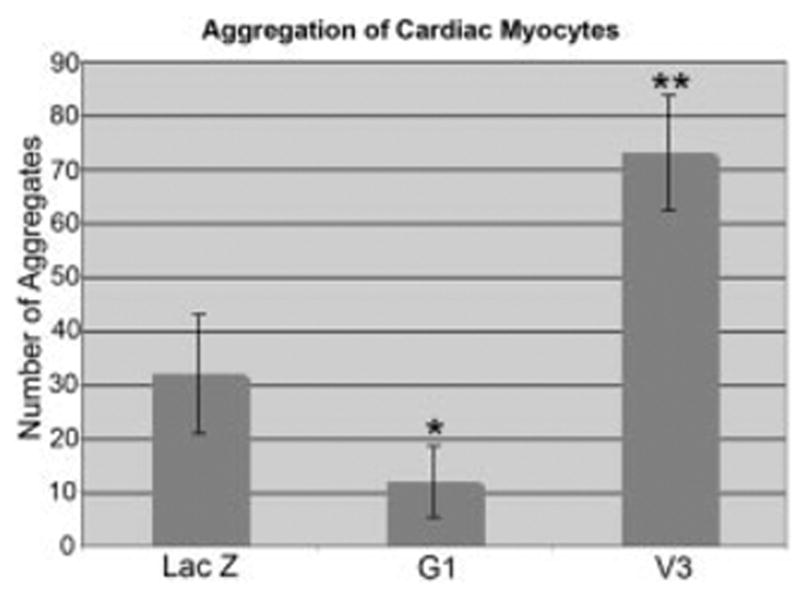
V3 promotes myocardial cell aggregation. Primary cardiac myocyte cultures were infected with adenovirus expressing LacZ, the G1 domain of versican, or the V3 versican variant. Nuclear condensations were counted 72 hr after infection. Values represent the average of three different experiments in which three separate optical fields of cell aggregates were counted. P values were calculated using an analysis of variance test coupled with the conservative Bonferroni’s analysis: the overall test results produced an F = 37.48 with a corresponding P = 0.0000; *pairwise analysis showed G1 and LacZ with a mean difference of 19.5, with a corresponding, P = 0.023, the G1 and V3 mean difference is 60.25 with a corresponding P = 0.000, the **V3 and LacZ mean difference is 40.75, with a corresponding P = 0.000.
AHF Injection of Versican-Expressing Adenovirus Led to Sustained Expression in the Cardiac Outlet Myocardium
In these studies, recombinant adenovirus was injected into the AHF (mesoderm of branchial arch 2) of chick embryos at Hamburger and Hamilton stage (HH) 18–20 (Fig. 6A). Approximately 18 hr after injection, virally expressed protein was evident in the branchial arch mesoderm (Fig. 6B). By stage 26, the viral recombinant protein was present in the proximal outlet (co-nus) of the developing heart (Fig. 6C). The findings indicate that injection of adenovirus into branchial arch 2 mesoderm resulted in myocardial cells of the developing OFT that expressed the virally produced recombinants. Previous mapping studies in chick and mouse demonstrate that the branchial arch mesoderm contributes to the myocardium of the developing OFT (Yamamura et al., 1997; Mjaatvedt et al., 1998, 2001; Kelly et al., 2001; Waldo et al., 2001). These present findings suggest adenoviral infection into the AHF may be a useful technique for ectopic expression of recombinant proteins in the developing OFT myocardium. Although other extra cardiac cell precursors are present in the branchial arch/AHF region at these time points, we did not observe adenovirally produced versican recombinants within any other cell population.
Fig. 6.
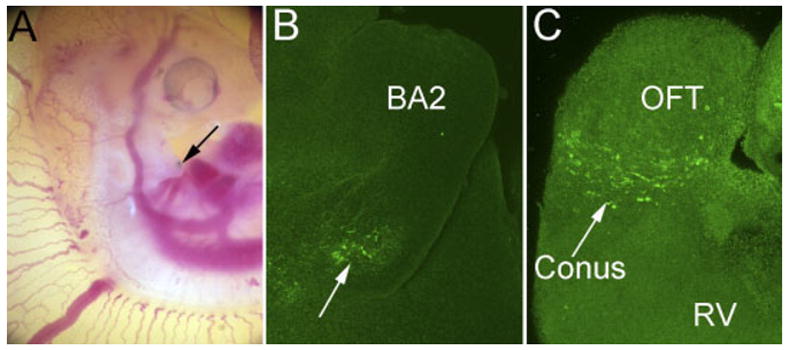
Injection of versican-expressing adenovirus into the anterior heart field leads to sustained expression of recombinant versican in the outflow tract. A: Photomicrograph showing the site of viral microinjection (arrow) in the anterior heart field (branchial arch 2) of a Hamburger and Hamilton stage (HH) 20 embryo. B: Whole-mount anti– hyaluronan (HA) -tag immunostaining detection virally expressed V3 versican in the branchial arch region 18 hr postinjection. C: Anti–HA-tag detection of recombinant V3 versican variant in the conus or proximal region of the OFT of the heart (arrow) 48 hr postinjection (HH26). BA2, branchial arch 2; OFT, cardiac outflow tract; RV, right ventricle.
Our in vitro studies revealed that myocardial cells in culture required at least 10-fold less viral particles per cell for infection (i.e., a lower multiplicity of infection [MOI]) as compared with mesenchymal cells. It is possible that in vivo adenoviral infection of mesenchymal cells including cardiac neural crest was below the necessary threshold (MOI) for infection with the titer (2 × 109 pfu/ml) of our injected constructs.
Expression of G1 in the OFT Myocardium Resulted in a Loss of Myocardium and an Increase in Mesenchymal Tissue After Adenoviral Delivery Into the AHF
After adenoviral injection into the AHF at HH20, recombinant G1 domain expression was detected in the myocardial cuff of the proximal outlet of infected embryos at HH29 (Fig. 7C). Expression of G1 correlated with a lack of continuity of the myocardial sleeve in the proximal OFT compared with the normally continuous band of α-sarcomeric actin–positive OFT myocardium, of control embryos (Fig. 7A). Breaks in the myocardial sleeve of G1-infected embryos were evidenced by discontinuity in α -sarcomeric actin staining (Fig. 7B,C, arrows). Mesenchymal cells, HA-positive (red) and α -sarcomeric actin–negative (blue), were found within the myocardial breaks and associated with the adjacent epicardium, which exhibited a nonuniform appearance. In addition, the epicardium of G1-infected embryos, appeared to have relatively higher levels of HA staining compared with the epicardial tissue in the control OFTs that contained little or no HA and had a uniform thickness around the myocardial wall (Fig. 7A).
Fig. 7.
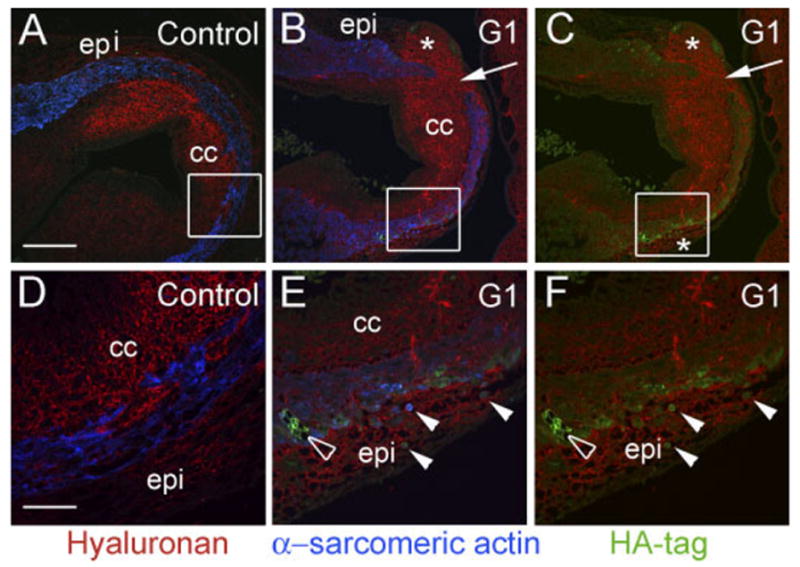
G1 causes a loss of myocardium in the proximal outflow tract (OFT). Avian embryos were injected in the anterior heart field (AHF) at Hamburger and Hamilton stage (HH) 20 with G1-expressing adenovirus and harvested at HH29. A and D show the normal myocardial wall (α -sarcomeric actin–positive cells, blue) surrounding the conal cushions of the proximal outlet in control embryos. B, C, E, and F show proximal OFTs from embryos injected with G1-expressing virus. G1 expression resulted in an abnormal loss of α -sarcomeric actin–positive myocardium (arrows in B and C) compared with controls. B and C boxes denote areas in which the myocardium appeared thin and there was a relatively high level of G1, as detected by HA-tag antibodies (green). D shows high magnification of the box in A. Normal myocardial cells exhibit a “fibroblastic” appearance. E shows high magnification of the box in B. F depicts high magnification of the box in C. In E and F, G1-infected cells demonstrate an abnormal rounded appearance (arrows). E and F show that G1-expressing, sarcomeric actin–positive cells are present in the epicardium (solid arrowheads). The open arrowhead shows G1-infected cells within the myocardial wall that appeared to have lost cell–cell contact. epi, epicardium; cc, conal cushions. The asterisk denotes an increase in hyaluronan staining within the epicardium of G1-infected hearts. Scale bars = 150 μm in A (applies to B,C), 50 μm in D (applies to E,F).
G1 Expression in the Outflow Tract Resulted in Rounding of Myocardial Cells That Correlated With a Thinner Myocardial Layer
Analysis of the OFT myocardium of G1-infected embryos revealed myocardial cells that exhibited a rounded morphology that was distinctly different from that seen in control embryos where myocardial cells have an extended cell body or stellate appearance. Rounded α -sarcomeric actin–positive (Fig. 7E blue, closed arrowheads), G1-expressing cells could be seen within the myocardial sleeve, and also in the adjacent epicardium (Fig. 7F, green, closed arrowheads). The myocardial cell layer also appeared thinner in the region where rounded myocardial cells, expressing G1, appeared in the epicardium. The epicardium of control embryos did not contain α -sarcomeric actin–positive cells (Fig. 7E,F). All myocardial cells of control embryos had a stellate or “fibroblastic” appearance and were associated with the myocardial sleeve. In normal embryos, myocardial cells within the proximal OFT, extended into the conal cushions (Fig. 7D); however, these cells retained their normal stellate appearance.
We examined the G1-infected hearts to determine whether there was an increase in apoptosis in the region surrounding the loss of myocardium or in the region where myocardial cells expressing G1 appeared in the epicardium. At this stage, we were unable to correlate apoptosis with HA-tag detection of the virally expressed G1 protein (data not shown). The fact that, at HH29, we were unable to detect an increase in apoptosis that correlated with G1 expression would not preclude apoptosis from contributing to the apparent loss of myocardium at earlier time points of development before our analysis.
Adenoviral Expression of the G1 Domain of Versican in Avian Hearts Resulted in Abnormal Rotation of the OFT and a Ventricular Septal Defect
Embryos were injected into the AHF at HH20 with G1-expressing adenovirus and harvested at HH29. G1-infected hearts were examined using whole-mount confocal microscopy (Miller et al., 2005). In normal HH29 hearts, the pulmonary artery was visible at a focal depth of approximately 50μm from the ventral surface (Fig. 8A), while the aortic branch was viewed at a focal depth of approximately 200 μm (Fig. 8C). Conversely, hearts infected with the G1 domain of versican (Fig. 8B,D) imaged at a depth of 175 μm showed both the pulmonary and aortic vessel in the same focal plane. This finding suggested that expression of the G1 domain of versican resulted in a delay or block in the normal rotation of the developing outlet. A pronounced VSD was also apparent in the G1-infected hearts (Fig. 8F, arrow) when compared with controls (Fig. 8E) that showed a complete interventricular septum. Further analysis revealed a normal arrangement of the aortic arch arteries in control and G1-infected hearts (data not shown). Thus, in addition to the loss of OFT myocardium in G1-expressing hearts, confocal analysis revealed a lack of rotation and VSD consistent with developmental delay.
Fig. 8.

G1 causes a lack normal rotation of the outflow tract (OFT) and a ventricular septal defect (VSD). Confocal analysis of hearts from embryos injected with adenovirus into the anterior heart field (AHF; branchial arch 2) at stage 20 and incubated until Hamburger and Ham-ilton stage (HH) 29. A: Control, LacZ-infected hearts display the pulmonary artery at a depth of 50 μm and the aorta positioned dorsally at a depth of 200 μm, due to the normal rotation of the pulmonary trunk. B–D: Different G1-infected embryos where the aortic and pulmonary arteries are within the same plane of section (175 μm depth). E,F: A pronounced ventricular septal defect is apparent in the G1-infected hearts (arrow in F) when compared with the intact interventricular septum of the control heart in E. PA, pulmonary artery; Ao, aorta; LA, left atrium; RA, right atrium; LV, left ventricle; A3, aortic arch artery 3; A6, aortic arch artery 6. Scale bar = 400 μm in A (applies to B–F).
Adenovirally Expressed Versican V3 in Primary Cultures of Myocardial Cells Caused an Increase in Cell–Cell Aggregation
In addition to examining the effect of the versican G1 domain, the effect of the versican V3 variant was also examined. V3 contains the N-terminal G1 domain and also the G3 domain where versican interacts with additional ECM molecules, such as fibulin-1 (Wu et al., 2005). Because V3 does not contain any GAG attachment domains, it is devoid of known MMP proteolytic cleavage sites. Therefore, the naturally occurring versican variant, V3, represents a “noncleavable” form of the intact versican variants. Using Western analysis, we routinely examined cultures expressing the viral V3 construct and never observed recombinant V3 below the predicted size (data not shown).
Infection of primary cardiomyocytes with V3 versican-expressing virus showed a dramatic increase in cell clustering (Figs. 5, 9B,D,F) compared with the LacZ-infected control cultures. The clustered cells stained positive for α -sarcomeric actin, this suggested that the cell aggregates were composed predominantly of myocardial cells that had increased cell–cell association with expression of the noncleavable V3 versican variant (Fig. 9C,D). Immunolocalization with an antibody that recognized the C-terminal HA-tag demonstrated recombinant V3 protein within the large myocyte aggregate and also deposited within the collagen matrix (Fig. 9D,F blue). Aggregates were quantified by counting areas of nuclear condensations (based on propidium iodide immunostaining) 72 hr after infection. This analysis revealed a significant difference in cell–cell aggregation of V3-expressing myocardial cell cultures compared with control cultures expressing LacZ (Fig. 5) and the antagonistic effect of G1, which showed a decrease in myocardial cell–cell aggregation.
Fig. 9.
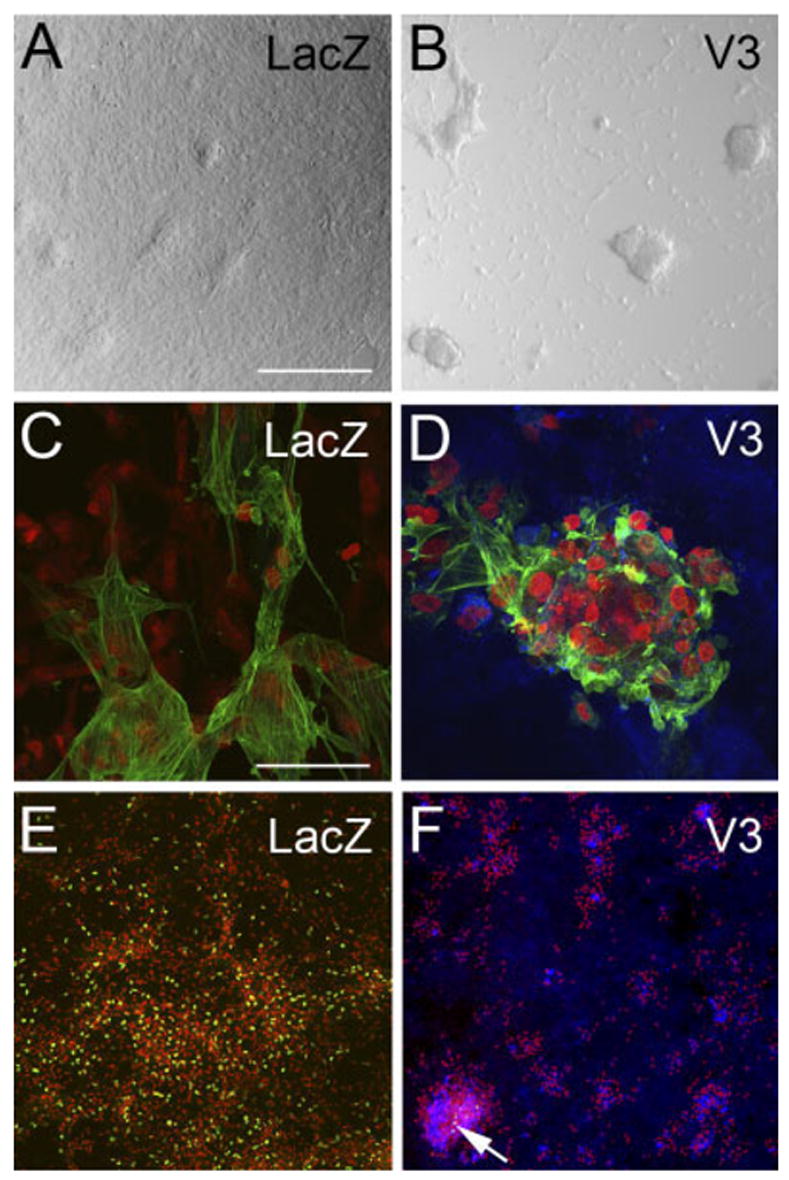
Expression of the noncleavable versican V3 variant in myocardial cells caused an increase in cell– cell aggregation. A,C,E: Primary cardiomyocyte cultures were infected with adenovirus expressing LacZ. B,D,F: Primary cardiomyocyte cultures infected with the versican variant V3. A,B: Cultures infected with V3 developed more cell aggregates (B) than control-infected cultures (A). C,D: Immunodetection using α-sarcomeric actin antibody (green). The results indicate that aggregates were composed of myocardial cells. D,F: Immunolabeling using anti– hyaluronan tag (blue). The results indicate that recombinant V3 is expressed within the aggregates as well as in the extracellular matrix (ECM) of cells throughout the culture. E,F: Bromodeoxyuridine (BrdU) labeling as a measure of cell proliferation (green nuclei). Propidium iodide was used to stain the nuclei of cells (red). Arrow in F shows BrdU-positive cells that represent a very small fraction of total cells in the dish. See Figure 5 for quantification of the myocardial cell aggregates. Scale bars = 300 μm in A (applies to B,E,F), 50 μm in C (applies to D).
To determine whether there was an increase in cell proliferation that may have contributed to the increase in myocardial aggregates, we compared the rate of proliferation using bromodeoxyuridine incorporation between LacZ-and V3-expressing primary cardiomyocyte cultures (Fig. 9E,F, arrow, green nuclei). We observed a dramatic decrease in cell proliferation in V3-expressing cultures compared with LacZ control cultures 72 hr postinfection.
Adenoviral Expression of Versican V3 in Avian Hearts Resulted in an Increase in the Thickness of Outflow Tract Myocardium
The effects of versican V3 on the OFT myocardium was also evaluated. The AHFs of HH20 embryos were injected with V3- or LacZ-expressing virus, and the OFT myocardium was examined at HH29. The virally expressed LacZ was detected using X-gal staining (Fig. 10A), and V3 was detected in the myocardium of the proximal OFT using the antibody to the C-terminal HA-tag (Fig. 10B). V3 expression caused an increase in thickness of the OFT myocardium (α -sarcomeric actin–positive cells) compared with the OFT myocardium of embryos injected with LacZ-expressing virus (Fig. 10C,D, brackets). Thickness of the OFT myocardium correlated directly with the relative levels of V3 expression. By contrast, the OFT myocardium of LacZ-infected hearts displayed a uniform thickness of α -sarcomeric actin–positive cells around the circumference of the OFT, even in areas in which the level of LacZ expression was relatively high (Fig. 10A,C).
Fig. 10.
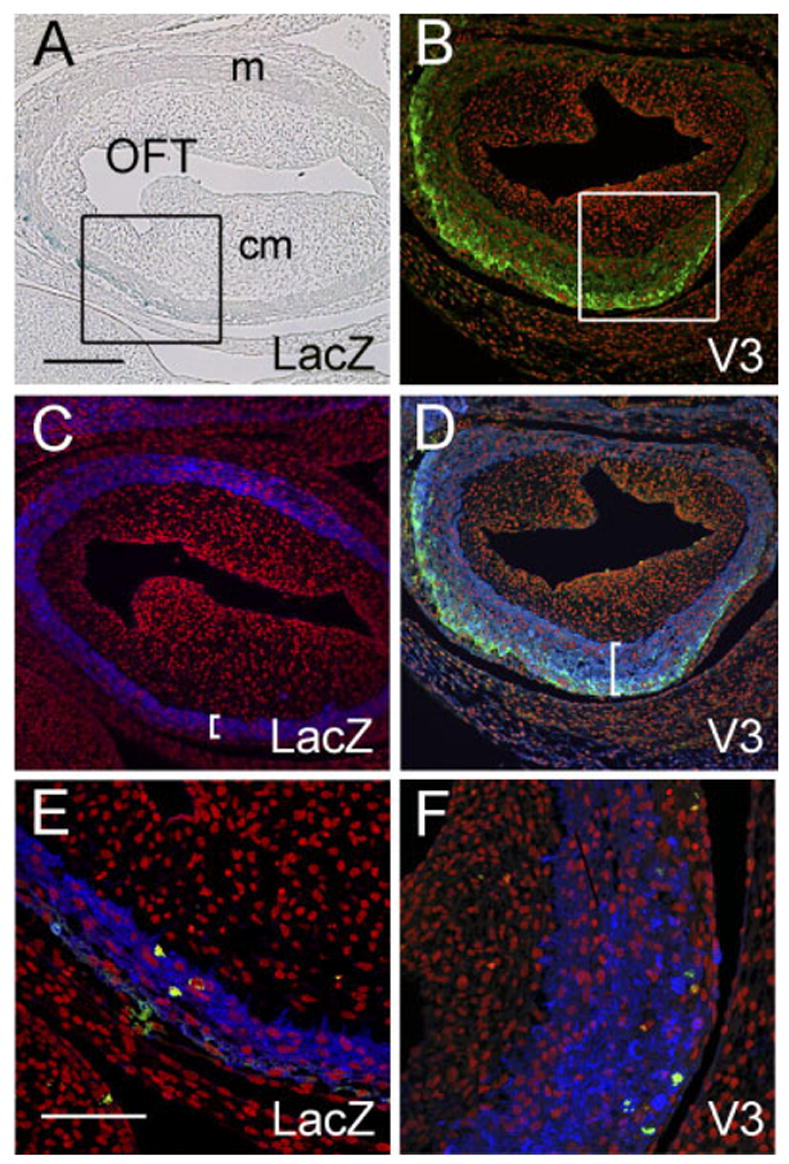
V3 causes an increase in thickness of the myocardium of the proximal cardiac outlet. Adenovirus expressing versican V3 or LacZ were injected into the anterior heart field (AHF) of Hamburger and Hamilton stage (HH) 19 chick embryos and evaluated at HH29. A: X-gal staining (blue) of the proximal outflow tract (OFT) of an embryo injected with LacZ-expressing virus. B: Anti– hyaluronantag staining (green) of the proximal OFT of an embryo injected with V3-expressing virus. C: Anti–α-sarcomeric actin staining (blue) of the same section shown in A demonstrates that LacZ-expressing cells are present within the myocardial sleeve of the OFT. D: Anti–α -sarcomeric actin staining (blue) of the same section shown in B demonstrates that V3-expressing cells are present within the myocardial sleeve of the OFT. Brackets in C and D indicate the thickness of the OFT myocardium. E and F show phosphohistone H3 immunodetection to assess cell proliferation within the OFT myocardium (green). Propidium iodide was used to stain the nuclei of cells (red). Scale bars = 150 μm in A (applies to B–D), 50 μm in E (applies to F).
The effect of V3 expression on proliferation in the OFT myocardium was assessed using phosphohistone H3 immunolabeling. The relative levels of phosphohistone H3 were similar in the OFT myocardium of both V3- and LacZ-expressing embryos (Fig. 10E,F, green nuclei). Therefore, the increase in thickness of the OFT myocardium of V3-infected embryos does not seem to be attributable to an increase in cell proliferation. This interpretation is consistent with our in vitro findings, which indicated that V3 did not increase proliferation of cultured cardiomyocytes (Fig. 10E,F).
Adenoviral Expression of Versican V3 in Avian Hearts Resulted in an Increase in the Thickness of the Ventricular Myocardium
In addition to evaluating the effects of versican V3 expression in the myocardium of the proximal OFT, we also assessed the effects of V3 expression in the ventricular myocardium. The pericardial sacs of HH19 embryos were injected with V3- or LacZ-expressing virus, and the compact layers of the right ventricular myocardium were examined at HH29. These hearts of V3-injected embryos showed an ~two-fold increase in the thickness of the compact layer of the ventricle compared with hearts of LacZ-infected embryos (Fig. 11A,C,E). The increased compact layer thickness was accompanied by a decrease in the overall size of the V3-infected hearts compared with LacZ-infected hearts (Fig. 11B,D,F).
Fig. 11.
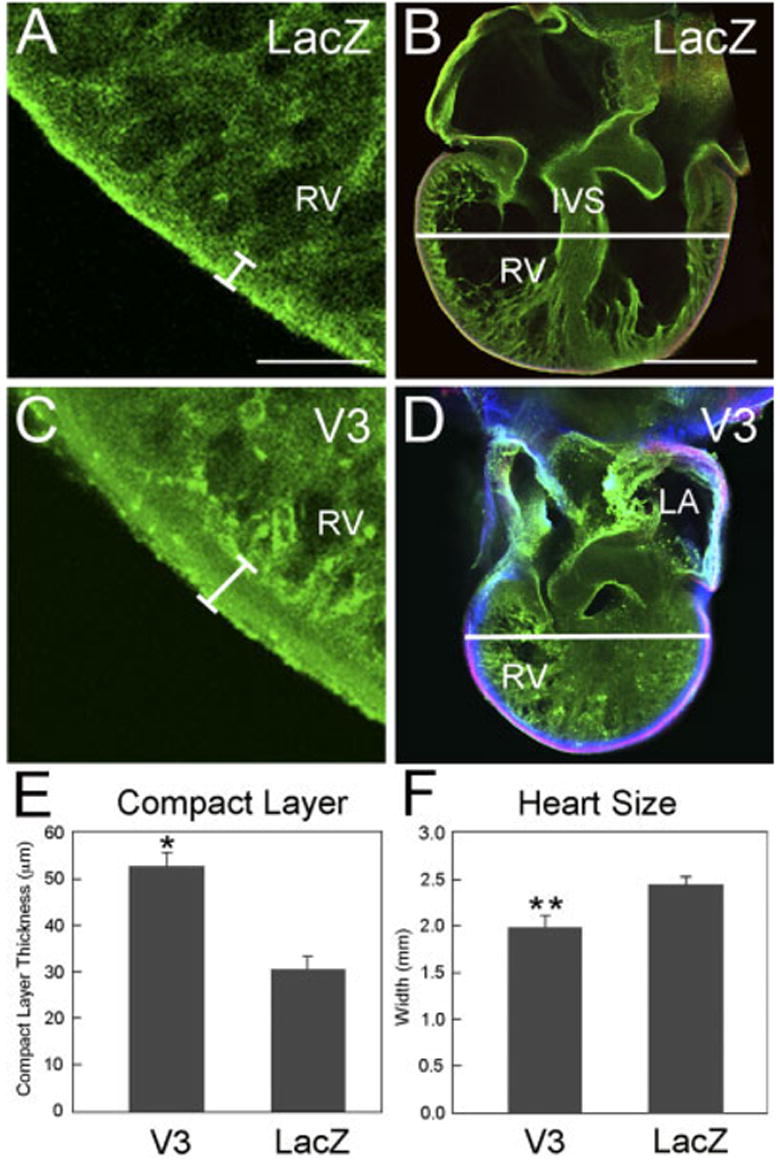
V3 causes an increase in the thickness of the compact layer of the right ventricle and reduces overall heart size. A–D: Whole-mount confocal analysis of embryonic avian hearts (Hamburger and Hamilton stage [HH] 29) that had been injected into the pericardial sac at HH19 with adenovirus and subsequently “endopaint” labeled. A and C show confocal images from the right ventricle of a LacZ-infected (A) and V3-infected (C) embryo. The brackets in A and C indicate the compact layer of the myocardium. B and D show low-magnification confocal images of hearts from a LacZ-infected (B) and V3-infected (D) embryo labeled with fluorescein isothiocyanate–poly-L-lysine (green), anti-α-ac-tinin (red), and anti– hyaluronan-tag (to detect virally expressed V3, blue). E: Graph of thickness of right ventricular compact layer of the hearts infected with LacZ and V3 adenovirus. F: Graph of the widths of hearts infected with LacZ and V3 adenovirus. The plotted values represent average width measurements (n = 4 hearts) from the widest point across both ventricles within the Z-series. P values were calculated using a two-tailed Student t-test with a variance of three. *P < 0.0003; **P < 0.003. Scale bars = 150 μm in A, 450 μm in B.
DISCUSSION
The mechanisms underlying the remodeling of the cardiac OFT that lead to the disappearance of myocardium from the distal cardiac outlet have not been elucidated. Several possible mechanisms have been investigated, including apoptosis, retraction, and transdifferentiation. However, apoptosis occurs primarily in the proximal outlet myocardium (conus) and cushions and appears related to remodeling of the inner curvature (Watanabe et al., 1998; van den Hoff et al., 2000; Cheng et al., 2002; Kubalak et al., 2002; Sugishita et al., 2004). There is also evidence that the conus becomes incorporated predominantly into the enlarging right ventricle through a retraction process (Thompson and Fitzharris, 1979). Several studies provide evidence that some of the myocardial cells in the distal truncus undergo transdifferentiation and directly contribute to the remodeling of OFT arterial walls (Ya et al., 1998a; Yang et al., 2004). Other evidence suggests that the proximal myocardium contributes to the adjacent cushions by a process called myocardialization to form the muscular outlet septum (van den Hoff et al., 1999). Although these studies offer morphogenetic insights, they do not reveal molecular mechanisms that contribute to the process of remodeling of the cardiac OFT.
Herein, we report that the loss of the distal-most myocardium of the cardiac outlet was preceded by a loss of intact versican and the appearance of the DPEAAE-containing versican cleavage fragment (Fig. 2). These findings suggest that proteolysis of versican may be involved in the remodeling of the cardiac outlet. In support of this possibility are the findings that the versican-cleaving proteinases AD-AMTS-1 and 9 (Jungers et al., 2005; Lee et al., 2005; Kern et al., 2006) as well as the ADAMTS cofactor fibulin-1 (Lee et al., 2005), are expressed in the heart coincident with the events of OFT remodeling. In addition to the ADAMTS family of MMPs, MMP-2 is also highly expressed in the OFT (Alexander et al., 1997; Cai et al., 2000; Ratajska and Cleutjens, 2002) and has been shown to degrade versican (Perides et al., 1995; Passi et al., 1999). Cardiac neural crest cells are at least one cell type in the developing OFT known to express MMP-2 (Cai et al., 2000). In the chick, versican expression is similar to the mouse, including a heterogeneity that the authors attribute to ECM proteolysis and led to the hypothesis that versican proteolysis was occurring during OFT development and may be initiated by the migration of cardiac neural crest cells (Capehart et al., 1999; Zanin et al., 1999).
The observed correlation between versican cleavage and loss of the distal myocardium in the truncus raised the possibility that proteolytic cleavage of versican may not only inactivate versican activity but also generate fragments with activities distinct from the intact versican molecule. It is possible that versican degradation may be generating cryptic activities residing in discrete versican domains. One such fragment is the G1 domain– containing aminoterminal fragment, which is generated as a result of ADAMTS and/or MMP2-mediated proteolysis. To investigate the significance of versican degradation on cardiac OFT development, we tested the effects of two recombinant adenoviruses, one expressing a naturally occurring “non-cleavable” versican variant V3 and a second expressing only the G1 domain similar to the N-terminal proteolytically cleaved fragment. As a result, we found that expression of G1 appeared to decrease myocardial cell– cell association both in vitro and in vivo. Myocardial cells appeared to interdigitate with mesenchymal cells (non–α-sarcomeric actin–positive cells) in primary cardiomyocyte cultures (Fig. 4), while in vivo G1 expression caused accelerated loss of myocardium from the OFT, independent of apoptosis (Fig. 6). In addition, a subset of G1-expressing myocardial cells were found within the epicardium, dissociated from the OFT myocardial epithelium. The observed reduction in myocardial cell association in the absence of a change in apoptosis are consistent with findings from in vitro studies reported by others (Ang et al., 1999; Yang et al., 1999; Zhang et al., 1999), suggesting a functional role for the G1 domain of versican distinct from the intact molecule.
The rounded morphology of OFT cardiomyocytes expressing the recombinant G1 domain in vivo is distinct from the stellate appearance of normal OFT myocardial cells. In support of the possibility that the G1 domain fragment of versican can elicit a “rounding effect” on cell morphology are findings showing that cushion cells subjacent to the endocardium are rounded and express the anti-DPEAAE–reactive, G1 domain–containing, versican fragment (Kern et al., 2006). The region of the cushion subjacent to the endocardium also displays relatively high levels of the MMP-2 (Cai et al., 2000; Song et al., 2000). These observations are in contrast to findings that mesenchymal cells located in the endocardial cushion core, a region with relatively high levels of intact versican (Kern et al., 2006) and low levels of MMP-2 (Cai et al., 2000), have a stellate morphology. Thus, the generation of G1-containing versican fragments may serve to promote changes in cell morphology that are a critical aspect of both the detachment of the newly formed mesenchymal cells from the endocardium during cushion morphogenesis as well as the loss of OFT myocardial cells from the distal region of the cardiac outlet.
In contrast to G1, adenoviral-mediated expression of the V3 variant of versican promoted aggregation of myocardial cells in vitro and increased the thickness of OFT myocardium and the ventricular compact layer in vivo. Given that the V3 variant lacks an ADAMTS cleavage site, these findings can be interpreted as a consequence of the overproduction of a form of versican that is both intact and noncleavable. In this way, V3 overexpression produced a dominant effect in milieus in which versican would otherwise have been cleaved. Based on the findings from our experiments, in which we speculate that the V3 variant serves as a surrogate for intact isoforms of versican, we conclude that intact versican promotes augmented cell survival, ECM biosynthesis and/or stabilization of the myocardial layer. Mechanistically, the “dominant” effects of V3 on myocardial tissue may relate to the persistence of signaling or lack thereof resulting from bivalent interactions by means of the HA-binding G1 domain and the G3 domain, which contains binding sites for numerous ECM proteins, including fibulin-1 and tenascin-C (Zhang et al., 1998; Day et al., 2004; Wu et al., 2005). Taken together with findings from our G1 expression studies, we conclude that the stability of the OFT myocardium is a function of the dynamics between persistence of the intact form of versican vs. its cleavage, which generates the aminoterminal, G1 domain–containing fragment.
On the basis of the findings presented here, together with findings from other studies, we have developed a model for the involvement of versican in the recruitment of mesoderm from the AHF to form the OFT myocardium and the subsequent removal of OFT myocardium during remodeling. Initially, the production of versican may be stimulated in the Isl-1–positive cells of the AHF possibly by means of expression of bone morphogenetic protein-4 and -7 and fibroblast growth factor-10 (Cai et al., 2003). Because intact versican functions as a bivalent link between HA and other ECM molecules such as fibulin-1 and tenascin (Aspberg et al., 1999; Olin et al., 2001), we speculate that the resulting ECM complex signals the differentiation and organization of AHF-derived cells into a stable myocardial epithelium, a hypothesis consistent with interpretations based on studies of the versican insertional mutant (hdf) phenotype (Yamamura et al., 1997; Mjaatvedt et al., 1998).
Our model further predicts that, following the formation of the primary OFT myocardium, the process by which the myocardium is remodeled (i.e., replacement of myocardium with smooth muscle) is promoted by the cleavage of versican to yield the amino-terminal G1 domain– containing fragment(s). In the case of overexpression of the noncleavable V3 isoform, bivalent signaling persists, sustaining OFT myocardial stabilization rather than the normal loss of myocardium facilitated by versican cleavage. Versican cleavage is presumably driven by versican-cleaving proteinases belonging to the ADAMTS and MMP family, although definitive studies of the expression of the complete repertoire of these proteases concomitant with OFT remodeling have yet to be performed. It is reasonable to propose that CNC cells mediate the proteolytic processing of versican, because changes in versican epitope detection (anti-DPEAAE–reactive and intact versican) correlate with CNC migration, and the fact that CNCs have been shown to express MMPs in the distal cardiac outlet (Alexander et al., 1997; Capehart et al., 1999; Zanin et al., 1999; Cai et al., 2000; Ratajska and Cleutjens, 2002). Consistent with the hypothesis that the G1 domain– containing fragment from versican acts to decrease myocardial cell– cell association required to initiate loss of distal myocardium, we showed that overexpression of the G1 domain accelerated the normal remodeling. Cells released from the myocardial layer through such a process could migrate, apoptose, or differentiate and perhaps contribute to other structures of the outlet (e.g., smooth muscle wall, septum).
Taken together, these studies demonstrate the critical involvement of versican in both the recruitment of myocardial progenitors from the AHF and the formation of a stable OFT myocardium. Furthermore, the studies reveal that versican proteolysis may mediate the loss of truncal myocardium during remodeling of the cardiac outlet.
EXPERIMENTAL PROCEDURES
Immunohistochemistry
Mouse embryos (CD1 strain) at different stages of development, were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 5 μm. Deparaffinized sections were rehydrated through a graded series of ethanols to phosphate buffered saline (PBS). Sections for immunostaining of versican (α-GAGβ, provided by Stan Hoffman), α-DPEAAE, an antibody that recognizes the neoepitope of V0 and V1 isoforms after cleavage with ADAMTS MMPs (Affinity BioRe-agents, Golden, CO), fibulin-1 (provided by Scott Argraves), and phosphohistone H3 (Upstate, Lake Placid, NY) were subjected to antigen unmasking based on a high-temperature citric acid formula (H-3300, Vector Laboratories, Burlingame, CA). Next, sections were blocked 1 hr at room temperature with blocking buffer (PBS [Sigma] containing 3% normal goat serum [NGS, Cappel, Malvern, PA] and 1% bovine serum albumin [catalog no. B4287, Sigma]) then incubated overnight at 4°C with primary antibody diluted in blocking buffer (anti-DPEAAE was diluted to 0.20 μg/ml; anti-GAGβ was diluted to 5 μg/ml). Following primary antibody incubations, specimens were washed 5 times in PBS and incubated at room temperature with fluorochrome conjugated secondary antibody (Jackson Laboratories, Bar Harbor, ME) diluted in blocking buffer. Nuclei were labeled with 1 μg/ml propidium iodide (Molecular Probes C-7590). Controls for immunohistochemical staining experiments included the use of the secondary antibody only. Immunostained sections were analyzed using a Leica TCS SP2 AOBS Confocal Microscope System (Leica Microsystems, Inc., Exton, PA).
Detection of HA in Embryonic Tissue Sections
Hyaluronan was detected in embryonic heart tissue sections using a modification of procedure described by Toole et al. (2001) previously published (Kern et al., 2006). Briefly, tissues sections were incubated with 2 μg/ml biotinylated HA binding protein (bHABP, Seikagaku America, East Falmouth, MA; a complex of aggrecan HA-binding domain and link protein) in 10% fetal calf serum for 1 hr at room temperature. To detect bound bHABP, sections were incubated for 1 hr in fluorescein avidin DCS (Vector Laboratories, Burlington, CA; at 20 μg/ml) in 0.1 M sodium bicarbonate buffer containing 0.85% sodium chloride and evaluated by confocal microscopy. Control experiments included incubation of tissue sections with 6 units/ml streptomyces hyaluronidase for 1 hr at 37°C before addition of bHABP and fluorescein avidin DCS.
Production of Replication-Defective, Recombinant Adenovirus
Recombinant adenovirus expressing the N-terminal G1 domain of versican were constructed using the Adeno-X vector system (Mizuguchi and Kay, 1998, 1999; Clontech, Palo Alto, CA). A portion of the mouse versican gene containing exons 2– 6, including the start site of translation and Kozak sequence, was cloned into the pShuttle donor vector. Expression of the versican G1 insert was driven by the cytomegalovirus (CMV) promoter. Control viral constructs contained an identical promoter and viral backbone containing the β-galactosidase gene.
The G1 construct was produced by polymerase chain reaction (PCR) using 154F: 5′-CAGTAAGCCGCCTT-TCAAGGAC-3′ and 1219HA: 5′-CTA-AGCGTAATCTGGTAC GTCGTATG-GGTATTTAAAGCAGTAGGCATC-3′. To construct the V3 isoform, 154F and 10857RHA: 5′CTAAGCGTAATCTG-GTACGTCGTAGCGCCTGGTTTCCT-GCCGCCGGC 3′ was used with a cDNA of versican serving as the PCR template for each recombinant. To facilitate detection of recombinant versican proteins V3 and G1, the reverse primer also contains the sequence encoding a synthetic hemagglutinin tag sequence, YPDVPDYA; HEK 293 cells were transfected with the versican adenoviral constructs, and recombinant virions were isolated, amplified, and purified by CsCl gradient centrifugation.
Infection of Recombinant Primary Cardiac Myocardial Cultures
Fertilized White Leghorn eggs (viral free; Spafas) were incubated at 37.5°C until HH18 –20. In cold PBS, ventricular myocardium was isolated from hearts and pooled on ice. Isolated myocardium was trypsinized for approximately 10 min at 37°C. Cells were counted and plated on collagen-coated four-well culture slides at approximately 3,000 cells/10 μl drop. After cells adhered to the culture slide during an overnight incubation at 37°C, they were infected with adenovirus at an MOI of approximately 50 for 1 hr at 37°C. Primary cardiac cultures were maintained in M199 medium (Gibco), supplemented with ITS (BD Biosciences) and 1% chick serum (Sigma). Approximately seven dozen embryonic chicken hearts were isolated per experiment. Results presented in this manuscript represent at least three separate infection experiments of primary cardiac myocardial cultures. To quantitate the aggregates of myocardial cells, clusters were defined as nuclei aggregates if 15 or more nuclei were clustered. Nuclei were identified using propidium iodide staining. Aggregates that appeared to be in close proximity to other aggregates were counted separately, because their nuclei clusters were distinct.
Microinjection of Recombinant Virus
Fertilized White Leghorn eggs (viral free; Spafas) were incubated at 37.5°C until HH21. Eggs were “windowed” by removing a small area of shell and underlying membrane.
V3-, G1-, or LacZ-expressing adenovirus was microinjected into HH18–20 embryos. As controls, embryos were injected with LacZ adenovirus alone. Viral microinjections were performed using a picospritzer II pressure regulator (Parker Hannifin Corp, Cleveland, OH). Embryos were injected in the second branchial arch region within the AHF. Each microinjection delivered approximately 2 × 103 pfu (titer, 2 × 109 pfu/ml) in a total 20- to 200-nl volume. More experimental embryos were injected than controls due to the greater rate of death resulting from infection with the V3 and G1 virus.
Microinjection into the AHF avoided the complication of secondary defects resulting from direct injection into the developing OFT. An additional advantage afforded by AHF microinjection was that predominantly the OFT myocardial cells were infected rather than ventricular myocardium that would result if microinjection into the pericardial cavity was performed. Additional populations of extra cardiac cell precursors are present in the branchial arch or AHF region at these time points; however, in these studies, we only observed myocardial cells expressing adenovirally produced versican recombinants. We have observed that myocardial cells in culture have a greater than10-fold decrease in the MOI compared with mesenchymal cells; it is possible that adenoviral infection of mesenchymal/neural crest cells in vivo was below a threshold necessary for infection with the titer (titer, 2 × 109 pfu/ml) obtained with our constructs.
After microinjection, the eggs were sealed with Parafilm and returned to the incubator. After approximately 4 days, the embryos (HH29) were removed and viral expression was assessed by confocal microscopy after HA-tag and/or X-gal staining.
The embryos used in this study were those that received a nonlethal injection of virus as defined by survival greater than 48 hr postinfection. Findings presented here are based on analysis of a minimum of seven experimental embryos that expressed LacZ in the myocardium of the conus as well as the HA-tag.
Whole Heart Confocal Imaging by Cardiac “Endopainting”
Whole heart confocal imaging was performed using a modification of the endocardial painting procedure as described previously (Miller et al., 2005). Briefly, hearts were placed in relaxation buffer (1% lidocaine, PBS) and perfused using pulled glass needles. Hearts were then perfused with fluorescein isothiocyanate–poly-L-lysine (Sigma Chemical Corp), which binds to the endothelium of vessels and internal heart structures. The embryos were fixed in 4% paraformaldehyde for 2 hr, and trimmed and mounted in a precise frontal plane. Dehydration was performed before incubation in Murray’s clear. Hearts were examined on a Leica TCS SP2 AOBS Confocal Microscope System (Leica Microsystems).
Whole-Mount β-Galactosidase Reactivity
Embryos were removed from eggs between HH28 –30, rinsed in PBS, and fixed in 4% paraformaldehyde. After fixation, embryos were washed (0.1 M NaH2PO4, 2 mM MgCl2, 0.1% sodium deoxycholate in PBS) extensively and stained solution (0.1% sodium deoxycholate, 2 mM MgCl2, 4 mM ferrocyanide, 5 mM ferricyanide, 0.2% NP40, 1 mg/ml X-gal in PBS) for approximately 2 hr at 37°C.
Acknowledgments
We thank Drs. Jeremy Barth and Keith Rodgers for the statistical analysis of these data and Joshua Spruill for his technical assistance. R.R.M., C.H.M., W.S.A., R.P.T., and S.H. were funded by the NIH, and S.H. was funded by DOD contracts.
Footnotes
Grant sponsor: NIH; Grant numbers: RR016434; HL52813 (R.R.M., C.H.M., W.S.A.); HL66231 (C.H.M.); HL50582 (R.P.T.); HL61873 (W.S.A.); HL37641 (S.H.); HL33756 (R.R.M.); Grant sponsor: DOD; Grant number: N6311601MD10004; Grant number: N6311600MDMOU01 (S.H.).
References
- Alexander SM, Jackson KJ, Bushnell KM, McGuire PG. Spatial and temporal expression of the 72-kDa type IV collagenase (MMP-2) correlates with development and differentiation of valves in the embryonic avian heart. Dev Dyn. 1997;209:261–268. doi: 10.1002/(SICI)1097-0177(199707)209:3<261::AID-AJA2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Anderson RH, Davies MJ, Becker AE. Atrioventricular ring specialized tissue in the normal heart. Eur J Cardiol. 1974;2:219 –230. [Google Scholar]
- Ang LC, Zhang Y, Cao L, Yang BL, Young B, Kiani C, Lee V, Allan K, Yang BB. Versican enhances locomotion of astrocytoma cells and reduces cell adhesion through its G1 domain. J Neuropathol Exp Neurol. 1999;58:597–605. doi: 10.1097/00005072-199906000-00004. [DOI] [PubMed] [Google Scholar]
- Arguello C, De La Cruz MV, Sanchez C. Ultrastructural and experimental evidence of myocardial cell differentiation into connective tissue cells in embryonic chick heart. J Mol Cell Cardiol. 1978;10:307–315. doi: 10.1016/0022-2828(78)90380-2. [DOI] [PubMed] [Google Scholar]
- Aspberg A, Adam S, Kostka G, Timpl R, Heinegard D. Fibulin-1 is a ligand for the C-type lectin domains of aggrecan and versican. J Biol Chem. 1999;274:20444 –20449. doi: 10.1074/jbc.274.29.20444. [DOI] [PubMed] [Google Scholar]
- Cai DH, Vollberg TM, Hahn-Dantona E, Quigley JP, Brauer PR. MMP-2 expression during early avian cardiac and neural crest morphogenesis. Anat Rec. 2000;259:168 –179. doi: 10.1002/(SICI)1097-0185(20000601)259:2<168::AID-AR7>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capehart AA, Mjaatvedt CH, Hoffman S, Krug EL. Dynamic expression of a native chondroitin sulfate epitope reveals microheterogeneity of extracellular matrix organization in the embryonic chick heart. Anat Rec. 1999;254:181–195. doi: 10.1002/(SICI)1097-0185(19990201)254:2<181::AID-AR4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Cheng G, Wessels A, Gourdie RG, Thompson RP. Spatiotemporal and tissue specific distribution of apoptosis in the developing chick heart. Dev Dyn. 2002;223:119–133. doi: 10.1002/dvdy.1244. [DOI] [PubMed] [Google Scholar]
- Day JM, Olin AI, Murdoch AD, Canfield A, Sasaki T, Timpl R, Hardingham TE, Aspberg A. Alternative splicing in the aggrecan G3 domain influences binding interactions with tenascin-C and other extracellular matrix proteins. J Biol Chem. 2004;279:12511–12518. doi: 10.1074/jbc.M400242200. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Li J, Lang D, Chen F, Brown CB, Jin F, Lu MM, Thomas M, Liu E, Wessels A, Lo CW. Migration of cardiac neural crest cells in Splotch embryos. Development. 2000;127:1869 –1878. doi: 10.1242/dev.127.9.1869. [DOI] [PubMed] [Google Scholar]
- Gao G, Westling J, Thompson VP, Howell TD, Gottschall PE, Sandy JD. Activation of the proteolytic activity of AD-AMTS4 (aggrecanase-1) by C-terminal truncation. J Biol Chem. 2002;277:11034 –11041. doi: 10.1074/jbc.M107443200. [DOI] [PubMed] [Google Scholar]
- Henderson DJ, Copp AJ. Versican expression is associated with chamber specification, septation, and valvulogenesis in the developing mouse heart. Circ Res. 1998;83:523–532. doi: 10.1161/01.res.83.5.523. [DOI] [PubMed] [Google Scholar]
- Ito K, Shinomura T, Zako M, Ujita M, Kimata K. Multiple forms of mouse PG-M, a large chondroitin sulfate proteoglycan generated by alternative splicing. J Biol Chem. 1995;270:958 –965. doi: 10.1074/jbc.270.2.958. [DOI] [PubMed] [Google Scholar]
- Jungers KA, Le Goff C, Somerville RP, Apte SS. Adamts9 is widely expressed during mouse embryo development. Gene Expr Patterns. 2005;5:609 –617. doi: 10.1016/j.modgep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell. 2001;1:435–440. doi: 10.1016/s1534-5807(01)00040-5. [DOI] [PubMed] [Google Scholar]
- Kern CB, Twal WO, Mjaatvedt CH, Fairey SE, Toole BP, Iruela-Arispe ML, Ar-graves WS. Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev Dyn. 2006;235:2238 –2247. doi: 10.1002/dvdy.20838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubalak SW, Hutson DR, Scott KK, Shannon RA. Elevated transforming growth factor beta2 enhances apoptosis and contributes to abnormal outflow tract and aortic sac development in retinoic X receptor alpha knockout embryos. Development. 2002;129:733–746. doi: 10.1242/dev.129.3.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NV, Rodriguez-Manzaneque JC, Thai SN, Twal WO, Luque A, Lyons KM, Argraves WS, Iruela-Arispe ML. Fibulin-1 Acts as a cofactor for the matrix metalloprotease ADAMTS-1. J Biol Chem. 2005;280:34796 –34804. doi: 10.1074/jbc.M506980200. [DOI] [PubMed] [Google Scholar]
- Margolis RU, Margolis RK. Aggrecan-versican-neurocan family proteoglycans. Methods Enzymol. 1994;245:105–126. doi: 10.1016/0076-6879(94)45008-0. [DOI] [PubMed] [Google Scholar]
- Miller CE, Thompson RP, Bigelow MR, Gittinger G, Trusk TC, Sedmera D. Confocal imaging of the embryonic heart: how deep? Microsc Microanal. 2005;11:216–223. doi: 10.1017/S1431927605050464. [DOI] [PubMed] [Google Scholar]
- Mizuguchi H, Kay MA. Efficient construction of a recombinant adenovirus vector by an improved in vitro ligation method. Hum Gene Ther. 1998;9:2577–2583. doi: 10.1089/hum.1998.9.17-2577. [DOI] [PubMed] [Google Scholar]
- Mizuguchi H, Kay MA. A simple method for constructing E1- and E1/E4-deleted recombinant adenoviral vectors. Hum Gene Ther. 1999;10:2013–2017. doi: 10.1089/10430349950017374. [DOI] [PubMed] [Google Scholar]
- Mjaatvedt CH, Yamamura H, Capehart T, Turner D, Markwald RR. The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev Biol. 1998;202:56 –66. doi: 10.1006/dbio.1998.9001. [DOI] [PubMed] [Google Scholar]
- Mjaatvedt CH, Nakaoka T, Moreno-Rodriguez R, Norris RA, Kern MJ, Eisenberg CA, Turner D, Markwald RR. The outflow tract of the heart is recruited from a novel heart-forming field. Dev Biol. 2001;238:97–109. doi: 10.1006/dbio.2001.0409. [DOI] [PubMed] [Google Scholar]
- Olin AI, Morgelin M, Sasaki T, Timpl R, Heinegard D, Aspberg A. The proteoglycans aggrecan and Versican form networks with fibulin-2 through their lectin domain binding. J Biol Chem. 2001;276:1253–1261. doi: 10.1074/jbc.M006783200. [DOI] [PubMed] [Google Scholar]
- Passi A, Negrini D, Albertini R, Miserocchi G, De Luca G. The sensitivity of versican from rabbit lung to gelatinase A (MMP-2) and B (MMP-9) and its involvement in the development of hydraulic lung edema. FEBS Lett. 1999;456:93–96. doi: 10.1016/s0014-5793(99)00929-1. [DOI] [PubMed] [Google Scholar]
- Perides G, Asher R, Lark M, Lane W, Robinson R, Bignami A. Glial hyaluronate-binding protein: a product of metalloproteinase digestion of versican? Biochem J. 1995;312:377–384. doi: 10.1042/bj3120377. Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajska A, Cleutjens JP. Embryogenesis of the rat heart: the expression of collagenases. Basic Res Cardiol. 2002;97:189–197. doi: 10.1007/s003950200011. [DOI] [PubMed] [Google Scholar]
- Russell DL, Doyle KM, Ochsner SA, Sandy JD, Richards JS. Processing and localization of ADAMTS-1 and proteolytic cleavage of versican during cumulus matrix expansion and ovulation. J Biol Chem. 2003;278:42330 –42339. doi: 10.1074/jbc.M300519200. [DOI] [PubMed] [Google Scholar]
- Sandy JD. Proteoglycan core proteins and catabolic fragments present in tissues and fluids. Methods Mol Biol. 2001;171:335–345. doi: 10.1385/1-59259-209-0:335. [DOI] [PubMed] [Google Scholar]
- Sandy JD, Westling J, Kenagy RD, Iruela-Arispe ML, Verscharen C, Rodriguez-Mazaneque JC, Zimmermann DR, Lemire JM, Fischer JW, Wight TN, Clowes AW. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem. 2001;276:13372–13378. doi: 10.1074/jbc.M009737200. [DOI] [PubMed] [Google Scholar]
- Somerville RP, Longpre JM, Jungers KA, Engle JM, Ross M, Evanko S, Wight TN, Leduc R, Apte SS. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J Biol Chem. 2003;278:9503–9513. doi: 10.1074/jbc.M211009200. [DOI] [PubMed] [Google Scholar]
- Song W, Jackson K, McGuire PG. Degradation of type IV collagen by matrix metalloproteinases is an important step in the epithelial-mesenchymal transformation of the endocardial cushions. Dev Biol. 2000;227:606 –617. doi: 10.1006/dbio.2000.9919. [DOI] [PubMed] [Google Scholar]
- Sugishita Y, Watanabe M, Fisher SA. Role of myocardial hypoxia in the remodeling of the embryonic avian cardiac out-flow tract. Dev Biol. 2004;267:294–308. doi: 10.1016/j.ydbio.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Thompson RP, Fitzharris TP. Morphogenesis of the truncus arteriosus of the chick embryo heart: tissue reorganization during septation. Am J Anat. 1979;156:251–264. doi: 10.1002/aja.1001560206. [DOI] [PubMed] [Google Scholar]
- Thompson RP, Abercrombie V, Wong M. Morphogenesis of the truncus arteriosus of the chick embryo heart: movements of autoradiographic tattoos during septation. Anat Rec 218:434 –. 1987;440:394 – 435. doi: 10.1002/ar.1092180411. [DOI] [PubMed] [Google Scholar]
- Toole BP, Yu Q, Underhill CB. Hyaluronan and hyaluronan-binding proteins. Probes for specific detection. Methods Mol Biol. 2001;171:479 – 485. doi: 10.1385/1-59259-209-0:479. [DOI] [PubMed] [Google Scholar]
- van den Hoff MJ, Moorman AF, Ruijter JM, Lamers WH, Bennington RW, Markwald RR, Wessels A. Myocardialization of the cardiac outflow tract. Dev Biol. 1999;212:477–490. doi: 10.1006/dbio.1999.9366. [DOI] [PubMed] [Google Scholar]
- van den Hoff MJ, van den Eijnde SM, Viragh S, Moorman AF. Programmed cell death in the developing heart. Cardiovasc Res. 2000;45:603–620. doi: 10.1016/s0008-6363(99)00401-0. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Kumiski DH, Wallis KT, Stadt HA, Hutson MR, Platt DH, Kirby ML. Conotruncal myocardium arises from a secondary heart field. Development. 2001;128:3179 –3188. doi: 10.1242/dev.128.16.3179. [DOI] [PubMed] [Google Scholar]
- Waller BR, III, McQuinn T, Phelps AL, Markwald RR, Lo CW, Thompson RP, Wessels A. Conotruncal anomalies in the trisomy 16 mouse: an immunohistochemical analysis with emphasis on the involvement of the neural crest. Anat Rec. 2000;260:279 –293. doi: 10.1002/1097-0185(20001101)260:3<279::AID-AR65>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Choudhry A, Berlan M, Singal A, Siwik E, Mohr S, Fisher SA. Developmental remodeling and shortening of the cardiac outflow tract involves myocyte programmed cell death. Development. 1998;125:3809 –3820. doi: 10.1242/dev.125.19.3809. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Jafri A, Fisher SA. Apoptosis is required for the proper formation of the ventriculoarterial connections. Dev Biol. 2001;240:274 –288. doi: 10.1006/dbio.2001.0466. [DOI] [PubMed] [Google Scholar]
- Wu Y, Chen L, Zheng PS, Yang BB. beta 1-Integrin-mediated glioma cell adhesion and free radical-induced apoptosis are regulated by binding to a C-terminal domain of PG-M/versican. J Biol Chem. 2002;277:12294 –12301. doi: 10.1074/jbc.M110748200. [DOI] [PubMed] [Google Scholar]
- Wu YJ, La Pierre DP, Wu J, Yee AJ, Yang BB. The interaction of versican with its binding partners. Cell Res. 2005;15:483–494. doi: 10.1038/sj.cr.7290318. [DOI] [PubMed] [Google Scholar]
- Ya J, van den Hoff M, de Boer P, Tesink-Taekema S, Franco D, Moorman A, Lamers W. The normal development of the outflow tract in the rat. Circ Res. 1998a;82:464 –472. doi: 10.1161/01.res.82.4.464. [DOI] [PubMed] [Google Scholar]
- Ya J, van den Hoff MJ, de Boer PA, Tesink-Taekema S, Franco D, Moorman AF, Lamers WH. Normal development of the outflow tract in the rat. Circ Res. 1998b;82:464 –472. doi: 10.1161/01.res.82.4.464. [DOI] [PubMed] [Google Scholar]
- Yamamura H, Zhang M, Markwald R, Mjaatvedt C. A heart segmental defect in the anterior-posterior axis of a transgenic mutant mouse. Dev Biol. 1997;186:58 –72. doi: 10.1006/dbio.1997.8559. [DOI] [PubMed] [Google Scholar]
- Yang BL, Zhang Y, Cao L, Yang BB. Cell adhesion and proliferation mediated through the G1 domain of versican. J Cell Biochem. 1999;72:210 –220. doi: 10.1002/(sici)1097-4644(19990201)72:2<210::aid-jcb5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Yang YP, Li HR, Jing Y. Septation and shortening of outflow tract in embryonic mouse heart involve changes in cardiomyocyte phenotype and alpha-SMA positive cells in the endocardium. Chin Med J (Engl) 2004;117:1240 –1245. [PubMed] [Google Scholar]
- Zako M, Shinomura T, Ujita M, Ito K, Kimata K. Expression of PG-M(V3), an alternatively spliced form of PG-M without a chondroitin sulfate attachment in region in mouse and human tissues. J Biol Chem. 1995;270:3914 –3918. doi: 10.1074/jbc.270.8.3914. [DOI] [PubMed] [Google Scholar]
- Zanin MK, Bundy J, Ernst H, Wessels A, Conway SJ, Hoffman S. Distinct spatial and temporal distributions of aggrecan and versican in the embryonic chick heart. Anat Rec. 1999;256:366 –380. doi: 10.1002/(SICI)1097-0185(19991201)256:4<366::AID-AR4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Cao L, Yang BL, Yang BB. The G3 domain of versican enhances cell proliferation via epidermal growth factor-like motifs. J Biol Chem. 1998;273:21342–21351. doi: 10.1074/jbc.273.33.21342. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Cao L, Kiani C, Yang BL, Hu W, Yang BB. Promotion of chondrocyte proliferation by versican mediated by G1 domain and EGF-like motifs. J Cell Biochem. 1999;73:445–457. [PubMed] [Google Scholar]